
CO2-Responsive polymer materials
Hanbin
Liu
b,
Shaojian
Lin
c,
Yujun
Feng
*ab and
Patrick
Theato
*c
aPolymer Research Institute, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, P. R. China. E-mail: yjfeng@scu.edu.cn
bChengdu Institute of Organic Chemistry, Chinese Academy of Sciences, Chengdu 610041, P. R. China
cInstitute for Technical and Macromolecular Chemistry, University of Hamburg, 20146 Hamburg, Germany. E-mail: theato@chemie.uni-hamburg.de
First published on 12th August 2016
Abstract
CO2-Responsive polymer materials have received enormous attention in recent years, since CO2 as a new trigger has many advantages such as abundant availability, low cost, energy-saving, environment-friendly, non-toxic, good reversibility as well as great biocompatibility. In this review, we first discuss the virtues of CO2-responsiveness by comparing with traditional stimuli-sensitive materials that respond to pH, light, or redox stimuli. Then, the chemical fundamentals of CO2-responsive polymer materials are revealed including recently discovered “unexpected” CO2-sensitive features. Recent progress of CO2-responsive polymer materials is highlighted followed by various CO2-responsive “smart” polymer systems. Finally, challenges and outlooks in this area are discussed.
 Hanbin Liu | Hanbin Liu received his PhD degree from the Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences in 2014 under the supervision of Prof. Yujun Feng, working on the synthesis and self-assembly of CO2-responsive polymers. After one year of post-doc research in Prof. Mary B. Chan-Park's team at Nanyang Technological University, Singapore, he joined the group of Prof. Philip G. Jessop and Prof. Michael Cunningham at Queen's University, Canada as a postdoctoral fellow. His present research interests involve the self-assembly of block copolymers, stimulus-responsive polymer materials as well as their potential applications. |
 Shaojian Lin | Shaojian Lin received his B.E. in Textile Engineering from Shaoxing University (China) in 2009, and then M.E. from Sichuan University (China) under the supervision of Prof. Jianwu Lan on poly(ester-ether) elastomers and fibers. Now he is a PhD student in the group of Prof. Patrick Theato at the University of Hamburg (Germany) focusing on gas-responsive polymers and fibers. |
 Yujun Feng | Yujun Feng is currently a Professor at the Polymer Research Institute and the State Key Laboratory of Polymer Materials Engineering, Sichuan University. After earning his PhD in applied chemistry from Southwest Petroleum University, China, in 1999, he moved to France to undertake his post-doctoral research at the Laboratoire de Physico-Chimie des Polymères, CNRS/Université de Pau, and at the Institut Français du Pétrole (IFP), respectively. In 2004, he joined the Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences, and has been acting as a team leader since then. In September 2012, he relocated to Sichuan University, where he is focusing on smart soft materials, in particular stimuli-responsive surfactants and polymers. Professor Feng is now serving as an associate editor for “RSC Advances” and “Journal of Surfactants and Detergents”. |
 Patrick Theato | Patrick Theato studied chemistry at the University of Mainz and the University of Massachusetts, Amherst, and obtained his Ph.D. degree from the University of Mainz in 2001. After postdoctoral studies at Seoul National University supported by a Feodor Lynen Postdoctoral Research Fellowship and at Stanford University, he completed his Habilitation at the University of Mainz in 2007. From 2009 to 2012 he held a joint appointment at Seoul National University within the WCU program. In 2011, he accepted a prize senior lectureship at the University of Sheffield. Shortly after he moved to the University of Hamburg, where he is currently an associate (W2) professor for polymer chemistry. His research interests are post-polymerization modifications, stimuli-responsive polymers, surface modifications, energy storage, functional nanoobjects, and nanofibers. |
1. Introduction
Stimuli-responsive materials, which possess the ability to respond to external or internal stimuli via transition behaviours, are one of the most exciting scientific areas of smart systems.1 Consequently, stimuli-responsive polymers have received enormous attention in recent years.2–8 These responsive polymers can reveal “smart” responses adapting to weak signal changes in their environment, such as temperature,9–11 pH,12,13 light,14 ion strength,15 enzyme,16,17 redox,18 magnetic field,19 mechanical force,20 just to name a few. The recent development of stimuli-responsive polymers leads to potential applications in biomedical delivery (such as drug and gene carriers),21 sensors,1 as well as “smart” surfaces,3 catalysts22 and adhesives.23Remarkably, among these stimuli-sensitive polymeric systems, CO2-responsive polymers have received much attention over the last decade24–27 due to the “green” characteristics endowed by the environmentally-benign trigger – carbon dioxide (CO2). Compared with the current triggers, CO2 shows some unique advantages. First of all, differing from the temperature-responsive polymers, which require heating or cooling to adjust the environmental temperature to reveal sensitive behaviors,24 CO2-responsive polymers just require simply addition or removal of CO2 gas to change the pH of the solution, resulting in their corresponding transition behaviours. Moreover, the use of CO2 gas is cost-effective since it is abundant in our environment, which also makes the utilization of CO2 a hot research topic.28–30 Second, stimuli-responsive polymers based on pH, ionic, enzyme or redox as triggers, usually need repeated addition of chemical agents to realize reversible responsive behaviours followed by the generation of by-products during recycling processes,28–31 while CO2-responsive polymers can show repeated and reversible responses via alternately purging CO2 and inert gases (argon or nitrogen) without contamination,31,32 and thus they can be repeated in more responsive cycles with low sensitivity depletion.33–36 Third, the stimulus from CO2 provides a good penetration depth due to water as a medium, allowing a CO2-responsive behaviour even deep inside the material, which makes a CO2-responsive polymer overcome the limitation of depth given sometimes from light, magnetic or mechanical-responsive polymers.24,37 Last but not least, CO2 is an important metabolite in human cells with good biocompatibility and membrane permeability, which endows the CO2-responsive polymers with great potential for bio-medical applications.33,38
Considering the appealing advantages mentioned above, CO2-responsive polymers have been widely and intensively investigated, involved in CO2-manipulated self-assemblies,33 CO2-triggered drug carriers,39 “smart” latexes,40 catalysts,22 CO2-switchable surfaces,41 fibers,42 and so on. Although some researchers highlighted the preparation, self-assembly as well as potential applications of CO2-responsive polymers in different reviews,24,37,43–45 considerably more progress and understanding have been made in the last several years. Here in this review, we start from recalling the chemical fundamentals including the CO2-responsive groups as well as some “unexpected” CO2-sensitiveness, to highlight the recent progress of CO2-responsive polymer materials in our teams including CO2-responsive self-assemblies and hybrids as well as “living” polymers formed by CO2-sensitive surfactants. Subsequently, some CO2-responsive polymer systems, such as CO2-responsive latexes and surfaces, as well as CO2 capturing materials and sensors using CO2-sensitive polymers, are also summarized. Finally, emerging trends and challenges as well as future prospects are outlined to close this review.
2. Chemical fundamentals of CO2-responsive polymers
2.1 CO2-Responsive functional groups
CO2-Responsiveness of polymers is realized by incorporation of CO2-sensitive functional groups along the polymer chains. The most widely reported CO2-responsive functional groups include amidines, amines and guanidines, which are organobases that can react with carbonic acid generated by CO2 in the presence of water or wet organic solvents (Scheme 1). Unlike the pH-responsive system, which needs alternative addition of an acid and a base, and thus would produce a by-product and deteriorate the sensitivity, CO2-responsive systems are free of such contamination and can keep good reversibility.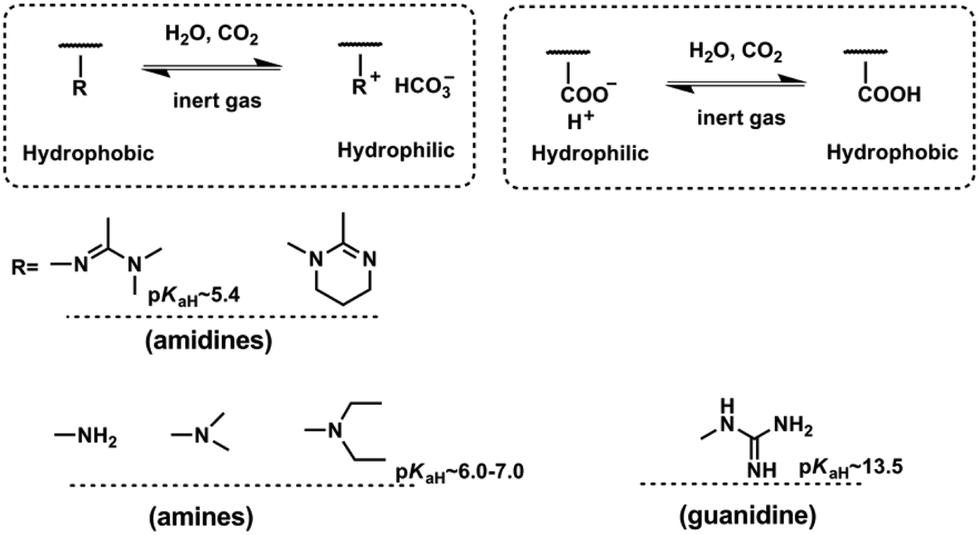 | ||
| Scheme 1 CO2-Responsive polymers based on different functional groups. | ||
The use of CO2 as a trigger in aqueous solutions can date back to the switchable surfactants containing an amidine group reported by Jessop/Cunningham's team in 2006.34 Subsequently, both the Yuan33 and Feng46 groups introduced the amidine groups into the skeleton of block copolymers to develop CO2-responsive soft materials. In Feng's team, the amidine functional group was “clicked” onto the backbone of a precursor poly(4-chloromethylstyrene) (PCMS), endowing it with a CO2-reactivity (Fig. 1a). In a CHCl3/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) biphasic mixture, the polymer could be dissolved in the lower organic phase CHCl3 in the absence of CO2. However, after streaming CO2 into the polymer dispersion, the polymer moved into the upper aqueous phase, demonstrating that the polymer was transformed from a hydrophobic to a hydrophilic state (Fig. 1b). The amidine has a pKaH of around 5.4,33 so it can be protonated by carbonic acid (pH ∼ 4.0–5.0) produced from the reaction of CO2 with water. As deduced from the equation shown in Fig. 1b, the hydrophobic amidine group reacts with carbonic acid and transforms into bicarbonate, thus becoming water-soluble. This hydrophobicity to hydrophilicity transition, being reversible or not, demonstrates the fundamental concept of a CO2-responsive “smart” system.
:1, v/v) biphasic mixture, the polymer could be dissolved in the lower organic phase CHCl3 in the absence of CO2. However, after streaming CO2 into the polymer dispersion, the polymer moved into the upper aqueous phase, demonstrating that the polymer was transformed from a hydrophobic to a hydrophilic state (Fig. 1b). The amidine has a pKaH of around 5.4,33 so it can be protonated by carbonic acid (pH ∼ 4.0–5.0) produced from the reaction of CO2 with water. As deduced from the equation shown in Fig. 1b, the hydrophobic amidine group reacts with carbonic acid and transforms into bicarbonate, thus becoming water-soluble. This hydrophobicity to hydrophilicity transition, being reversible or not, demonstrates the fundamental concept of a CO2-responsive “smart” system.
 | ||
| Fig. 1 (A) Synthesis of a CO2-responsive polymer containing amidine groups and (B) its transformation from hydrophobic to hydrophilic. Reprinted and adapted from ref. 46 with permission of the Royal Society of Chemistry. | ||
An interesting and curious question is why some CO2-responsive transitions are switchable but others are not. To unveil the reason, Yin et al.47 attempted to correlate the CO2-switchability and basicity using a series of melamine derivatives with different amine groups. They found that the CO2-switchability should be considered separately as protonation and deprotonation. The easy deprotonation process is sometimes called a good reversibility. Higher pKaH implies a stronger base and produces a higher protonation degree. However, for the deprotonation process, it becomes much more complicated. Higher pKaH would make the charged base more difficult to deprotonate. Furthermore, deprotonation is also impacted by the number of base groups in solution as well as the environmental temperature. As shown in Scheme 1, the pKaH of the amidine group in the polymer backbone lies at around 5.4, which is a weak base and thus can only be partially protonated with a CO2-stimulus.33 In contrast, the guanidine group whose pKaH is as high as 13.548 is a super base and difficult to deprotonate as expected. This may be the reason why Lowe and his co-workers49 synthesized a polymer containing guanidine groups with a post-modification strategy but did not find any tunable solubility with CO2/N2 cycling. Recently, Theato's team found an opposite switching behaviour under CO2-stimulation of a polymer containing guanidine groups. Namely, the lower critical solution temperature (LCST) of this polymer decreased and thereby presented a much more hydrophobic character than the polymer before passing CO2.36,50 Furthermore, polymers using the amidine or guanidine sensitive moieties are difficult to synthesize.49,51 That is the reason why the main interests of CO2-responsive polymers transfer to those possessing tertiary amine groups.
Zhao and his co-workers firstly discovered the CO2-responsiveness of polymers containing tertiary amine groups, poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA) and poly(N,N-diethylaminoethyl methacrylate) (PDEAEMA). The pKaH of the tertiary amine groups is around 6.0–7.0,52,53 which means that it is a moderate base and expected to have good switchability, i.e. it is easy to protonate and has good reversibility. In fact, the tertiary amine, especially PDEAEMA, is demonstrated as a perfect CO2-switchable moiety to date and widely used in various applications.24 Besides, the DEAEMA monomer is a commercial monomer free of a tedious synthesis procedure, which endows another advantage over other CO2-responsive moieties.54
In contrast to organobases, the weak acid–carboxylic acid group can also realize CO2-responsiveness. However, it is a different story. The polymers bearing carboxylic acid groups show a reverse transition, i.e. from hydrophilicity to hydrophobicity under CO2 stimulation, compared with that containing base groups. Under neutral conditions, the carboxyl acid moieties are in the negative charge state and thus appear to be hydrophilic. After treatment with CO2, these negative charges combine with the hydrogen ion and thus are naturalized into a hydrophobic pendant. In view of this unique characteristic, the groups of Zhao,55 Espí56 and Feng57 employed carboxylic acid groups in their synthesis of the corresponding CO2-responsive gels, latexes and worm-like micelles, respectively.
2.2 “Unexpected” CO2-responsive features
As mentioned above, usually CO2-responsive polymers containing guanidine groups are expected to show a similar responsive behavior to polymers based on amidine or amine groups after reaction with CO2, namely, they become more hydrophilic upon CO2 exposure. However, some CO2-responsive polymers containing guanidine groups showed an inverted responsive behavior.36,50 Theato and co-workers used poly(pentafluorophenyl acrylate) (PPFPA) as a precursor polymer for a subsequent post-modification with different amines, resulting in poly(L-arginine methyl ester acrylamide-co-N-cyclopropyl acrylamide) (poly(AME-co-CPAM)) bearing guanidine groups (Fig. 2a). The reversible LCSTs of this polymer can be switched by addition and removal of CO2. Interestingly, its CO2-responsive behavior revealed an opposite transition compared to other reported CO2-resposnive polymers based on amidine and amine groups. Namely, the LCST of poly(AME-co-CPAM) decreased after CO2 stimulation, resulting in a much more hydrophobic state.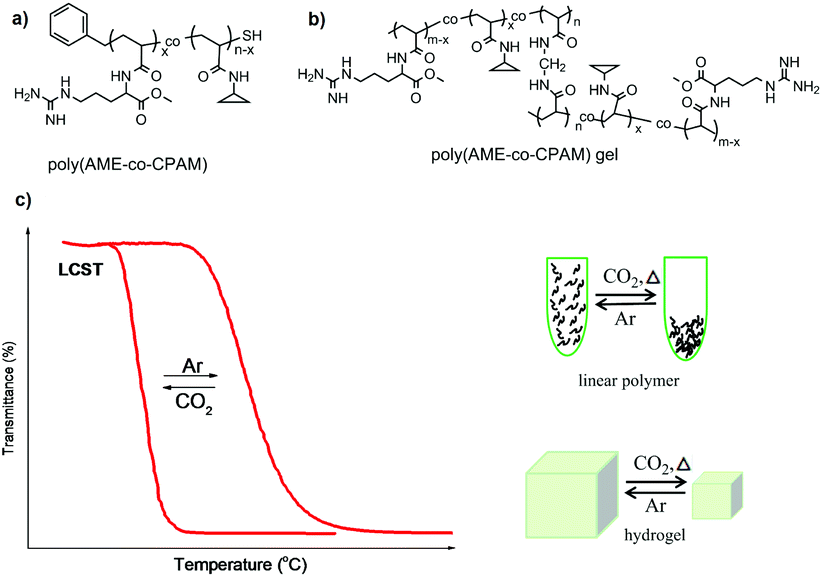 | ||
| Fig. 2 The chemical structures of linear polymers (a) and corresponding gels (b), and illustration of their transitions upon CO2 stimulation (c) reprinted and adapted from ref. 36 with permission of American Scientific Publishers and ref. 50 with permission of Elsevier. | ||
For further investigation of this “unusual” CO2-responsive behaviour, a series of poly(AME-co-CPAM) polymers and corresponding hydrogels (Fig. 2b) with different compositions were synthesized.36 It was found that all linear poly(AME-co-CPAM) copolymers appeared much more hydrophobic after purging CO2 into solution compared with their original states. Their LCST transition decreased to about 12 °C to 3 °C corresponding to the contents of AME in the polymers of 3–15%, respectively, after the reaction with CO2. Similar transitions also occurred in the corresponding hydrogels, which revealed a dramatic shrinkage upon exposure to CO2 and recovered after the removal of CO2 (Fig. 2c). This series of “unusual” CO2-responsive polymers can act as potential smart materials for CO2 capture and drug release.
The mechanism that is responsible for this “unexpected” CO2-responsive behavior is under an ongoing investigation. Noteworthily, other CO2-responsive polymers based on amidine or amine groups prepared from PPFPA via post-polymerization modification, e.g., poly(AME-co-CPAM), poly(3-N′,N′-dimethylaminopropyl acrylamide-co-N-isopropyl acrylamide) (poly(DMPA-co-NIPAM)), showed a normal switchability.50 Their corresponding LCST increased upon exposure to CO2 and recovered after passing argon through the solution. Similarly, Lowe and co-workers49 prepared parent block polymers based on PPFPA, which were converted into the corresponding CO2-responsive block polymers by post-polymerization modification with histamine dihydrochloride (HIS) and L-arginine methyl ester dihydrochloride (ARG), respectively. The poly(HIS-b-DMA) aggregated into micelles that can transform into unimers upon stimulation of CO2 and recover to micelles upon purging N2. Another work using PPFPA as a precursor polymer to prepare a series of mechanical and CO2-responsive homo- and block copolymers was done by Roth and Lowe.58 The polymers were soluble or dispersed in supersaturated aqueous CO2 solutions to form various nanostructures, which can be changed via an external mechanical force to remove CO2 in solution due to the deprotonation of CO2-responsive groups. From these examples,49,50,58 it is demonstrated that the above-mentioned “unexpected” CO2-responsive behaviors found by Theato's group were not caused by the polymerization method, i.e., post-polymerization modification of PPFPA. Further explorations are still on the way.
In this section, the chemical fundamentals of CO2-responsive polymers have been discussed. In the following part, we will look into various typical CO2-responsive systems.
3. CO2-Responsive polymer self-assemblies
Polymer self-assembled structures are mostly derived from amphiphilic copolymers; hence the development of CO2-responsive self-assembled polymer structures relies on the synthesis of CO2-responsive amphiphilic block copolymers. For example, an amphiphilic diblock copolymer containing a segment with amidine groups was first demonstrated by Yuan and her co-workers,33 who used it for the preparation of CO2-responsive vesicles. They found that the block copolymer formed vesicles that expanded under the stimulus of CO2, and shrunk back to their original state after removing CO2 by purging with N2 gas. Interestingly, this transformation is reversible, reminiscent of a “breathing” behaviour. Furthermore, it was demonstrated that these CO2-responsive vesicles have the capability for a size-selective release, separation and reaction.25Other than diblock copolymers, triblock copolymers were also studied as a platform for self-assembled structures, since they provide access to a broader range of possible self-assembly morphologies, including tubes, worms as well as compartmentalized large-compound sacs which can also be manipulated with CO2 as it has been demonstrated by Zhao's team.35,53 Except for these general geometries, CO2 can also fulfill a more complicated mission, i.e., manipulation of the morphology of a higher level self-assembly–multicompartment micelles (MCMs), which are regarded as the mimics of eukaryotic cells in nature. Recently, Liu et al.52 reported CO2-switchable MCMs with a segregated corona that self-assembled from a linear ABC triblock copolymer, which is composed of a hydrophilic segment of poly(ethylene oxide) (O), a fluorocarbon block of poly(2,2,3,4,4,4-hexafluorobutyl methacrylate) (F), and a CO2-responsive segment of poly(2-(diethylamino)ethyl methacrylate) (E), abbreviated as O113F110E212 (Fig. 3a). This triblock copolymer aggregated into uniform spherical micelles in the absence of CO2, and could be switched to MCMs with a segregated corona in the presence of CO2 (Fig. 3b). Noteworthily, the MCMs appeared in different variations including “hamburgers”, “reverse hamburgers”, “clovers”, and “footballs” (Fig. 3b). More interestingly, the MCMs can be reversed back to spheres (Fig. 3c) after removal of CO2 by purging with N2 gas, which was also proved by dynamic light scattering (DLS, Fig. 3d). In this case, the protonation–deprotonation transition of the tertiary amine groups of the “E” block can be accounted for this reversible morphological transition.
 | ||
| Fig. 3 (a) Schematic representation of CO2-switchable multicompartment micelles (MCMs) and (b) TEM images under treatment with CO2 (“hamburgers” (1), “reverse hamburgers” (2), “clovers” (3), “footballs” (4) and more complex structures (5)) and (c) after removing CO2 and (d) DLS data in each situation. Reprinted and adapted from ref. 52 with permission of the Royal Society of Chemistry. | ||
With the same triblock copolymer, O113F110E212, Liu et al.59 discovered CO2-responsive worm-like micelles that are driven by adjusting the composition of the solvent (Fig. 4). The polymer was subjected to a mixed solvent of ethanol and water and kept at 75 °C for one week after the removal of oxygen. The polymer then gradually dissolved and formed a clear micellar solution. Interestingly, by increasing the volume ratio of water, the aggregates appear as spheres, rods, cylinders and finally worm-like micelles, indicating a significantly solvent driven trend to worm-like micelles. Besides, under the stimulus of CO2, the worm-like micelles partially recover as spherical micelles (Fig. 4). This incomplete shape alternation may account for the closely-packed arrangement of the “F” block in the presence of water, which formed a so-called super strong segregation regime (SSSR) that is difficult to re-arrange the polymer chain. In spite of lacking perfect CO2-sensitivity, this solvent driven strategy may pave a novel route to fabricate polymer worm-like micelles, which is difficult to achieve because of a narrow composition window.60
 | ||
| Fig. 4 Solvent driven formation of a worm-like micelle and its shape alternation under the stimulus of CO2. Reprinted and adapted from ref. 59 with permission of the American Chemistry Society. | ||
Apart from the well-defined block copolymers, block-random segmented copolymers, distinguished from the traditional classical block structure, which bear at least one random copolymer as a building block,61 are also explored for the fabrication of CO2-responsive self-assemblies.27 By replacing the homo-polymer block with a random one, further tailoring of the polymer properties can be targeted. Furthermore, multiple properties can be combined into one random segment in one single polymerization step from diverse components, escaping from multi-step copolymerization and tedious post-treatment. Feng and his co-workers27 observed a vesicle-to-spherical micelle transformation (Fig. 5a) using a block-random segmented copolymer of the CO2-responsive monomer 2-(diethylamino)ethyl methacrylate (DEAEMA) and hydrophobic monomer styrene (St) from a macromolecular chain transfer agent (CTA) of poly(ethylene oxide) (PEO), i.e., PEO45-b-(DEAEMA90-r-St66) (Pr). This morphological transition occurs very fast, which can finish in about 2 min through monitoring transmittance of the micellar solution. Interestingly, a triblock copolymer counterpart, i.e., PEO45-b-DEAEMA93-b-St66 (Pb), with a similar degree of polymerization showed no significant morphological change except for an expansion (Fig. 5a). The vesicle-to-sphere shape transition can be ascribed to the accumulated interfacial energy caused by restricted hydration of the CO2-responsive moieties in the random structure. On the other hand, the CO2-sensitive chain in the triblock copolymer can be hydrated freely, thus being free of interfacial energy increasing.
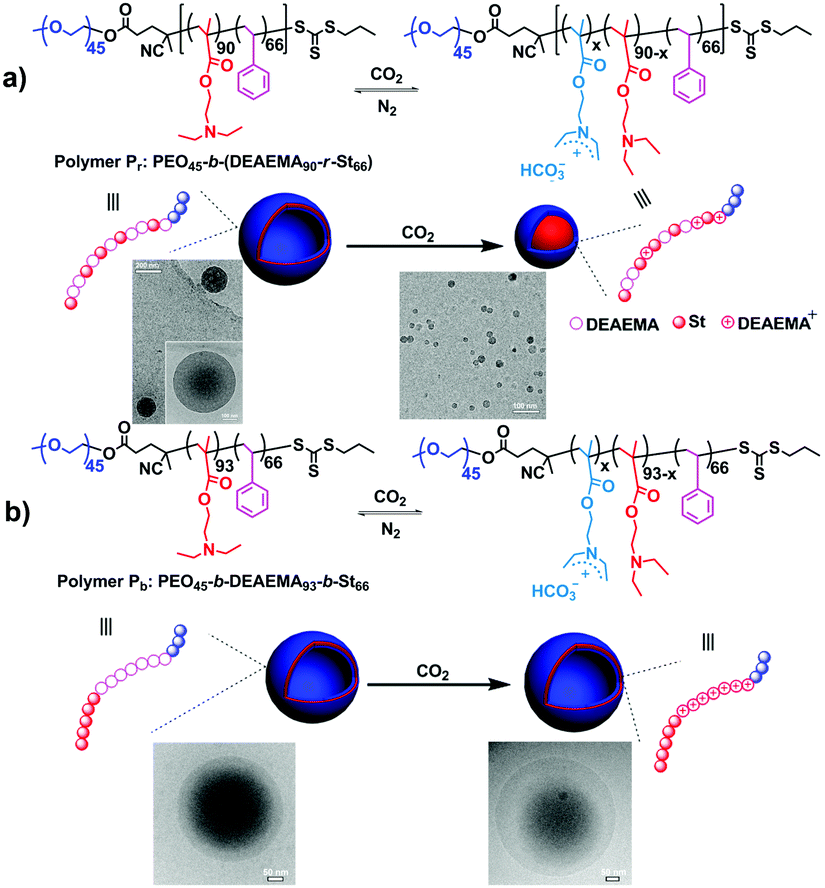 | ||
| Fig. 5 CO2-Responsive block-random segmented copolymer (a) and its triblock counterpart (b), as well as schematic illustration of the different morphological changes upon CO2 stimulation with the corresponding TEM images. Reprinted and adapted from ref. 27 with permission of the Royal Society of Chemistry. | ||
Following a similar strategy to target precise morphological manipulation, Wang et al.62 developed a reversible morphology transition (Fig. 6) from giant worms to polymersomes from a block-random segmented copolymer composed of a hydrophilic poly(ethylene oxide) block and a random hydrophobic block copolymerized from 2-(diethylamino)ethyl methacrylate (DEAEMA) and 4-vinyl pyridine (4VP), i.e., PEO113-b-P(4VP90-r-DEAEMA30). The copolymer self-assembled into vesicles in water, which then quickly fused into giant worms. After treatment with CO2 gas, the giant worms transformed into vesicles, which can be converted back to aggregate like a necklace after the removal of CO2 by bubbling N2. This serial shape variation was also caused by the hydrophobic–hydrophilic transition of the CO2-responsive moieties DEAEMA (the 4VP moieties here cannot be protonated by CO2 because the pH in the CO2 saturated solution is higher than the pKaH of 4VP). Furthermore, the hydrogen bonding between 4VP moieties is believed to be helpful for the stabilization of the giant worms from which the morphological transition benefits too.
 | ||
| Fig. 6 Schematic illustration of the transformation from giant worms to vesicles to necklace aggregates from block-random segmented copolymers PEO113-b-P(4VP90-r-DEAEMA30) upon alternating treatment with CO2 and N2. Reprinted from ref. 62 with permission of the Royal Society of Chemistry. | ||
Even though CO2-responsive self-assembled polymer structures show promising potential as “smart” materials for biotherapy, there are still some challenges to be resolved in the future. First of all, a method should be developed to identify the exact concentration of CO2 in solution that can trigger the morphological transformation, which could help precisely control the aggregates formed with CO2. Furthermore, one question should be answered, namely whether the variations in CO2 concentration in the human body (including in blood or cells) are sufficient to trigger the morphological transition of polymer self-assemblies, which is a prerequisite for any possible application as a CO2-triggered release system. Another question is whether we can control the actual response time, meaning how long the morphological transition would take place after the stimulation with CO2. It is noteworthy that these challenges offer a lot of opportunities for future research.
4. CO2-Responsive polymer hybrids
Since CO2 is regarded as a “green” trigger, various applications using CO2-responsive compounds as well as polymers have been investigated. After conjugation of a CO2-sensitive polymer to another material, a CO2-responsive hybrid can be obtained. Guo et al.,26 for instance, developed “smart” single-walled carbon nanotubes (SWCNTs), the solubility of which in different solvents can be reversibly switched by CO2 (Fig. 7). In a two-phase mixture of dichloromethane (DCM) and water, SWCNTs coated with a CO2-responsive polymer could originally be dispersed in organic DCM without CO2-stimulation. After treatment with CO2, the polymer moved into the water phase. In pure water, the “smart” SWCNTs cannot be dispersed and appeared as a precipitate, which can only be dispersed after bubbling CO2 into water. These “smart” SWCNTs were demonstrated as a sensitive sensor to detect CO2 in water by monitoring the transmittance of water (Fig. 7). | ||
| Fig. 7 CO2-Responsive “smart” SWCNTs that can work as a sensitive sensor to detect trace amounts of CO2. Reprinted from ref. 26 with permission of Wiley-VCH. | ||
Apart from stimuli-responsive CNTs, “smart” graphene has also received considerable attention due to its promising applications.63–66 Feng's group65 fabricated CO2-responsive graphene based on strongly attaching N2,N4,N6-tris(3-(dimethylamino)propyl)-1,3,5-triazine-2,4,6-triamine (MET) to the surfaces of reduced graphene oxide through van der Waals interaction. Such complexes can be stably dispersed in water via steric hindrance or electrostatic repulsive forces among them. Then, graphene could aggregate upon bubbling CO2 into the aqueous solution. The reason can be ascribed to the protonated tertiary amine groups from MET that are detached from the graphene surface resulting in separation between MET molecules and graphene. However, graphene could be dispersed again after removal of CO2 by bubbling N2 due to the deprotonation of tertiary amine groups. Noteworthily, such graphene can reversible aggregate and disperse via alternately purging CO2 and N2, respectively. He et al.66 also developed CO2 responsive graphene using a dispersant containing a tertiary amine and a pyrene group. The modified graphene shows a fast response (within 1.5 min) and a low recovery temperature (40 °C).
In addition, more CO2-responsive hybrid materials have been reported as well, including a CO2-switchable catalyst and surface developed by Zhao's group.22 CO2-Responsive magnetic particles were reported by Yuan and co-workers,67,68 and Yung's group reported functionalized gold nanoparticles as a candidate to detect CO2 gas.69 Clearly, such kinds of hybrid “smart” materials will find application in numerous upcoming areas in the near future, in particular considering the types of CO2-responsive polymers that have already been developed.
5. CO2-Responsive “living” polymers
Similar to amphiphilic block copolymers, surfactants are another important class of amphiphiles that can form numerous self-assembled structures,2,70 including vesicles, micelles as well as worm-like micelles (WLMs). The surfactant-based WLMs can entangle with each other and produce a dynamic reversible network, which constantly breaks and reforms, so that they can be regarded as “living” or “equilibrium” polymers.71By introducing stimulus-responsive moieties into the surfactant WLM system, stimulus-sensitive “living” polymers can be easily targeted.72 Based on this idea, Zhang et al.73 fabricated CO2-switchable WLMs by adding a CO2-sensitive small molecule N,N,N′,N′-tetramethyl-1,3-propanediamine (TMPDA) into the commercially available anionic surfactant sodium dodecyl sulphate (SDS) (mole ratio of 1:2). The two compounds are mixed in water; no interaction exists between them with only spherical micelles formed by the surfactant SDS in the absence of CO2. However, when CO2 is bubbled into this mixture, a dramatic change occurs immediately: the solution becomes a viscoelastic fluid caused by the formation of WLMs (Fig. 8). The production of WLMs is attributed to the protonation of TMPDA after the reaction with CO2, which acts as a “bridge” between two SDS molecules through electrostatic attraction, resulting in a change of the packing parameter (P) of SDS from P = 0.11 to P = 0.45, and this ultimately determines the morphology of surfactant assemblies.
 | ||
| Fig. 8 CO2-switchable “living” polymer based on a pseudogemini surfactant. Reprinted and adapted from ref. 73 with permission of the American Chemistry Society. | ||
In an effort to develop a new kind of surfactant that can produce such “living” polymers, Feng's group74 synthesized a long-chain surfactant, octadecyl dipropylene triamine (ODPTA), which self-assembled into CO2-switchable WLMs in aqueous solution (Fig. 9). In the absence of CO2, ODPTA is neutral in water and it self-assembles into small vesicles, whereas after treatment with CO2, the two amine groups of this surfactant are protonated and transformed into cationic species as well as an anionic by-product after conjugation with CO2, which results in the self-assembly of WLMs. This one-component “living” polymer may find application in biomedicine, microfluidics, and water-alternating-gas flooding in tertiary oil recovery, etc., because it could be resistant against phase-segregation in two-component systems.
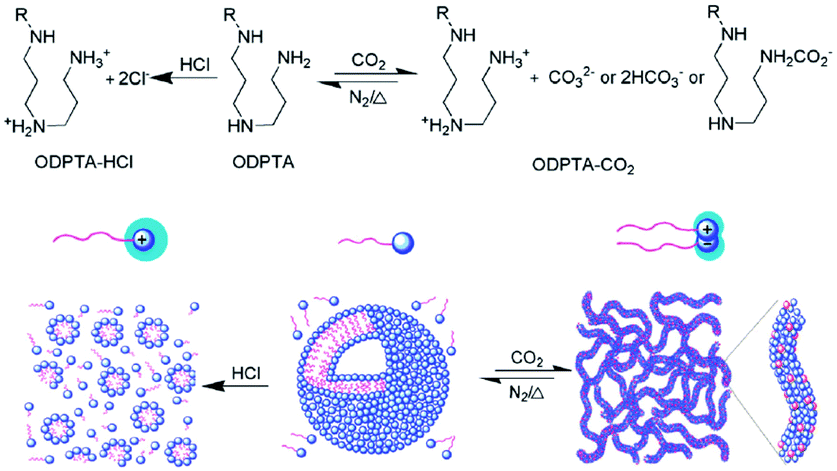 | ||
| Fig. 9 CO2-switchable “living” polymer from a C18-tailed polyamine surfactant. Reprinted from ref. 74 with permission of the Royal Society of Chemistry. | ||
The above-mentioned CO2-responsive “living” polymers can be turned “off” (switched back to spherical micelles or free surfactants) by bubbling inert gases (including N2 or Ar) to replace the CO2 gas. Some polymers require additionally an increase of the temperature. This raises an interesting question, namely whether the CO2-responsiveness can be achieved at room temperature with air rather than an inert gas. Feng and co-workers75 clarified this confusion by fabricating WLMs with a surfactant N-erucamidopropyl-N,N-dimethylamine (UC22AMPM), as shown in Fig. 10. In aqueous solution, UC22AMPM does not form any self-assembled aggregates due to poor solubility (cmc ∼0.0085 mM) caused by the long hydrophobic tail. However, after exposure to CO2, the tertiary amine head was converted into an ammonium bicarbonate, making UC22AMPM a cationic surfactant, which has the capability to self-assemble into micelles. These micelles then gradually grew into WLMs in solution (after 1 minute of CO2 streaming at a flow rate of 0.1 L min−1). When this “living” polymer was exposed to air (rather than an inert gas), the surfactant gradually lost its charge and returned to the original neutral structure, which resulted in disruption of the WLMs. This special CO2-responsive system shows potential to thicken and separate specific fluids, such as water alternative-gas oil recovery processes in the oil and natural-gas industry, because of the cost-effective use of air rather than an inert gas or heating.
 | ||
| Fig. 10 “Living” polymer switched by CO2 and air. Reprinted and adapted from ref. 75 with permission of the Royal Society of Chemistry. | ||
6. Other CO2-responsive polymeric systems
Except for the above-mentioned types of CO2-responsive polymers, numerous other examples utilizing CO2-responsive polymers were reported in the past few years owing to the fact that CO2 is a green trigger, which results in their potential application in various fields, including CO2-responsive latexes, CO2-responsive surfaces, CO2-responsive polymers for CO2 capture, CO2-responsive polymers as monitors and so on. These types of CO2-responsive polymeric systems will be discussed in the following section to take a panoramic view of the whole research field of CO2-sensitive polymer materials.6.1 CO2-Responsive latexes
Compared to traditional and other-responsive latexes, CO2-responsive latex possesses switchable surfactants to realize a repeated coagulation and re-dispersion without forming by-products during cycle stimulated processes via alternately purging with CO2 or an inert gas. These advantages contribute to CO2-responsive latexes that can easily transport in the coagulated state in the absence of water, and they then can be re-dispersed via purging CO2 in water without an additional stabilizer. In the area of CO2-responsive latexes, both Zhu's group32,76,77 and Cunningham/Jessop's group40,78,79 have independently conducted numerous studies in the past few years. Cunningham and Jessop et al.,78 first exploited CO2-responsive polystyrene (PS) latexes based on amidine groups to implement a reversible aggregation and re-dispersion upon purging N2 and CO2 (Fig. 11a). Subsequently, CO2-responsive latexes based on amidine groups were developed by Zhu77 and Cunningham/Jessop's40 groups. Then, they employed commercially available materials to prepare CO2-responsive latexes which provide the possibility for large scale production.31,32 Furthermore, several CO2-responsive latexes that can be much easily re-dispersed using ultrasonic treatment or heating rather than addition of extra caustic soda were also developed.51 However, as mentioned above, amidine groups are not stable in water because they tend to hydrolyze over time. Therefore, a series of CO2-responsive poly(methyl methacrylate) (PMMA) or PS latexes based on amine moieties were investigated, which showed good stability against hydrolysis. In these studies, 2-(dimethylamino)ethyl methacrylate) (DMAEMA)32,51 or 2-(diethylamino)ethyl methacrylate) (DEAEMA)31,80 was utilized to prepare CO2-switchable surfactants, which were then employed in the synthesis of CO2-responsive latexes that can be reversibly and steadily coagulated and re-dispersed under CO2 stimulation. In addition, Espí's group56 prepared carboxyl group functionalized CO2-responsive PS latexes that can be coagulated by purging CO2 and re-dispersed by removal of CO2. | ||
| Fig. 11 The reversible coagulation and re-dispersion of CO2-responsive latexes based on amidine groups (a), and the CO2-responsive surface for reversible capture and release of protein (b). (a) Reprinted from ref. 77 with permission of the American Chemistry Society. (b) Reprinted from ref. 41 with permission of the Royal Society of Chemistry. | ||
6.2 CO2-Responsive surfaces
CO2-Responsive surfaces can switch their hydrophilicity or hydrophobicity in response to CO2. Therefore, such surfaces find potential applications in various fields such as protein absorption and release, nanoparticle separation or as non-fouling surfaces. Zhao's group41 prepared a CO2-responsive surface via immobilizing an initiator for an atom transfer radical polymerization (ATRP) on silica wafer, and then grafting PDEDEMA polymer brushes from the surface. The obtained PDEDEMA brushes can reversibly switch from a hydrophobic to a hydrophilic state by addition or removal of CO2, which leads to an absorption and release of proteins owing to the switchable hydrophilicity of the surfaces (Fig. 11b). Similarly, Wang's group81 firstly prepared N-(2-aminoethyl)-5-(1,2-dithiolan-3-yl)pentanamide (NADP), which then reacted with N,N-dimethylacetamide dimethyl acetal (DMADMA) to result in an amidine end-modified NADP (NADP-amidine). Then, amidine functionalized NADP was fixed on the surface of a gold substrate to obtain CO2-responsive surfaces, which allowed a controlled change in the wettability of the surface by CO2 because of reversible transition from charged to neutral amidine moieties under CO2 stimulation. Based on the change of hydrophilicity on the surface, it can realize selective absorption of hydrophilic or hydrophobic molecules. Additionally, Yuan et al.42 fabricated CO2-responsive electrospun membranes from copolymers derived from methylmethacrylate (MMA) and N,N-diethylaminoethyl methacrylate (DEAEMA), which are able to switch the surface oil/water wettability under CO2 stimulation. Originally, oil in the oil/water mixture could pass the hydrophobic membrane to separate from water. After treatment with CO2, the membrane switched from the hydrophobic to the hydrophilic state, and hence, only water passed through the membrane. This CO2-switchable membrane may be used as a smart valve in different applications, such as water treatment or micro-fluidics. Recently, Feng and Billon's group82 prepared CO2-responsive honeycomb porous films with switchable surface wettability from polystyrene-b-poly(N,N-dimethylamino ethyl methacrylate) (PS-b-PDMAEMA). They pointed out that PDMEAEMA as a hydrophilic segment plays an essential role in determining porous sizes and arrays on surfaces. The film surface showed a reversible transition between hydrophilic and hydrophobic states upon purging CO2 or heating up to 60 °C, respectively, attributing to the protonation/deprotonation of tertiary amine groups under CO2 stimulation or heating to remove CO2. These CO2-responsive porous films are demonstrated to serve as good candidates for cell attachment and spreading.6.3 CO2-Responsive polymers for CO2 capture
With the increment of CO2 in the atmosphere from industrial pollution and automobile exhaust, efficient materials for capturing CO2 from air are expected. Among the different materials that are under investigation, polymers for CO2 capture feature a couple of advantages: polymers are much easier to process and model compared with other CO2-capturing materials such as ammonia water,83,84 inorganic materials85,86 and ionic liquids.87,88 With the development of other CO2-capturing polymers, Endo's group89,90 synthesized a series of polymers containing amidine moieties to enable the reaction between CO2 and amidine groups, resulting in reversible capture and release of CO2. With further investigation, Lu et al.91 developed a much more effective polymer, poly(N-heterocyclic carbenes-co-styrene), to capture CO2. The results showed that this polymer can capture CO2 faster at a relative low CO2 concentration and can also release CO2 more quickly at a high temperature. In addition, recently, Rieger's group92 synthesized an acylated polyethylenimine that contained amine groups, featuring thermal-and CO2 responsive behaviors for CO2 capture and release. The results revealed that the thermal-responsive properties of the polymer contributes to CO2 release because more protons were formed during the phase transition (LCST) while heating that induced decomposition of ammonium bicarbonate moieties, and thereby released CO2 easily from acylated polyethylenimine solution.6.4 CO2-Responsive polymers as a CO2 sensor
The preparation of CO2-responsive polymers to monitor, detect and quantify CO2 has received immense attention recently. Kang's group93 introduced a fluorophore to PDMAEMA to prepare a CO2-responsive polymer that revealed a reversible color change under protonation or deprotonation of amine moieties in PDMAEMA via addition or removal of CO2, resulting in switching the fluorescence emission strength. Consequently, the color of the solution reversibly changed from dark red to orange with increasing amounts of CO2 in solution. Thus, this CO2-responsive polymer was potentially applied to monitor CO2 in aqueous solutions. Following this, Yung's group69 simply mixed gold nanoparticles and random copolymer poly(dimethyl acrylamide-co-(N-amidino)ethyl acrylamide) (P(DMA-co-NAEAA)) containing amidine groups, which resulted in a system that can be used to detect the dissolved CO2 level. Upon addition of different amounts of NaHCO3 into the mixture, the amidine groups on the polymer chains were protonated by the protons from the dissociation of dissolved CO2 and switched from a neutral to a positive charged state, which triggered the negatively-charged gold nanoparticles to coagulate due to the electrostatic interactions between polymer chains and gold nanoparticles. As a result, the plasmonic changes could be tracked by the naked eye and UV according to the level of dissolved CO2 in solution. Besides, as mentioned above, the CO2-responsive CNTs developed by Guo can also detect CO2 in water solution.267. Challenges and outlook
The development of CO2-responsive polymers has gained rapid momentum over the last few years because CO2 is an environmentally-friendly “green” trigger compared with other traditional stimuli-responsive polymers (temperature, light, pH, redox, etc.). Strictly speaking, the CO2-response is a kind of pH responsive behaviour because the CO2-functional groups are protonated or deionized according to the pH of aqueous solutions that are controlled by the concentration of CO2. The shortcoming of a CO2 responsive polymer might be the limited pH range, usually from 4.0 to around 7.6.27 In addition, higher viscosity can have an impact on the diffusion of CO2, but to our knowledge, no detailed study has been conducted yet. However, unlike traditional pH-responsive polymers, CO2-responsive polymers realize their response behaviours via addition or removal of CO2 as an external stimulus without any acid or base chemical compounds to accumulate and contaminate the system. Therefore, CO2-responsive polymers can undergo repeated stimulation via alternately purging with CO2 or an inert gas without significant decay. What is more important, CO2 is a metabolite of cells, thus possessing good biocompatibility and permeability. Given these advantages, CO2-responsive polymers have a bright prospect as “smart” devices, biosensors as well as drug vehicles. In addition, CO2-responsive polymers can also work as CO2 capturing materials to reduce the CO2 content in the atmosphere and reuse it as a carbon resource. However, CO2-responsive polymers still face some challenges. As yet, CO2-responsive polymers can only present their responsive behaviours under high CO2 concentrations in aqueous solutions, which might be a bottleneck for their biomedical applications. Taking drug delivery as an example, it requires a sensitive response under a very low concentration of CO2 in the human body. A multi-responsive system arose very recently, coupling with pH-,94,95 temperature-,38,95–99 O2-sensitive100–102 polymers or magnetic-particles,67,103 which may pave the way to overcome this problem. As potential CO2 capture materials, the realization of CO2-responsive polymers to enable a highly effective capture and release at room temperature also requires further investigations. Last but not least, the industrialization of CO2-responsive polymers also remains a major challenge. To date, CO2-responsive latexes have a rosy future to achieve this goal. Other efforts were put into the scaled-up utilization of CO2-responsive polymers as well.104 Nevertheless, although achieving further progress is necessary to overcome the above-mentioned challenges, CO2-responsive polymers still have a promising prospect in the future in various applications.Acknowledgements
YF would like to thank the financial support from the National Natural Science Foundation of China (21273223), and the open funding of the State Key Laboratory of Polymer Materials Engineering (sklpme 2014-2-06). SL gratefully acknowledges the China Scholarship Council (CSC, grant 201306240132) for financial support.Notes and references
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- P. Brown, C. P. Butts and J. Eastoe, Soft Matter, 2013, 9, 2365–2374 RSC.
- P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2512–2529 RSC.
- A. E. Smith, X. W. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef CAS.
- H. I. Lee, J. Pietrasik, S. S. Sheiko and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 24–44 CrossRef CAS.
- S. K. Ahn, R. M. Kasi, S. C. Kim, N. Sharma and Y. X. Zhou, Soft Matter, 2008, 4, 1151–1157 RSC.
- Z. L. Chu, C. A. Dreiss and Y. J. Feng, Chem. Soc. Rev., 2013, 42, 7174–7203 RSC.
- M. Xia, Y. Cheng, Z. Meng, X. Jiang, Z. Chen, P. Theato and M. Zhu, Macromol. Rapid Commun., 2015, 36, 477–482 CrossRef CAS PubMed.
- L. E. Bromberg and E. S. Ron, Adv. Drug Delivery Rev., 1998, 31, 197–221 CrossRef CAS.
- H. K. Ju, S. Y. Kim and Y. M. Lee, Polymer, 2001, 42, 6851–6857 CrossRef CAS.
- Q. Yang, S. Wang, P. Fan, L. Wang, Y. Di, K. Lin and F.-S. Xiao, Chem. Mater., 2005, 17, 5999–6003 CrossRef CAS.
- S. Liu, J. V. Weaver, Y. Tang, N. C. Billingham, S. P. Armes and K. Tribe, Macromolecules, 2002, 35, 6121–6131 CrossRef CAS.
- J. M. Schumers, C. A. Fustin and J. F. Gohy, Macromol. Rapid Commun., 2010, 31, 1588–1607 CrossRef CAS PubMed.
- S. Sumalekshmy and C. J. Fahrni, Chem. Mater., 2010, 23, 483–500 CrossRef.
- C. Park, H. Kim, S. Kim and C. Kim, J. Am. Chem. Soc., 2009, 131, 16614–16615 CrossRef CAS PubMed.
- R. V. Ulijn, J. Mater. Chem., 2006, 16, 2217–2225 RSC.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- S. Medeiros, A. Santos, H. Fessi and A. Elaissari, Int. J. Pharm., 2011, 403, 139–161 CrossRef CAS PubMed.
- S. Lu, Y.-H. Sun, R. Shi, C. Clark, L. Li and V. L. Chiang, Plant Cell, 2005, 17, 2186–2203 CrossRef CAS PubMed.
- M. S. Aw, J. Addai-Mensah and D. Losic, J. Mater. Chem., 2012, 22, 6561–6563 RSC.
- J. M. Zhang, D. H. Han, H. J. Zhang, M. Chaker, Y. Zhao and D. L. Ma, Chem. Commun., 2012, 48, 11510–11512 RSC.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- S. Lin and P. Theato, Macromol. Rapid Commun., 2013, 34, 1118–1133 CrossRef CAS PubMed.
- Q. Yan, J. Wang, Y. Yin and J. Yuan, Angew. Chem., Int. Ed., 2013, 52, 5070–5073 CrossRef CAS PubMed.
- Z. R. Guo, Y. Feng, S. He, M. Z. Qu, H. L. Chen, H. B. Liu, Y. F. Wu and Y. Wang, Adv. Mater., 2013, 25, 584–590 CrossRef CAS PubMed.
- H. Liu, Z. Guo, S. He, H. Yin, C. Fei and Y. Feng, Polym. Chem., 2014, 5, 4756–4763 RSC.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- J. C. Hicks, J. H. Drese, D. J. Fauth, M. L. Gray, G. Qi and C. W. Jones, J. Am. Chem. Soc., 2008, 130, 2902–2903 CrossRef CAS PubMed.
- N. Du, H. B. Park, G. P. Robertson, M. M. Dal-Cin, T. Visser, L. Scoles and M. D. Guiver, Nat. Mater., 2011, 10, 372–375 CrossRef CAS PubMed.
- J. Pinaud, E. Kowal, M. Cunningham and P. Jessop, ACS Macro Lett., 2012, 1, 1103–1107 CrossRef CAS.
- Q. Zhang, G. Yu, W.-J. Wang, B.-G. Li and S. Zhu, Macromol. Rapid Commun., 2012, 33, 916–921 CrossRef CAS PubMed.
- Q. Yan, R. Zhou, C. Fu, H. Zhang, Y. Yin and J. Yuan, Angew. Chem., Int. Ed., 2011, 50, 4923–4927 CrossRef CAS PubMed.
- Y. X. Liu, P. G. Jessop, M. Cunningham, C. A. Eckert and C. L. Liotta, Science, 2006, 313, 958–960 CrossRef CAS PubMed.
- Q. Yan and Y. Zhao, Angew. Chem., Int. Ed., 2013, 52, 9948–9951 CrossRef CAS PubMed.
- S. Lin, P. Schattling and P. Theato, Sci. Adv. Mater., 2015, 7, 948–955 CrossRef CAS.
- Q. Yan and Y. Zhao, Chem. Commun., 2014, 50, 11631–11641 RSC.
- A. Feng, C. Zhan, Q. Yan, B. Liu and J. Yuan, Chem. Commun., 2014, 50, 8958–8961 RSC.
- N. Che, S. Yang, H. Kang, R. Liu, Z. Li, Z. Liu, P. Li, X. Qu and Y. Huang, Polym. Chem., 2014, 5, 7109–7120 RSC.
- X. Su, P. G. Jessop and M. F. Cunningham, Macromolecules, 2012, 45, 666–670 CrossRef CAS.
- S. Kumar, X. Tong, Y. L. Dory, M. Lepage and Y. Zhao, Chem. Commun., 2013, 49, 90–92 RSC.
- H. Che, M. Huo, L. Peng, T. Fang, N. Liu, L. Feng, Y. Wei and J. Yuan, Angew. Chem., Int. Ed., 2015, 54, 8934–8938 CrossRef CAS PubMed.
- Q. Yan and Y. Zhao, Polym. Mater. Sci. Eng., 2014, 2, 032 CrossRef.
- M. Cunningham and P. Jessop, Eur. Polym. J., 2016, 76, 208–215 CrossRef CAS.
- A. Darabi, P. Jessop and M. Cunningham, Chem. Soc. Rev., 2016, 45, 4391–4436 RSC.
- Z. R. Guo, Y. Feng, Y. Wang, J. Y. Wang, Y. F. Wu and Y. M. Zhang, Chem. Commun., 2011, 47, 9348–9350 RSC.
- H. Yin, Y. Feng, H. Liu, M. Mu and C. Fei, Langmuir, 2014, 30, 9911–9919 CrossRef CAS PubMed.
- J. Y. Quek, T. P. Davis and A. B. Lowe, Chem. Soc. Rev., 2013, 42, 7326–7334 RSC.
- J. Y. Quek, P. J. Roth, R. A. Evans, T. P. Davis and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 394–404 CrossRef CAS.
- P. Schattling, I. Pollmann and P. Theato, React. Funct. Polym., 2014, 75, 16–21 CrossRef CAS.
- Q. Zhang, G. Yu, W.-J. Wang, H. Yuan, B.-G. Li and S. Zhu, Macromolecules, 2013, 46, 1261–1267 CrossRef CAS.
- H. Liu, Y. Zhao, C. A. Dreiss and Y. Feng, Soft Matter, 2014, 10, 6387–6391 RSC.
- Q. Yan and Y. Zhao, J. Am. Chem. Soc., 2013, 135, 16300–16303 CrossRef CAS PubMed.
- D. Han, X. Tong, O. Boissière and Y. Zhao, ACS Macro Lett., 2011, 1, 57–61 CrossRef.
- D. Han, O. Boissiere, S. Kumar, X. Tong, L. Tremblay and Y. Zhao, Macromolecules, 2012, 45, 7440–7445 CrossRef CAS.
- V. Fischer, K. Landfester and R. Muñoz-Espí, ACS Macro Lett., 2012, 1, 1371–1374 CrossRef CAS.
- Y. Zhang, H. Yin and Y. Feng, Green Mater., 2014, 2, 95–103 CrossRef.
- P. J. Roth, J. Y. Quek, Y. Zhu, B. M. Blunden and A. B. Lowe, Chem. Commun., 2014, 50, 9561–9564 RSC.
- H. Liu, W. Wang, H. Yin and Y. Feng, Langmuir, 2015, 31, 8756–8763 CrossRef CAS PubMed.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- C. Tsitsilianis, G. Gotzamanis and Z. Iatridi, Eur. Polym. J., 2011, 47, 497–510 CrossRef CAS.
- W. Wang, H. Liu, M. Mu, H. Yin and Y. Feng, Polym. Chem., 2015, 6, 2900–2908 RSC.
- J. Q. Liu, L. Tao, W. R. Yang, D. Li, C. Boyer, R. Wuhrer, F. Braet and T. P. Davis, Langmuir, 2010, 26, 10068–10075 CrossRef CAS PubMed.
- D. Ager, V. A. Vasantha, R. Crombez and J. Texter, ACS Nano, 2014, 8, 11191–11205 CrossRef CAS PubMed.
- H. Y. Yin, H. B. Liu, W. Wang and Y. J. Feng, Langmuir, 2015, 31, 12260–12267 CrossRef CAS PubMed.
- S. He, M. Z. Qu and Y. J. Feng, ChemNanoMat, 2015, 6, 438–444 CrossRef.
- J. Guo, N. Wang, J. Wu, Q. Ye, C. Zhang, X.-H. Xing and J. Yuan, J. Mater. Chem. B, 2014, 2, 437–442 RSC.
- H. Che, M. Huo, L. Peng, Q. Ye, J. Guo, K. Wang, Y. Wei and J. Yuan, Polym. Chem., 2015, 6, 2319–2326 RSC.
- Y. Ma and L.-Y. L. Yung, Anal. Chem., 2014, 86, 2429–2435 CrossRef CAS PubMed.
- X. Su, T. Robert, S. M. Mercer, C. Humphries, M. F. Cunningham and P. G. Jessop, Chem. – Eur. J., 2013, 19, 5595–5601 CrossRef CAS PubMed.
- Z. Chu, C. A. Dreiss and Y. Feng, Chem. Soc. Rev., 2013, 42, 7174–7203 RSC.
- X. Su, M. F. Cunningham and P. G. Jessop, Chem. Commun., 2013, 49, 2655–2657 RSC.
- Y. M. Zhang, Y. J. Feng, Y. J. Wang and X. L. Li, Langmuir, 2013, 29, 4187–4192 CrossRef CAS PubMed.
- Y. M. Zhang, Y. Feng, J. Y. Wang, S. He, Z. R. Guo, Z. L. Chu and C. A. Dreiss, Chem. Commun., 2013, 49, 4902–4904 RSC.
- Y. M. Zhang, Z. L. Chu, C. A. Dreiss, Y. J. Wang, C. H. Fei and Y. J. Feng, Soft Matter, 2013, 9, 6217–6221 RSC.
- Q. Zhang, W. J. Wang, Y. Y. Lu, B. G. Li and S. P. Zhu, Macromolecules, 2011, 44, 6539–6545 CrossRef CAS.
- Q. Zhang, G. Yu, W.-J. Wang, H. Yuan, B.-G. Li and S. Zhu, Langmuir, 2012, 28, 5940–5946 CrossRef CAS PubMed.
- M. Mihara, P. Jessop and M. Cunningham, Macromolecules, 2011, 44, 3688–3693 CrossRef CAS.
- J. Pinaud, E. Kowal, M. Cunningham and P. Jessop, ACS Macro Lett., 2012, 1, 1103–1107 CrossRef CAS.
- P. Liu, W. Lu, W.-J. Wang, B.-G. Li and S. Zhu, Langmuir, 2014, 30, 10248–10255 CrossRef CAS PubMed.
- N. Li, L. Thia and X. Wang, Chem. Commun., 2014, 50, 4003–4006 RSC.
- H. Y. Yin, A. L. Bulteau, Y. J. Feng and L. Billon, Adv. Mater. Interfaces, 2016, 3 DOI:10.1002/admi.201500623.
- H. Bai and A. C. Yeh, Ind. Eng. Chem. Res., 1997, 36, 2490–2493 CrossRef CAS.
- J. T. Yeh, K. P. Resnik, K. Rygle and H. W. Pennline, Fuel Process. Technol., 2005, 86, 1533–1546 CrossRef CAS.
- R. Banerjee, H. Furukawa, D. Britt, C. Knobler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 3875–3877 CrossRef CAS PubMed.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- J. E. Bara, D. E. Camper, D. L. Gin and R. D. Noble, Acc. Chem. Res., 2009, 43, 152–159 CrossRef PubMed.
- B. Ochiai, K. Yokota, A. Fujii, D. Nagai and T. Endo, Macromolecules, 2008, 41, 1229–1236 CrossRef CAS.
- T. Endo, D. Nagai, T. Monma, H. Yamaguchi and B. Ochiai, Macromolecules, 2004, 37, 2007–2009 CrossRef CAS.
- H. Zhou, W.-Z. Zhang, Y.-M. Wang, J.-P. Qu and X.-B. Lu, Macromolecules, 2009, 42, 5419–5421 CrossRef CAS.
- J. Kainz, P. D. L. Werz, C. Troll and B. Rieger, RSC Adv., 2015, 5, 9556–9560 RSC.
- L. Q. Xu, B. Zhang, M. Sun, L. Hong, K.-G. Neoh, E.-T. Kang and G. D. Fu, J. Mater. Chem. A, 2013, 1, 1207–1212 CAS.
- B. A. Abel, M. B. Sims and C. L. McCormick, Macromolecules, 2015, 48, 5487–5495 CrossRef CAS.
- Z. Song, K. Wang, C. Gao, S. Wang and W. Zhang, Macromolecules, 2016, 49, 162–171 CrossRef CAS.
- W. Yuan, J. Shen and H. Zou, RSC Adv., 2015, 5, 13145–13152 RSC.
- H. Zou and W. Yuan, Polym. Chem., 2015, 6, 2457–2465 RSC.
- W. Yuan, H. Zou and J. Shen, Carbohydr. Polym., 2016, 136, 216–223 CrossRef CAS PubMed.
- B. Liu, H. Zhou, S. Zhou, H. Zhang, A. Feng, C. Jian, J. Hu, W. Gao and J. Yuan, Macromolecules, 2014, 47, 2938–2946 CrossRef CAS.
- Q. Zhang and S. Zhu, Macromol. Rapid Commun., 2014, 35, 1692–1696 CrossRef CAS PubMed.
- Q. Zhang and S. Zhu, ACS Macro Lett., 2014, 3, 743–746 CrossRef CAS.
- L. Lei, Q. Zhang, S. Shi and S. Zhu, Langmuir, 2015, 31, 2196–2201 CrossRef CAS PubMed.
- H. Che, M. Huo, L. Peng, Q. Ye, J. Guo, K. Wang, Y. Wei and J. Yuan, Polym. Chem., 2015, 6, 2319–2326 RSC.
- D. Li, B. Ren, L. Zhang, J. Ezekiel, S. Ren and Y. Feng, Chem. Eng. Res. Des., 2015, 102, 234–243 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |