
Smart polymers in drug delivery: a biological perspective
A. Christy
Hunter
*a and
S. Moein
Moghimi
b
aManchester Pharmacy School, University of Manchester, Stopford Building, Oxford Road, Manchester, M13 9PT, UK. E-mail: Christy.hunter@manchester.ac.uk
bSchool of Medicine, Pharmacy and Health, Durham University, Queen's Campus, Stockton-on-Tees, TS17 6BH, UK
First published on 17th May 2016
Abstract
Key to the widespread application of smart polymers in drug delivery is understanding the mechanistic interplay, as well as consequence, of the presence of these macromolecules within living systems. This review looks at these interactions in terms of host response to macromolecular structure and subsequent clinical implications. In order to highlight this, three distinct routes of drug delivery are discussed, enabling a journey from the outside of the body in to the cell. This is used to contrast the need for different scientific approaches replete to successful drug delivery in these physiologically diverse areas. The discussion initiates with the application of smart polymers to the oral route of drug delivery, followed by macromolecular fate within the systemic circulation and finally intracellular delivery. The advantages, in terms of biological performance, as well as the challenges of using smart polymers within this multifaceted arena are delineated.
 A. Christy Hunter | Dr Christy Hunter is a Senior Lecturer in Pharmaceutical Nanoscience at the University of Manchester and is a Fellow of both The Royal Pharmaceutical Society and the Royal Society of Chemistry. He received his first degree in pharmacy from the University of Brighton and following pre-registration training in hospital qualified as a pharmacist in 1993. He undertook post graduate studies in bioorganic chemistry (1993–1996) at the University of Sussex. He was appointed to faculty in the School of Pharmacy at the University of Brighton where he undertook a range of roles within the division of pharmaceutics over the following 14 years. He joined the University of Manchester in July 2012. His primary research focus is in understanding the behaviours and mechanistic basis of synthetic polymer interaction within biological milieu particularly in reference to experimental and currently used nanomedicines. |
 S. Moein Moghimi | Moein Moghimi is Professor and Chair in Pharmaceutics at Durham University, UK. He is also a full affiliate member/professor at the Division of Translational Imaging, Houston Methodist Research Institute (Houston, Texas), and an Adjoint Professor at the Department of Pharmaceutical Sciences, University of Colorado Medical Center. His research interests lies in nanomedicine design and engineering as well as safety aspects of nanopharmaceuticals. |
1. Introduction: outside-in
Smart polymers have huge potential for use in medicine, this technology has been applied to a diverse number of areas such as insulin delivery,1 anti-cancer drug delivery2 and gene delivery.3,4 These macromolecules have also been used in a range of delivery systems (oral and topical), based on hydrogels,5 as well as novel drug delivery nanostructures (e.g. nanofibers) or as coatings for nanoparticles6–8 for parenteral use. Research emphasis on smart polymers for application in medicine has had significant focus on the elegant chemistry behind the design of new and improved macromolecules with interesting physicochemical characteristics. However, an in depth understanding of the interplay of these materials at the interface of biology, chemistry and medicine are less well delineated. For example, synthetic polymers have a range of actions within biological systems that may not initially be considered, or be apparent in the initial design of the smart systems. Examples of biological activity observed with a range of polymers include antitumor, antibiotic, antiviral, antithrombotic, pro-apoptotic, immune modulatory and inhibition of efflux pumps.9 Polymers used within the pharmaceutical industry have tended to be presumed inert, yet increasing evidence9–12 does not support this view. It is not possible at present to accurately predict precisely what type of biological activity a specific polymer will possess, as there are so few structure activity studies available. This in part is hampered by the need for a multi-disciplinary approach, but is further restricted by a lack of straightforward standardized methodologies available to the scientific community that are readily available and do not require a specialist skills set to use.1.1 Host defense and synthetic polymers
A key problem in nanomedicine using polymers either alone or as surface coatings, is the activation of our host defense system resulting in recognition and removal of the macromolecule either alone or as a construct.10,12,13 Unfortunately these types of interactions may also trigger anaphylactic type responses which have significant clinical impact.12 As smart polymer systems change their 3 dimensional structure in response to change in environment (such as temperature) they present new surfaces to the immune system. This results in distinctly different macromolecular architectures being revealed to the immune system by the same molecule, significantly increasing the difficulty of making these smart materials stealthy in the biological environment. At present there is also a relative paucity of knowledge with respect to the types of interaction of smart polymers within biological systems in terms of both tolerance and toxicity.14 Understanding of the mechanistic basis of these interactions is the key to the future of using these macromolecular/supramolecular materials in medicine, as it would guide researchers on to efficient knowledge reinforced design paths as opposed to a ‘hit and hope’ approach.9This review will look at three distinct drug delivery routes and enable a physiological journey from the outside of the body to the inside of a cell, starting with the role of smart polymers in oral drug delivery via the gastrointestinal tract. This will be followed by a discussion of macromolecular fate in the systemic circulation where immune focus for foreign entity recognition and removal is the key challenge in parenteral medicine. Finally, current knowledge of the of the interaction of smart polymers used in intracellular delivery will be determined, especially in terms of interaction with cellular organelles and the significant associated challenges. These three areas of drug delivery will highlight the key advantages as well as challenges in this arena. Smart polymers have been applied to other routes such as topical drug delivery and these have been covered elsewhere and are beyond the scope of this discussion.15–17
2. Smart polymers in oral drug delivery
2.1 The gastrointestinal tract
Oral administration of drugs takes advantage of integrative physiological processes in the gut including passive diffusion, active transport as well as endocytosis and exocytosis.18 The gastrointestinal tract (GIT) has a large mucosal interface (300–400 m2), which is also designed for physiological and immunological protection of this external environment.19 The detailed scrutiny of foreign entities is achieved by a complex interaction between epithelial cells and a variety of immunocompetent cells and may be modulated by gut microbiota. Indeed, numerous bacteria are present in the intestinal mucosa (up to 1014) and represent a diverse number of species (500+).20The gastrointestinal system has high levels of tolerance to foreign entities, yet it has been revealed that during infection tolerance to commensal organisms is lost.21 The oral route is the most widely used and most accepted route of drug administration in the adult population.22,23 The most popular dosage form is the tablet which is designed to carry an accurate drug dose and release it at an appropriate site within the intestine. The primary rationale for application of polymer technologies to oral drug delivery is the inherent flexibility of the carrier's physicochemical characteristics to control bioavailability and hence the pharmacokinetics of the incorporated drug molecules.24 This is can be readily achieved by using a protective smart polymeric coating over a tablet core (which may also be polymer based) containing the active drug.
The gastric acid resident in the stomach is composed primarily of hydrochloric acid that is produced from oxyntic cells (pH 0.8) in the gastric glands, and generates a pH range of 1.5 to 3.5 in the lumen of the stomach.25 This degradative environment is further enhanced by the release of (inactive) pepsinogen and rennin from gastric chief cells. The hydrochloric acid converts the pepsinogen in to the enzyme pepsin breaks down (proteolysis) bonds linking amino acids. The highly acidic environment is also an important physiological barrier, in that it kills many potential pathogenic organisms. This drives carrier design to protect the drug payload through the hostile environment present in the stomach and release the drug in a site-specific fashion in the upper intestine. To achieve this, copolymers of pH sensitive methacrylic monomers (e.g., methacrylic acid) and hydrophobic methacrylate monomers (e.g., methyl methacrylate) are used as tablet coatings. Another coating strategy is based on a cellulosic polymer backbone where some of the secondary alcohols are esterified with phthalic anhydride.26 The flexibility of these water soluble coatings facilitates their use in controlled release of the drug, an example of such a combination is ethylcellulose/poly(vinyl-alcohol)-poly(ethylene-glycol) graft co-polymer.27 Equally, varying the thickness of this or other polymer coatings over the tablet core will control rate of drug release. On entrance in to the basic environment of the upper intestine, surface coatings on tablets become soluble releasing the drug.
Smart polymers have also been developed with a combination of pH and temperature responsiveness for use as oral matrix systems28 using N-isopropyl acrylamide and acrylic acid. The advantage of such a system is that a small change in pH results in sharp volume changes at a constant temperature. Another strategy is to incorporate biodegradable polymers as the tablet matrix to control drug release rates. Examples of these polymers include polyanhydrides, polyesters and polylactic acid.29–31 Accordingly, the drug absorption, distribution and elimination following oral administration are not only determined by the drug molecule, but also by the physicochemical properties of the carrier. A variety of characteristics can be used to tune the polymers for optimal drug release, such as variation in carrier composition, molecular weight of the matrix, morphology, associated charge and hydrophile–lipophile balance.24,32–34 This flexibility has enabled polymer based carrier systems to be used in a wide range of clinically driven strategies that include systemic delivery (polymeric nanoparticles),35,36 high velocity drug dispersal in to the small intestine (self-micro and nanoemulsifying drug delivery systems)37,38 all of which can profoundly improve drug bioavailability.
2.2 Mucoadhesion
Smart polymer technology has also been applied for mucoadhesion in the GIT, for example, the application of chitosan and hydroxypropylmethyl cellulose (HPMC) for buccal delivery39 in the soft palate of the oral cavity to deliver antiemetics and for pain control. These dosage forms are advantageous in that they avoid initial metabolism by the liver associated with absorption from the upper intestine and that they can be used in patients who cannot swallow. Hydrogels [e.g. poly(nitrophenyl methacrylate-co-methacrylic acid)] have been designed for controlled drug release in the basic environment of the upper intestine40 with other smart polymers and that specifically target the GIT mucus layer and exploit mucoadhesion to retain the drug delivery device for prolonged periods, which enhances sustained release profiles.41–43 Site-specific drug delivery to the colon has been achieved with smart polymers by exploiting the properties of the colonic microflora. This contains 1010–1012 bacteria per gram gut contents in humans,44,45 that are absent from the rest of the GIT. This location also contains a range of enzymes (β-glycosidase, β-glucuronidase, nitroreductase, nitrate reductase) however; it is the azoreductases that have been utilized46 for colonic drug delivery. Here a pro-drug approach is taken where the polymer backbone has azo-linker-attached drug molecule.47 These linkers are stable in the acidic environment of the gastric acid yet can release drug in basic environment of the colon following the action of the bacterially derived azo reductases. This has facilitated the design of a range of smart polymer based systems for the treatment of colonic diseases such as Crohn's, ulcerative colitis, inflammatory bowel disease and colon cancer. Pro-drugs utilizing azo linkages include sulfasalazine, ipsalazine, basalazine and osalazine47 which have been used to reduce inflammation. Other examples include polyphosphazene-anticancer drug conjugates that have been applied to the delivery of methotrexate and gemcitabine48 for treatment of colon cancer.The gastrointestinal environment is the perfect physiological platform to apply a wide range of smart polymer technology, and there is plenty of opportunity with the myriad of new drugs being produced, which require sustained release profiles. There is also an important gap in the application to site specific smart polymer targeting of areas within the GIT for immunological benefit, such as Payer's patches.18 Importantly, this is a safe environment to use these polymer technologies as millions of tablets using smart polymer coating are used on a daily basis worldwide in a safe and effective fashion.
3. Complement-a potent biological barrier in parenteral medicine
Parenteral drug administration refers to any non-oral route but is generally related to direct injection in to the body by passing the skin or mucus membranes. Examples of some of these include intravenous (into a vein), subcutaneous (under the skin) intra-arteriole (in to an artery) and intrathecal (injection in to the spinal canal). There are significant advantages to using the intravenous route of administration in that 100% of the drug is available. This is in contrast to the oral route where drugs are absorbed across the GIT and undergo significant first-pass metabolism in the liver, resulting in reduction of drug concentration before entering the post-hepatic systemic circulation. Hence, the initial quantity of the same drug delivered orally will always be much greater, with onset of action taking longer than the equivalent effective dose if administered intravenously, which has near immediate onset of pharmacological effect. When using synthetic macromolecules either alone or as part of a construct within the systemic circulation, recognition of these materials by the immune system (and possible anaphylactic responses) becomes a primary barrier to their use in medicine. This is reflected by the relatively low number of polymer based dosage forms in parenteral medicine available for use in the clinic.3.1 The complement system
Evolution has equipped humans with a system for recognition of foreign entities called complement. This system is an ancient component of innate immunity and is not only present in man, but also in a range of invertebrate species including Drosophila, horseshoe crab and mosquitoes.49 It is made up of three distinct primary activation pathways (classical, lectin and alternative) and composed of more than 30 soluble membrane bound proteins. It is found primarily in the blood and other biological fluids such as lymph and tear, as well as in compartments such as the brain and the lungs and even solid tumors. Complement is arguably one of the most complex defense systems for drug delivery and material scientists to circumvent. It has a primary role in host defense ranging from non-clinically significant to overwhelming infections such as those leading to sepsis.50 It can also identify synthetic macromolecules as well as micro- and nano-particles.12,34,51,52 It is thought to have a role in a wide variety of diseases such as pathogenesis of psoriasis, adult respiratory distress syndrome, bullous pemphigoid, rheumatoid arthritis, ischemia-reperfusion injury, glomerulopathy, systemic lupus erythematosus and in trauma where presence of anaphylatoxins significantly increases risk of mortality rate.53–58 Although a fundamental part of innate host defense, over activation (by release of the anaphylatoxins C3a and C5a (Fig. 1) as well as the membrane attack complex) can lead to host damage (vide supra) which results, for example, in worsening of prognosis for patients, as observed with septicemia.50 Consideration of the balance between helpful and harmful response must be determined in the development of any smart synthetic system, as the equilibrium reaction response has the potential to be the key rate-limiting step to the design in vivo due to the potential of patient harm.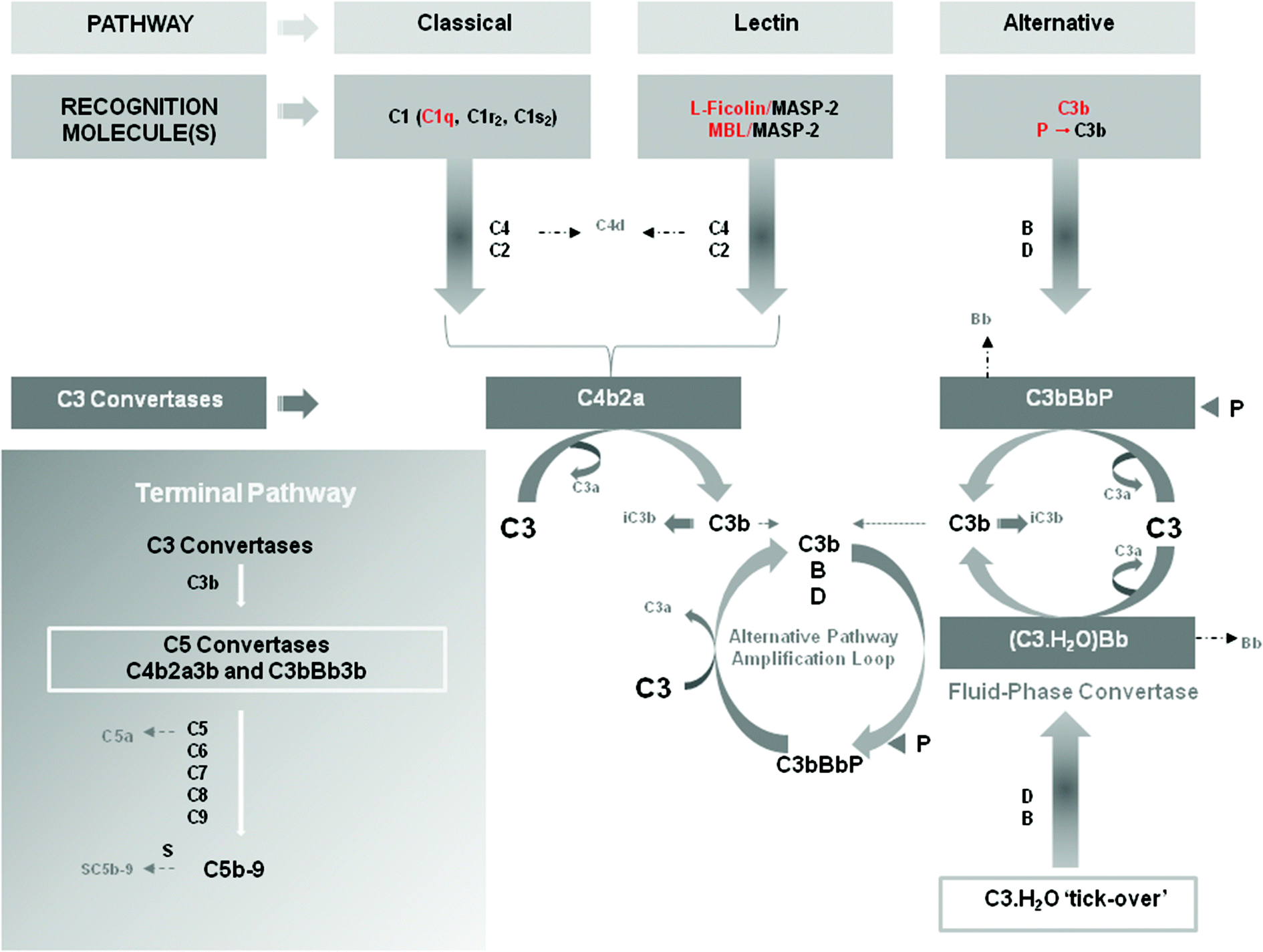 | ||
| Fig. 1 Complement recognition molecules and complement activation pathways. Key complement recognition molecules are shown in red. The cartoon shows sequential activation of complement components for each pathway as well as measurable pathway-dependent complement activation products (e.g., C4d, Bb, iC3b, C3a, C5a, SC5b-9). Properdin (P) has dual roles. It acts not only as a pattern recognition molecule capable of activating the alternative pathway but also as a stabilizer of the alternative pathway convertase C3bBb, preventing its rapid dissociation by complement regulatory proteins. The inset represents the terminal pathway of the complement system and sequential activation of the complement proteins leading to the assembly of the lytic complex C5b-9 (also known as the membrane attack complex). In the fluid phase, S-protein (vitronectin) binds to C5b-9 and prevents interaction with membranes. Natural complement inhibitors and complement regulatory proteins are not shown in the scheme. MBL = mannan-binding lectin; MASP-2 = mannose-binding protein associated serine protease-2. Reproduced with permission from ref. 61. | ||
3.2 Complement and molecular recognition
Complement is highly attuned to chemical structure and can recognize molecular motifs 6 atoms in length12 as well as highly defined synthetic polymers, such as poly(ethylene glycol) (PEG)59,60 in soluble form and PEG coated nanoparticles.53,60,61 In terms of bacterial or viral presence, the complement system recognizes these infective entities through pattern recognition. A similar mechanism is thought to underline complement activation through macromolecular structure either alone or as part of particle surface coverage.10,11,13 Nano-stealth technology can circumvent rapid recognition of the particles following surface coating with polymers such as PEG or by careful control of structural architecture (such as curvature) to facilitate prolonged circulation within the blood compartment. An example used in the clinic is Doxil®, the PEGylated liposomal formulation of doxorubicin. This is used in the treatment of HIV related Karposi’ sarcoma, ovarian cancer and advanced metastatic breast cancer. This formulation for parenteral administration, as well as others has been associated with activation of the complement system. In the case of regulatory approved nanopharmaceuticals up to 45% of patients can be reactive13 following parenteral administration. Further, a wide variety of symptoms can present and frequently include back pain, chest pain, chills, dyspnea, facial swelling, fever, flushing and skin rash. Between individuals presentation of symptoms can range from very minor to fatal. Recent studies in pigs indicate that the cardiopulmonary distress caused by Doxil® and other nanoparticles strongly correlate with complement activation.62 In general, complement responses occur upon first exposure to nanopharmaceuticals without prior sensitization, but paradoxically may decrease or even disappear on further treatment.3.3 The complement cascade
Initiation of the complement cascade (Fig. 1) starts with triggering of one (or more) of the three distinct primary pathways, namely classical, lectin and alternative. They facilitate a highly potent defense system based on recognition of non-self that is analogous to a military unit. The classical pathway is akin to the intelligence core and recognizes past threats through antibody mediation. The lectin pathway may be compared to a Special Forces unit, in that it can recognize specific targets such as bacterial cell walls and viral coats. The alternative pathway is the sentry on guard duty, always monitoring potential threats. This pathway activates in the presence of nucleophiles such as hydroxyl and amino groups. This can be highly problematic to smart polymers as they frequently contain these functional groups. Activation of one or more of the pathways results in a proteolytic cascade with each step generating bioactive fragments. Each of the fragments generated has a specific role, which fits into the tasks of recognition of foreign entities, flagging its presence to the immune system, attraction and activation of host defense cells, destruction of microbes and finally removal of debris to the liver for destruction.3.4 Recognition of polymeric nanoparticles
In the case of polymeric nanoparticles intravenously injected in to the systemic circulation their surfaces will undergo opsonization, this process is driven by cleavage of the C3 fragment which generates C3b and iC3b.63–66 This facilitates recognition of the particle and results in rapid clearance by neutrophils, monocytes and macrophages of the reticuloendothelial system bearing complement receptors. The physicochemical nature of polymer or particle will determine the opsonic process involved. Indeed, not all cases of surface opsonization result in clearance.66,67 This may be due to the presence of ‘dysopsonic’ molecules, surface topology and in the case of particles’ surface structures, which generate steric constraints. Thus macromolecular architecture has a significant effect on the fate of nanoparticles in vitro65,68,69 and is a fundamentally important consideration in smart polymer use.For example, intravenous injection of uncoated and poloxamine coated polystyrene nanoparticles (50–200 nm in diameter) with polyethylene oxide (PEO) chains in a “mushroom” as opposed to “brush” configuration (lying over the surface of the particle as opposed to projecting outwards respectively) results in rapid sequestration by Kupffer cells.68 In comparison polystyrene particles of similar size ranges transitional between “mushroom” and “brush” configuration, have increased resistance to clearance by Kupffer cells. This results in these particles remaining in the blood for extended periods of time.68,70,71 For larger particles (200–300 nm) with PEO chains on the surface in either mushroom-brush or brush conformations results in clearance from the blood pool by the spleen72,73 (Fig. 2). This splenic filtration of these particles (Fig. 3) from blood is due to the physiological size of the interendothelial slits in the (rat) spleen. It is important to note that any particle following contact with blood will increase in size (also as a result of nanoparticle aggregation) due to attachment of opsonic molecules as well as non-specific plasma protein deposition. It is therefore imperative that size determination is performed on the construct of interest following contact with the biological milieu. This will ensure that the designed particle is not so large that it is removed from the systemic circulation by splenic filtration or that it can block capillaries in the lung which, for example, may result in pulmonary embolism. Red blood cells despite their relatively large size (≈7 μm) can deform their structure and pass through the interendothelial slits however, rigid synthetic particles of equal or greater size than these cell slits (200 nm) are prone to splenic filtration. This can however be used as a strategy for spenotropic targeting of drugs.73
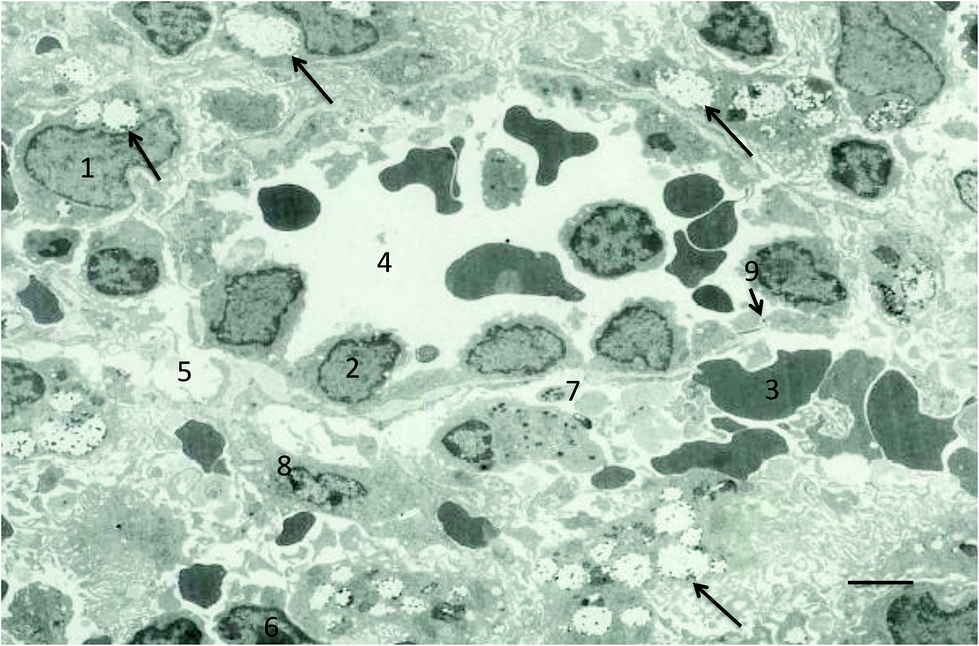 | ||
| Fig. 2 Electron micrograph of the red-pulp region of the rat spleen. The micrograph shows the presence of a typical venous sinus and accumulation of nanospheres (220 nm in diameter) in macrophages of the red-pulp. Significant accumulation of nanospheres in the red pulp macrophages (1) (arrows); endothelial cell lining sinuses (2), red blood cell (3), venous sinus (4), interstitial space (5), lymphocyte (6), platelet (7), reticular cell (8) and position (arrow) of interendothelial cell slits (9). Bar = 5 μm. Reproduced with permission from ref. 93. | ||
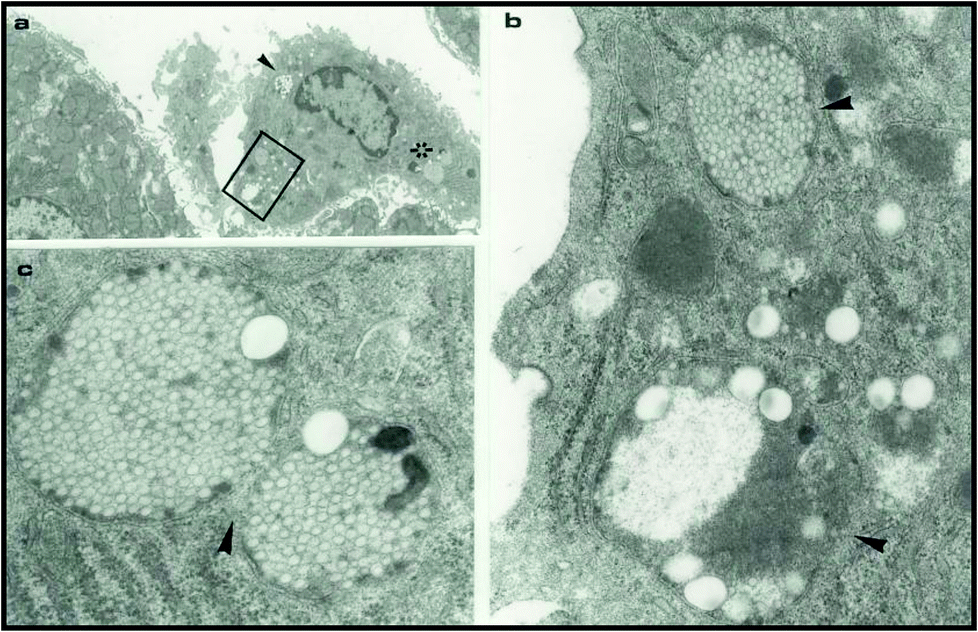 | ||
| Fig. 3 Electron micrographs of a rat liver 24 h after intravenous injection of surface engineered 220 nm and 60 nm polystyrene nanoparticles. Panel (a) shows a sinusoidal Kupffer cell with ingested nanoparticles. Panels (b) and (c) are magnified views of lysosomal compartments (arrows) of the Kupffer cell in (a) containing the ingested nanoparticles (the box and the star marked regions). Reproduced with permission from ref. 93. | ||
Poloxamer 407 is a smart non-ionic block co-polymer composed of polyethylene oxide (PEO) and polypropylene oxide that has reversible thermogelation properties. This polymer has been used in a wide range of sustained release applications as well as being proposed as a vascular occludant for use in surgery.74–82 Recently it has been demonstrated that poloxamer 407 can activate complement through lectin and alternative pathways.52 This activation is further exacerbated following gel–sol transition. Remarkably high-density lipoprotein (HDL) and low-density lipoprotein (LDL) demonstrated a protective role against complement activation. A potential mechanism for this is the formation of new structures following interaction of the poloxamer with the hydrophobic areas of these lipoproteins such as apoAI and apoB-100.
Working hand-in-hand with complement is the reticuloendothelial system, also known as the mononuclear phagocytic system. These cells form an important system for engulfment of foreign entities as well as their presentation to the immune system. This will trigger further host response and generation of cytokines to the specific threat. Any entity (e.g. cellular debris, foreign substances, microorganisms, cancer cells) that does not have healthy body cell specific proteins on their surface will be on their radar. Monocytes, neutrophils, eosinophils are white blood cells (leukocytes) and are also part of the innate immune system. Monocytes make up 2–10% of all leukocytes in humans and have two primary functions. These are based around their ability to differentiate in to macrophages (and dendritic cells) to elicit immune response. Functionally they replenish macrophages in normal physiological state and in response to inflammatory signals (such as those from complement fragments) can migrate to (8–12 hours) sites of infection to trigger immune response. Macrophages exist in heterogeneous populations that are present in a wide variety of physiologically important areas. Examples include the Kupffer cells of the liver, macrophages of the lungs, splenic marginal zone and red pulp, neural tissue, bone and connective tissues. All of which have the potential to sequester smart polymers alone or those used on surfaces of micro- and nano-constructs.
3.5 Complement activation
Over activation of the complement system can result in damage to surrounding host tissues.11,64,83 Mechanistically this is due to the generation of the terminal part of the complement pathway where the membrane attack complex is generated (C5b-9). This forms holes in biomembranes; in the case of bacteria this results in destruction of the cell by osmosis. But equally, in over activation local healthy cells can be damaged examples of which have been seen following myocardial infarction, adverse pregnancy outcomes and also resulted in alteration of blood brain barrier integrity as well as damage to organs such as lung, liver, kidney.84–88 Induction of this level of complement activation by smart polymers deserves serious consideration and highlights the need for exhaustive structure activity/immune response studies.A range of strategies can be applied in the clinic to control the deleterious effects of anaphylaxis. This may include reduction of the rate of infusion in to the venous system as well as administration of drugs (antihistamines, bronchodilators and corticosteroids) that will prevent complement activation-related allergic-like reactions in the majority of subjects.13,53 However, these are non-specific approaches and potentially fatal reactions still occur in some individuals. In order to target specific points in the complement pathways, inhibitors have been developed. The first antibody approved (2007) for human, use by the food and drug administration was eculizumab which is active against C5.89 The license for the use of this product was granted exclusively for the treatment of paroxysmal nocturnal hemoglobinuria and as yet has not been applied to combination with smart macromolecules or nanopharmaceuticals.90 Ideally, molecules, which specifically inhibit C3, thus inhibiting the generation of the anaphylatoxins C3a and C5a as well as the membrane attack complex, would be the drug molecule of choice.51,91 Development of these types of pharmacophores would have wide reaching implications for the use of synthetic materials within the vascular compartment by overcoming the primary obstacle. However overuse of such an inhibitor could shift the immunological equilibrium to make individuals more prone to infections. As many individuals reduce in their pseudoallergic response to intravenously administered nanomedicines, this approach could have significant merit if applied on first dose.
To date, clinical success has been dogged by these complement inhibiting molecules having low potency, specificity as well as short half-lives. Covalent conjugation of naturally occurring complement inhibitors and regulators to nanoparticles has been problematic.10,92,93 This has included challenging synthetic procedures for the coupling chemistry, complexities associated with the generated surface design and topology as well as poor stability and high cost.94
4. Intracellular compartment
pH sensitive smart polymers contain weakly acidic or basic functional groups that will either accept or loose protons depending on the local pH. This may be taken advantage of where disease state or intracellular differences afford variation in pH for targeted release of pharmacophores.95,96 This strategy has been extensively applied to gene therapy, in part to overcome the inherent problems associated with viral gene vectors. Although viral vectors have had some remarkable successes, for example in the treatment of children with severe combined immunodeficiency (SCID) X1 disease,97,98 advancement of the field has been problematic due to induction of toxicity and fatality in some cases.99 In order to surmount the problems associated with using viral vectors, a wide range of smart synthetic polymer systems have been developed for generation of cationic polymer/DNA complexes for gene delivery. These include the ‘gold standard’ poly(ethylenimine) as well as poly(L-lysine), N-Ac-poly(L-histidine)-graft-poly(L-lysine), poly(2-alkylacrylic acid), poly(phosphazines), poly(amidomines), trimethylchitosans/methacrylic acid and poly(L-histidine).100–108 The most mechanistically studied polycations are poly(ethylenimine) (PEI) and poly(L-lysine) (PLL). In terms of efficiency for gene transfection branched PEI (25 kDa) is the most efficient. Condensed PEI and DNA form torroidal and globular nanostructures that can gain entrance in to a cell via endocytic and phagocytic routes.109 It is generally thought that the nanovector system, following internalization in to the cell, ends up in a lysosome. Until recently110 the mechanistic basis resulting in release of the gene in to the intracellular space was based on the proton sponge hypothesis.111 This mechanism was driven by unprotonated amines of PEI buffering the internal environment of the lysosome by taking up protons as they are pumped in to the lysosome. This resulted in more protons being pumped in causing an increase in intracellular chloride ions and water. Consequently, a combination of osmotic swelling and increases in the size of the protonated PEI that was believed to result in the rupture of the endolysosomal membrane and release of the contents in to the cytoplasm. However, the latter mechanistic view has been refuted by new evidence that demonstrates no change in internal lysosomal pH following uptake of the polyplex.110 Evidence from electron microscopy indicates that the polyplexes escape from the lysosomes through pores in the membrane.112 Whether this is the primary mechanism remains to be revealed.The fundamental drawback in using cationic polymers for gene therapy has been the limited expression time of the genes of interest. The gold standard synthetic polymer for gene therapy has been PEI, which has achieved the most robust expression profiles. A mechanistic study113 in to the intracellular interactions of branched, and linear PEI either alone or complexed with DNA have revealed the precise mechanistic basis of low gene expression, namely the induction of apoptosis, necrosis and autophagy modes of cell death.114 For instance, both branched (25 kDa) and linear (750 kDa) PEI were tested in three human cell lines (Jurkat T cells, umbilical vein endothelial cells and THLE3 hepatocyte-like cells) and were found to induce a two-phase cytotoxicity. Phase 1 was relatively rapid with onset at 30 minutes after PEI incubation with early necrotic-like changes observed due to disrupted membrane integrity. This was revealed by detection of significant lactate dehydrogenase release as well as phosphatidylserine (PS) translocation from the inner plasma membrane to the outer cell surface. Rate of PS externalization was PEI dose dependent. Rapid externalization occurred at 20 μg mL−1 in contrast to lower PEI concentrations (10 μg mL−1) with significant translation observed after 24 h. Phase 2 cytotoxicity was observed initially at 24 h post polycation incubation and represented mitochondrially mediated induction of apoptosis. Interaction of the PEI with the outer membrane of the mitochondrion resulted in release in pro-apoptotic cytochrome c that ultimately activated release of the cell ‘executioner’ caspase-3 resulting in cell death. Use of the caspase-3 inhibitor Ac-DEVD-CHO demonstrated inhibition of apoptosis in these cell lines, confirming this mechanistic route to cell death. The absolute delineation of the precise causative factors with the DNA–PEI complex was not possible. This was due to the mechanism of apoptosis that results in generation of activated caspases, which also induce PS “scrambling” followed by loss of membrane integrity.115,116 Further, DNA–PEI complex preparations may contain up to 85% free PEI molecules and these may contribute to PEI-mediated cytotoxicity.117 Indeed, recent studies have shed more light on the mechanisms of PEI-architecture- and size-dependent cytotoxicity, which involve perturbations of integrated cellular metabolomics through simultaneous destabilization of plasma membrane and mitochondrial membranes with consequences on mitochondrial oxidative phosphorylation, glycolytic flux and redox homeostasis.118,119
PEI is not biodegradable but a large number of biodegradable derivatives using two low molecular weight PEIs and 24 bi and oligo-acrylate macromolecules120 have demonstrated reduced toxicity and high transfection ability.121–123 There are new developments in design and engineering of PEI derivatives with better and improved functionalities and safety profiles.124,125 These types of study demonstrated that improved smart polymer design is potentially advantageous, but must be combined with thorough testing of cellular response.
A similar mechanistic investigation to that of PEI126 has been undertaken to look at the interaction of PLLs, which have been used for DNA compaction and transfection methodologies with a range of results.127 PLLs demonstrated a similar pattern of programmed cell death to that observed with PEI with early plasma membrane damage followed by late phase (24 h) apoptosis in human cell lines.126 Interestingly isolated mitochondria experiments revealed that high molecular weight PLL was able to release cytochrome c and induce atrial mitochondrial depolarization, yet the low molecular weight did not. This underlines the complexity of the mechanistic basis of these polymer–cell interactions which, can vary so widely depending on molecular weight of the polymer alone. Again, these differences may be related to the impact of PLL size on cellular bioenergetics processes.127 Other nanovehicle smart polymers used in drug delivery such as poly(amidoamine) dendrimers have also triggered mitochondrially mediated apoptosis.128,129 Collectively, these observations suggest the need for future development of smart synthetic gene carriers, which could undergo charge reversal on cellular internalization (from cationic to neutral or anionic), thus avoiding intracellular interactions that may promote programmed cell death (Fig. 4).
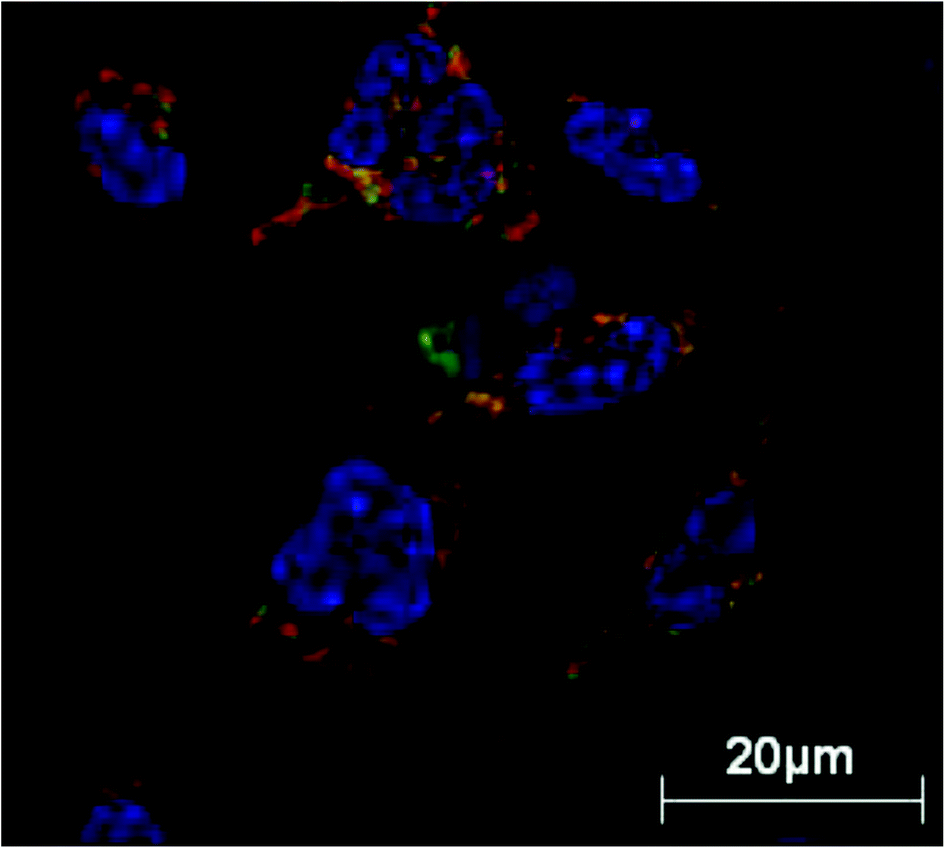 | ||
| Fig. 4 Live-cell microscopy of rat peritoneal macrophages treated with newly engineered PEGylated polymer micelles for 1 hour at 37 °C. Polymer micelles are dynamic structures and are in equilibrium with monomers with the concentration of monomers being equal to the critical micelle concentration. The results demonstrate translocation of internalized monomer chains (covalently labelled with rhodamine B) in to the mitochondrial network with mitochondria stained with Mitotracker Green with H33342 nuclear stain, confirming mitochondrial translocation of micelles. Reproduced with permission from ref. 128. | ||
5. Conclusion
The factors that govern macromolecule recognition as well as biological and cellular interplay are significant for the development of this field and highly complex in nature. This complexity is further enhanced by the wide range of disciplines required to collaborate in this area, as well as the requirement for application of cutting edge technologies. It is clear that pH sensitive smart polymers such as cellulosic polymer backbones esterified with phthalic anhydride are ideally suited to use in the biologically tolerant environment of the gastrointestinal tract. However, parenteral administration of smart polymers will only reach their full potential through the understanding of macromolecular behavior (e.g. biodegradability, polydispersity, shape)9 in biological systems. Furthermore, precise structural control of macromolecular architecture will enhance proposed biological action and avoid side interactions (e.g. complement activation).References
- Z. Li, Y. Zhang, D. Lu and Z. Liu, J. Appl. Polym. Sci., 2015, 132, 42596 Search PubMed.
- X. Guo, C. L. Shi, G. Yang, J. Wang, Z. H. Cai and S. B. Zhou, Chem. Mater., 2014, 26, 4405–4418 CrossRef CAS.
- H. Chen, Y. Zhao, S. Cui, D. Zhi, S. Zhang and P. Xiaojun, J. Appl. Polym. Sci., 2015, 132, 42469 Search PubMed.
- J. M. Hu, G. Y. Zhang, Z. S. Ge and S. Y. Liu, Prog. Polym. Sci., 2014, 39, 1096–1143 CrossRef CAS.
- A. Vashist, A. Vashist, Y. K. Gupta and S. Ahmad, J. Mater. Chem. B, 2014, 2, 147–163 RSC.
- S. Demirci, A. Celebioglu, Z. Aytac and T. Uyar, Polym. Chem., 2014, 5, 2050–2056 RSC.
- Q. G. He, J. Liu, C. Y. Huang and Z. H. Wu, Sci. Adv. Mater., 2014, 6, 387–398 CrossRef CAS.
- W. de Groot, S. Demarche, M. G. Santonicola, L. Tiefenauer and G. J. Vancso, Nanoscale, 2014, 6, 2228–2237 RSC.
- A. C. Hunter and S. M. Moghimi, Drug Discovery Today, 2002, 7, 998–1001 CrossRef CAS PubMed.
- S. M. Moghimi, A. J. Anderson, D. Ahmadvand, P. P. Wibroe, T. L. Andresen and A. C. Hunter, Adv. Drug Delivery Rev., 2011, 63, 1000–1007 CrossRef CAS PubMed.
- S. M. Moghimi, A. J. Andresen, S. H. Hashemi, B. Lettiero, D. Ahmadvand, A. C. Hunter, T. L. Andresen, I. Hamad and J. Szebeni, J. Controlled Release, 2010, 146, 175–181 CrossRef CAS PubMed.
- S. M. Moghimi, A. C. Hunter, C. M. Dadswell, S. Savay, C. R. Alving and J. Szebeni, Biochim. Biophys. Acta, Mol. Basis Dis., 2004, 1689, 103–113 CrossRef CAS PubMed.
- J. Szebeni, F. Muggia, A. Gabizon and Y. Barenholz, Adv. Drug Delivery Rev., 2011, 63, 1020–1030 CrossRef CAS PubMed.
- E. Cabane, X. Zhang, K. Langowska, C. G. Palivan and W. Meier, Biointerphases, 2012, 7, 9, DOI:10.1007/s13758-011-0009-3.
- A. Vashist, A. Vashist, Y. K. Gupta and S. Ahmad, J. Mater. Chem. B, 2014, 2, 147–166 RSC.
- F. Rafie, Y. Javadzadeh, A. R. Javadzadeh, L. A. Ghavidel, B. Jafari, M. Moogooee and S. Davaran, Curr. Eye Res., 2010, 35, 1081–1089 CrossRef CAS PubMed.
- S. Venkatesh, S. P. Sizemore and M. E. Byrne, Biomaterials, 2007, 28, 717–724 CrossRef CAS PubMed.
- A. C. Hunter, J. Elsom, P. P. Wibroe and S. M. Moghimi, Nanomedicine, 2012, 8, S5–S20 CAS.
- C. Manichanh, N. Borruel, F. Casellas and F. Guarner, Nat. Rev. Gasteroenterol. Hepatol., 2012, 9, 599–608 CrossRef CAS PubMed.
- A. J. Macpherson and N. J. Harris, Nat. Rev. Immunol., 2004, 4, 478–485 CrossRef CAS PubMed.
- T. W. Hand, L. M. D. Santos, N. Bouladoux, M. J. Malloy, A. J. Pagán, M. Pepper, C. L. Maynard, C. O. Elson and Y. Belkaid, Science, 2012, 337, 553–556 CrossRef PubMed.
- E. Roger, F. Lagarce, E. Garcion and J. P. Benoit, Nanomedicine, 2010, 5, 287–306 CrossRef CAS PubMed.
- G. Ponchel and J. M. Irache, Adv. Drug Delivery Rev., 1998, 34, 191–219 CrossRef CAS PubMed.
- S. D. Li and L. Huang, Mol. Pharmacol., 2008, 5, 496–504 CrossRef CAS PubMed.
- A. C. Guyton and J. E. Hall, Textbook of medical physiology, Elsevier Saunders, Philadelphia, 11th edn, 2006, p. 797 Search PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2013, 65, 10–16 CrossRef CAS PubMed.
- C. Gendre, M. Gentry, J. C. da Silva, A. Tafayli, M. Boiret, O. Lecoq, M. Baron, P. Chaminade and J. M. Péan, Eur. J. Pharm. Biopharm., 2012, 81, 657–665 CrossRef CAS PubMed.
- L. C. Dong and A. S. Hoffman, J. Controlled Release, 1991, 15, 141–152 CrossRef CAS.
- Z. X. Cui, Y. Y. Peng, K. Li, J. Peng, H. B. Zhao, L. S. Turng and C. Y. Shen, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2013, 28, 793–797 CrossRef CAS.
- J. Heller and K. J. Himmelstein, Methods Enzymol., 1985, 112, 422–436 CAS.
- C. S. Proikakis, P. A. Tarantili and A. G. Andreopoulos, Eur. Polym. J., 2006, 42, 3269–3276 CrossRef CAS.
- K. Sachs-Barrable, S. D. Lee, E. K. Wasan, S. J. Thornton and K. M. Wasan, Adv. Drug Delivery Rev., 2008, 60, 692–701 CrossRef CAS PubMed.
- H. Laroui, D. S. Wilson, G. Dalmasso, K. Salaita, N. Murthy, S. V. Sitaraman and D. Merlin, Am. J. Physiol. Gastrointest. Liver Physiol., 2010, 300, G371–G383 CrossRef PubMed.
- S. M. Moghimi, A. C. Hunter and T. L. Andresen, Annu. Rev. Pharmacol. Toxicol., 2012, 52, 481–503 CrossRef CAS PubMed.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, FASEB J., 2005, 19, 311–330 CrossRef CAS PubMed.
- M. P. Desai, V. Labhasetwar, G. L. Amidon and R. J. Levy, Pharm. Res., 1996, 13, 1838–1845 CrossRef CAS.
- V. Venkateswarlu and K. Manjunath, J. Controlled Release, 2004, 95, 627–638 CrossRef CAS PubMed.
- A. Hanafy, H. Spahn-Langguth, G. Vergnault, P. Grenier, M. T. Grozdanis, T. Lenhardt and P. Langguth, Adv. Drug Delivery Rev., 2007, 59, 419–426 CrossRef CAS PubMed.
- N. V. S. Madhav, R. Semwal, D. K. Semwal and R. B. Semwal, Expert Opin. Drug Delivery, 2015, 9, 629–647 CrossRef PubMed.
- F. Li, Y. C. Zhu, B. You, D. H. Zhao, Q. C. Ruan, Y. Zeng and C. X. Ding, Adv. Funct. Mater., 2010, 20, 669–676 CrossRef CAS.
- N. Shrestha, S. Mohammad-Ali, A. Francisca, M. Ermel, J. Raula, E. I. Kauppinen, J. Salonen, J. Sarmento, B. Sarmento, J. Santos and A. Helder, Biomaterials, 2015, 68, 9–20 CrossRef CAS PubMed.
- A. Ahuja, R. K. Khar and J. Ali, Drug Dev. Ind. Pharm., 1997, 23, 489–515 CrossRef CAS.
- H. Takeuchi, J. Thongborisute, Y. Matsui, H. Sugihara, H. Yamamoto and Y. Kawashima, Adv. Drug Delivery Rev., 2005, 57, 1583–1594 CrossRef CAS PubMed.
- A. Rubinstein, Biopharm. Drug Depos., 1990, 11, 465–475 CrossRef CAS.
- H. W. Smith, J. Pathol. Bacteriol., 1965, 89, 95–122 CrossRef CAS PubMed.
- M. Chourasia and S. K. Jain, J. Pharm. Pharm. Sci., 2003, 6, 33–66 CAS.
- A. Jain, Y. Gupta and S. K. Jain, Crit. Rev. Ther. Drug Carrier Syst., 2006, 23, 349–399 CrossRef CAS PubMed.
- R. Sharma, R. K. Rawal, M. Manav, A. K. Sharma and T. R. Bhardwaj, Bioorg. Med. Chem., 2014, 22, 1104–1114 CrossRef CAS PubMed.
- P. Kopacek, O. Hajdusek and V. Buresova, Adv. Exp. Med. Biol., 2012, 710, 83–93 CrossRef CAS PubMed.
- M. M. Markiewski, R. A. DeAngelis and J. D. Lambris, J. Cell. Mol. Med., 2008, 12, 2245–2254 CrossRef CAS PubMed.
- D. Ricklin and J. P. Lambris, Nat. Biotechnol., 2007, 25, 1265–1275 CrossRef CAS PubMed.
- I. Hamad, A. C. Hunter and S. M. Moghimi, J. Controlled Release, 2013, 170, 167–174 CrossRef CAS PubMed.
- J. Szebeni, P. Bedöcs, R. Urbanics, R. Bünger, L. Rosivall, M. Tóth and Y. Barenholz, J. Controlled Release, 2012, 160, 382–387 CrossRef CAS PubMed.
- T. V. Arumugam, I. A. Shiels, T. M. Woodruff, D. N. Granger and S. M. Taylor, Shock, 2004, 21, 401–409 CrossRef CAS PubMed.
- J. P. M. Melis, K. Strumane, S. R. Ruuls, F. J. Beurskens, J. Schuurman and P. W. H. I. Parren, Mol. Immunol., 2015, 67, 117–130 CrossRef CAS PubMed.
- P. F. Zipfel, C. Skerka, Q. Chen, T. Wiech, T. Goodship, S. Johnson, V. Fremeaux-Bacchi, C. Nester, S. R. de Córdoba, M. Noris, M. Pickering and R. Smith, Mol. Immunol., 2015, 67, 21–30 CrossRef CAS PubMed.
- S. D. Mahajan, N. U. Parikh, T. M. Woodruff, J. N. Jarvis, M. Lopez, T. Hennon, P. Cunningham, R. J. Quigg, S. A. Schwartz and J. J. Alexander, Immunology, 2015, 146, 130–143 CrossRef CAS PubMed.
- M. T. Ganter, K. Brohi, M. J. Cohen, L. A. Shaffer, M. C. Walsh, G. L. Stahl and G. L. Pittet, Shock, 2007, 28, 29–34 CrossRef CAS PubMed.
- I. Hamad, A. C. Hunter, J. Szebeni and S. M. Moghimi, Mol. Immunol., 2008, 46, 225–232 CrossRef CAS PubMed.
- I. Hamad, A. C. Hunter, K. J. Rutt, Z. Liu, H. Dai and S. M. Moghimi, Mol. Immunol., 2008, 45, 3797–3803 CrossRef CAS PubMed.
- A. J. Andersen, J. T. Robinson, H. Dai, A. C. Hunter, T. L. Andresen and S. M. Moghimi, ACS Nano, 2013, 7, 1108–1119 CrossRef CAS PubMed.
- J. Szebeni, L. Baranyi, S. Savay, M. Bodo, J. Milosevits, C. R. Alving and R. Bunger, Am. J. Physiol. Heart Circ. Physiol., 2006, 290, H1050–H1058 CrossRef CAS PubMed.
- M. J. Walport, N. Engl. J. Med., 2001, 344, 1140–1144 CrossRef CAS PubMed.
- D. Ricklin, G. Hajishengallis, K. Yang and J. D. Lambris, Nat. Immunol., 2010, 11, 785–797 CrossRef CAS PubMed.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol. Rev., 2001, 53, 283–318 CAS.
- S. M. Moghimi, I. Hamad, T. L. Andresen, K. Jørgensen and J. Szebeni, FASEB J., 2006, 20, 2591–2593 CrossRef CAS PubMed.
- N. Dos Santos, C. Allen, A. M. Doppen, K. A. K. Cox, R. C. Galleher, G. Karlsson, K. Edwards, G. Kenner, L. Samuells, M. S. Webb and M. B. Bally, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 1367–1377 CrossRef CAS PubMed.
- S. Stolnik, B. Daudali, A. Arien, J. Whetstone, C. R. Heald, M. C. Garnett, S. S. Davis and L. Illum, Biochim. Biophys. Acta, Biomembr., 2001, 1514, 261–279 CrossRef CAS.
- I. Hamad, O. Al-Hanbali, A. C. Hunter, K. J. Rutt, T. L. Andresen and S. M. Moghimi, ACS Nano, 2010, 4, 6629–6638 CrossRef CAS PubMed.
- O. Al-Hanbali, K. J. Rutt, D. K. Sarker, A. C. Hunter and S. M. Moghimi, J. Nanosci. Nanotechnol., 2006, 6, 3126–3133 CrossRef CAS PubMed.
- S. M. Moghimi, Biochim. Biophys. Acta, Gen. Subj., 1997, 1336, 1–6 CrossRef CAS.
- S. M. Moghimi, H. Hedeman, I. S. Muir, L. Illum and S. S. Davis, Biochim. Biophys. Acta, 1993, 1157, 233–240 CrossRef CAS.
- S. M. Moghimi, Adv. Drug Delivery Rev., 1995, 17, 103–115 CrossRef CAS.
- J. Juhasz, V. Lenaerts, P. Raymond and H. Ong, Biomaterials, 1989, 10, 265–268 CrossRef CAS PubMed.
- M. L. Vetries, G. Couarraze, S. Geiger, F. Agnely, L. Massias, B. Kunzli and F. Faurisson, Int. J. Pharm., 1999, 192, 183–193 CrossRef.
- T. Liu and B. Chu, J. Appl. Crystallogr., 2000, 33, 727–730 CrossRef CAS.
- S. M. Moghimi and A. C. Hunter, Trends Biotechnol., 2000, 18, 412–420 CrossRef CAS PubMed.
- T. Moore, S. Croy, S. Mallapregada and N. Pandit, J. Controlled Release, 2000, 67, 191–202 CrossRef CAS PubMed.
- G. Dumortier, J. L. Grossiord, F. Agnely and J. C. Chaumeil, Pharm. Res., 2006, 23, 2709–2728 CrossRef CAS PubMed.
- A. Steinleitner, H. Lambert, C. Kazensky and B. Cantor, Obstet. Gynecol, 1991, 77, 48–52 CAS.
- J. Raymond, A. Metcalfe, I. Salazakin and A. Schwarz, Biomaterials, 2004, 25, 3983–3989 CrossRef CAS PubMed.
- M. Boodhwani, J. Feng, S. Mieno, B. Ramlawi, N. Sodha, R. Clements and F. W. Sellke, Eur. J. Cardiothor. Surg., 2006, 29, 736–741 CrossRef PubMed.
- A. J. Andersen, A. H. Hashemi, T. L. Andresen, A. C. Hunter and S. M. Moghimi, J. Biomed. Nanotechnol., 2009, 5, 364–372 CrossRef CAS PubMed.
- A. F. Fordjour, Y. Wang, Y. Shi, K. Agyemang, M. Akinyi, Q. Zhang and G. Fan, Eur. J. Pharmacol., 2015, 760, 72–80 CrossRef PubMed.
- J. F. Regal, J. S. Gilbert and R. M. Burwick, Mol. Immunol., 2015, 67, 56–70 CrossRef CAS PubMed.
- S. D. Mahajan, N. U. Parikh, T. M. Woodruff, J. N. Jarvis, M. Lopez, T. Hennon, P. Cunningham, R. J. Quigg, S. A. Schwartz and J. J. Alexander, Immunology, 2015, 146, 130–143 CrossRef CAS PubMed.
- N. F. Russkamp, R. Ruemmier, J. Roewe, B. B. Moore, P. A. Ward and M. Bosmann, FASEB J., 2015, 29, 3762–3772 CrossRef CAS PubMed.
- T. L. Laursen, T. D. Sandhal, S. Stoy, F. V. Schiodt, W. M. Lee, H. Vilstrup, S. Thiel and H. Gronbaek, Liver Int., 2015, 35, 756–763 CrossRef CAS PubMed.
- R. Cofiell, A. Kukreja, K. Bedard, Y. Yan, A. P. Mickle, M. Ogawa, C. L. Bedrosian and S. J. Faas, Blood, 2015, 125, 3253–3262 CrossRef CAS PubMed.
- R. P. Rother, S. A. Rollins, C. F. Mojcik, R. A. Brodsky and L. Bell, Nat. Biotechnol., 2007, 25, 1256–1264 CrossRef CAS PubMed.
- H. Qu, D. Ricklin and J. D. Lambris, Mol. Immunol., 2009, 47, 185–195 CrossRef CAS PubMed.
- Y. Wu, H. C. Qu, G. Sfyroera, A. Tzekou, K. K. Kay, B. Nilsson, K. N. Ekdahl, D. Ricklin and J. D. Lambris, J. Immunol., 2011, 186, 4269–4277 CrossRef CAS PubMed.
- H. Chen, D. Ricklin, M. Hammel, B. L. Garcia, W. J. McWhorter, G. Sfyroera, Y. Wu, A. Tzekou, S. Li, B. V. Geisbrecht, V. L. Woods and J. D. Lambris, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17621–17626 CrossRef CAS PubMed.
- S. M. Moghimi and Z. S. Farhangrazi, Nanomedicine, 2013, 9, 458–460 CAS.
- F. Heath, A. O. Saeed, S. S. Pennadam, S. Sivanand, K. J. Thurecht and C. Alexander, Polym. Chem., 2010, 1, 1252–1262 RSC.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- M. Cavazzana-Calvo, S. Hacein-Bay, G. de Saint Basile, F. Gross, E. Yvon, P. Nusbaum, F. Selz, C. Hue, J.-L. Casanova, P. Bousso, F. le Deist and A. Fisher, Science, 2000, 288, 669–672 CrossRef CAS PubMed.
- M. Schmidt, S. Hacein-Bay-Abina, M. Wissler, F. Carlier, A. Lim, C. Prinz, H. Glimm, I. Andre-Schmutz, C. Hue, A. Garrigue, F. le Deist, C. Lagresle, M. Fisher, M. Cavazzana-Calvo and C. von Kalle, Blood, 2005, 105, 2699–2706 CrossRef CAS PubMed.
- S. Hacein-Bay-Abina, C. von Kalle, M. Schmidt, M. P. McCcormack, N. Wulffraat, P. Leboulch, A. Lim, C. S. Osborne, R. Pawliuk, E. Morillion, R. Sorensen, A. Forster, P. Fraser, J. I. Cohen, G. de Saint Basile, I. Alexander, U. Wintergerst, T. Frebourg, A. Aurias, D. Stoppa-Lyonnet, S. Romana, I. Radford-Weiss, R. Gross, F. Valensi, E. Delabesse, E. Macintyre, F. Sigaux, J. Soulier, L. E. Leiva, M. Wissler, C. Prinz, T. H. Rabbits, F. Le Deist, A. Fisher and M. Cavazzana-Calvo, Science, 2003, 302, 415–419 CrossRef PubMed.
- A. C. Hunter, Adv. Drug Delivery Rev., 2006, 58, 1523–1531 CrossRef CAS PubMed.
- A. C. Hunter and S. M. Moghimi, Biochim. Biophys. Acta, Bioenerg., 2010, 1797, 1203–1209 CrossRef CAS PubMed.
- J. M. Benns, J. S. Chol, R. Mahato, J. S. Park and S. W. Kim, Bioconjugate Chem., 2000, 11, 637–645 CrossRef CAS PubMed.
- R. A. Jones, C. Y. Cheung, F. E. Black, J. K. Zia, P. S. Stayton, A. S. Hoffman and M. R. Wilson, Biochem. J., 2003, 372, 65–75 CrossRef CAS PubMed.
- C. Y. Cheung, N. Murthy, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 2001, 12, 906–910 CrossRef CAS PubMed.
- J. Luten, J. H. van Steenis, R. van Someren, J. Kemmink, N. M. E. Schuurmans-Nieuwenbroek, G. A. Koning, D. J. A. Crommelin, C. F. van Nostrum and W. E. Hennik, J. Controlled Release, 2003, 89, 483–497 CrossRef CAS PubMed.
- R. Jain, S. M. Standley and J. M. J. Frechet, Macromolecules, 2007, 40, 452–457 CrossRef CAS.
- V. Dehousse, N. Garbacki, A. Colige and B. Evard, Biomaterials, 2010, 31, 1839–1849 CrossRef CAS PubMed.
- J. J. Gu, X. Wang, X. Y. Jiang, Y. Z. Chen, L. C. Chen, X. L. Fang and X. Y. Sha, Biomaterials, 2012, 33, 644–658 CrossRef CAS PubMed.
- A. Kichler, J. Gene Med., 2003, 9, S3–S10 Search PubMed.
- R. V. Benjaminsen, M. A. Mattebjerg, J. R. Henriksen, S. M. Moghimi and T. L. Andresen, Mol. Ther., 2013, 21, 149–157 CrossRef CAS PubMed.
- J. Behr, Chimia, 1997, 51, 34–36 CAS.
- T. Bieber, W. Meissner, S. Kostin, A. Niemann and H. P. Elsasser, J. Controlled Release, 2002, 82, 441–454 CrossRef CAS PubMed.
- S. M. Moghimi, P. Symonds, J. C. Murray, A. C. Hunter, D. Debska and A. Szewczyk, Mol. Ther., 2005, 11, 990–995 CrossRef CAS PubMed.
- L. Parhamifar, H. Andersen, L.-P. Wu, A. Hall, D. Hudzech and S. M. Moghimi, Adv. Genet., 2014, 88, 353–398 Search PubMed.
- S. J. Martin, D. M. Finucane, G. P. Amarantemendes, G. A. O'Brien and D. A. Green, J. Biol. Chem., 1996, 271, 28753–28756 CrossRef CAS PubMed.
- S. J. Martin, C. P. Reutelingsperger, A. J. McGahon, J. A. Rader, R. C. A. A. Vanschie, D. M. LaFace and D. R. Green, J. Exp. Med., 1995, 182, 1545–1556 CrossRef CAS PubMed.
- J. P. Clamme, G. Krishnamoorthy and Y. Mély, Biochim. Biophys. Acta, Biomembr., 2003, 1617, 52–61 CrossRef CAS.
- A. Hall, L. Parhamifar, M. K. Lange, K. D. Meyle, M. Sanderhoff, M. Roursgaard, A. K. Larsen, H. Andersen, P. B. Jensen and J. Bartek, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 328–342 CrossRef CAS PubMed.
- A. Hall, A. K. Larsen, L. Parhamifar, K. D. Myele, L.-P. Wu and S. M. Moghimi, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1213–1225 CrossRef CAS PubMed.
- M. Thomas, J. J. Lu, C. C. Zhang, J. Z. Chen and A. M. Klibanov, Pharm. Res., 2007, 24, 1564–1571 CrossRef CAS PubMed.
- A. Zintchenko, A. Phillipp, A. Dehshahri and E. Wagner, Bioconjugate Chem., 2008, 19, 1448–1455 CrossRef CAS PubMed.
- M. R. Park, H. W. Kim, C. S. Hwang, K. O. Han, Y. J. Choi, S. C. Song, M. H. Cho and C. S. Cho, J. Gene Med., 2008, 10, 198–207 CrossRef CAS PubMed.
- Y. X. Sun, W. Xiao, S. X. Cheng and X. Z. Zhuo, J. Controlled Release, 2008, 128, 171–178 CrossRef CAS PubMed.
- U. Lachelt, P. Kos, F. M. Mickler, A. Hermann, E. E. Salcher, N. Badgujar, C. Brauchle and E. Wagner, Nanomedicine, 2014, 10, 35–44 Search PubMed.
- C. Scholz, P. Kos, L. Leclercq, X. Y. Jin, H. Cottet and E. Wagner, ChemMedChem, 2014, 9, 2104–2110 CrossRef CAS PubMed.
- P. Symonds, J. C. Murray, A. C. Hunter, G. Debska and A. Szewczyk, FEBS Lett., 2005, 579, 6191–6198 CrossRef CAS PubMed.
- A. Hall, L.-P. Wu, L. Parhamifar and S. M. Moghimi, Biomacromolecules, 2015, 16, 2119–2126 CrossRef CAS PubMed.
- J.-H. Lee, K. E. Cha, M. S. Kim, H. W. Hong, D. J. Chung, G. Ryu and H. Myung, Toxicol. Lett., 2009, 190, 202–207 CrossRef CAS PubMed.
- S. M. Moghimi, L. Parhamifar, D. Ahmadvand, P. P. Wibroe, T. L. Andresen, Z. S. Farhangrazi and A. C. Hunter, J. Innate Immun., 2012, 4, 509–528 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |