
The use of electrostatic association for rapid RAFT synthesis of histamine polyelectrolyte in aqueous solutions at and below 25 °C†
Tao
Huang
,
Zhigang
Cui
,
Yi
Ding
,
Xinhua
Lu
and
Yuanli
Cai
*
Jiangsu Key Laboratory of Advanced Functional Polymer Design and Application, Suzhou Key Laboratory of Macromolecular Design & Precision Synthesis, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: ylcai@suda.edu.cn; Fax: +86-512-65884419; Tel: +86-512-65884419
First published on 16th October 2015
Abstract
Aqueous synthesis of a well-defined polyion or polyelectrolyte via reversible degenerative radical polymerisation (RDRP) in dilute solution at or below room temperature is increasingly imperative for emerging biological applications, such as in situ grafting of polyelectrolytes onto heat-sensitive proteins and DNA/RNA. However, the polymerisation generally suffers from a time-consuming and less effective process because of electrostatic repulsion. This problem can be circumvented by media-adjustable electrostatic association. Herein we report the use of electrostatic association for rapid and quantitative aqueous RAFT of histamine-based ionic monomers in dilute solutions under visible light irradiation at and below 25 °C. We demonstrate that electrostatic interactions dictate polymerisation rates, including the initialization period and chain growth. The use of bulky counter-anions and the addition of methanol and sodium chloride can enhance electrostatic association, and thus promote the chain-growth rate. More importantly, abnormal chain-growth acceleration can be achieved in a cold solution at 8 °C, which proceeds faster than at 25 °C, because of ion-pairing association. These external regulations over aqueous RDRP show general implications for rapid and quantitative aqueous synthesis of well-defined polyelectrolytes in dilute solutions at and below room temperature.
Introduction
Aqueous synthesis of a well-defined polyion or polyelectrolyte in dilute solution at or below room temperature is increasingly imperative in emerging biological fields,1–4 as a cell-penetrating agent,5 for bacteria detection/inhibition6 and for controlled drug/gene delivery and bio-imaging systems, via the in situ grafting of polyelectrolytes onto heat-sensitive proteins and DNA/RNA. To achieve this goal, a challenge has to be tackled, i.e. electrostatic repulsion interactions,7–9 which impede or even prevent ionic monomers from chain growth as observed in conventional free radical polymerisation.10–14 The majority of the current syntheses involve the use of reversible degenerative radical polymerisation (RDRP) in the absence of additional metal ions, reversible addition-fragmentation chain transfer (RAFT)15,16 polymerisation, of ionic monomers in hot solutions at 60–80 °C.17–19 In contrast, room-temperature aqueous synthesis is less exploited because it is rather time-consuming and labile to loss of living character.17 Basically, it is indispensable to properly understand the variability of electrostatic interactions upon chain growth, for exploration of a new approach for rapid aqueous synthesis of well-defined polyelectrolytes in dilute solution at or below room temperature.Herein we report the use of electrostatic association for rapid and quantitative aqueous RAFT synthesis of polyelectrolytes in dilute solutions at and below 25 °C. Histamine acrylamide (HA) was synthesized and acidified in situ using different acids (HCl, HBF4, CF3SO3H or TfOH) into cationic monomers (HA-HX, Chart 1). As is well known, the ionic polypeptide moieties play crucial roles in hierarchical structures and living functions of proteins, in which histidine units (precursor of histamine) are chargeable peptide motifs that dictate enzymatic hydrolysis,20 oxidation,21 and acid–base catalysis.22 These inspirations gave rise to many biomedical products23 and materials for solar cells,24 CO2 capture,25 and selective anion recognition.26 Zhong and co-workers27 prepared poly(histamine acrylamide) (PHA) via a thiol-mediated radical polymerization in methanol at 60 °C for 2 days. Lowe and co-workers28 obtained PHA by RAFT of an activated ester monomer in acetonitrile at 70 °C and then post reaction with histamine. With respect to biological applications, it is imperative to explore a new approach for rapid aqueous synthesis of histamine-polymer at or below room temperature.
 | ||
| Chart 1 Chemical structures of the ionized histamine acrylamide (HA-HX; HX = HCl, HBF4, CF3SO3H or TfOH) monomers, 4-cyano-4-ethylsulfanylthiocarbonylsulfanylpentanoic acid (CEP) chain transfer agent, and sodium phenyl-2,4,6-trimethylbenzoylphosphinate (SPTP) photo-initiator that were utilized in this article. | ||
Visible light irradiation was used for this synthesis to promote the room-temperature aqueous RAFT polymerisation. Our results have demonstrated that visible light irradiation is sufficient to activate aqueous RAFT and thus induce an ultrafast polymerisation of non-ionic monomers at 25 °C.29 Moreover, the chain growth can be induced and suspended immediately by turning on and off visible light,30,31 such that the reaction rate can be measured by suspending the reaction at a predetermined time.32–34 Fu and co-workers35,36 revealed the effectiveness of visible light irradiation for activating cobalt-mediated polymerization. Boyer and co-workers37–39 exploited photo-induced electron transfer-RAFT (PET-RAFT) polymerisation via visible light irradiation at room temperature. In contrast, our method is more straightforward in terms of elucidation of the electrostatic association on the room-temperature aqueous RAFT synthesis of polyelectrolytes owing to the absence of additional metal ions. In addition, the living character can be maintained by the use of the fast reaction under such aqueous conditions. Different from as-reported aqueous RAFT synthesis,29–34 the present work focuses on the polymerisation in dilute aqueous solution to ensure significant and adjustable electrostatic interactions. The effects of the electrostatic repulsion-association on the room-temperature aqueous RAFT of HA-HX monomers were studied by differing counter-anions and concentrations, and the addition of methanol or sodium chloride. Moreover, the reaction in a cold solution at 8 °C was studied to explore the low-temperature reaction for the as-mentioned biological applications. Quantitative synthesis of a structure-tuned polyelectrolyte was conducted to illustrate the synthesis efficiency.
Experimental
Materials
Acryloyl chloride, histamine dihydrochloride, deuterium oxide and deuterchloric acid (DCl) were purchased from Sigma-Aldrich; trifluoromethanesulfonic acid (TfOH), fluoroboric acid (HBF4), hydrochloric acid (HCl), triethylamine (TEA), dichloromethane (DCM), N,N-dimethylformamide (DMF), methanol, NaOH and NaCl were from Sinopharm. Acryloyl chloride was distilled; methanol was distilled over Mg/I2. Other agents were used as received. 4-Cyano-4-ethylsulfanylthiocarbonylsulfanylpentanoic acid (CEP)40 chain transfer agent and sodium phenyl-2,4,6-trimethylbenzoylphosphinate (SPTP)41 initiator were synthesized according to literature procedures. Deionized water (R > 18.2 MΩ cm−1) was used for the synthesis and characterization.Visible light source
A mercury lamp was used as an initial light source. UV light (λem < 400 nm) was filtered by JB400 filters. The light intensity was decreased to I420 nm = 0.20 mW cm−2 (ca. 5% sunlight intensity in May in Suzhou), as determined by a UV-A radiometer equipped with a λ = 420 nm detector.Synthesis of non-ionic histamine acrylamide (HA) monomer
Histamine dihydrochloride (25.00 g, 0.136 mol) and NaOH (16.30 g, 0.408 mol) were dissolved in water (200 mL) in a 500 mL flask. Acryloyl chloride (13.54 g in 100 mL DCM, 0.150 mol) was added dropwise under stirring at −8 °C overnight. After removal of DCM, the solution was lyophilized in a Labconco Free-zone 2.5 L freeze-drier. The crude product was extracted using 2-propanol and passed through a silica column. Drying under vacuum afforded a white solid product. Yield: 12.62 g, 56%. 1H NMR (in DCl/D2O, δ, ppm): 8.48, 7.13 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C
C![[H with combining low line]](https://www.rsc.org/images/entities/b_char_0048_0332.gif) –NHCNH+ in imidazoliums, 2H); 6.05, 5.61 (C2C, 3H); 3.46 (CONHC2, 2H); 2.85 (CONHCH2C2, 2H).
–NHCNH+ in imidazoliums, 2H); 6.05, 5.61 (C2C, 3H); 3.46 (CONHC2, 2H); 2.85 (CONHCH2C2, 2H).
Aqueous RAFT under visible light irradiation at 25 °C
Typically, HA monomer (0.62 g, 3.76 mmol) was dissolved in water (4.3 g) in a 10 mL round-bottom flask. CEP chain transfer agent (3.3 mg in 0.56 g methanol, 13 μmol) and SPTP photo-initiator (0.97 mg in 96 mg water, 3.13 μmol) were added to the flask. The solution was diluted to [HA-HCl]0 = 0.6 M, and adjusted to pH 3.1 using 5.0 M hydrochloric acid. The flask was immersed into a water bath at 25 °C, bubbled with argon gas for 60 min, and irradiated with visible light. Solution samples were taken at predetermined times, and the reaction was ceased by exposure to air and adding hydroquinone for 1H NMR and SEC studies.Quantitative synthesis of a structure-tuned polyelectrolyte
HA monomer (0.508 g, 3.08 mmol) and water (3.1 g) were added to a 25 mL flask. The CEP chain transfer agent (2.7 mg in 0.28 g methanol, 10.3 μmol) and SPTP photo-initiator (0.794 mg in 80 mg water, 2.58 μmol) were added to the flask. The solution was adjusted, by adding water and TfOH (50% in water) under stirring for complete dissolution, to [HA-TfOH]0 = 0.6 M at pH 3.1. The solution was bubbled with argon gas for 60 min and irradiated at 25 °C for 80 min. Thereafter, the visible light was turned off, and argon gas-saturated monomer solution (0.485 g HA-TfOH in 2.08 g water, 1.54 mmol) was added in the dark, and the solution was irradiated for 80 min. Finally, the monomer solution (0.485 g HA-TfOH in 2.08 g water, 1.54 mmol) was added after shielding visible light, and the solution was irradiated for 80 min. The reaction was ceased by exposure to air and the addition of traces of hydroquinone.Characterization
1H NMR spectroscopy was performed on an Inova 400 MHz NMR instrument. Size exclusion chromatography (SEC) was carried out on a PL-GPC220 integrated system fitted with a refractive index detector and a set of SEC columns (2 × PLGel MIXED-B + 1 × PLGel MIXED-D). The eluent was DMF that contained 10.0 mM LiBr. PMMA standards (Agilent, 7.36–2136.0 kDa) were used for calibration. Calibration and analysis were conducted at a flow rate of 1.0 mL min−1 at 80 °C. Samples were protected by di-tert-butyldicarbonate according to the literature procedures,42 such that the absorption of ionic motifs onto SEC column materials could be circumvented. Solution conductivity was measured on a Bibby Scientific JENWAY 4520 conductivity meter at controlled temperatures within ± 0.1 °C. UV-vis spectra were recorded on SHIMADZU UV-3150 instrument. Acid–base titration was conducted on a Denver UB-7 digital pH meter. The laser light scattering intensity was measured on a Brookhaven BI-200SM setup at an angle of 90°.Results and discussion
Conditions for aqueous RAFT of the ionic monomers at 25 °C
The non-ionic HA monomer was charged into ionic monomers in water using HCl, HBF4 and TfOH, in which the electrostatic interactions can be mediated by the use of different counter-anions. The solution was adjusted to pH 3.1 before irradiation, in which monomer salts were molecularly dissolved (as judged by discernible signals of as-lyophilized monomer salts by 1H NMR) with negligible free acids according to the acid–base titration results (Fig. S1†).Solution conductivity was measured to evaluate electrostatic interactions since electrostatic association is correlated with the conductivity decrease.43 As shown in Fig. 1, the κ value depends on the monomer concentration and the counter-anion, in that the κ values with bulkier counter-anions (BF4−, TfO−) are lower than those with Cl− at constant concentrations, in good agreement with the electrostatic association capacity in the order of TfO− > BF4− > Cl−.44–46 Moreover, the ion-paired monomers dissociated into free ions43 upon diluting to a critical concentration, [HA-TfOH]crit = 0.09 M, [HA-HBF4]crit = 0.11 M, [HA-HCl]crit = 0.18 M, as judged by an abrupt increase of the molar conductivities (Λ, κ/[HA-HX]). Ion-pair dissociation increases the electrostatic repulsion, and thus hampers the chain growth as observed in traditional free radical polymerization.11,12 Accordingly, it is indispensable to elucidate the effects of electrostatic interactions, in order to achieve fast and efficient aqueous RAFT.
 | ||
| Fig. 1 The solution conductivity (κ, solid) and molar conductivity (Λ, hollow) of HA-HCl (square), HA-HBF4 (circle), and HA-TfOH (triangle) as a function of concentration. | ||
The water-soluble SPTP photo-initiator (Chart 1) was synthesized and used herein because of its high efficiency for initialization under visible light irradiation in aqueous solution.47 In addition, the CEP chain transfer agent (CTA) was synthesized according to a literature procedure40 and used to mediate the aqueous RAFT of the acrylamide monomers.17 It should be mentioned that, minor fractions of methanol (10 wt%) were added, such that CEP was dissolved in the reaction solution and electrostatic association was enhanced before initiation of polymerisation.48,49 The reactions proceeded at a target degree of polymerization DPtarget = 300, i.e. [HA-HX]0/[CEP]0 = 300. A ratio of [HA-HX]0/[SPTP]0 = 1200 was used to ensure a sufficient initialization rate under weak visible light irradiation at an I420 nm = 0.20 mW cm−2. Both the monomer salts and ionic polymers were molecularly dissolved as judged by 1H NMR and laser light scattering (ESI, Fig. S2†). Thus, monomer conversions were precisely determined by 1H NMR.
Electrostatic repulsion induces chain growth slowing-down
As shown in Fig. 2a, high conversions (97%) have been achieved for [HA-TfOH]0 = 0.9 M with irradiation for 1 h, roughly comparable to HA-HBF4 but faster than HA-HCl. The reaction rates differed clearly in the dilute solutions for [HA-HX]0 = 0.60 M (Fig. 2b). Moreover, for [HA-HX]0 = 0.9 M, HA-TfOH polymerized in a typical pseudo first-order kinetics to above 97% conversions, but the others showed a downward curvature of linear semi-logarithmic kinetics plots after a critical conversion (91% HA-HBF4 and 87% HA-HCl). The downward curvature is more significant in the dilute solutions for [HA-HX]0 = 0.60 M (Fig. 2b), as observed at conversions of 90% HA-TfOH, 87% HA-HBF4, and 72% HA-HCl. The critical concentrations of un-reacted monomers were determined: 0.06 M HA-TfOH, 0.08 M HA-HBF4 and 0.16 M HA-HCl. Considering the electrostatic contribution of growing chains,50 the above-assessed un-reacted monomer concentrations are consistent with the corresponding critical concentrations for ion-pair dissociation (Fig. 1). Thus, the electrostatic repulsion was induced by ion-pair dissociation, leading to chain growth slowing-down. More importantly, the electrostatic association of bulky anions can circumvent this chain-growth slowing-down. | ||
| Fig. 2 Kinetics plots of RAFT of HA-HCl (square), HA-HBF4 (circle) and HA-TfOH (triangle) that started at [HA-HX]0 = 0.9 M (a) and 0.6 M (b). | ||
As illustrated in the inserts of Fig. 3, in both cases, SEC traces are unimodal and symmetrical, and shift towards high molecular weights with conversion. The molecular weight (Mn) increased linearly and parallel to the theoretical plots (Mn,theo, solid lines) with conversion, and the polydispersity (PDI) decreases to Mw/Mn ≈ 1.15. These well-controlled behaviours suggest the high fidelity of CEP chain-ends. These molecular weights were overestimated in comparison to the theoretical values owing to BOC-protection and the use of PMMA calibration standards. The high fidelity of CTA chain-ends were also confirmed by the invariable characteristic band (λmax,CEP = 427 nm) in the reaction solution for [HA-HCl]0 = 0.6 M (Fig. S3†). These indicate that electrostatic repulsion (rather than loss of CEP chain-ends) slowed down the chain growth after peculiar critical conversions.
 | ||
| Fig. 3 Molecular weight (Mn) and polydispersity (PDI, Mw/Mn) as a function of conversion in reactions for [HA-HX]0 = 0.90 M (a) and 0.60 M (b). Insert: the shift of representative SEC traces for [HA-TfOH]0 = 0.90 M (a) and [HA-HCl]0 = 0.60 M (b), respectively. | ||
As shown in Table 1, the (κt − κ0)/κ0 values, as indicators of conductivity variability with polymerisation, follows HA-HCl > HA-HBF4 > HA-TfOH, suggesting that electrostatic association follows Cl− < BF4− < TfO− during polymerisation. On one hand, the as-formed polymers molecularly dissolved in the solutions. On the other hand, the chain growth led to the simultaneous dilution of un-reacted monomers and an increase in the polymer chain length. These effects promoted the ion-paired monomers to dissociate into free ions and their counter-anions to enrich surrounding ionic growing-chain coils. In contrast, the bulky anions associated strongly with cationic monomer ions, which minimized repulsion interactions, thus promoting chain growth. These results are consistent with the observations by Amajjahe et al.,12 in which conventional free radical polymerisation of vinylimidazolium monomer was highly sensitive to the counteranion complexation.
Monomer dilution induces chain growth slowing-down
As discussed above (Fig. 1), chloride anions tended to dissociate into free ions in comparison to BF4− and TfO−. The electrostatic interactions of HA-HCl monomers should thus be highly sensitive to the concentrations and conversions. Clarifying the electrostatic interactions of chloride ions with oppositely charged monomer ions on the polymerisation is important because chloride ions are ubiquitous in physiological environments. Accordingly, the HA-HCl monomer was used to elucidate the concentration-variable electrostatic interactions on aqueous RAFT polymerisation.As shown in Fig. 4a, chain growth proceeded rapidly in the solutions for [HA-HCl]0 = 2.0–1.2 M (>95% in 1 h) but slowly for [HA-HCl]0 = 0.7–0.5 M (4 h of irradiation led to 63% conversion for [HA-HCl]0 = 0.5 M). As shown in Fig. 4b, the perfect linear kinetics plots were observed until high conversion (>95%) above 1.2 M. While a downward curvature of the kinetics plots occurred and became significant when diluted from 1.10 to 0.50 M (Fig. 4b).
 | ||
| Fig. 4 Kinetics plots of aqueous RAFT polymerisation of HA-HCl monomer at the predetermined concentrations. | ||
More specifically, apparent propagation rate constants were determined according to the plot gradients in Fig. 4b. The gradients of the fast-reaction parts were named as kapp,1, and those of slow parts were termed as kapp,2. As shown in Fig. 5, the kapp,1 values decreased linearly with dilution from 2.0 M to 1.2 M but sharply from 1.1 to 0.50 M. A downward curvature of semi-logarithmic plots was observed over 1.1–0.5 M, in which the kapp,2 values decrease linearly with initial monomer concentrations. Moreover, the un-reacted monomer concentrations at critical conversions are comparable to the critical concentration of ion-pair dissociation (0.09 M, Fig. 1), indicating that the electrostatic repulsion impeded the chain growth after the critical conversions. Electrostatic repulsion in dilute reaction solutions hampered the reaction of monomer radicals with CTA, as judged by the relatively long initialisation period.53,54 The SEC traces shift toward high molecular weights (Fig. 6a). The Mn values increased linearly with the same variability for [HA-HCl]0 = 0.60, 0.90, and 1.50 M (Fig. 6b). These results indicate that the electrostatic repulsion of ionized monomers dictated the as-observed propagation slowing-down.
 | ||
| Fig. 5 Apparent propagation rate constant (kapp,1 and kapp,2) versus initial HA-HCl monomer concentrations. | ||
 | ||
| Fig. 6 (a) The shift of SEC trace with polymerisation for [HA-HCl]0 = 1.50 M (see Fig. 4); (b) variation of molecular weight (Mn) and polydispersity (Mw/Mn) in the reactions for [HA-HCl]0 = 0.60 (black), 0.90 (red), and 1.50 M (blue). | ||
Media-sensitive electrostatic association promotes chain growth
The addition of a non-ionic organic substance decreases the solution polarity, and thus strengthens the electrostatic association of the monomer cations with Cl− in water.51,52 Indeed, the polymerisation proceeded rapidly and in a well-controlled manner (Fig. S4†) in 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 methanol/water at a [HA-HCl]0 = 0.6 M (Fig. 7a). 93% conversions were achieved with irradiation for 95 min, which was more rapid than in 10:90 methanol/water (just 75% conversions in 100 min), albeit the initialization period53,54 (25 min) was longer than the latter (15 min). The critical conversion (at 89%) was also higher than the latter (at 71%). The enhanced electrostatic association was confirmed by the solution conductivity (18.8 mS m−1) being lower than the latter (21.8 mS m−1). Therefore, the addition of a non-ionic organic substance can increase electrostatic association, and thus increase the chain-growth rate.
:70 methanol/water at a [HA-HCl]0 = 0.6 M (Fig. 7a). 93% conversions were achieved with irradiation for 95 min, which was more rapid than in 10:90 methanol/water (just 75% conversions in 100 min), albeit the initialization period53,54 (25 min) was longer than the latter (15 min). The critical conversion (at 89%) was also higher than the latter (at 71%). The enhanced electrostatic association was confirmed by the solution conductivity (18.8 mS m−1) being lower than the latter (21.8 mS m−1). Therefore, the addition of a non-ionic organic substance can increase electrostatic association, and thus increase the chain-growth rate.
 | ||
| Fig. 7 Kinetics plots of polymerization of HA-HCl in dilute solutions at a [HA-HCl]0 = 0.6 M on addition of (a) 10% (black) and 30% (red) methanol, and (b) 0 M (black), 0.5 M (blue) and 1.5 M (red) of sodium chloride. | ||
Similarly, the addition of a strong electrolyte, e.g. sodium chloride, can strengthen the electrostatic association.13,14,55–57 Indeed, the polymerisation rates increased with NaCl concentration (Fig. 7b), in which kapp,1 and kapp,2 increased with salt concentration. The critical conversion increased from 70 to 77 and 85% upon the increase of NaCl to 0.5 and 1.5 M, respectively. Different from the prolonged initialization period due to the addition of methanol, the initialization periods remained roughly constant, suggesting that the monomer radical addition to CEP53,54 was less sensitive to ionic strength than the media polarity. These results suggest that the aqueous RAFT of ionic monomers can be regulated on demand simply by adjusting the electrostatic association by adjusting the solution media before starting polymerisation.
Cooling induces abnormal chain-growth acceleration
Our recent results30 have demonstrated that visible light can activate efficient aqueous RAFT of a nonionic monomer, and thus polymerisation rates remained constant on cooling from 25 °C to 7 °C. This unique behaviour facilitates biological applications because proteins, DNA and RNA are generally heat-sensitive.2 This requirement encouraged us to explore the aqueous synthesis of a well-defined polyelectrolyte below room temperature. The HA-HCl monomer was used because Cl− ions are ubiquitous in physiological environments and their electrostatic association is sensitive to temperature.48,58As shown in Fig. 8a, the initialization period prolonged from 11 to 48 min upon cooling the reaction solution from 25 to 8 °C. These results indicate that the small-molecule reaction of the monomer radicals with CEP53,54 was slowed down by cooling. More importantly, the chain growth proceeded more rapidly at 8 °C than at 25 °C. 175 min of irradiation gave rise to 94% conversion at 8 °C but just 82% at 25 °C, despite the former suffering from a significantly longer initialization period before the chain growth. More precisely, the apparent propagation rate constants at 8 °C are roughly two-fold higher than at 25 °C, kapp,1 = 2.06 h−1 and kapp,2 = 0.66 h−1 at 8 °C, kapp,1 = 1.19 h−1 and kapp,2 = 0.24 h−1 at 25 °C. The former critical conversion (80% at 8 °C) was also higher than the latter (71% at 25 °C). 1H NMR and SEC results confirmed the well-controlled RAFT process at 8 °C (Fig. S5†). Thus, the aqueous RAFT polymerisation shows a cooling-induced abnormal acceleration in the chain-growth reaction, which is unprecedented in the as-exploited RDRP family.
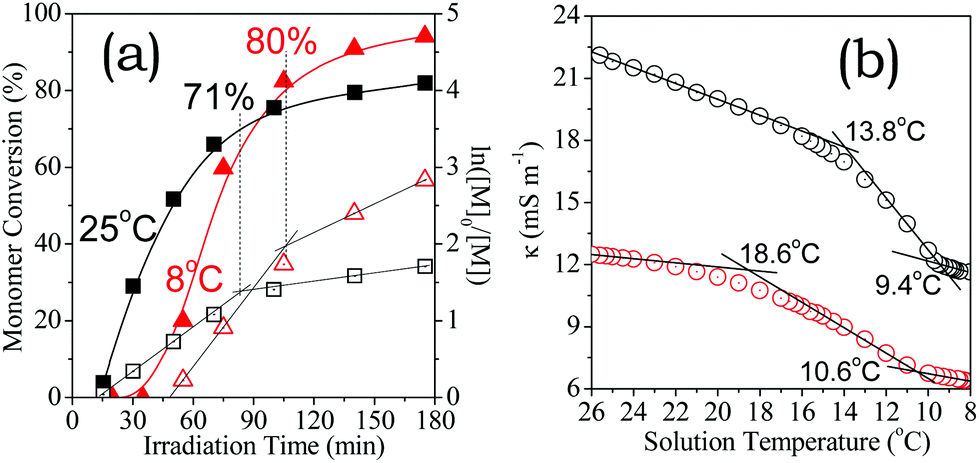 | ||
| Fig. 8 (a) Kinetics plots of aqueous RAFT for [HA-HCl]0 = 0.6 M at 25 °C (black) and 8 °C (red); (b) the solution conductivity variability (black: the monomer, red: the polyelectrolyte at DP = 300 and PDI = 1.13) with temperature. | ||
To elucidate the peculiar cooling-induced aqueous chain-growth acceleration, the as-obtained resultant solution (>98% conversion) was dialyzed using a membrane with MWCO = 1.0 kDa, and then lyophilized. 1H NMR and SEC revealed a DP of 300 and a PDI of 1.13 for the as-purified polyelectrolyte. This polyelectrolyte and HA-HCl monomer were dissolved in methanol/water (10:90). The solutions were adjusted to [HA-HCl]0 = 0.6 M and pH 3.1. Both conductivity (κ) values were inspected upon cooling the solutions from 26 °C to 8 °C.
As shown in Fig. 8b, the κ value decreased with temperature. More importantly, a sharp decrease was observed at a critical temperature, owing to ion-pairing.43 The decrease in the κ value becomes moderate upon cooling to another critical temperature, indicating an equilibrium of ion-pairing. As expected, the polyelectrolyte exhibits κ values lower than the monomer since the electrostatic association is stronger than in the corresponding ionic monomer. These observations are consistent with less free chloride anions in the positively charged polyelectrolyte solution than in the corresponding monomer solution.46 In both cases, chloride anions are maintained in the equilibrated ion-pairing state at 8 °C as judged by the temperature lower than low critical temperatures at 9.4 °C and 10.6 °C, respectively, but are essentially in a free-ion state at 25 °C as judged by the temperature higher than high critical temperatures at 13.8 °C and 18.6 °C, respectively. Polymerisations beyond these critical zones thus show inverse electrostatic regulation over chain growth.
On one hand, the ion-pairing in the cold solution at 8 °C improved electrostatic association. It inevitably increased the accessibility of growing-chain radicals to the ion-paired monomers, and thus promoted the chain growth. On the other hand, this aqueous polymerisation was dictated by visible light irradiation.30 Both effects led to cooling-induced chain-growth acceleration. To our awareness, the cooling-induced polymerisation acceleration is unprecedented in the current RDRP family, although it has been thoroughly surveyed in hot aqueous solutions.15,17,59,60 More importantly, a cooling-promoted rapid synthesis holds potential for low temperature-demanded applications.
Quantitative synthesis of a structure-tuned polyelectrolyte
As discussed above (Fig. 2), aqueous RAFT of ionized monomers containing bulky counter-anions under visible light irradiation can reach high conversions due to the electrostatic association. Thus, the iterative synthesis via RAFT exploited by Perrier,61 Cu(0)-mediated radical polymerization exploited by Whittaker62 and Haddleton,63 and PET-RAFT by Boyer,64 can be explored for the rapid quantitative synthesis of structure-tuned polyelectrolyte in dilute solution at 25 °C. The HA-TfOH monomer was chosen due to its strong electrostatic association. A quantitative synthesis proceeded via subsequent addition of monomer after the former was completely consumed, and visible light irradiation gave successive chain extension.As shown in Fig. 9a, >97% conversions have been achieved by step-wise irradiation after the addition of new HA-TfOH monomers, in which the reactions started up subsequently at [HA-TfOH]0 = 0.60 M, 0.20 M and 0.15 M. SEC traces of the as-generated polymers (Fig. 9b) are unimodal and symmetrical, and shift towards high molecular weights. SEC results revealed a series of well-defined polyelectrolytes with a varied MW from P(HA-TfOH) with Mn = 80.0 kDa and PDI = 1.15, to P(HA-TfOH)-b-P(HA-TfOH) with Mn = 107.9 kDa and PDI = 1.19), finally to P(HA-TfOH)-b-P(HA-TfOH)-b-P(HA-TfOH) with Mn = 149.8 kDa and PDI = 1.21.
 | ||
| Fig. 9 The evolution of (a) 1H NMR spectrum and (b) SEC trace on iterative reaction under irradiation for 80 min (spectrum 1), followed by two monomer additions and irradiated for 80 min (spectra 2, 3). Top: schematic illustration of the one-pot synthesis. | ||
In short, simply by means of electrostatic association through adjusting the solution media, rapid quantitative aqueous synthesis of well-defined polyelectrolytes was achieved in dilute solution at 25 °C. More importantly, aqueous RAFT in a cold solution at 8 °C gave rise to an abnormal chain-growth acceleration. Such a unique performance meets the threshold of rapid and quantitative synthesis of a well-defined polyelectrolyte at or below room temperature that might be indispensable for modification of heat-sensitive proteins65,66 and the preparation of drug/gene delivery systems.67–70
Conclusions
This article reported the use of electrostatic association for fast aqueous RAFT of histamine-based cationic monomers (HA-HX) in dilute aqueous solutions at and below 25 °C. To this end, HA was charged using different acids to give counter-anion variable HA-HX monomers. Polymerisation proceeded under visible light irradiation at 25 °C or 8 °C. Kinetic characters were studied by using different counter-anions (Cl−, BF4− and TfO−), adding methanol and sodium chloride, and carrying out the reaction in cold solution at 8 °C. Electrostatic interactions in the reaction solutions were studied by solution conductivity analysis. The versatile feature was illustrated by a rapid and quantitative aqueous synthesis of a structure-adjustable polyelectrolyte.The results demonstrated that the electrostatic interactions dictate the effectiveness of the aqueous RAFT synthesis. For instance, the electrostatic repulsion was promoted during chain growth due to the dissociation of monomer ion pairs into free ions, which slowed down the chain-growth reaction. On the contrary, an increase of electrostatic association, by the use of bulky counter-anions, the addition of methanol and sodium chloride, promoted chain-growth rate. Moreover, an abnormal chain-growth acceleration was achieved in a cold solution at 8 °C, which proceeded faster than at 25 °C due to ion-pairing association. These external regulations were highly efficient in dictating the aqueous RAFT. Therefore, a well-defined and structure-tuned polyelectrolyte could be synthesized on demand even in dilute aqueous solution at or below room temperature, by means of electrostatic association via adjusting the solution media. This approach meets the requirements for the direct aqueous synthesis of a well-defined polyelectrolyte and thus holds potential for controlled drug/gene delivery and bio-imaging systems.67–70
Acknowledgements
This research work was supported by National Natural Science Foundation of China (21274097, 21474069, and 21334004) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.Notes and references
- H. Cabral, N. Nishiyama and K. Kataoka, Acc. Chem. Res., 2011, 44, 999–1008 CrossRef CAS PubMed
.
- X. Li, L. Wang, G. Chen, D. M. Haddleton and H. Chen, Chem. Commun., 2014, 50, 6506–6508 RSC
- S. Chuanoi, Y. Anraku, M. Hori, A. Kishimura and K. Kataoka, Biomacromolecules, 2014, 15, 2389–2397 CrossRef CAS PubMed
- K. Hayakawa, S. Uchida, T. Ogata, S. Tanaka, K. Kataoka and K. Itaka, J. Controlled Release, 2015, 197, 1–9 CrossRef CAS PubMed
- X. Hu, G. Liu, Y. Li, X. Wang and S. Liu, J. Am. Chem. Soc., 2015, 137, 362–368 CrossRef CAS PubMed
- Y. Li, H. Yu, Y. Qian, J. Hu and S. Liu, Adv. Mater., 2014, 26, 6734–6741 CrossRef CAS PubMed
- S. Forster and M. Schmidt, Adv. Polym. Sci., 1995, 120, 51–133 CrossRef
- J. Bohrisch, C. D. Eisenbach, W. Jaeger, H. Mori, A. H. E. Müller, M. Rehahn, C. Schaller, S. Traser and P. Wittmeyer, Adv. Polym. Sci., 2004, 165, 1–41 CrossRef CAS
- S. Forster, V. Abetz and A. H. E. Müller, Adv. Polym. Sci., 2004, 166, 173–210 CrossRef
- H. Ohno, Electrochim. Acta, 2001, 46, 1407–1411 CrossRef CAS
- S. Beuermann, M. Buback, P. Hesse, S. Kukučková and I. Lacık, Macromol. Symp., 2007, 248, 23–32 CrossRef CAS
- S. Amajjahe and H. Ritter, Macromolecules, 2008, 41, 716–718 CrossRef CAS
- K. S. Anseth, R. A. Scott and N. A. Peppas, Macromolecules, 1996, 29, 8308–8312 CrossRef CAS
- R. Losada and C. Wandrey, Macromol. Rapid Commun., 2008, 29, 252–257 CrossRef CAS
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS
- W. Jaeger, J. Bohrisch and A. Laschewsky, Prog. Polym. Sci., 2010, 35, 511–577 CrossRef CAS
- M. H. Allen Jr., S. T. Hemp, A. E. Smith and T. E. Long, Macromolecules, 2012, 45, 3669–3676 CrossRef
- X. Zhou, T.-F. Chou, B. E. Aubol, C. J. Park, R. Wolfenden, J. Adams and C. R. Wagner, Biochemistry, 2013, 52, 3588–3600 CrossRef CAS PubMed
- S. Nagano, M. Tanaka, K. Ishimori, Y. Watanabe and I. Morishima, Biochemistry, 1996, 35, 14251–14258 CrossRef CAS PubMed
- S. Mendel, A. Arndt and T. D. H. Bugg, Biochemistry, 2004, 43, 13390–13396 CrossRef CAS PubMed
- L. Zhang, X.-M. Peng, G. L. V. Damu, R.-X. Geng and C.-H. Zhou, Med. Res. Rev., 2014, 34, 340–437 CrossRef CAS PubMed
- S. Li, L. Qiu, C. Shi, X. Chen and F. Yan, Adv. Mater., 2014, 26, 1266–1271 CrossRef CAS PubMed
- J. E. Bara, D. E. Camper, D. L. Gin and R. D. Noble, Acc. Chem. Res., 2010, 43, 152–159 CrossRef CAS PubMed
- Z. Xu, S. K. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 1457–1466 RSC
- S. Luo, R. Cheng, F. Meng, T. G. Park and Z. Zhong, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3366–3373 CrossRef CAS
- J. Y. Quek, P. J. Roth, R. A. Evans, T. P. Davis and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 394–404 CrossRef CAS
- Y. Shi, H. Gao, L. Lu and Y. Cai, Chem. Commun., 2009, 1368–1370 RSC
- Y. Shi, G. Liu, H. Gao, L. Lu and Y. Cai, Macromolecules, 2009, 42, 3917–3926 CrossRef CAS
- G. Liu, H. Shi, Y. Cui, J. Tong, Y. Zhao, D. Wang and Y. Cai, Polym. Chem., 2013, 4, 1176–1182 RSC
- J. Tong, Y. Shi, G. Liu, T. Huang, N. Xu, Z. Zhu and Y. Cai, Macromol. Rapid Commun., 2013, 34, 1827–1832 CrossRef CAS PubMed
- Y. Jiang, N. Xu, J. Han, Q. Yu, L. Guo, P. Gao, X. Lu and Y. Cai, Polym. Chem., 2015, 6, 4955–4965 RSC
- H. Shi, K. Zhou, Q. Yu, Z. Cui, Y. Jiang, X. Lu and Y. Cai, Polym. Chem., 2015, 6, 7455–7463 RSC
- Y. Zhao, M. Yu, S. Zhang, Y. Liu and X. Fu, Macromolecules, 2014, 47, 6238–6245 CrossRef CAS
- Y. Zhao, S. Zhang, Z. Wu, X. Liu, X. Zhao, C.-H. Peng and X. Fu, Macromolecules, 2015, 48, 5132–5139 CrossRef CAS
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed
- S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9988–9999 CrossRef CAS PubMed
- S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS PubMed
- A. J. Convertine, D. S. W. Benoit, C. L. Duvall, A. S. Hoffman and P. S. Stayton, J. Controlled Release, 2009, 133, 221–229 CrossRef CAS PubMed
-
B. Bronstert, A. Henne, A. Hesse, M. Jacobi and G. Wallbillich, Patent US4719297, 1988 Search PubMed
- W. Liu, A. B. Greytak, J. Lee, C. R. Wong, J. Park, L. F. Marshall, W. Jiang, P. N. Curtin, A. Y. Ting, D. G. Nocera, D. Fukumura, R. K. Jain and M. G. Bawendi, J. Am. Chem. Soc., 2009, 132, 472–483 CrossRef PubMed
- Y. Marcus and G. Hefter, Chem. Rev., 2006, 106, 4585–4621 CrossRef CAS PubMed
- C.-L. Wong, A. N. Soriano and M.-H. Li, Fluid Phase Equilib., 2008, 271, 43–52 CrossRef CAS
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC
- J. Yuan and M. Antonietti, Polymer, 2011, 52, 1469–1482 CrossRef CAS
- B. D. Fairbanks, M. P. Schwartz, C. N. Bowman and K. S. Anseth, Biomaterials, 2009, 30, 6702–6707 CrossRef CAS PubMed
- W. J. Li, Z. F. Zhang, B. X. Han, S. Q. Hu, Y. Xie and G. Y. Yang, J. Phys. Chem. B, 2007, 111, 6452–6456 CrossRef CAS PubMed
- W. Liu, L. Cheng, Y. Zhang, H. Wang and M. Yu, J. Mol. Liq., 2008, 140, 68–72 CrossRef CAS
- P. Jia, Q. Yang, Y. Gong and J. Zhao, J. Chem. Phys., 2012, 136, 084904 CrossRef PubMed
- H. T. Xu, D. C. Zhao, P. Xu, F. Q. Liu and G. Gao, J. Chem. Eng. Data, 2005, 50, 133–135 CrossRef CAS
- F. Hallberg, I. Furo and P. Stilbs, J. Am. Chem. Soc., 2009, 131, 13900–13901 CrossRef CAS PubMed
- J. B. McLeary, J. M. McKenzie, M. P. Tonge, R. D. Sanderson and B. Klumperman, Chem. Commun., 2004, 1950–1951 RSC
- J. B. McLeary, F. M. Calitz, J. M. McKenzie, M. P. Tonge, R. D. Sanderson and B. Klumperman, Macromolecules, 2004, 37, 2383–2394 CrossRef CAS
- C. L. McCormick and L. C. Salazar, J. Macromol. Sci., Pure Appl. Chem., 1992, 29, 193–205 CrossRef
- V. F. Kurenkov, I. N. Nadezhdin, O. A. Antonovich and F. I. Lobanov, Russ. J. Appl. Chem. Commun., 2004, 77, 804–808 CrossRef CAS
- A. Dobrynin and M. Rubinstein, Prog. Polym. Sci., 2005, 30, 1049–1118 CrossRef CAS
- O. Zech, A. Stoppa, R. Buchner and W. Kunz, J. Chem. Eng. Data, 2010, 55, 1774–1778 CrossRef CAS
- S. Yusa, Y. Shimada, Y. Mitsukami, T. Yamamoto and Y. Morishima, Macromolecules, 2003, 36, 4208–4215 CrossRef CAS
- M. Barsbay, O. Gueven, T. P. Davis, C. Barner-Kowollik and L. Barner, Polymer, 2009, 50, 973–982 CrossRef CAS
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4 Search PubMed
- C. Boyer, A. H. Soeriyadi, P. B. Zetterlund and M. R. Whittaker, Macromolecules, 2011, 44, 8028–8033 CrossRef CAS
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef CAS PubMed
- J. Xu, K. Jung and C. Boyer, Macromolecules, 2014, 47, 4217–4229 CrossRef CAS
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS PubMed
- J. Xu, K. Jung, N. A. Corrigan and C. Boyer, Chem. Sci., 2014, 5, 3568–3575 RSC
- J. Liu, V. Bulmus, D. L. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Angew. Chem., Int. Ed., 2007, 46, 3099–3103 CrossRef CAS PubMed
- J. K. Pokorski, K. Breitenkamp, L. O. Liepold, S. Qazi and M. G. Finn, J. Am. Chem. Soc., 2011, 133, 9242–9245 CrossRef CAS PubMed
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS PubMed
- J. D. Wallat, K. A. Rose and J. K. Pokorski, Polym. Chem., 2014, 5, 1545–1558 RSC
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and UV-vis spectra of HA, HA-HX and reaction solutions; variation of solution conductivity; SEC traces and Mn & PDI variation. See DOI: 10.1039/c5py01524c |
| This journal is © The Royal Society of Chemistry 2016 |