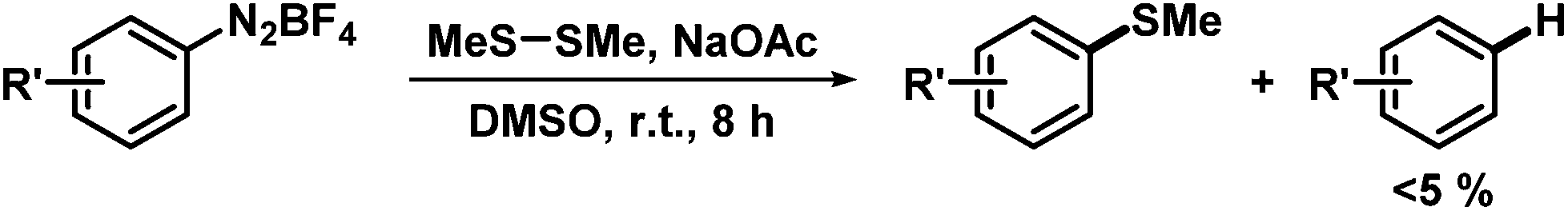DOI:
10.1039/C6OB02276F
(Communication)
Org. Biomol. Chem., 2016,
14, 11347-11352
Metal-free radical thiolations mediated by very weak bases†
Received
28th October 2016
, Accepted 16th November 2016
First published on 24th November 2016
Abstract
Aromatic thioethers and analogous heavier chalcogenides were prepared by reaction of arene-diazonium salts with disulfides in the presence of the cheap and weak base NaOAc. The mild and practical reaction conditions (equimolar reagents, DMSO, r.t., 8 h) tolerate various functional groups (e.g. Br, Cl, NO2, CO2R, OH, SCF3, furans). Mechanistic studies indicate the operation of a radical aromatic substitution mechanism via aryl, acetyloxyl, thiyl, and dimsyl radicals.
Introduction
Sulfur-substituted aromatics constitute a versatile class of building blocks which find numerous applications in the synthesis of bioactive compounds, materials and fine chemicals (Scheme 1).1 Besides electrophilic aromatic substitutions with conc. sulfuric acid, various synthetic procedures have been reported for the construction of aryl–S bonds from aryl electrophiles and sulfur nucleophiles. The most prominent methods are transition metal-catalyzed thiolations of aryl halides2 and Sandmeyer-type3 radical substitutions of arenediazonium salts4 (Scheme 2, top). The latter procedure can be performed under metal-free basic conditions but affords only moderate yields and bears significant hazard potential due to the intermediacy of explosive azosulfides.5 An elegant alternative is the use of disulfides in radical aromatic substitutions which are initiated by single-electron transfer (SET) and operate under mild conditions.6 Base-free aromatic thiolations with disulfides were recently realized by organic photoredox-catalysis.7 However, light-mediated processes require the presence of a suitable photo-sensitizer and irradiation with a powerful light source, operate at low concentrations over long reaction times, and generally suffer from the attenuation of light intensity in solution (Lambert–Beer law).
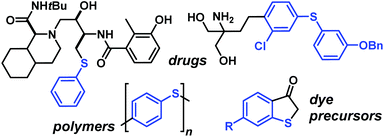 |
| | Scheme 1 Selected compounds containing aryl–S entities. | |
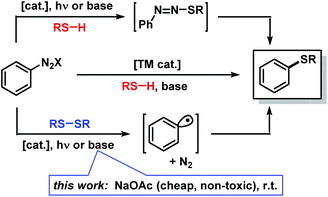 |
| | Scheme 2 Thiolation methods of arenediazonium salts. | |
Powerful metal-free electron donors are mostly strong bases8 and nucleophiles which are incompatible with sensitive functional groups and protic solvents. However, even weak bases and nucleophiles can trigger radical aromatic substitutions (SRAr) at electrophilic aryl–X if the elimination of X was irreversible or a facile bond homolysis can occur.9,10 Following observations on mechanistically related photo-redox substitutions,7 we directed our attention at SET-mediated reactions of disulfides with arenediazonium salts.11 Here, we present an operationally facile protocol which utilizes the cheap, environmentally benign, and weak base sodium acetate (NaOAc) in radical aromatic substitutions of arenediazonium salts with disulfides under mild reaction conditions (Scheme 2, bottom).
Method development
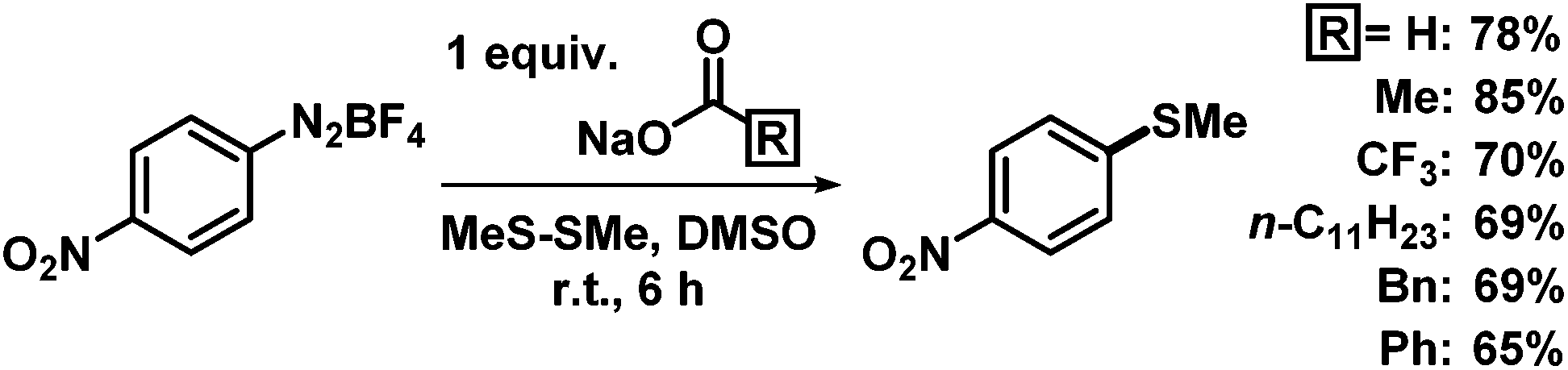
Simple inorganic bases are attractive due to their low price, low toxicity, and facile removal from organic products while organic bases are mostly more expensive, environmentally problematic, and less facile to remove from organic phases. We studied the model reaction of 4-nitrobenzenediazonium tetrafluoroborate, containing two π-electrophilic sites, with dimethyldisulfide (Table 1). Various simple bases were competent in this thiolation protocol. N,N-Dimethylhydrazine was most active;12 however, we refrained from further studies due to the toxicity and hazard potential as carcinogen, explosive, and groundwater pollutant.13 Strong bases (e.g. KOtBu, KHMDS, n-BuLi) gave lower selectivities and many by-products. Polar solvents (acetonitrile, methanol, dimethylsulfoxide (DMSO)) exhibited high reagents solubility and good yields. Identical reactions in N,N-dimethylformamide (DMF) underwent competing hydrodediazotation (25%); in benzene 4-nitrobiphenyl was formed (15%). Significantly higher selectivities were observed in the absence of air and moisture. Gratifyingly, no excess of reagents was required. The optimized conditions (equimolar ArN2BF4, NaOAc, Me2S2, 0.2 M in DMSO, 18 °C, 8 h) enabled clean conversion to the arylthioether with minimal formation of anisole as reduction by-product (<3%). Similar yields were obtained when using other simple carboxylates (Scheme 3).
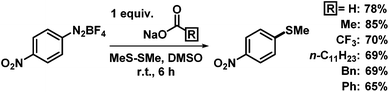 |
| | Scheme 3 Similar activity of various sodium carboxylates. | |
Table 1 Selected optimization experimentsa
|

|
| Entry |
Equiv. Me2S2 |
Base (equiv.) |
Conditions |
Yieldb [%] |
|
Optimized conditions: 4-Nitrobenzenediazonium tetrafluoroborate (0.6 mmol), NaOAc (0.6 mmol) under N2, DMSO (3 mL), r.t., 8 h.
GC yields vs. internal 1-dodecanenitrile.
Individual reactions in one solvent.
80% isolated yield.
After 60 h.
60 °C, 1 h.
|
| 1 |
5 |
— |
|
0 |
| 2 |
1 |
n-Bu4NI (1.5) |
|
66 |
| 3 |
5 |
KOtBu (5) |
|
65 |
| 4 |
5 |
NaOAc (5) |
|
85 |
| 5 |
1.5 |
NaOAc (5) |
|
85 |
| 6 |
1.5 |
NaOAc (1) |
|
83 |
| 7 |
1.5 |
NaOAc (0.5) |
16 h |
56 |
| 8 |
1.5 |
NaOAc (0.2) |
16 h |
29 |
| 9 |
1 |
KOAc (1) |
DMSO, r.t., 8 h |
80 |
| 10 |
0.5 |
NaOAc (1) |
DMSO, r.t., 8 h |
42 |
| 11 |
1 |
NaOAc (1) |
H2O/THF/AcMec |
66/31/73 |
| 12 |
1 |
NaOAc (1) |
MeOH/DMF/MeCNc |
77/62/75 |
|
13
|
1
|
NaOAc (1)
|
DMSO, r.t., 8 h
|
85
(90)
(83)
|
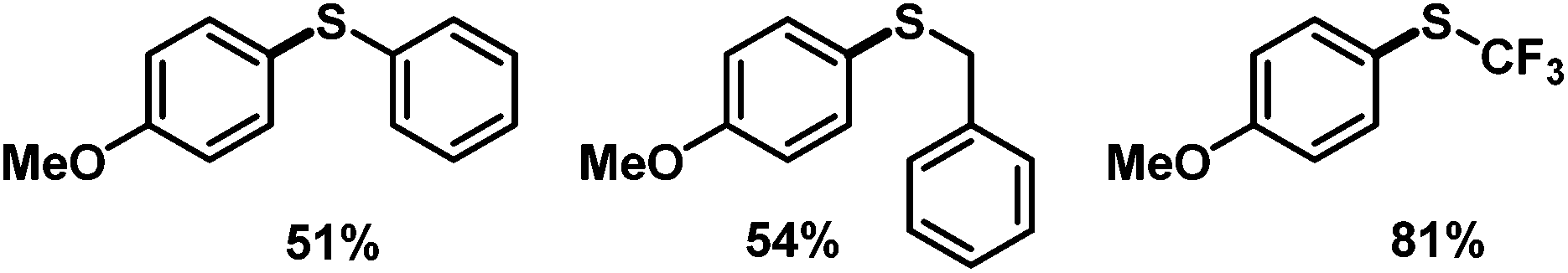
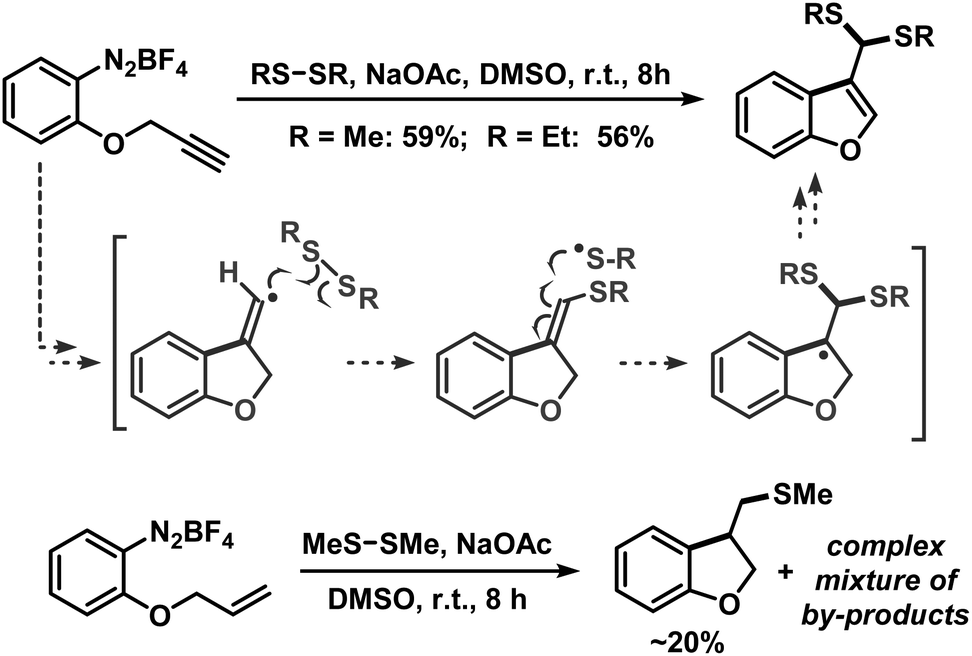
We then applied the optimized conditions to a series of diversely substituted arenediazonium salts (Table 2). The protocol proved to be rather general with regard to substituents at the arene and the disulfide, respectively. Chloro, bromo, ester, nitro, hydroxy, and naphthyl moities within the diazonium salts were tolerated. Alkyl disulfides afforded slightly higher yields than aryl disulfides (Scheme 4). The trifluoromethylsulfanyl moiety, which attracts great interest from pharmaceutical programs due to their lipophilic properties, could also be introduced by reaction with commercial bis(trifluoromethyl) disulfide in the same (Scheme 4).14 Sequential radical 5-exo-dig cyclization and thiolation was observed with 2-(propynyloxy)benzenediazonium tetrafluoroborate to give 3-thioketal benzofurans (Scheme 5).15 The attack of the S-centered radical on the newly formed double bond is facilitated by the stabilization of the resulting benzyl radical. On the other hand, the corresponding 2-allyloxybenzenediazonium salt failed to undergo a similarly effective domino reaction but afforded a different thioether skeleton and many by-products (Scheme 5, bottom).
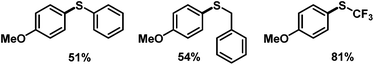 |
| | Scheme 4 Preparation of other unsymmetrical aryl thioethers. | |
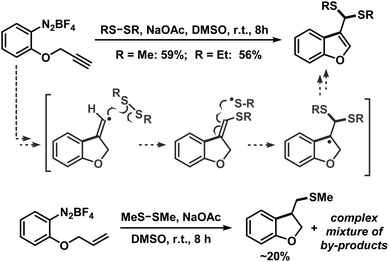 |
| | Scheme 5 Base-mediated radical cyclization-thiolation. | |
Table 2 NaOAc-mediated methylthiolation of arenediazonium saltsa
|
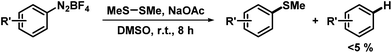
|
| R′ |
Yield [%] |
R′ |
Yield [%] |
|
Conditions: arenediazonium tetrafluoroborate (0.6 mmol), dimethyl disulfide (0.6 mmol), NaOAc (0.6 mmol), DMSO (2 mL), 20 °C, 8 h.
|
| 4-OMe |
89 |
3-NO2 |
77 |
| 4-Cl |
70 |
2-NO2 |
80 |
| 4-NO2 |
85 |
2-Br |
69 |
| 4-OH |
87 |
2-SMe |
73 |
| 4-Ph |
58 |
2-CO2Me |
71 |
| 2,4,6-Cl3 |
56 |
1-Naphthyl |
53 |
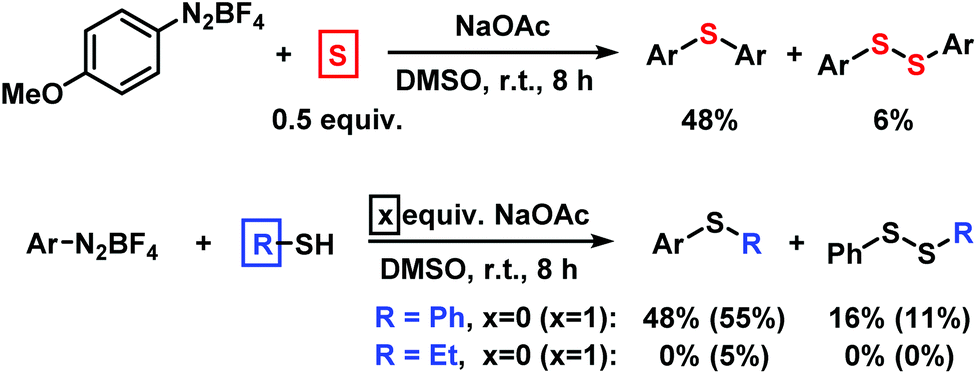
We employed elemental sulfur as alternative thiolation reagents which indeed led to the formation of symmetrical diaryl sulfides in moderate to good yields (Scheme 6, top). While thiols are easy available, they result in the formation of explosive azosulfide intermediates and under our conditions gave only moderate to low yields of the desired thioethers. The direct thiolation with benzenethiol proceeded with moderate yields in the absence of base. With ethanethiol, mostly hydrodediazotation was observed (Scheme 6, bottom).
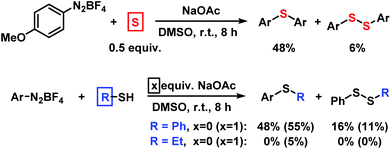 |
| | Scheme 6 Procedures with alternative sulfur sources (Ar = 4-MeOC6H4). | |
Mechanistic studies
Reaction progress analysis documented an immediate onset of reactivity within less than 10 s, more than 50% conversion within 1 h, and a significantly slower turnover after 1–2 h. This behaviour could be a consequence of two competing reaction mechanisms: a rapid base-induced thiolation and a slow radical chain propagation.16 The operation of the latter is also supported by experiments with catalytic amounts of NaOAc (20 mol% and 50 mol%) which afforded 29% and 56% yield, respectively, after 16 h. Further mechanistic insight has already been gained by the initial optimization experiments (see Table 1). The stoichiometry of the reaction (equimolar ArN2BF4, R2S2, and NaOAc) indicates a loss channel of one half of the disulfide molecule which is not electron transfer in nature (RS˙ → RS+ + e−)16 as such a scenario could operate with catalytic amounts of base.
We postulate a mechanism that involves homolysis of the initially formed diazoacetate (Scheme 7). The resultant aryl and acetyloxyl radicals engage in orthogonal onward reactions. The former reacts with the disulfide upon release of a thiyl radical which is trapped by a dimsyl radical derived from H atom transfer (HAT) with acetyloxyl. The presence of the aryl radical intermediate Ar˙ was confirmed by radical trapping with TEMPO (2,2,6,6-tetramethylpiperidin-1-yl)-oxyl in a standard reaction. The aryl radical intermediate attacks the disulfide in an SH2 fashion by homolysis of the S–S bond in a single step.17 The resulting DMSO-SR adducts (R = Me, Ph) were detected by ESI-MS (Scheme 7, bottom left).7a,18 The generation of acetic acid as major byproduct was monitored indirectly by the continuous decrease of the pH value over the course of the reaction (pH 8 → 4, after aqueous quench) despite the buffering effect of the weak acid/base pair and the solvent. Crossover-experiments with dimethyl-disulfide and diphenyldisulfide resulted in the formation of the mixed methylphenyl disulfide (MeS-SPh) as byproduct by recombination of free thiyl radicals.
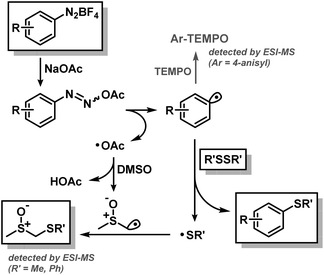 |
| | Scheme 7 Postulated weak base-mediated SRAr mechanism. | |
Other mechanistic scenarios could be excluded based on the following instructive experiments and considerations: (i) The operation of the acetate anion as single-electron transfer reagent19 is discouraged by the lack of any detectable CO2 formation by head-space MS analyses and gas phase absorptions into aqueous Ba(OH)2. (ii) A direct disulfide-mediated electron transfer was already disproven in entry 1 of Table 1. Consistently, a solution of arenediazonium salt and NaOAc in DMSO underwent significant hydrodediazotation after 30 min in the absence of disulfides; deuterium incorporation from DMSO-d6 was also observed (Scheme 8, top). (iii) Base-mediated aryl radical formation from arene-diazonium salts could in principle proceed by SET from the diazotate anion.20 On thermodynamic grounds, the diazotate anion/radical appears to be a reasonable 1e-redox couple under weakly basic conditions.21 However, this mechanism requires the operation of a highly selective H atom transfer (HAT) from a suitable H-donor (DMSO) to the azotate radical19 but not to the aryl radical intermediate. A DFT-based analysis excludes such mechanism because the HAT to the aryl radical is thermodynamically and kinetically highly favoured (Scheme 8, bottom).20,22 (iv) The operation of a dimsyl anion-mediated SRN1 mechanism can be excluded by the large pKa difference of acetic acid (∼12) and DMSO (∼35) in DMSO.23
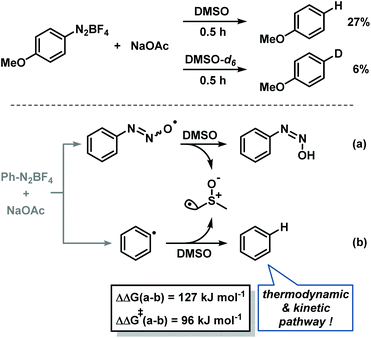 |
| | Scheme 8 Experimental and calculated H atom transfer (HAT). | |
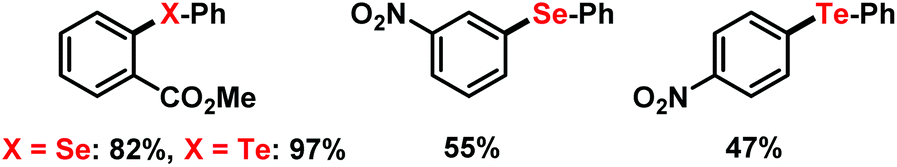
Under identical conditions, the general protocol was applied to the synthesis of arylselenoethers and aryltelluroethers (Scheme 9).24 Employment of the diphenyl dichalcogenides afforded very good yields of the methyl 2-phenylcarboxylate derivatives and moderate yields of nitrobenzene derivatives. Mechanistic studies on related systems were performed by Pandey and coworkers which reported the facile SET oxidation of diselenides in the presence of suitable acceptors and subsequent mesolysis to a phenylselenyl radical and phenylselenyl cation.6b,25
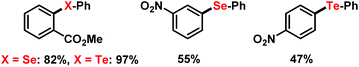 |
| | Scheme 9 Selenations and tellurations under similar conditions. | |
Conclusion
We have developed a new metal-free thiolation method for synthesis of arylsulfides which employs the cheap, weak, and environmentally benign base sodium acetate. Experimental and theoretical studies indicated the operation of an initial homolysis of diazoacetates and exclude alternative base-, dimsyl- and diazotate-mediated electron transfer mechanisms. Applications to the synthesis of various functionalized thioethers, selenides, and tellurides demonstrate the scope of this mild base-mediated protocol. Further applications of similar activation mechanisms in radical substitution reactions are currently being investigated.
Experimental section
General procedure for the synthesis of arenediazonium salts
The parent aniline (10 mmol) was dissolved in glacial acetic acid (6 mL) and 32% aqueous tetrafluoroboric acid (1.6 mL) at r.t. Then, a solution of t-butyl nitrite (1.2 mL) in glacial acetic acid (2 mL) was slowly added at r.t. over 5 min. Diethylether (15 mL) was added, and the mixture was cooled to −30 °C in order to induce crystallization. The crystals were filtered off, washed with cold diethylether (2 × 10 mL) and dried on air to give analytically pure arenediazonium salts.
General procedure for base-induced thiolation, selenylation and telluration
A vial (5 mL) was charged with a magnetic stir bar, the arenediazonium salt (0.5 mmol), the disulfide (0.5 mmol) and sodium acetate (0.5 mmol) and capped with a rubber septum. The vial was purged with N2 (5 min). Dry DMSO (2.5 mL) was added. After 8 h of stirring, water (5 mL) was added to give an emulsion, which was extracted with diethylether (3 × 5 mL). The organic phases were washed with brine (5 mL) and dried (MgSO4). The solvent was evaporated in vacuo; the residue was purified by SiO2 flash column chromatography (pentane/ethyl acetate).
Calculations
Geometries and energies were calculated using Gaussian G09W.21a DFT calculations were performed using M06-2X functional,21b and 6-31+G(d,p) basis set. Geometry optimizations and single point calculations were done using the polarized continuum model to account for solvation effects.
Acknowledgements
D. K. thanks the Bayhost program of the State of Bavaria (2014–2015) and the Fonds der Chemischen Industrie (FCI, 2015–2017) for doctoral fellowships. M. M. is a fellow of the Graduate School on “Chemical Photocatalysis” (GRK 1626) of the Deutsche Forschungsgemeinschaft (DFG).
Notes and references
-
(a)
Patai's Chemistry of Functional Groups: The Thiol Group, ed. S. Patai, Wiley-VCH, 1974, vol. 1 and 2 Search PubMed;
(b)
Patai's Chemistry of Functional Groups: The Ether Linkage, ed. S. Patai, Wiley-VCH, 1967, ch. 13 Search PubMed;
(c)
D. N. Jones, in Comprehensive Organic Chemistry, ed. D. H. R. Barton and D. W. Ollis, Pergamon, New York, 1979, vol. 3 Search PubMed.
-
(a) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS PubMed;
(b) C. C. Eichman and J. P. Stambuli, Molecules, 2011, 16, 590–608 CrossRef CAS PubMed;
(c) V. Gomez-Benitez, O. Baldovino-Pantaleon, C. Herrera-Alvarez, R. A. Toscano and D. Morales-Morales, Tetrahedron Lett., 2006, 47, 5059–5062 CrossRef CAS;
(d) O. Baldovino-Pantaleon, S. Hernandez-Ortega and D. Morales-Morales, Inorg. Chem. Commun., 2005, 8, 955–959 CrossRef CAS;
(e) O. Baldovino-Pantaleon, S. Hernandez-Ortega and D. Morales-Morales, Adv. Synth. Catal., 2006, 348, 236–242 CrossRef CAS;
(f) N. Taniguchi, J. Org. Chem., 2004, 69, 6904–6906 CrossRef CAS PubMed;
(g) S.-I. Fukuzawa, D. Tanihara and S. Kikuchi, Synlett, 2006, 2145–2147 CrossRef CAS;
(h) P.-S. Luo, M. Yu, R.-Y. Tang, P. Zhong and J.-H. Li, Tetrahedron Lett., 2009, 50, 1066–1070 CrossRef CAS;
(i) W. Fu, T. Liu, Z. Fang, Y. Ma, X. Zheng, W. Wang, X. Ni, M. Hu and T. Tang, Chem. Commun., 2015, 51, 5890–5893 RSC;
(j) N. Taniguchi and T. Onami, J. Org. Chem., 2004, 69, 915–920 CrossRef CAS PubMed;
(k) S. Kumar and L. Engman, J. Org. Chem., 2006, 71, 5400–5403 CrossRef CAS PubMed;
(l) N. Taniguchi, J. Org. Chem., 2007, 72, 1241–1245 CrossRef CAS PubMed.
-
(a) H. H. Hodgson, Chem. Rev., 1947, 40, 251–277 CrossRef CAS PubMed;
(b) R. Leuckart, J. Prakt. Chem., 1890, 41, 179–224 CrossRef CAS.
- F. Mo, G. Dong, Y. Zhang and J. Wang, Org. Biomol. Chem., 2013, 11, 1582–1593 CAS.
-
(a) J. Laquidara, Chem. Eng. News, 2001, 79, 6 CrossRef CAS;
(b) H. Spencer, Chem. Brit., 1977, 13, 240 CAS.
- Selected examples:
(a) T. Zincke, Chem. Ber., 1911, 44, 769 CrossRef CAS;
(b) A. Luxen and L. Christiaens, Tetrahedron Lett., 1982, 23, 3905 CrossRef CAS;
(c) G. Petrillo, R. Novi, G. Garbarino and C. Dellerba, Tetrahedron, 1986, 42, 4007–4016 CrossRef CAS;
(d) F. Effenberger and H. Isak, Chem. Ber., 1989, 122, 545 CrossRef CAS;
(e) J. A. Burns, J. C. Butler, J. Moran and G. M. Whitesides, J. Org. Chem., 1991, 56, 2648 CrossRef CAS;
(f) P. J. Hogg, Trends Biochem. Sci., 2003, 28, 210 CrossRef CAS PubMed;
(g) M. Erlandsson and M. Hällbrink, Int. J. Pept. Res. Ther., 2005, 11, 261 CrossRef CAS;
(h) O. Dmitrenko, C. Thorpe and R. D. Bach, J. Org. Chem., 2007, 72, 8298 CrossRef CAS PubMed;
(i) D. Witt, Synthesis, 2008, 2491 CrossRef CAS;
(j) D. Kundu, S. Ahammed and C. R. Brindabad, Green Chem., 2012, 14, 2024 RSC.
-
(a) M. Majek and A. Jacobi von Wangelin, Chem. Commun., 2013, 49, 5507–5509 RSC;
(b) X. Wang, G. D. Cuny and T. Noël, Angew. Chem., Int. Ed., 2013, 52, 7860–7864 CrossRef CAS PubMed.
- J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, L. E. A. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402–7410 CrossRef CAS PubMed.
-
(a) L. C. Schmidt, V. Rey and A. B. Penenory, Eur. J. Org. Chem., 2006, 2210–2214 CrossRef CAS;
(b) M. E. Buden, J. F. Guastavino and R. A. Rossi, Org. Lett., 2013, 15, 1174–1177 CrossRef CAS PubMed;
(c) A. Dewanji, S. Murarka, D. P. Curran and A. Studer, Org. Lett., 2013, 15, 6102–6105 CrossRef CAS PubMed. For applications of NaOAc in radical substitution reactions, see:
(d) U. M. V. Basavanag, A. Dos Santos, L. El Kaim, R. Gamez-Montano and L. Grimaud, Angew. Chem., Int. Ed., 2013, 52, 7194–7197 CrossRef CAS PubMed;
(e) Y. Sawama, R. Nakatani, T. Imanishi, Y. Fujiwara, Y. Monguchi and H. Sajiki, RSC Adv., 2014, 4, 8657–8660 RSC;
(f) KOAc-mediated Meerwein alkenylation: C. Molinaro, J. Mowat, F. Gosselin, P. D. O'Shea, J.-F. Marcoux, R. Angelaud and I. W. Davies, J. Org. Chem., 2007, 72, 1856–1858 CrossRef CAS PubMed.
-
(a) R. Francke and D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC;
(b) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed.
- Base-mediated thiolations: A. Kumar, B. S. Bhakuni, Ch. D. Prasad, S. Kumar and S. Kumar, Tetrahedron, 2013, 69, 5383–5392 CrossRef CAS; N. Mukherjee, T. Chatterjee and B. C. Ranu, J. Org. Chem., 2013, 78, 11110–11114 CrossRef PubMed; R.-Y. Tang, P. Zhong and Q.-L. Lin, Synthesis, 2007, 85–91 Search PubMed.
- S. Shaaban, A. Jolit, D. Petkova and N. Maulide, Chem. Commun., 2015, 51, 13902–13905 RSC.
- K. G. Back and A. A. Thomas, Am. Ind. Hyg. Assoc. J., 1963, 24, 23–27 CrossRef CAS PubMed.
- G. Landelle, A. Panossian and F. R. Leroux, Curr. Top. Med. Chem., 2014, 14, 941–951 CrossRef CAS PubMed.
-
(a) A. L. J. Beckwith, C. J. Easton and A. K. Serelis, J. Chem. Soc., Chem. Commun., 1980, 482 RSC;
(b) A. L. J. Beckwith, Tetrahedron, 1981, 37, 3073 CrossRef CAS;
(c) A. L. J. Beckwith and C. H. Schiesser, Tetrahedron, 1985, 41, 3925 CrossRef CAS;
(d) J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734 RSC;
(e) A. Srikrishna, J. Chem. Soc., Chem. Commun., 1987, 587 RSC.
-
(a) F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef PubMed;
(b)
M. P. Bertrand and C. Ferreri, in Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, 2001, pp. 485–504 Search PubMed.
- E. H. Krenske, W. A. Pryor and K. N. Houk, J. Org. Chem., 2009, 74, 5356–5360 CrossRef CAS PubMed.
- ESI-MS spectra also documented the formation of TEMPO-CH3 and TEMPO-CD3 adducts from standard reactions performed in DMSO and DMSO-d6, respectively.
- Electron affinity of the acetyloxy radical: X.-B. Wang, H.-K. Woo, L.-S. Wang, B. Minofar and P. Jungwirth, J. Phys. Chem. A, 2006, 110, 5047–5050 CrossRef CAS PubMed.
-
(a) A comprehensive review of radical reactions with arenediazonium salts: C. Galli, Chem. Rev., 1988, 88, 765–792 CrossRef CAS. Diazotate radicals in the presence of weak carboxylate bases:
(b) R. Huisgen and G. Horeld, Ann. Chem., 1949, 562, 137 CrossRef CAS;
(c) R. Huisgen and H. Nakaten, Ann. Chem., 1951, 573, 181 CrossRef CAS;
(d)
G. H. Williams, Homolytic Aromatic Substitution, Pergamon, London, 1960 Search PubMed;
(e) T. Kauffmann, H. O. Friestad and H. Henkler, Ann. Chem., 1960, 634, 64 CrossRef;
(f) C. Rüchardt and E. Merz, Tetrahedron Lett., 1964, 5, 2431 CrossRef;
(g) C. Rüchardt, B. Freudenberg and E. Merz, Spec. Publ. Chem. Soc., 1965, 154 Search PubMed;
(h) J. Besse and H. Zollinger, Helv. Chim. Acta, 1981, 64, 529 CrossRef CAS;
(i) R. M. Elofson, N. Cyr and J. K. Laidler, Tetrahedron Lett., 1990, 31, 7205 CrossRef CAS;
(j) S. Kindt, K. Wicht and M. R. Heinrich, Org. Lett., 2015, 17, 6122–6125 CrossRef CAS PubMed.
-
(a) This mechanism would require the deprotonation of the intermediate arenediazohydroxide by NaOAc which is very unlikely based on the reported pKa value of 6–8 (H2O): E. S. Lewis and M. P. Hanson, J. Am. Chem. Soc., 1967, 89, 6268–6272 CrossRef CAS;
(b) For diazotates in acidic/basic water, see: O. Macháčková and V. Štěrba, Collect. Czech. Chem. Commun., 1972, 37, 3313–3327 CrossRef.
-
(a)
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev. E.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed;
(b) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–251 CrossRef CAS.
-
(a) J. F. Bunnett, Acc. Chem. Res., 1978, 11, 413–420 CrossRef CAS;
(b) C. L. Øpstad, T.-B. Melø, H.-R. Sliwka and V. Partali, Tetrahedron, 2009, 65, 7616–7619 CrossRef;
(c) K. L. Handoo and A. Kaul, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1992, 31, 561 Search PubMed;
(d) R. A. Alonso and R. A. Rossi, Tetrahedron Lett., 1985, 26, 5763–5764 CrossRef CAS;
(e) N. Kornblum and A. S. Erickson, J. Org. Chem., 1981, 46, 1037–1039 CrossRef CAS.
- For selected works on dichalcogenide reactions, see:
(a) A. L. Stein, F. N. Bilheri and G. Zeni, Chem. Commun., 2015, 51, 15522 RSC;
(b) G. Sartori, J. S. S. Neto, A. P. Pesarico, D. F. Back, C. W. Nogueiraa and G. Zeni, Org. Biomol. Chem., 2013, 11, 1199 RSC;
(c) A. R. Rosario, K. K. Casola, C. E. S. Oliveira and G. Zeni, Adv. Synth. Catal., 2013, 355, 2960–2966 CrossRef CAS.
-
(a) G. Pandey, K. S. Sesha Poleswara Rao, D. K. Palit and J. P. Mittal, J. Org. Chem., 1996, 61, 6799–6804 CrossRef CAS PubMed;
(b) G. Pandey and S. R. Gadre, Acc. Chem. Res., 2004, 37, 201–210 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data of all new compounds. See DOI: 10.1039/c6ob02276f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a