
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Open-resorcinarenes, a new family of multivalent scaffolds†
Marco M. D.
Cominetti
a,
David L.
Hughes
b and
Susan E.
Matthews
*a
aSchool of Pharmacy, University of East Anglia, Norwich Research Park, Norwich, Norfolk NR4 7TJ, UK. E-mail: Susan.matthews@uea.ac.uk
bSchool of Chemistry, University of East Anglia, Norwich Research Park, Norwich, Norfolk NR4 7TJ, UK
First published on 6th October 2016
Abstract
A new family of multivalent ligand platforms, the open-resorcinarenes, has been prepared in a straightforward two-step reaction. Modification of the core gives a range of topologically diverse scaffolds; functionalisation confirms the versatility of this approach, as shown through the formation of an octacalixarene array.
Multivalency, in which biological responses are enhanced through several independent interactions from a single multiply-functionalised molecule is of considerable interest in the development of new biologically active molecules and therapeutic agents.1–6 Whilst original exploitation focused on the role of multi-functionalised platforms in carbohydrate–lectin interactions5–7 and condensation of DNA,4,8 their use in other systems such as the inhibition of enzymes9,10 is now gaining prominence.
Platforms functionalised with phenols have proven particularly attractive, as they can be readily adapted through O-alkylation, either directly, to prepare ligand platforms, or to serve as dendrimer cores8,11–13 for the formation of highly functionalised multivalent systems. From our studies using calixarenes 1,6,14 resorcin[4]arenes 25 and pillararenes 37 to investigate carbohydrate–lectin interactions, it is clear that topology is just as important as valency for high activity and selectivity between biological targets. Extending the available architectures through a new family of scaffolds will thus offer additional tools to develop selective and strongly binding ligands as well as providing dendrimer cores with new geometries.
We have now designed and synthesised a family of new platform molecules 6 (Fig. 1), which can be viewed as “open” analogues of resorcin[4]arenes. Through varying both the regiochemistry and nature of the core, we envisaged that scaffolds with different topologies and valency could be obtained. Whilst Nangia and co-workers15 have described a series of H-shaped tectons 4, and similar catechol compounds (5) have been patented for applications as photo-resists,16 resorcinol-based scaffolds have only been postulated by Nishikubo to be intermediates in the formation of more complex waterwheel17,18 and triple-ringed15 structures.
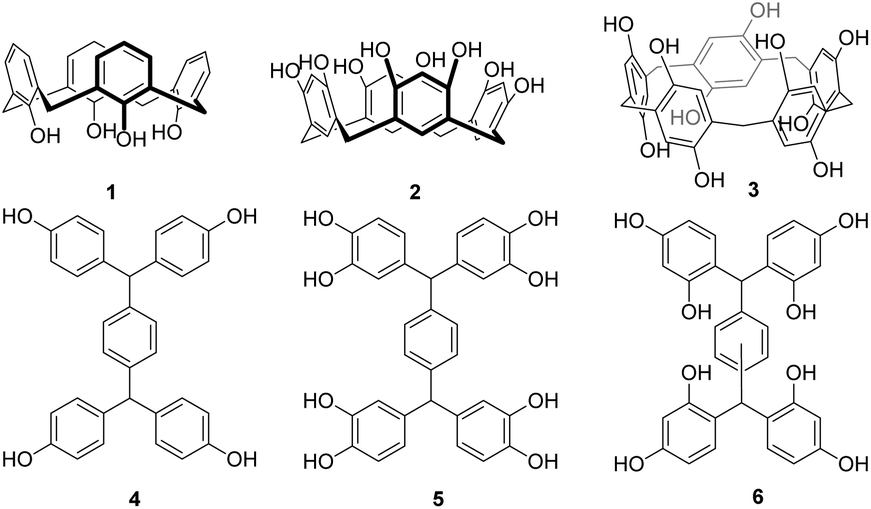 | ||
| Fig. 1 Phenol-based platforms. | ||
Here, we disclose the readily scalable synthesis of a new family of open-resorcinarenes 6, the crystal structures of these platforms and their facile functionalisation towards novel dendrimers.
As expected, access to open-resorcinarene 6via a simple reaction of resorcinol with benzene-1,4-dicarboxaldehyde proved impossible, owing to the formation of polymeric condensation products. Thus a two-step procedure was developed, where halogen atoms are first used to block the additional reactive site(s) during condensation and are subsequently removed to yield the desired platforms. Our initial studies focused on the preparation of 7a–c incorporating a phenyl bridge, through reaction with the three regioisomeric benzenedicarboxaldehydes. Two methods were considered: the classical resorcin[4]arene synthesis (aq. HCl (37%)/ethanol 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 70 °C (Method 1))19 and a milder alternative, (8% methanesulfonic acid in 1:1 diethyl ether/dichloromethane (1:1) (Method 2)).20
:1 at 70 °C (Method 1))19 and a milder alternative, (8% methanesulfonic acid in 1:1 diethyl ether/dichloromethane (1:1) (Method 2)).20
Condensation of 4-chlororesorcinol and benzene-1,4-dicarboxaldehyde gave the desired core featuring eight phenols 7a (Scheme 1) in excellent yield using both approaches (Method 1 67%, Method 2 87%). However, Method 2 was more attractive as the product could be obtained in high yields by simple crystallisation from the reaction mixture. Conversely, for the meta-analogue 7b (Scheme 1) Method 1 (91%) was preferred, yielding the scaffold without recourse to chromatography. Preparation of the ortho-analogue 7c (Method 1 80%, Method 2 94%) led to the identification of an unexpected material which was not the postulated intermediate aldehyde 8 (Scheme 2). Isolation, full NMR investigation, HRMS and single-crystal analysis confirmed that the product was a previously unreported bicyclic structure, acetal 9, as shown in Fig. 2/Scheme 2.
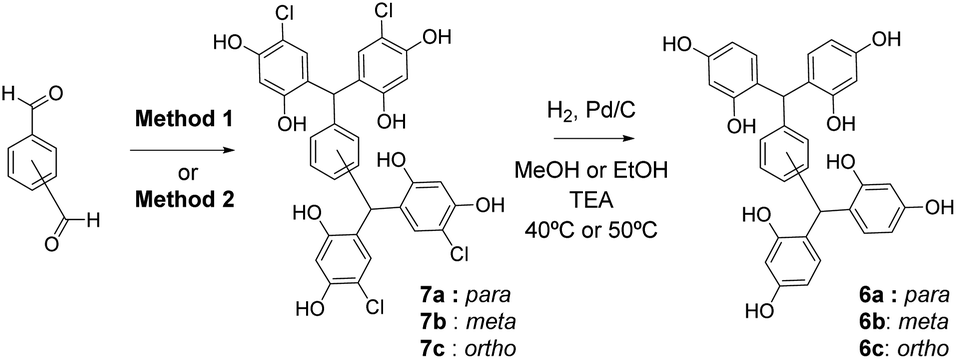 | ||
| Scheme 1 Synthesis of open-resorcinarenes 6a–c. Method 1: HCl (37%)/ethanol 1:1, 70 °C. Method 2: 8% MeSO3H in 1:1 diethyl ether/dichloromethane 1:1. | ||
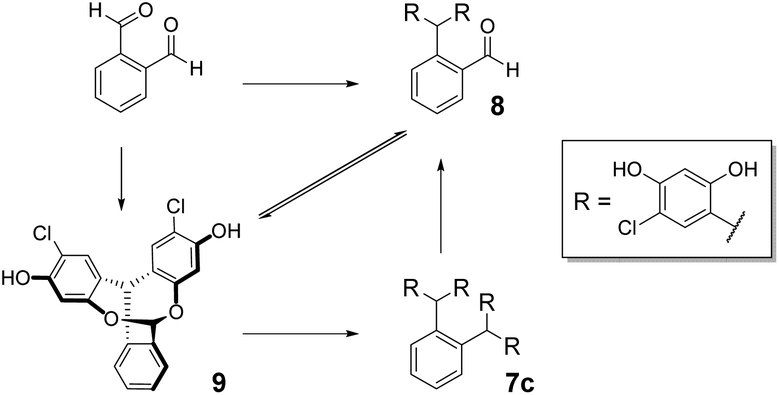 | ||
| Scheme 2 Synthesis of open-resorcinarene 7c and equilibrium between aldehyde 8 and its closed acetal 9. | ||
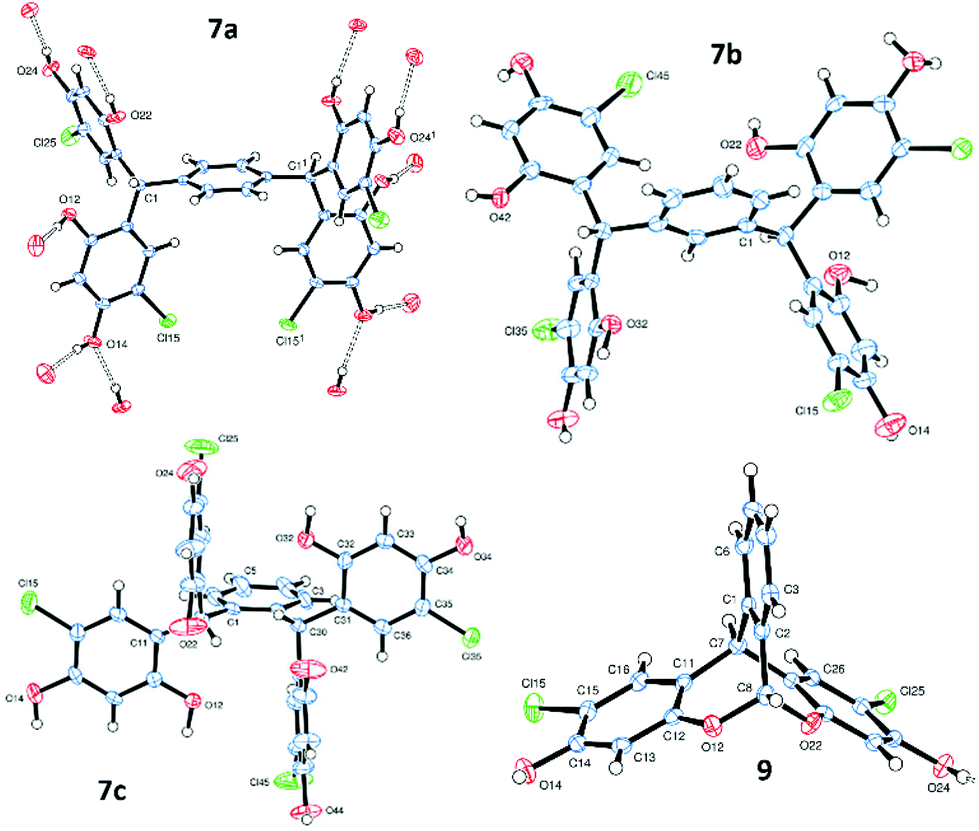 | ||
| Fig. 2 ORTEP views from single crystals of 7a, 7b, 7c and 9. | ||
To investigate the formation of this unexpected product further, benzene-1,2-dicarboxaldehyde was treated with only two equivalents of 4-chlororesorcinol. 1H NMR spectroscopy revealed that 8 and 9 were present, in a stable 1.6:1 ratio, after both 6 and 24 h (ESI Fig. 9†). Moreover, when the isolated acetal was treated with an excess of 4-chlororesorcinol, it gave tetramer 7c as the single product and 1H NMR spectroscopy during the reaction showed the presence of the open aldehyde 8 in the reaction mixture (ESI Fig. 10†). These results indicate that 9 is involved in an equilibrium with the intermediate aldehyde and that there is free interconversion between the two forms under Method 2 reaction conditions.
Single crystals of 7a–c, suitable for X-ray diffraction,21 were obtained directly from the reaction work-up (Fig. 2 and ESI†). All three platforms display extensive intermolecular hydrogen bonding between the units, which are arranged in ladder-type structures, similar to those observed for other H-shaped tectons,15 indicating their potential for crystal engineering applications.22 In the case of 7b, two ladders are H-bonded together about centres of symmetry (therefore running in opposite directions). Additionally, an extensive network of H-bonded solvent (diethyl ether and/or methanol) surrounds each individual ladder.
Subsequently, dehalogenation of the core structures 7, to give the open-resorcinarenes 6, was achieved by hydrogenolysis in methanol or ethanol, with Pd/C and triethylamine (Scheme 1). Triethylamine was essential to neutralise the generated HCl, to avoid fragmentation by acid-catalysed reversal of the scaffold-forming condensation.
Elaboration of platform 6a was achieved by propargylation under standard conditions, to give the octa-alkyne 10 (Scheme 3).
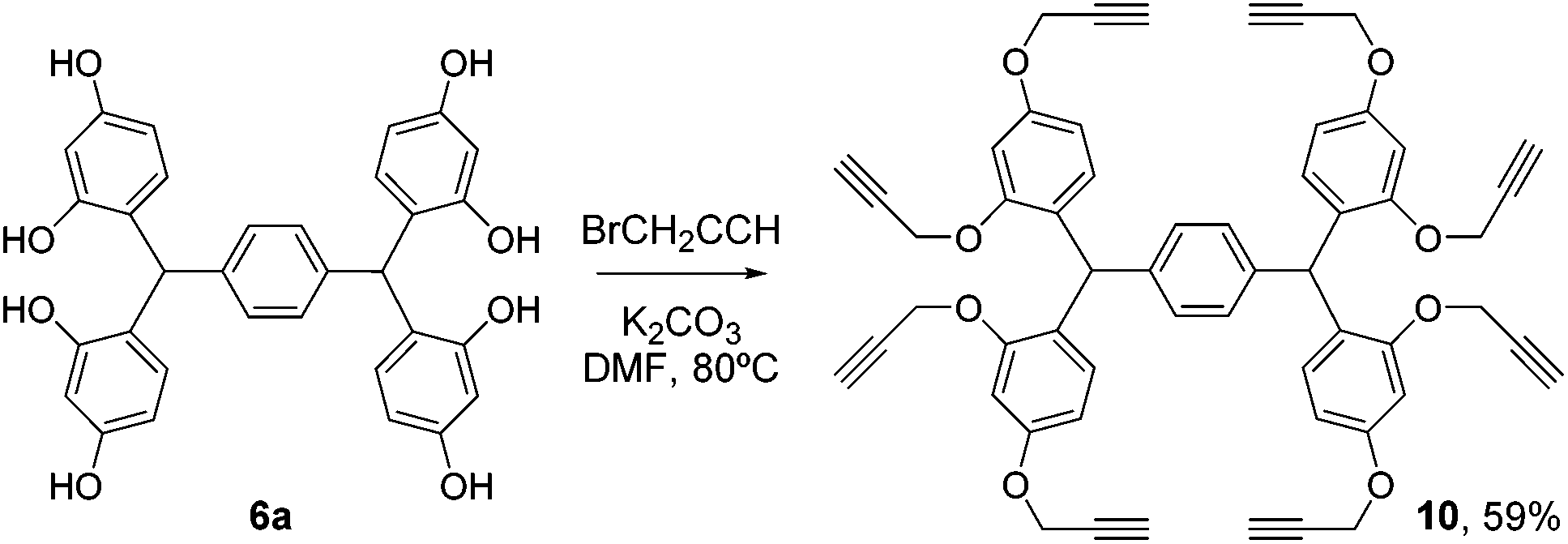 | ||
| Scheme 3 Elaboration of 6a to give the clickable scaffold 10. | ||
In order to demonstrate the versatility of this approach to preparation of diverse platforms and vary the ligand topology further, Suzuki couplings23 were used to prepare biphenyl dialdehydes 11 and 12. Using Method 2, these gave rise to the desired tetramers 13 and 14 after crystallisation, which were subsequently dehalogenated to give stretched open-resorcinarenes 15 and 16 (Scheme 4).
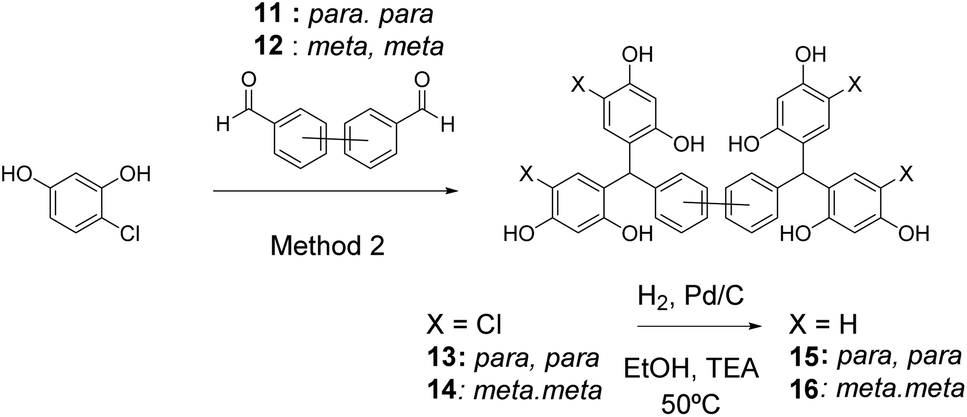 | ||
| Scheme 4 Synthesis of biaryl-core open-resorcinarenes 14 and 15. | ||
Alternatively, the scaffolds can be isolated in their functionalised form where crystallisation does not yield pure polyphenols. Thus the rigid dodecavalent platform 18 was prepared through condensation of benzene-1,3,5-tricarboxaldehyde with 4-chlororesorcinol (Method 2) to give the expected hexamer 17, which was subsequently dehalogenated and directly propargylated, to yield the functionalised core 18, which was easily purified by flash chromatography (Scheme 5).
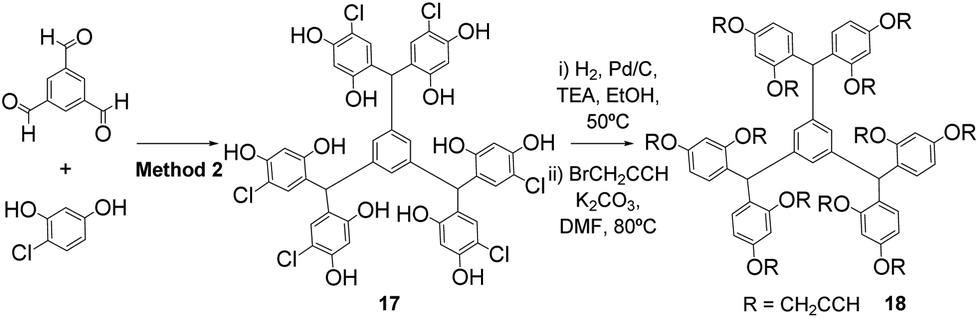 | ||
| Scheme 5 Synthesis of dodecavalent open-resorcinarene 18. | ||
To demonstrate the suitability of our new scaffolds as multivalent platforms, 6a was used in the preparation of a functionalised octacalixarene dendrimer, featuring thirty-two surface amines, through a Cu-catalysed azide–alkyne cycloaddition (CuAAC). Octa-yne 10 reacted readily with sixteen equivalents of azidopropoxy-calixarene 21 (ESI†) in the presence of CuSO4 and sodium ascorbate (Scheme 6 and ESI†) to give a high yield of the octacalixarene dendrimer, featuring eight calixarene units, after column chromatography.
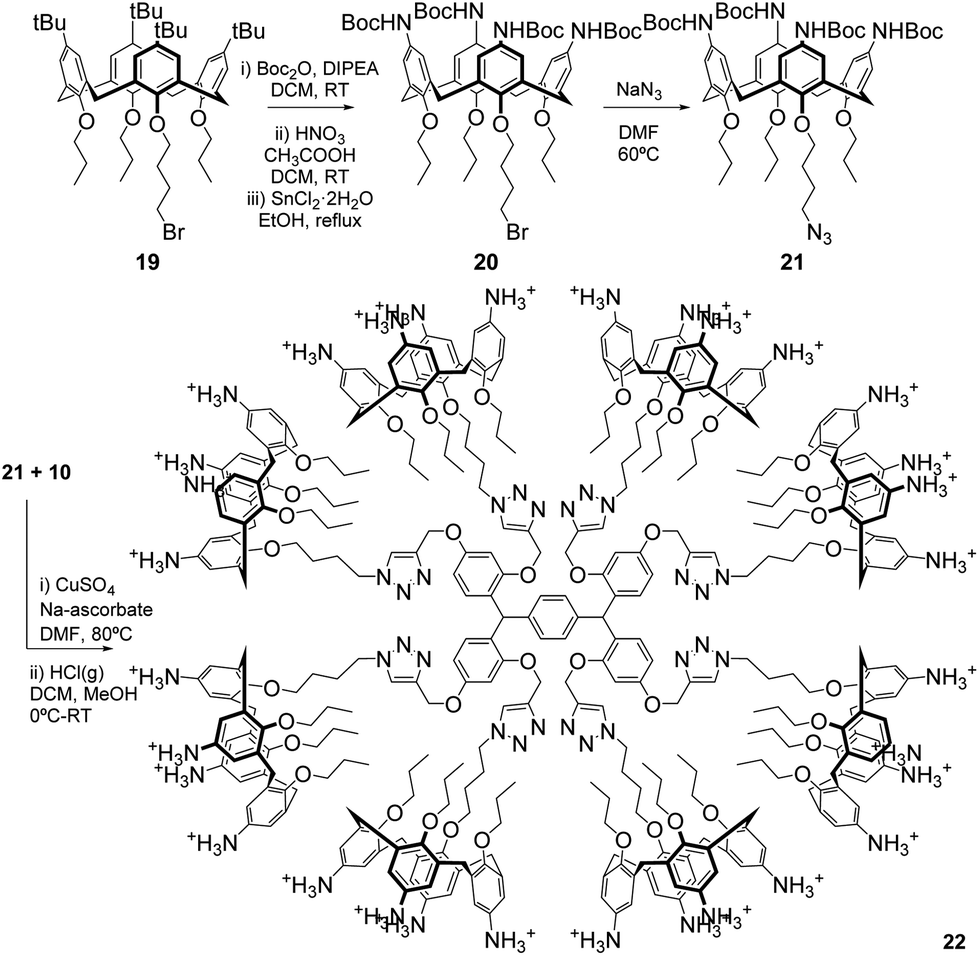 | ||
| Scheme 6 Synthesis of azidopropoxy-calix[4]arene 21 and octacalixarene 22. | ||
Careful control of the temperature during deprotection with HCl led to the target array 22 in 79% yield after RP-HPLC purification, showing that deprotection in acidic conditions can be achieved, despite the apparent acid-sensitivity of the core.
In conclusion, a straightforward two-step synthesis to access a novel family of phenolic scaffolds has been developed. By use of a halogen protection strategy to control condensation, open-resorcinarene analogues with fixed geometries and valences can be prepared in a scalable manner, without chromatographic purifications, from readily accessible and cheap starting materials. Their versatility for exploitation in multivalent biological applications has been demonstrated through the preparation of a novel octacalixarene dendrimer. Additionally, single-crystal analysis demonstrates that the molecules form long-range ladder structures in the solid state, suggesting potential in crystal engineering.
The authors would like to thank the EPSRC UK National Mass Spectrometry Facility at Swansea University for provision of high resolution mass spectrometry.
Notes and references
- A. Dondoni and A. Marra, Chem. Rev., 2010, 110, 4949 CrossRef CAS PubMed.
- F. Sansone and A. Casnati, Chem. Soc. Rev., 2013, 42, 4623 RSC.
- R. Joseph, A. Naugolny, M. Feldman, I. M. Herzog, M. Fridman and Y. Cohen, J. Am. Chem. Soc., 2016, 138, 754 CrossRef CAS PubMed.
- L. Baldini, A. Casnati, F. Sansone and R. Ungaro, Chem. Soc. Rev., 2007, 36, 254 RSC.
- Z. H. Soomro, S. Cecioni, H. Blanchard, J.-P. Praly, A. Imberty, S. Vidal and S. E. Matthews, Org. Biomol. Chem., 2011, 9, 6587 CAS.
- S. Cecioni, R. Lalor, B. Blanchard, J.-P. Praly, A. Imberty, S. E. Matthews and S. Vidal, Chem. – Eur. J., 2009, 15, 13232 CrossRef CAS PubMed.
- K. Buffet, I. Nierengarten, N. Galanos, E. Gillon, M. Holler, A. Imberty, S. E. Matthews, S. Vidal, S. P. Vincent and J.-F. Nierengarten, Chem. – Eur. J., 2016, 22, 2955 CrossRef CAS PubMed.
- R. Lalor, J. L. DiGesso, A. Mueller and S. E. Matthews, Chem. Commun., 2007, 46, 4907 RSC.
- S. G. Gouin, Chem. – Eur. J., 2014, 20, 11616 CrossRef CAS PubMed.
- P. Compain and A. Bodlenner, ChemBioChem, 2014, 15, 1239 CrossRef CAS PubMed.
- R. Lalor, A. P. Gunning, V. J. Morris and S. E. Matthews, Chem. Commun., 2010, 46, 8665 RSC.
- G. Gattuso, G. Grasso, N. Marino, A. Notti, A. Pappalardo, S. Pappalardo and M. F. Parisi, Eur. J. Org. Chem., 2011, 5696 CrossRef CAS.
- Y. Rudzevich, V. Rudzevich, C. Moon, G. Brunklaus and V. Bohmer, Org. Biomol. Chem., 2008, 6, 2270 CAS.
- A. M. Boukerb, A. Rousset, N. Galanos, J.-B. Méar, M. Thépaut, T. Grandjean, E. Gillon, S. Cecioni, C. Abderrahmen, K. Faure, D. Redelberger, E. Kipnis, R. Dessein, S. Havet, B. Darblade, S. E. Matthews, S. de Bentzmann, B. Guéry, B. Cournoyer, A. Imberty and S. Vidal, J. Med. Chem., 2014, 57, 10275 CrossRef CAS PubMed.
- R. Thakuria, B. Sarma and A. Nangia, New J. Chem., 2010, 34, 623 RSC.
- M. Kawada, M. Kashiwagi and K. Koito, JPN Patent04301851A, 1992 Search PubMed.
- H. Kudo, R. Hayashi, K. Mitani, T. Yokozawa, N. C. Kasuga and T. Nishikubo, Angew. Chem., Int. Ed., 2006, 45, 7948 CrossRef CAS PubMed.
- N. Niina, H. Kudo, H. Oizumi, T. Itani and T. Nishikubo, Thin Solid Films, 2013, 534, 459 CrossRef CAS.
- E. Bouthenet, K.-B. Oh, S. Park, N. K. Nagi, H.-S. Lee and S. E. Matthews, Bioorg. Med. Chem. Lett., 2011, 21, 7142 CrossRef CAS PubMed.
- J. P. Bacci, A. M. Kearney and D. L. Van Vranken, J. Org. Chem., 2005, 70, 9051 CrossRef CAS PubMed.
- X-ray analyses of compounds 7a–c and 9 are described in the ESI† and the results have been deposited at the CCDC, codes 1497840–1497843.
- C. B. Aakeroy, S. Panikkattu, P. D. Chopade and J. Desper, CrystEngComm, 2013, 15, 3125 RSC.
- N. Kuhnert, C. Patel and F. Jami, Tetrahedron Lett., 2005, 46, 7575 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Methods, characterization data and crystallographic data. CCDC 1497840–1497843. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob02164f |
| This journal is © The Royal Society of Chemistry 2016 |