
Dehydrative glycosylation with cyclic phosphonium anhydrides†
Rajendar
Dyapa
,
Lance T.
Dockery
and
Maciej A.
Walczak
 *
*
Department of Chemistry and Biochemistry, University of Colorado, Boulder, CO 80309, USA. E-mail: maciej.walczak@colorado.edu
First published on 5th September 2016
Abstract
Cyclic phosphonium anhydrides generated from bis-phosphine oxides and trifluoromethanesulfonic anhydride are shown as general coupling reagents in a dehydrative glycosylation reaction of C1-hemiacetals. This reaction protocol is characterized by a broad substrate scope and high yields, including reactions of O-, C-, N-, and S-based nucleophiles with furanose, pyranose, and deoxysugar donors.
The glycosidic bond is one of the most abundant structural elements found in natural biopolymers,1–3 and efficient and general methods for the synthesis of oligosaccharides and glycoconjugates are critical for realizing the full potential of saccharides in biology and medicine.4 Among a gamut of methodologies for stereoselective preparation of saccharides, dehydrative glycosylation5 plays a special role due to the unique properties of anomeric alcohols, such as the ease of preparation6,7 and the mild activation protocols often compatible with other glycosyl donors.8–16 Despite these seemingly useful properties of C1-hemiacetals, established dehydrative methods often lead to variable yields and selectivities. Direct conversion of the hydroxyl functionality using Brønsted or Lewis acid conditions (Fischer glycosylation, Scheme 1) or in situ activation with electrophilic reagents such as pyridinium salts,17–19 sulfoxonium triflates,20–24 carboxylic acid anhydrides,25–27 triflic anhydride,28,29 Appel reagents,30 or transition metals (Ti,31,32 Sn,33,34 Cu,35 Bi36) result in variable yields and selectivities (Scheme 1). Moreover, use of in situ activating agents is often limited to O-based nucleophiles. With the above considerations in mind, we explored the utility of electrophilic phosphorus reagents37 in dehydrative glycosylation reactions. Herein, we describe an efficient method applicable to the preparation of O-, C-, N-, and S-substituted glycosides using cyclic phosphonium anhydrides as coupling reagents. These reagents promoted the synthesis of glycosides in high yields and selectivities, and, unlike previous methods, the glycosyl donors and the coupling reagent can be recovered and reused.
 | ||
| Scheme 1 Selected dehydrative glycosylation reactions with C1-hemiacetals. | ||
Our interest in phosphonium salts was sparked by a report from Mukaiyama,38 who investigated the use of the Hendrickson's ‘POP’ reagent (Ph3P+OP+Ph3·2TfO−)39–42 in a glycosylation reaction of furanoses. More recently, the use of the Hendrickson's reagent was reported in reactions of glucopyranoses, indicating that the utility of the phosphonium anhydrides can be extended to pyranose donors.43 Yet, there were limitations to this protocol including a major side reaction from dimerization of the glycosyl donor and diminished reactivity of substrates substituted with deactivating protective groups. Moreover, the Hendrickson's reagent failed to promote glycosylation of a ribose donor with a moderately reactive acceptor.44 We reasoned that cyclic phosphonium anhydrides45 could be more suitable for glycosylations due to their increased electrophilic character (as measured by the Gutman–Beckett method)46,47 and the ease of activation of the anomeric alcohol. Because of their broad functional group tolerance, convenient methods of formation, and facile modulation of structural and electronic properties, this class of reagents offers advantages over other dehydrative reagents. Furthermore, activation of the anomeric alcohol by formation of a mixed anhydride would lead to an intermediate with a cationic nature, thus diminishing its propensity to undergo unwanted dimerization because exchange at the phosphonium centre with a glycosyl acceptor would be disfavored.48
To test the above hypothesis, we prepared cyclic phosphonium anhydrides (Scheme 2). Bis-phosphine oxides were treated with Tf2O at 0 °C, cleanly generating the corresponding cyclic anhydrides 7–16 within minutes, as determined by 31P NMR measurements.45 The anhydrides 7 and 9 were also isolated by precipitation with Et2O and analysed by NMR spectroscopy, and the material obtained via isolation was spectroscopically identical to the intermediates generated in situ. For practical reasons, however, the phosphonium salts in this study were freshly prepared and used as a solution in CH2Cl2.
 | ||
| Scheme 2 Generation of cyclic phosphonium anhydrides from bis-phosphine bis-oxides. | ||
With a series of phosphonium salts in hand, we started our methodological study by investigating glycosylation reactions of 2,3,4,6-tetra-O-benzyl-D-glucopyranose 17 (α/β 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with benzyl or cyclohexyl alcohol as a model system (Table 1). Glycosyl donor 17 was treated with cyclic phosphonium anhydrides 7–16 in the presence of 2,4,6-tri-tert-butylpyridine (TTBP), and, after 20 minutes at 0 °C, reacted with an alcohol acceptor. We found that the cyclic alkyl chain reagents successfully promoted the glycosylation reactions, and salts 7 and 9 gave the best overall yields. As the size of the phosphonium salt had little impact on the diastereoselectivity, we wondered if the use of chiral phosphonium anhydrides might lead to preferential formation of a given glycosyl anomer. To test this hypothesis, we chose two phosphonium salts 15 and 16, which were prepared under the standard conditions from the corresponding bis-oxides (entries 12–15). The yields for the glycosylation reactions were largely dependent on the type of a reagent used (either enantiomer of 16 gave better yields than 15) rather than their absolute configuration, and the chiral phosphonium salts had little impact on the diastereoselectivity of these reactions. To compare the reactivity of the cyclic phosphonium anhydrides with an acyclic alkyl analogue, we generated a salt from n-Bu3PO/Tf2O, but this salt failed to provide 18 under the standard conditions. Based on these optimization studies we elected reagent 9 (generated from 1,4-bis(diphenylphosphino)butane bis-oxide, DPPBO2) as the optimal reagent. Reaction of 1 equiv. of glycosyl donor with 1.5 equiv. of DPPBO2, 1.25 equiv. of Tf2O, and 3 equiv. of TTBP cleanly afforded the glycoside 18b in an excellent yield (91%) and 1:1.2 α/β diastereselectivity (entry 5).21 In order to improve the anomeric selectivity, we hypothesized that the putative phosphonium intermediate can undergo anomerization in the presence of an iodide or bromide salt and can result in the formation of a more reactive β anomeric halide, similar to the protocol by Lemieux.49
:1) with benzyl or cyclohexyl alcohol as a model system (Table 1). Glycosyl donor 17 was treated with cyclic phosphonium anhydrides 7–16 in the presence of 2,4,6-tri-tert-butylpyridine (TTBP), and, after 20 minutes at 0 °C, reacted with an alcohol acceptor. We found that the cyclic alkyl chain reagents successfully promoted the glycosylation reactions, and salts 7 and 9 gave the best overall yields. As the size of the phosphonium salt had little impact on the diastereoselectivity, we wondered if the use of chiral phosphonium anhydrides might lead to preferential formation of a given glycosyl anomer. To test this hypothesis, we chose two phosphonium salts 15 and 16, which were prepared under the standard conditions from the corresponding bis-oxides (entries 12–15). The yields for the glycosylation reactions were largely dependent on the type of a reagent used (either enantiomer of 16 gave better yields than 15) rather than their absolute configuration, and the chiral phosphonium salts had little impact on the diastereoselectivity of these reactions. To compare the reactivity of the cyclic phosphonium anhydrides with an acyclic alkyl analogue, we generated a salt from n-Bu3PO/Tf2O, but this salt failed to provide 18 under the standard conditions. Based on these optimization studies we elected reagent 9 (generated from 1,4-bis(diphenylphosphino)butane bis-oxide, DPPBO2) as the optimal reagent. Reaction of 1 equiv. of glycosyl donor with 1.5 equiv. of DPPBO2, 1.25 equiv. of Tf2O, and 3 equiv. of TTBP cleanly afforded the glycoside 18b in an excellent yield (91%) and 1:1.2 α/β diastereselectivity (entry 5).21 In order to improve the anomeric selectivity, we hypothesized that the putative phosphonium intermediate can undergo anomerization in the presence of an iodide or bromide salt and can result in the formation of a more reactive β anomeric halide, similar to the protocol by Lemieux.49
|
|
||||
|---|---|---|---|---|
| Entry | Anhydrideb | ROH | Yieldc | α:βd |
| a General reaction optimization protocol: 17 (1 equiv.), ROH (1.5 equiv.), bis-phosphine oxide (1.5 equiv.), Tf2O (1.25 equiv.), TTBP (3 equiv.), CH2Cl2, 4 Å MS, 0 °C. b Phosphonium anhydrides were generated in situ. c Yields refer to isolated, analytically pure material. d Determined by 1H NMR of the crude reaction mixture. e TBAI (1 equiv.) was added and (CH2Cl)2 was used as a solvent. | ||||
| 1 | 7 | CyOH | 56% | 1.8:1 |
| 2 | 7 | BnOH | 69% | 1.2:1 |
| 3 | 8 | CyOH | <5% | N.D. |
| 4 | 9 | CyOH | 83% | 1:1.9 |
| 5 | 9 | BnOH | 91% | 1:1.2 |
| 6e | 9 | CyOH | 50% | α only |
| 7 | 10 | CyOH | 56% | 1:2 |
| 8 | 11 | CyOH | 72% | 1:2 |
| 9 | 12 | CyOH | <5% | N.D. |
| 10 | 13 | CyOH | <5% | N.D. |
| 11 | 14 | CyOH | <5% | N.D. |
| 12 | (R)-15 | CyOH | 36% | 1:1.5 |
| 13 | (S)-15 | CyOH | 23% | 1:2.2 |
| 14 | (R)-16 | CyOH | 54% | 1:1.8 |
| 15 | (S)-16 | CyOH | 75% | 1:1.7 |

Thus, to fine-tune the reaction conditions, a solution of 1 equiv. of TBAI (tetra-n-butylammonium iodide) was added to the activated anomeric phosphonium intermediate together with the alcohol acceptor. Although the reaction yield was diminished, exclusive α selectivity of 18a was observed (entry 6). Other activation modes of DPPBO2 (e.g., TMSOTf, Ms2O) as well as the use of bis-phosphine monoxide (DPPE monoxide) in combination with Tf2O and TTBP resulted in trace amounts of the expected glycosides 18. Due to the limited solubility of the phosphonium anhydrides, solvents such as PhMe, THF, Et2O, and MeCN were not compatible with the glycosylation reactions, and 18 was formed in significantly lower yields. The nature of the base is also important – exclusion of the base altogether as well as the use of smaller bases (Et3N, pyridine) led to diminished yields of 18, rendering TTBP the reagent of choice for this reaction. The progress of the glycosylation reaction can be easily monitored visually because a very polar phosphine bis-oxide by-product (DPPBO2) precipitates once the reaction is complete. Furthermore, to demonstrate the scalability of the dehydrative protocol, a glycosylation reaction of 17 with Cy–OH was conducted with 1.2 g of DPPBO2 and 2,3-di-tert-butyl-4-methylpyridine as an inexpensive alternative to TTBP to afford the product 18a in 83% isolated yield using the optimized conditions. The phosphine bis-oxide DPPBO2 was recovered from the reaction mixture in 91% yield via simple precipitation with hexane and filtration prior to chromatographic purification and reused in subsequent reactions without any loss of activity.
Having established a general glycosylation protocol, the scope of the reaction was investigated (Table 2). We found that deoxysugars (fucose, entry 1; arabinose, entries 2 and 3) and ribofuranose50 (entry 4) are excellent substrates for the glycosylation reactions with primary and secondary alcohol acceptors. Furthermore, when the benzylidene mannose donor 27 was activated under the optimized conditions, preferential formation of the β-anomer (entry 5) was observed,51–58 whereas 2-O-benzoyl-mannose gave the expected α-anomer (entries 7 and 8). Similarly, phenolic acceptor was also tolerated in this reaction giving rise to the α anomer 33 in 98% yield. However, 2-O-benzyl mannose resulted in a mixture of anomers (entry 10). Curiously, the sulfoxide donor was also smoothly activated by the phosphonium salt (entry 11),59 whereas the anomeric thioether was tolerated in this protocol (entry 7) opening a possibility of a sequential one-pot glycosylation via orthogonal activation with C1-hemiacetals.60 In all examples tested under the dehydrative conditions, we were unable to detect homocoupling products, and the excess of the acceptor could be recovered.


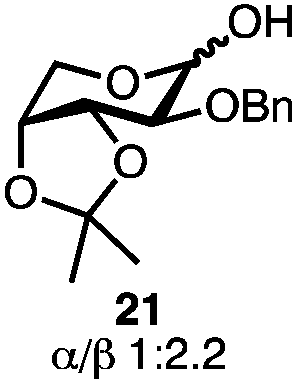







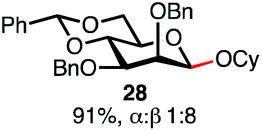





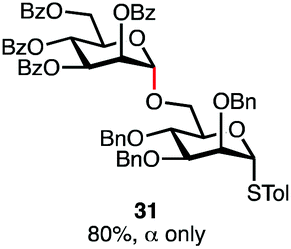






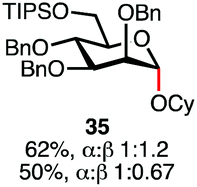
Table 3 lists reactions of C1-hemiacetals with sulfur-, nitrogen- and carbon-based nucleophiles promoted by dicationic phosphonium salt 9. Excellent to good selectivities were observed even without a participating group at C2 (entries 1 and 2), giving primarily the α-anomers for small nucleophiles and β-anomers for aromatic acceptors (entries 3 and 4). These reactions represent a rare example where anomeric alcohols can be directly converted into unnatural glycosides in excellent yields and selectivities. The reaction with a thiol is of interest as the majority of chemical methods to install the anomeric thioether rely on a participating group at C2 resulting in predominantly β anomers.
| Entry | Donor | Product | Yieldb | α:β |
|---|---|---|---|---|
| a Reactions were carried out using the optimized protocol from Table 1 (entry 5). b Yields refer to isolated material. c TolSH used as a nucleophile. d TMSN3 used as a nucleophile. e Indole used as a nucleophile. f Benzotriazole used as a nucleophile. | ||||
| 1c |

|

|
96% | 6:1 |
| 2d |

|

|
94% | 12:1 |
| 3e |

|

|
88% | β only |
| 4f |

|
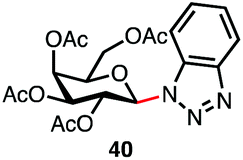
|
75% | β only |
To ascertain the reactivity of cyclic phosphonium salts, the progress of the dehydrative glycosylation reaction was monitored by 31P NMR spectroscopy.61 Addition of alcohol 17 to the cyclic salt 9 resulted in the formation of two species, which were assigned as the phosphonium anomers (see, ESI†). The ratio of α/β anomers in this reaction remained unchanged (CD2Cl2, rt, 24 h), and external additives such as phosphine oxides (Ph3PO) had no influence on the anomer distribution. Addition of cyclohexanol led to immediate formation of the expected glycoside 18a and phosphine oxide (DPPBO2). Based on the above observations, the proposed mechanism of glycosylation with cyclic phosphonium anhydrides is presented in Scheme 3. Addition of a glycoside hemiacetal 41 to cyclic phosphonium anhydride 42 generates a mixture of anomeric intermediates 43. The 31P NMR also supports the notion that 43 exists in a cyclic form as we were unable to detect any signals corresponding to the putative phosphine oxide 45. Addition of the alcohol acceptor R′-OH leads then to the formation of the glycoside 44. The formation of the glycosidic bond can occur through a direct displacement at the anomeric carbon atom and release of DPPBO2 (low temperature experiments did not reveal the presence of anomeric triflate). Alternatively, in the presence of a C2 participating substituent, the delivery of nucleophile will be directed by the neighbouring group according to an established mechanism. On the other hand, reactions of C1-hemiacetals with acyclic phosphonium anhydrides 46 proceed via formation of the intermediate 47, which can react with alcoholic acceptor in a fashion similar to the cyclic analogs to furnish glycoside 44. However, the activated glycosidic acceptor 47 may undergo a competitive reaction with R′-OH to generate 48 and 41, which can engage in a reaction with 47 to form a glycoside dimer product. The use of a cyclic salt is therefore advantageous as it prevents undesired glycoside homocoupling reactions by minimizing the exchange reactions at the phosphonium centre in the activated glycosyl donor.
 | ||
| Scheme 3 Proposed mechanism of dehydrative glycosylation. | ||
To summarize, we have developed a dehydrative glycosylation method using phosphonium anhydrides and demonstrated its utility in a range of anomeric alcohol substrates. In contrast to previous methods, cyclic phosphonium anhydrides are versatile reagents for the preparation of O-, N-, C-, and S-glycosides in excellent yields, can tolerate deactivated glycosyl donors and the undesired dimerization of glycosyl substrates previously observed with Hendrickson's reagent was suppressed. This first application of cyclic phosphonium anhydrides in a glycosylation reaction indicates that the dicationic nature of these reagents suppresses undesired exchange reactions and opens up possibilities for the future use of the phosphonium salts in the synthesis of oligosaccharides. Moreover, unlike previous dehydrative methods, the glycosyl donor can be recovered unreacted and the coupling reagent (phosphine bis-oxide) reused. Further studies on the mechanism and scope of the dehydrative method will be reported in due course.
This work was generously supported by the University of Colorado at Boulder.
Notes and references
- P. H. Seeberger, Chem. Soc. Rev., 2008, 37, 19–28 RSC.
- X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS PubMed.
- C.-H. Hsu, S.-C. Hung, C.-Y. Wu and C.-H. Wong, Angew. Chem., Int. Ed., 2011, 50, 11872–11923 CrossRef CAS PubMed.
- A. V. Demchenko, Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance, Wiley-VCH Verlag, Weinheim, Germany, 2008 Search PubMed.
- D. Gin, J. Carbohydr. Chem., 2002, 21, 645–665 CrossRef CAS.
- M. M. Sim, H. Kondo and C. H. Wong, J. Am. Chem. Soc., 1993, 115, 2260–2267 CrossRef CAS.
- K. C. Nicolaou, S. A. Snyder, A. Z. Nalbandian and D. A. Longbottom, J. Am. Chem. Soc., 2004, 126, 6234–6235 CrossRef CAS PubMed.
- S. Hanessian and B. Lou, Chem. Rev., 2000, 100, 4443–4464 CrossRef CAS PubMed.
- J. D. C. Codée, L. H. Hossain and P. H. Seeberger, Org. Lett., 2005, 7, 3251–3254 CrossRef PubMed.
- J. P. Issa and C. S. Bennett, J. Am. Chem. Soc., 2014, 136, 5740–5744 CrossRef CAS PubMed.
- J. M. Nogueira, S. H. Nguyen and C. S. Bennett, Org. Lett., 2011, 13, 2814–2817 CrossRef CAS PubMed.
- J. P. Issa, D. Lloyd, E. Steliotes and C. S. Bennett, Org. Lett., 2013, 15, 4170–4173 CrossRef CAS PubMed.
- K. Shinkiti, H. Hisamitsu, S. Sonoko, H. Motoko, N. Teiko, M. Chika, F. Misuzu, G. Ayano, S. Tomoko, O. Masato, Z. Shonosuke, Y. Kazuo and T. Fumiya, Bull. Chem. Soc. Jpn., 1995, 68, 2331–2348 CrossRef.
- K. Shinkiti, H. Yoshio and Z. Shonosuke, Chem. Lett., 1975, 4, 587–588 CrossRef.
- J. M. Nogueira, M. Bylsma, D. K. Bright and C. S. Bennett, Angew. Chem., Int. Ed., 2016, 55, 10088–10092 CrossRef CAS PubMed.
- K. A. D'Angelo and M. S. Taylor, J. Am. Chem. Soc., 2016, 138, 11058–11066 CrossRef PubMed.
- T. Mukaiyama, S.-I. Shoda, T. Nakatsuka and K. Narasaka, Chem. Lett., 1978, 7, 605–608 CrossRef.
- T. Mukaiyama, Angew. Chem., Int. Ed. Engl., 1979, 18, 707–721 CrossRef.
- T. Mukaiyama, Y. Hashimoto, Y. Hayashi and S.-I. Shoda, Chem. Lett., 1984, 13, 557–560 CrossRef.
- B. A. Garcia and D. Y. Gin, J. Am. Chem. Soc., 2000, 122, 4269–4279 CrossRef CAS.
- B. A. Garcia, J. L. Poole and D. Y. Gin, J. Am. Chem. Soc., 1997, 119, 7597–7598 CrossRef CAS.
- H. M. Nguyen, Y. Chen, S. G. Duron and D. Y. Gin, J. Am. Chem. Soc., 2001, 123, 8766–8772 CrossRef CAS PubMed.
- T. A. Boebel and D. Y. Gin, Angew. Chem., Int. Ed., 2003, 42, 5874–5877 CrossRef CAS PubMed.
- T. A. Boebel and D. Y. Gin, J. Org. Chem., 2005, 70, 5818–5826 CrossRef PubMed.
- K. S. Kim, D. B. Fulse, J. Y. Baek, B.-Y. Lee and H. B. Jeon, J. Am. Chem. Soc., 2008, 130, 8537–8547 CrossRef CAS PubMed.
- K. S. Kim, Y. J. Lee, H. Y. Kim, S. S. Kang and S. Y. Kwon, Org. Biomol. Chem., 2004, 2, 2408–2410 CAS.
- J. Y. Baek, B.-Y. Lee, R. Pal, W.-Y. Lee and K. S. Kim, Tetrahedron Lett., 2010, 51, 6250–6254 CrossRef CAS.
- J. Leroux and A. S. Perlin, Carbohydr. Res., 1976, 47, C8–C10 CrossRef CAS.
- G. Chen, Q. Yin, J. Yin, X. Gu, X. Liu, Q. You, Y.-L. Chen, B. Xiong and J. Shen, Org. Biomol. Chem., 2014, 12, 9781–9785 CAS.
- Y. Nishida, Y. Shingu, H. Dohi and K. Kobayashi, Org. Lett., 2003, 5, 2377–2380 CrossRef CAS PubMed.
- S. Suda and T. Mukaiyama, Chem. Lett., 1991, 20, 431–434 CrossRef.
- S. Suda and T. Mukaiyama, Bull. Chem. Soc. Jpn., 1993, 66, 1211–1215 CrossRef CAS.
- T. Mukaiyama, K. Matsubara and S. Suda, Chem. Lett., 1991, 20, 981–984 CrossRef.
- N. Shimomura and T. Mukaiyama, Chem. Lett., 1993, 22, 1941–1944 CrossRef.
- T. Suzuki, S. Watanabe, T. Yamada and K. Hiroi, Tetrahedron Lett., 2003, 44, 2561–2563 CrossRef CAS.
- T. Yamanoi, R. Inoue, S. Matsuda, K. Iwao, Y. Oda, A. Yoshida and K. Hamasaki, Heterocycles, 2009, 77, 445–460 CrossRef CAS.
- T. S. Mansour, S. Bardhan and Z.-K. Wan, Synlett, 2010, 1143–1169 CAS.
- M. Teruaki and S. Shinji, Chem. Lett., 1990, 19, 1143–1146 CrossRef.
- J. B. Hendrickson and S. M. Schwartzman, Tetrahedron Lett., 1975, 16, 277–280 CrossRef.
- A. Atle, G. Thor and H. Steinar, Tetrahedron Lett., 1979, 20, 2263–2264 CrossRef.
- J. B. Hendrickson and M. S. Hussoin, J. Org. Chem., 1987, 52, 4137–4139 CrossRef.
- Z. Moussa, ARKIVOC, 2012, 432–490 CAS.
- M. Mossotti and L. Panza, J. Org. Chem., 2011, 76, 9122–9126 CrossRef CAS PubMed.
- G. J. van der Heden van Noort, H. S. Overkleeft, G. A. van der Marel and D. V. Filippov, Org. Lett., 2011, 13, 2920–2923 CrossRef CAS PubMed.
- K. E. Elson, I. D. Jenkins and W. A. Loughlin, Aust. J. Chem., 2004, 57, 371–376 CrossRef CAS.
- C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374–1377 CrossRef CAS PubMed.
- M. H. Holthausen, R. R. Hiranandani and D. W. Stephan, Chem. Sci., 2015, 6, 2016–2021 RSC.
- E. V. Jennings, K. Nikitin, Y. Ortin and D. G. Gilheany, J. Am. Chem. Soc., 2014, 136, 16217–16226 CrossRef CAS PubMed.
- R. U. Lemieux, K. B. Hendriks, R. V. Stick and K. James, J. Am. Chem. Soc., 1975, 97, 4056–4062 CrossRef CAS.
- N. Oka, R. Kajino, K. Takeuchi, H. Nagakawa and K. Ando, J. Org. Chem., 2014, 79, 7656–7664 CrossRef CAS PubMed.
- D. Crich and S. Sun, J. Org. Chem., 1996, 61, 4506–4507 CrossRef CAS PubMed.
- D. Crich and S. Sun, J. Org. Chem., 1997, 62, 1198–1199 CrossRef CAS.
- D. Crich and S. Sun, J. Am. Chem. Soc., 1997, 119, 11217–11223 CrossRef CAS.
- D. Crich and S. Sun, J. Am. Chem. Soc., 1998, 120, 435–436 CrossRef CAS.
- D. Crich and S. Sun, Tetrahedron, 1998, 54, 8321–8348 CrossRef CAS.
- D. Crich and M. Smith, J. Am. Chem. Soc., 2002, 124, 8867–8869 CrossRef CAS PubMed.
- D. Crich and N. S. Chandrasekera, Angew. Chem., Int. Ed., 2004, 43, 5386–5389 CrossRef CAS PubMed.
- D. Crich, J. Org. Chem., 2011, 76, 9193–9209 CrossRef CAS PubMed.
- D. Kahne, S. Walker, Y. Cheng and D. Van Engen, J. Am. Chem. Soc., 1989, 111, 6881–6882 CrossRef CAS.
- J. D. C. Codée, L. J. van den Bos, R. E. J. N. Litjens, H. S. Overkleeft, J. H. van Boom and G. A. van der Marel, Org. Lett., 2003, 5, 1947–1950 CrossRef PubMed.
- T. G. Frihed, M. Bols and C. M. Pedersen, Chem. Rev., 2015, 115, 4963–5013 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data for new compounds and copies of NMR spectra. See DOI: 10.1039/c6ob01812b |
| This journal is © The Royal Society of Chemistry 2017 |