
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorinated diazoalkanes – a versatile class of reagents for the synthesis of fluorinated compounds
Lucas
Mertens
and
Rene M.
Koenigs
 *
*
RWTH Aachen University, Institute of Organic Chemistry, Landoltweg 1, D-52074 Aachen, Germany. E-mail: rene.koenigs@rwth-aachen.de
First published on 21st September 2016
Abstract
Although diazoalkanes find regular application in organic synthesis, the synthesis and application of their fluorinated homologues was long neglected. First described in 1943, trifluorodiazoethane found its way into the organic synthesis repertoire only in the past decade for atom-economical and practical synthesis of fluorinated building blocks and currently emerges as a versatile reagent. The synthetic applicability of this reagent is currently being investigated in great detail and many interesting new reactivities, and procedures for the synthesis of fluoroalkyl-substituted building blocks have been developed. In this context, a range of new fluoroalkyl substituted diazoalkanes have been introduced in the past few years, ranging from perfluorinated diazoalkanes towards highly reactive difluorodiazoethane which was first described in 2015. This tutorial review covers historic milestones of fluoroalkyl substituted diazoalkanes and highlights recent examples which underscore their vast potential to the synthesis of fluorinated compounds.
1. Introduction
Diazomethane, first described in 1894 by von Pechmann, represents the shortest purely aliphatic homologues of alkyl diazo compounds.1 Since then the synthetic application of diazomethane has been investigated to a great extent.2,3 More importantly, the use of state-of-the-art synthesis technologies such as continuous-flow chemistry has made significant progress with respect to lab safety and scalability.4–6 Regarding the toxicity of diazomethane, von Pechmann already stated in his seminal report:“…besitzt es höchst giftige Eigenschaften, welche zunächst Athemnoth […] hervorrufen und das Arbeiten damit äusserst unangenehm machen.”
“…it possesses highly toxic properties, which evoke respiratory depression […] and make handling particularly unpleasant.”
Although chemical synthesis using diazomethane requires strict safety regulations, applications of the smallest diazoalkane have become indispensable for modern organic synthesis. Similarly, substituted diazomethanes have found widespread application across the whole field of organic synthesis.
Interestingly, fluorinated diazoalkanes 1–4 (Fig. 1) are investigated to a much lower extent, although they represent a very practical and versatile class of reagents that enable chemists to quickly introduce fluorinated side chains from simple and readily available precursors.
 | ||
| Fig. 1 Fluoroalkyl substituted diazoalkanes. | ||
Trifluorodiazoethane (1) is probably the best-studied fluorinated diazoalkane. First described in 1943 by Gilman and Jones through reaction of trifluoroethylamine and nitrous acid,7 this reagent only found regular application in organic synthesis in the past decade as a versatile reagent yielding versatile fluorinated building blocks in a very efficient and atom-economical fashion.
Application of fluorinated diazoalkanes is of particular importance for chemists from all fields. Long perfluoroalkyl groups are of high interest in the development of materials, such as OLEDs.8 The short-chain analogues are of high importance to the pharmaceutical and agrochemical industry and allow the efficient construction of small fluorinated building blocks that find regular application in discovery programs.9,10
This tutorial review will concentrate on the organic chemistry of fluoroalkyl-substituted diazoalkanes (1–3), covering the range from historic milestones towards a detailed discussion of the current synthetic methodology. Technical aspects, such as the preparation of diazo compounds will be discussed as part of the synthesis application described herein.
2. Early applications
In their seminal report Gilman and Jones reported on the deca-gram scale synthesis of trifluoroethylamine hydrochloride (8·HCl) from commercially available sodium trifluoro-carboxylate (5). The free base (8) readily reacts in the presence of sulphuric acid with sodium nitrite in water under the formation of trifluorodiazoethane (1) that can be distilled as an ethereal solution with no apparent decomposition and 65–67% yield on the multi-gram scale (Scheme 1). Gilman and Jones also reported the synthesis of the neat reagent, which can be distilled at a boiling point of 13 to 13.5 °C. From a synthetic perspective, the reaction with iodine is the only application described within this manuscript.7 | ||
| Scheme 1 Synthesis of trifluoroethyl amine and trifluorodiazoethane by Gilman and Jones. | ||
Although Gilman and Jones reported on the synthesis of trifluorodiazoethane in detail, synthetic applications of this reagent were not reported until 1964. Fields and Haszeldine investigated decomposition and polymerization reactions of trifluorodiazoethane and heptafluoropropyl diazomethane, yet no applications for the synthesis of small molecules have been described within this manuscript.11 It should be noted that the authors describe serious risk hazards and severe explosions when working with these reagents.
In further reports Atherton and Fields reported proof of principle studies on cycloaddition reactions of trifluorodiazoethane with olefins yielding cyclopropanes12 (9) or pyrazolines (10 and 11) (Fig. 2),13 and on insertion reactions providing trifluoroethyl substituted alkanes, silanes, ethers and amines.14
 | ||
| Fig. 2 Reaction products obtained from trifluorodiazoethane. | ||
Branched fluoroalkyl-substituted diazo compounds were investigated by Stone and co-workers and their application as ligands in transition metal complexes.15
Meese reported the reaction of trifluorodiazoethane with organic acids in 1984. Interestingly, no reaction was observed upon treatment of trifluorodiazoethane with carboxylic acids, whereas the reaction with sulfonic acids yielded the corresponding sulfonates (13) in high yields. This is a remarkable observation and it highlights the unique nature of this reagent, as the typical esterification reaction of diazo compounds with carboxylic acids, such as acetic acid or benzoic acids is not taking place.16 This very unique transformation was applied to the preparation of sulpho protected glycosaminoglycans (15) by Linhardt and co-workers (Scheme 2).17
 | ||
| Scheme 2 Reaction of trifluorodiazoethane with carboxylic acids and sulfonic acids. | ||
In further investigations, the reaction of trifluorodiazoethane with alcohols,18 glyoxylate,19 chloral20 and aldehydes21 was studied. Additionally, trifluorodiazoethane was applied in the synthesis of pyrazoles22 and in ring expansion reactions to yield 7-membered rings.23 Although all of these transformations represent early examples of the applicability of this reagent to organic synthesis, they have in common that only a very limited substrate scope has been investigated.
At this point it should be noted that the correct IUPAC name of 1 is 2,2,2-trifluoro diazoethane. In the recent literature, the term trifluoromethyl diazomethane has been introduced, and it is important to know that both terms are used in the current literature.
The challenge when working with fluoroalkyl-substituted diazoalkanes is the significantly reduced nucleophilicity at the diazo-carbon atom, due to the strongly electron-withdrawing nature of the fluoroalkyl group. This review will outline the latest strategies and methods that have been developed to utilize this class of reagents in organic synthesis.
2. Applications of trifluorodiazoethane
2.1 Cyclopropanation reactions
Undoubtedly, cyclopropanation reactions are one of the most important synthetic applications of diazo compounds.24–26 After the very first report on trifluoromethyl cyclopropanes by Atherton and Fields in 1968, it took almost 40 years until Simonneaux and co-workers reported on iron(III) catalysed enantioselective cyclopropanation using trifluorodiazoethane with a broad range of different styrenes. A chiral homogeneous iron porphyrin complex 18 (Fig. 3) served for chiral induction. | ||
| Fig. 3 Iron catalysts used for cyclopropanation reactions with trifluorodiazoethane. | ||
Although the authors observed high diastereoselectivity, only moderate enantiomeric excess (17–75% ee) and limited yield (24–96%) could be obtained (Scheme 3). Still, this report marks the beginning of modern trifluorodiazoethane chemistry.27
 | ||
| Scheme 3 Iron catalysed enantioselective cyclopropanation by Simonneaux and co-workers. | ||
A critical drawback of the method described by Simonneaux and co-workers is the preparation of the diazo compound. Similarly to the early reports on trifluorodiazoethane, Simonneaux and co-workers prepared the diazo compound in a neat form by cold distillation for subsequent cyclopropanation reactions, which is far from being practical.
In 2007, Davies and co-workers achieved a remarkable improvement in cyclopropanation reactions using 2,2,2-trifluoroethane hydrazones (19) that can be readily oxidized to the diazoalkanes with MnO2. Application of 1-aryl-2,2,2-trifluorodiazoethanes (4) and chiral Rh(II) catalysts proved to be superior in cyclopropanation reactions with styrenes and the desired trifluoromethyl substituted cyclopropanes (20) could be obtained with high diastereoselectivity (>94%), high enantiomeric excess (88–94% ee) and good chemical yield (61–76%). However, aliphatic olefins provided the desired reaction products only with significantly reduced yield (Scheme 4).28
 | ||
| Scheme 4 Rhodium(II) catalysed enantioselective cyclopropanation of styrenes. | ||
Shortly after these reports, Komarov and co-workers reported on general Rh(II) and Cu(I) catalysed cyclopropanation reactions using trifluorodiazoethane. For the first time, Komarov and co-workers were able to demonstrate dihydropyran, enol ethers, dehydro amino acids and cyclohexene to be suitable substrates for cyclopropanation reactions using trifluorodiazoethane as the carbene precursor.29 Additionally, trifluoromethyl substituted proline analogues have been reported.30
In 2010, Carreira and co-workers achieved a remarkable break-through in cyclopropanation reactions using trifluorodiazoethane. Application of an achiral Fe(III) porphyrin catalyst 22 (Fig. 3) and in situ generation of trifluorodiazoethane in water were key to this method. This result is remarkable as it is the first example describing the utilization of trifluoroethyl amine and the direct reaction of in situ generated fluorinated diazoalkane with an olefin, which significantly facilitates the synthetic applicability of this versatile reagent (Scheme 5).31
 | ||
| Scheme 5 Cyclopropanation reactions on water. | ||
Carreira and co-workers reported on extensions of this work in 2011. Application of dienes and enynes in this cyclopropanation reaction furnished valuable vinyl- and alkynyl-substituted cyclopropanes. Carreira and co-workers used a similar protocol as in their initial report and were able to obtain these building blocks in high yield with excellent diastereoselectivities.32
Using a chiral salen-based Co(II)-catalyst (25) highly diastereoselective and enantioselective cyclopropanation reactions of styrenes on water could be achieved. Interestingly additional organic co-solvents resulted in reduced conversion to the desired cyclopropane (Scheme 5).33
Moreover, Carreira and co-workers reported on Rh(II) catalysed cyclopropanation reactions. Different aliphatic and aromatic alkynes could be converted to the corresponding cyclopropene with high yield using Rh2esp2 as a catalyst (Scheme 12).34
These ground breaking proof of principle reports on the application of trifluorodiazoethane in organic synthesis set the stage for a rich chemistry that followed in the next years.
Further applications of trifluorodiazoethane in cyclopropanation reactions concentrated on extensions of the substrate scope towards electron-rich olefins. Mykhailiuk reported [x.1.0]-bicycles (x = 2,3,4) that are versatile building blocks for applications in pharmaceutical and agrochemical research programs. In particular, the authors report on Cu(I) catalysed cyclopropanation reactions using cyclic enamides which provide direct access to building blocks that can be used for bioisosteric replacement of substituted trifluoromethyl substituted cyclohexanes (Scheme 6).35
 | ||
| Scheme 6 Cyclopropanation reactions with enamides. | ||
The reaction sequence of [2 + 3] cycloaddition and subsequent cycloreversion under extrusion of nitrogen provides an alternative and efficient pathway towards cyclopropanes, the advantage being that this approach does not require the use of expensive metal catalysts.
Mykhailiuk and co-workers first reported on this approach for the synthesis of trifluoromethyl cyclopropanes. Using maleimides, which readily react in [2 + 3] cycloaddition, the authors first prepared the corresponding pyrazolines, which then release nitrogen upon heating to 150 °C (Scheme 7).36
 | ||
| Scheme 7 Cycloaddition–cycloreversion for the synthesis of cyclopropanes. | ||
Lu, Xiao and co-workers reported the application of oxindoles in this particular transformation. In a metal-free transformation, trifluorodiazoethane was generated in situ from trifluoro ethylamine hydrochloride and then added to oxoindolin 31 resulting in spirocyclic pyrazoline 32 with high yield. In a next step this pyrazoline undergoes cycloreversion under thermal conditions yielding the desired trifluoromethyl substituted cyclopropane 33 in good yield and excellent diastereoselectivity.37
In 2015, Ma and coworkers reported an interesting cyclopropanation reaction of trifluorodiazoethane and azlactones. While typical cyclopropanation catalysts like CuOTf, FeTPPCl or Rh2esp2 proved inefficient, heating of the reaction mixture without a catalyst in the presence of brine afforded the desired cyclopropane in good yield and excellent diastereoselectivity. According to the authors, brine stabilizes the diazoalkane and thus leads to increased yield. The authors assume that this cyclopropanation may proceed either by [2 + 1] cycloaddition of the carbene or alternatively by an initial [2 + 3] cycloaddition yielding a pyrazole that liberates nitrogen under the formation of the cyclopropane (Scheme 8).38
 | ||
| Scheme 8 Brine stabilized synthesis of cyclopropanes. | ||
An extension of this work was reported by Ma and Cahard towards benzofuran-2(3H)-ones.39
Trifluoromethyl-substituted cyclopropanes find application as TRPV1 antagonists. Duncton and co-workers reported on the cyclopropanation reaction of vinyl dibutyl borate with trifluorodiazoethane and subsequent application of this boronic ester in the preparation of the corresponding trifluoroborate potassium salt. This salt can be coupled to different aromatic esters and serves as an entry into highly potent human TRPV1 antagonists (Fig. 4).40
 | ||
| Fig. 4 TRPV1 antagonists reported by Duncan and co-workers. | ||
2.2 Dipolar cycloadditions
As outlined in the last paragraph on cyclopropanation reactions, substantial progress has been made with regard to dipolar cycloaddition reactions with electron-poor olefins.41 This section will focus on applications of [2 + 3] cycloaddition reactions with the aim of preparing pyrazoles and pyrazolines.[2 + 3] cycloaddition reactions are one of the most commonly applied reactions of diazo compounds. Their dipolar nature makes them formidable substrates for the efficient synthesis of pyrazoles and pyrazolines, both being highly desired heterocycles for research in pharmaceutical and agrochemical industries.
Fluoroalkyl-substituted pyrazoles undoubtedly belong to one of the most important classes of heterocycles that find many applications in industrial research. Among the most prominent examples of fluoroalkyl substituted pyrazoles are COX inhibitors, such as celecoxib (38), mavacoxib (40) or deracoxib (39).42,43 Similarly, this class of compounds finds regular application in Factor Xa discovery programs, which led to the discovery of razaxaban (41) (Fig. 5).44 Typically, the preparation of these heterocycles requires multi-step synthesis and suffers from regioselectivity problems during preparation.
 | ||
| Fig. 5 Fluoroalkyl substituted pyrazoles. | ||
Initial reports by Fields and co-workers towards [2 + 3] cycloaddition reactions of trifluorodiazoethane remained unpractical due to long and harsh reaction conditions and limited substrate scope.13,22
Recently, Ma and co-workers reported significant progress with regard to this versatile transformation. In their report on dipolar cycloaddition reactions the authors investigated the effect of Lewis acids in the reaction of trifluorodiazoethane with terminal alkynes (42). Ag(I) salts proved to be superior for this transformation and the highly interesting trifluoromethylated pyrazoles (43) could be obtained in a single reaction step with high chemical yield with exquisite regioselectivity (Scheme 9).45
 | ||
| Scheme 9 Silver catalysed synthesis of pyrazoles. | ||
In an additional report on [2 + 3] cycloaddition Mykhailiuk reported an important improvement in the practical study when using trifluorodiazoethane.46 Inspired by the work of Carreira and co-workers,31 the authors described the synthesis and direct synthetic application of this reagent using a biphasic aqueous/organic solvent mixture. The advantage of the new protocols, described by Carreira and Mykhailiuk, is that there is no need for purification of the diazo compound and that hazards can be minimized as the diazo compound is directly consumed.
With regard to the preparation of the diazo compound, it should be noted that application of trifluoroethyl amine hydrochloride and sodium nitrite serves as a source for the on-water synthesis of trifluorodiazoethane and subsequent reactions can be either conducted on water, in a bi-phasic mixture by taking the organic layer and followed by addition to the reaction solution. Using this methodology, Mykhailiuk and co-workers were able to demonstrate that a broad range of olefins and alkynes bearing electron-withdrawing substituents could be converted to the corresponding pyrazoles with excellent yield (Scheme 10).
 | ||
| Scheme 10 Synthesis of pyrazolines by Mykhailiuk and Koenigs. | ||
Koenigs and co-workers further investigated this transformation using a continuous-flow protocol, which relies on the preparation of trifluorodiazoethane from the free base trifluoroethyl amine and tert butyl nitrite in the presence of catalytic amounts of acetic acid. This protocol thus serves as an option for the water-free preparation of this important reagent and allows an operationally simple handling of toxic diazo compounds (Scheme 10).47
Ma and co-workers recently reported an extension of this protocol towards the reaction of trifluorodiazoethane with allenes (47a and 47b)48 and to benzanellated pyrazoles (48) through reaction of trifluorodiazoethane with arynes.49
[2 + 3] cycloaddition reactions not only provide convenient access towards pyrazoles and pyrazolines, but can also be applied to the synthesis of a range of different other 5-membered heterocycles. Carreira and co-workers were first to report trifluorodiazoethane in the synthesis of trifluoromethylated benzofurans (49).50 Molander and co-workers reported on the synthesis of isoxazolidines (50)51 and Ma and co-workers were able to demonstrate the applicability of trifluorodiazoethane in the synthesis of oxazolines (51) (Fig. 6).52
 | ||
| Fig. 6 Trifluoromethyl substituted heterocycles prepared with trifluorodiazoethane. | ||
From a mechanistic perspective, these three reports differ significantly. The reaction of salicylaldehydes with trifluorodiazoethanes proceeds through Lewis-acid catalysed homologation of the aldehyde and subsequent hemi-acetal formation and dehydration to yield trifluoromethylated benzofurans. This homologation reaction will be discussed in detail in a separate paragraph of this review.
Contrarily, the synthesis of isoxazoles and oxazoles proceeds via [2 + 3] cycloaddition reactions. Here, we discuss this transformation for the synthesis of oxazoles exemplarily. Coordination to Cu(I) furnishes electrophilic intermediate 52, that undergoes reaction with a nitrile and formation of a 1,2-dipole 54, which last undergoes stepwise [2 + 3] cycloaddition with an aldehyde (Scheme 11).
 | ||
| Scheme 11 Mechanism of the oxazol synthesis. | ||
2.3 Insertion reactions
Insertion reactions are another very important application of diazo compounds. In the past a great variety of different catalytic C–H and heteroatom-H insertion reactions has been developed allowing efficient and sustainable synthesis of important building blocks.53–55This section will outline the achievements that have been made in the past few years utilizing fluoroalkyl-substituted diazoalkanes. These transformations are particularly important as they provide direct access towards fluorinated small molecules with exquisite synthetic efficiency.
In 2012, Ma and co-workers reported a protocol that is complementary to the cyclopropanation reaction described by Carreira and co-workers. Using catalytic amounts of Cu(I), Ma and co-workers have been able to demonstrate that terminal alkynes undergo C–H insertion reactions yielding trifluoroethyl-substituted alkynes (58).56 This is remarkable as Rh2esp2 provides cyclopropanes (59)34 and is an interesting example of catalyst controlled transformations (Scheme 12).
 | ||
| Scheme 12 Catalyst controlled reactions of trifluorodiazoethane. | ||
In addition to previously described C–H insertion reactions, Wang and co-workers investigated Ag(I) catalysed insertion reactions into N–H bonds.57 While Ag(I) catalysed insertion reactions into the N–H bond of anilines yielded trifluoroethyl-substituted secondary amines (61), the Ag(I) catalysed reaction with benzamides gave O-trifluoroethyl imidates selectively. Competitive experiments with olefins did not provide cyclopropane products, which the authors attribute to the following postulated reaction mechanism (Scheme 13).
 | ||
| Scheme 13 Insertion reaction into the N–H bond. | ||
Recently, Gouverneur and co-workers reported further extension of the insertion reactions of trifluorodiazoethane into heteroatom-H bonds. Application of a Cu(I) catalyst provided the insertion product into B–H, Si–H, S–H, P–H and N–H bonds (Scheme 14).58
 | ||
| Scheme 14 Insertion into different X–H bonds by Gourverneur and co-workers. | ||
Molander and co-workers reported further applications of trifluorodiazoethane in insertion reactions. Reaction of this diazo compound with aromatic boronic acids/esters or trifluoroborate potassium salts 69 provides the corresponding C–B bond insertion products (72) with excellent yield (Scheme 15).59 This reaction is particularly useful, as valuable trifluoromethyl substituted boron compounds can be easily obtained, which are useful intermediates for further functionalization, e.g. in Suzuki couplings, addition reactions to olefins or radical chemistry to name a few. In another report, Molander reported on the double homologation reaction of boroxines. For this transformation pinacol is a crucial additive to allow the second homologation step to proceed.60
 | ||
| Scheme 15 Mechanism of the homologation reaction of boronic acids. | ||
Depending on the work-up conditions, Molander and co-workers obtained the gem-difluoro vinyl compound 74 after elimination of fluoride. Wu and co-workers took up this observation and developed a protocol that allows the conversion of aromatic boronic acids to either trifluoroethyl or difluorovinyl substituted aromatic compounds, depending on the reaction conditions (Scheme 16).61
 | ||
| Scheme 16 Reactions of trifluorodiazoethane with boronic acids. | ||
2.4 Lewis acid catalyzed transformations
The homologation reaction of carbonyl compounds with diazo compounds is another very important application of this class of reagents.62,63 The Tiffeneau-Demjanov,64 Roskamp65 and Buchner–Curtius–Schlotterbeck66,67 reactions are among the best-studied reactions in this context. Fluoroalkyl substituted diazo compounds possess outstanding potential for these transformations and allow the direct and facile introduction of fluoroalkyl groups to common organic building blocks.Mock and co-workers reported in 1977 on the reaction of diazo compounds with ketones.68,69 Yet, the scope of this particularly useful transformation with trifluorodiazoethane was not investigated. In 1981, Wakselman and co-workers reported on Lewis-acid catalysed rearrangement reactions of the adduct of trifluorodiazoethane and aldehydes, yielding epoxides or ketones depending on the reaction conditions and the Lewis acid.21 In the presence of BF3·Et2O the rearrangement of benzaldehyde to the corresponding homologous fluorinated hydratropic aldehyde was observed, while SbCl5 provided the epoxide. Although, this transformation is particularly useful for the synthesis of fluorinated ketones and epoxides, further investigations regarding the Tiffeneau-Demjanov and Roskamp reaction of carbonyl groups and trifluorodiazoethane were reported only recently by Carreira and co-workers.
Careful mechanistic investigations revealed ZrCl4 to be the best Lewis acid for the Roskamp reaction of aldehydes 75 and trifluorodiazoethane 1 to yield the corresponding ketones with high yield. Interestingly, in the case of aromatic aldehydes, Carreira and co-workers observed a reaction product that is generated after an initial aryl migration yielding hydratropic aldehyde 77, which undergoes a second addition–rearrangement reaction with trifluorodiazoethane to yield ketone 78. Notably, in this case the hydride migration is preferred (Scheme 17).70
 | ||
| Scheme 17 Homologation of ketones. | ||
Cyclic ketones (80) react with trifluorodiazoethane in a Tiffeneau-Demjanov reaction affording α-trifluoromethyl ring-expanded ketones (81).70 In a similar way acetals (82) can be homologated to the corresponding α-branched acetals (83)71 and imines (84) undergo reaction to yield the corresponding aziridines (85).72 Salicylaldehydes (86) provide the corresponding benzofuranols (49) (Scheme 18).50
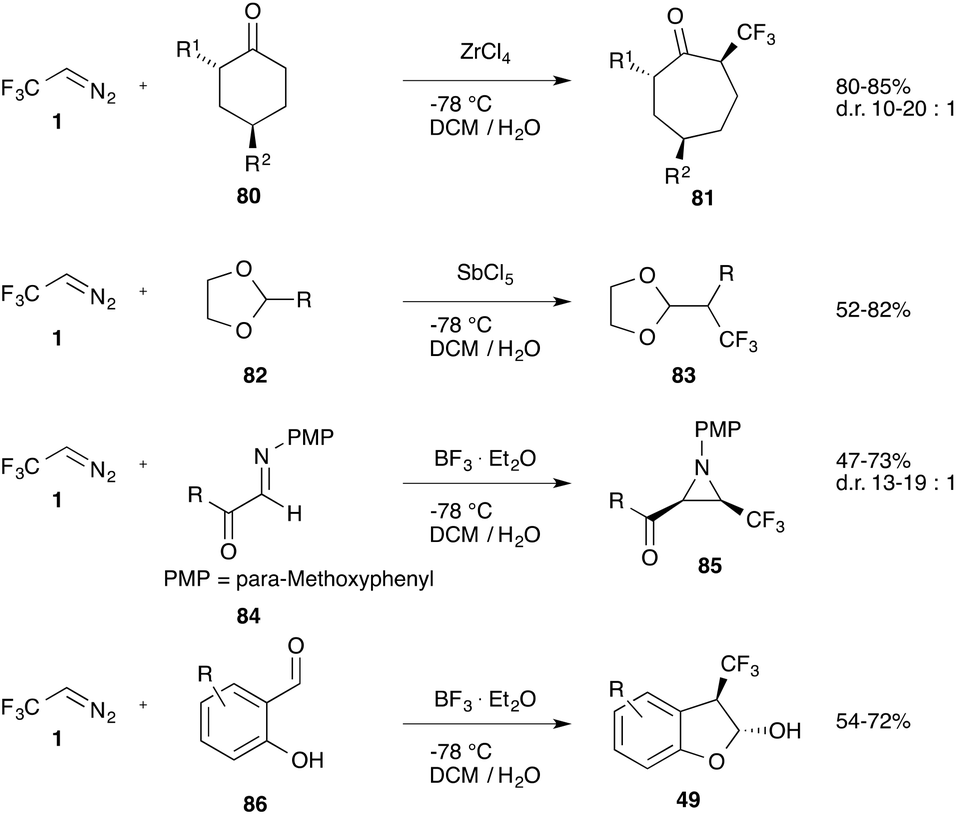 | ||
| Scheme 18 Reactions of different carbonyl compounds. | ||
Recently, Kappe and co-workers reported on the addition of trifluorodiazoethane to a range of different aldehydes and ketones using a continuous-flow protocol for the generation of trifluorodiazoethane. A tube-in-tube reactor was applied to transfer the diazo compound from the aqueous layer to the organic layer. Subsequent addition reaction of the stream of the diazo compound to a stream of the carbonyl compound in the presence of a base yielded the addition product in high yield and no subsequent rearrangement was observed.73
2.5 Further transformation
In the previous sections the reactivity of trifluorodiazoethane was discussed and different cycloaddition and nucleophilic addition reactions of this reagent have been discussed.Recently, Mykhailiuk and co-workers reported on an unexpected transformation when using trifluorodiazoethane. Treatment of this diazo compound with nucleophiles such as β-ketoesters or other 1,3-dicarbonyl groups yields the addition product of the nucleophile to the terminal nitrogen of the diazo compound, that is hydrazone 89. In the presence of an ortho-fluoro substituent in the proximity of the hydrazone, subsequent ring closure provides fluoroalkyl substituted cinnolinones (90). This observation is remarkable, as typically diazo compounds do react under extrusion of nitrogen and form a new bond at the diazo-carbon atom (Scheme 19).74
 | ||
| Scheme 19 N-Terminal electrophilic reactivity of trifluorodiazoethane. | ||
3. Perfluorinated diazoalkanes
While many applications on trifluorodiazoethane have been reported to date, longer chain perfluorinated diazoalkanes have been investigated to a much lower extent. Additionally, Fields and Haszeldine reported severe explosions when working with these diazo compounds.11 After the initial reports by Fields and co-workers synthetic applications of this class of diazo compounds have been scarce. Mykhailiuk was first to report on 2,2,3,3,3-pentafluorodiazopropane (3b) in 2014 and its application in dipolar cycloadditions yielding pyrazolines.75 Quickly after this report, Mykhailiuk showed that different short-perfluorinated diazo compounds (3a–b) can be prepared in batch and applied in the synthesis of pyrazolines using the one-pot biphasic protocol (Scheme 20).76 | ||
| Scheme 20 Perfluorinated diazoalkanes reported by Mykhailiuk and Koenigs, BPR = back pressure regulator. | ||
Koenigs and co-workers further investigated the synthesis of perfluoroalkyl-substituted diazoalkanes (3b–f) in continuous-flow. A range of different perfluorinated amines was applied in a continuous-flow protocol using tert butyl nitrite and catalytic amounts of acetic acid for the preparation of perfluorinated diazoalkanes with different chain lengths (Scheme 20).47
4. Difluorodiazoethane
In the previous sections, synthetic applications of trifluorodiazoethane or longer-chain perfluorinated analogues have been described in detail. From a drug discovery point of perspective, the difluoromethyl group finds regular application as a substitute for the trifluoromethyl group. The advantage is that difluoromethyl is not as lipophilic as trifluoromethyl and it may interact as a hydrogen bond donor. Additionally, the difluoromethyl group is slightly protic, it can interact with a target protein as a hydrogen bond donor and thus finds regular application as an isostere of thiols and alcohols.77,78Interestingly, difluorodiazoethane (2) was not reported in the literature until 2015, when Mykhailiuk reported the first synthesis and application in the synthesis of pyrazoles (95) in batch reactions. Mykhailiuk reported difluorodiazoethane to be highly sensitive to water and that the application of organic nitrites is crucial for its synthesis.79 Shortly after the initial report of Mykhailiuk, Koenigs and co-workers reported on the continuous-flow synthesis of difluorodiazoethane and its application to pyrazole and pyrazoline synthesis (96) (Scheme 21).47
 | ||
| Scheme 21 Reactions with difluorodiazoethane. | ||
5. Summary and outlook
After the first description of trifluorodiazoethane in 1943, this highly versatile reagent was applied only to a very limited extent in organic synthesis until it gained significant interest in the organic synthetic methodology. Over the past 6 years about 40 different publications have reported on novel synthetic procedures for fluorinated building blocks, which are of fundamental interest to chemists, ranging from drug discovery to materials sciences. The recent advent of synthetic applications of this small and valuable reagent clearly outlines that in the next years this reagent will find its way into standard organic synthesis protocols that can be utilized, from both an academic and an industrial perspective. These new protocols do not require the hazardous isolation of this special reagent and rely on phase-transfer reactions, continuous-flow chemistry and simple one-pot protocols and enables chemists to better understand reactivity and to find novel and interesting reactivities. Noteworthily, this reagent does not react with carboxylic acids under the formation of an ester and it can react as an electrophile at the diazo group terminal nitrogen, underlining the unique chemical versatility of this reagent.Recently, the corresponding difluoromethyl substituted analogue was reported and new reactions with this reagent are of current interest to organic chemists as it opens up new avenues to difluoromethyl-substituted building blocks.
References
- C. B. H. von Pechmann, Chem. Ber., 1894, 27, 1888–1891 CrossRef.
- A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10090 CrossRef CAS PubMed.
- T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091–1160 CrossRef CAS.
- S. T. R. Müller and T. Wirth, ChemSusChem, 2015, 2, 245–250 CrossRef PubMed.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed.
- B. J. Deadman, S. G. Collins and A. R. Maguire, Chem. – Eur. J., 2015, 21, 2298–2308 CrossRef CAS PubMed.
- H. Gilman and R. G. Jones, J. Am. Chem. Soc., 1943, 65, 1458–1460 CrossRef CAS.
- M. A. Omary and I. W. H. Oswald, WO2015084906, 2015 Search PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- R. Fields and R. N. Haszeldine, J. Chem. Soc., 1964, 1881–1889 RSC.
- J. H. Atherton and R. Fields, J. Chem. Soc. C, 1967, 1450–1454 RSC.
- J. H. Atherton and R. Fields, J. Chem. Soc. C, 1968, 1507–1513 RSC.
- J. H. Atherton and R. Fields, J. Chem. Soc. C, 1968, 2276–2278 RSC.
- J. Cooke, W. R. Cullen, M. Green and F. G. A. Stone, Chem. Commun., 1968, 170–171 RSC.
- C. O. Meese, Synthesis, 1984, 1041–1042 CrossRef CAS.
- N. A. Karst, T. F. Islam and R. J. Linhardt, Org. Lett., 2003, 5, 4839–4842 CrossRef CAS PubMed.
- D. T. Loehr, D. Armistead, J. Roy and H. C. Dorn, J. Fluorine Chem., 1988, 39, 283–287 CrossRef CAS.
- C. Wakselman and M. Tordeux, J. Fluorine Chem., 1982, 21, 99–106 CrossRef CAS.
- R. Fields and J. F. Tomlinson, J. Fluorine Chem., 1979, 13, 19–28 CrossRef.
- M. Tordeux and C. Wakselman, J. Fluorine Chem., 1981, 17, 299–304 CrossRef CAS.
- R. Fields and J. P. Tomlinson, J. Fluorine Chem., 1979, 13, 147–158 CrossRef CAS.
- H.-D. Martin, R. Iden, F.-J. Mais, G. Kleefeld, A. Steigel, B. Fuhr, O. Rümmele, A. Oftring and E. Schwichtenberg, Tetrahedron Lett., 1983, 24, 5469–5472 CrossRef CAS.
- H. Lebel, J. F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977–1050 CrossRef CAS PubMed.
- J. Hansen and H. M. L. Davies, Coord. Chem. Rev., 2008, 252, 545–555 CrossRef CAS PubMed.
- H. M. L. Davies and J. R. Denton, Chem. Soc. Rev., 2009, 38, 3061–3071 RSC.
- P. Le Maux, S. Juillard and G. Simonneaux, Synthesis, 2006, 1701–1704 CAS.
- J. R. Denton, D. Sukumaran and H. M. L. Davies, Org. Lett., 2007, 9, 2625–2628 CrossRef CAS PubMed.
- P. K. Mykhailiuk, S. Afonin, A. S. Ulrich and I. V. Komarov, Synthesis, 2008, 1757–1760 CAS.
- P. K. Mykhailiuk, S. Afonin, G. V. Palamarchuk, O. V. Shishkin, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2008, 47, 5765–5767 CrossRef CAS PubMed.
- B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 938–941 CrossRef CAS PubMed.
- B. Morandi, J. Cheang and E. M. Carreira, Org. Lett., 2011, 13, 3080–3081 CrossRef CAS PubMed.
- B. Morandi, B. Mariampillai and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 1101–1104 CrossRef CAS PubMed.
- B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 4294–4296 CrossRef CAS PubMed.
- O. S. Artamonov, E. Y. Slobodyanyuk, D. M. Volochnyuk, I. V. Komarov, A. A. Tomachev and P. K. Mykhailiuk, Eur. J. Org. Chem., 2014, 3592–3598 CrossRef CAS.
- O. S. Artamonov, E. Y. Slobodyanyuk, O. V. Shishkin, I. V. Komarov and P. K. Mykhailiuk, Synthesis, 2013, 225–230 CAS.
- T. R. Li, S. W. Duan, W. Ding, Y. Y. Liu, J. R. Chen, L. Q. Lu and W. J. Xiao, J. Org. Chem., 2014, 79, 2296–2302 CrossRef CAS PubMed.
- C.-L. Zhu, L.-J. Yang, S. Li, Y. Zheng and J.-A. Ma, Org. Lett., 2015, 17, 3442–3445 CrossRef CAS PubMed.
- C. L. Zhu, J. A. Ma and D. Cahard, Asian J. Org. Chem., 2016, 5, 66–69 CrossRef CAS.
- M. A. J. Duncton, L. Ayala, C. Kaub, S. Janagani, W. T. Edwards, N. Orike, K. Ramamoorthy, J. Kincaid and M. G. Kelly, Tetrahedron Lett., 2010, 51, 1009–1011 CrossRef CAS.
- A. Padwa, 1,3-Dipolar Cycloaddition Chemistry, John Wiley & Sons, New York, 1984 Search PubMed.
- T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Doctor, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347–1365 CrossRef CAS PubMed.
- S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. H. Doctor, M. J. Granets, I. K. Khanna, J. W. Malecha, J. M. Miyashiro, T. D. Penning, R. S. Rogers, D. J. Rogier Jr., J. J. Talley and and S. S. Yu, WO9515316, 1995 Search PubMed.
- M. L. Quan, P. Y. S. Lam, Q. Han, D. J. P. Pinto, M. Y. He, R. Li, C. D. Ellis, C. G. Clark, C. A. Teleha, J.-H. Sun, R. S. Alexander, S. Bai, J. M. Luettgen, R. M. Knabb, P. C. Wong and R. R. Wexler, J. Med. Chem., 2005, 48, 1729–1744 CrossRef CAS PubMed.
- F. Li, J. Nie, L. Sun, Y. Zheng and J. A. Ma, Angew. Chem., Int. Ed., 2013, 52, 6255–6258 CrossRef CAS PubMed.
- E. Y. Slobodyanyuk, O. S. Artamonov, O. V. Shishkin and P. K. Mykhailiuk, Eur. J. Org. Chem., 2014, 2487–2495 CrossRef CAS.
- L. Mertens, K. J. Hock and R. M. Koenigs, Chem. – Eur. J., 2016, 22, 9542–9545 CrossRef CAS PubMed.
- F.-G. Zhang, Y. Wei, Y.-P. Yi, J. Nie and J.-A. Ma, Org. Lett., 2014, 16, 3122–3125 CrossRef CAS PubMed.
- L. Sun, J. Nie, Y. Zheng and J. A. Ma, J. Fluorine Chem., 2015, 174, 88–94 CrossRef CAS.
- B. Morandi and E. M. Carreira, Org. Lett., 2011, 13, 5984–5985 CrossRef CAS PubMed.
- G. A. Molander and L. N. Cavalcanti, Org. Lett., 2013, 15, 3166–3169 CrossRef CAS PubMed.
- A.-J. Cai, Y. Zhenga and J.-A. Ma, Chem. Commun., 2015, 51, 8946–8949 RSC.
- H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC.
- H. M. L. Davies and S. J. Hedley, Chem. Soc. Rev., 2007, 36, 1109–1119 RSC.
- D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918–4931 RSC.
- C.-B. Liu, W. Meng, F. Li, S. Wang, J. Nie and J.-A. Ma, Angew. Chem., Int. Ed., 2012, 51, 6227–6230 CrossRef CAS PubMed.
- H. Q. Luo, G. J. Wu, Y. Zhang and J. B. Wang, Angew. Chem., Int. Ed., 2015, 54, 14503–14507 CrossRef CAS PubMed.
- S. Hyde, J. Veliks, B. Liegault, D. Grassi, M. Taillefer and V. Gouverneur, Angew. Chem., Int. Ed., 2016, 55, 3785–3789 CrossRef CAS PubMed.
- O. A. Argintaru, D. Ryu, I. Aron and G. A. Molander, Angew. Chem., Int. Ed., 2013, 52, 13656–13660 CrossRef CAS PubMed.
- G. A. Molander and D. Ryu, Angew. Chem., Int. Ed., 2014, 53, 14181–14185 CrossRef CAS PubMed.
- G. J. Wu, Y. F. Deng, C. Q. Wu, X. Wang, Y. Zhang and J. B. Wang, Eur. J. Org. Chem., 2014, 4477–4481 CrossRef CAS.
- W. Kirmse, Eur. J. Org. Chem., 2002, 2193–2256 CrossRef CAS.
- N. R. Candeias, R. Paterna and P. M. P. Gois, Chem. Rev., 2016, 116, 2937–2981 CrossRef CAS PubMed.
- P. W. Marc Tiffeneau and B. Tchoubar, Comptes Rendus, 1937, 205, 54–56 Search PubMed.
- C. R. Holmquist and E. J. Roskamp, J. Org. Chem., 1989, 54, 3258–3260 CrossRef CAS.
- E. Buchner and T. Curtius, Ber. Dtsch. Chem. Ges., 1885, 18, 2371–2377 CrossRef.
- F. Schlotterbeck, Ber. Dtsch. Chem. Ges., 1826, 40, 1826–1827 CrossRef.
- W. L. Mock and M. E. Hartman, J. Org. Chem., 1977, 42, 466–472 CrossRef CAS.
- W. L. Mock and M. E. Hartman, J. Org. Chem., 1977, 42, 459–465 CrossRef.
- B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 9085–9088 CrossRef CAS PubMed.
- J. Y. Hamilton, B. Morandi and E. M. Carreira, Synthesis, 2013, 1857–1862 CAS.
- S. A. Kunzi, B. Morandi and E. M. Carreira, Org. Lett., 2012, 14, 1900–1901 CrossRef CAS PubMed.
- B. Pieber and C. O. Kappe, Org. Lett., 2016, 18, 1076–1079 CrossRef CAS PubMed.
- A. V. Arkhipov, V. V. Arkhipov, J. Cossy, V. O. Kovtunenko and P. K. Mykhailiuk, Org. Lett., 2016, 18, 3406–3409 CrossRef CAS PubMed.
- P. K. Mykhailiuk, Chem. – Eur. J., 2014, 20, 4942–4947 CrossRef CAS PubMed.
- P. K. Mykhailiuk, Org. Biomol. Chem., 2015, 13, 3438–3445 CAS.
- J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465–7478 RSC.
- J. Hu and C. Ni, Sci. Synth., C-1 Build. Blocks Org. Synth., 2014, 409–458 Search PubMed.
- P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2015, 54, 6558–6561 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |