DOI:
10.1039/C6OB00593D
(Review Article)
Org. Biomol. Chem., 2016,
14, 5875-5893
Recent applications in natural product synthesis of dihydrofuran and -pyran formation by ring-closing alkene metathesis
Received
18th March 2016
, Accepted 13th April 2016
First published on 25th April 2016
Abstract
In the past two decades, alkene metathesis has risen in prominence to become a significant synthetic strategy for alkene formation. Many total syntheses of natural products have used this transformation. We review the use, from 2003 to 2015, of ring-closing alkene metathesis (RCM) for the generation of dihydrofurans or -pyrans in natural product synthesis. The strategies used to assemble the RCM precursors and the subsequent use of the newly formed unsaturation will also be highlighted and placed in context.
 From left to right, Reece Jacques, Ritashree Pal, Nicholas A. Parker, Claire E. Sear and Peter W. Smith | From left to right, Reece Jacques (MChem, University of Liverpool), Ritashree Pal (BSc, University of Calcutta, and MSc, Indian Institute of Technology Kharagpur), Nicholas A. Parker (MSci and BA, University of Cambridge), Claire E. Sear (MChem, University of Warwick) and Peter W. Smith (MSci, University of Glasgow) are all currently postgraduate students at the University of Oxford and part of the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine. |
 Aubert Ribaucourt | Aubert Ribaucourt (Diplôme D'ingénieur in Chemistry, National Institute of Applied Sciences of Rouen), is currently working as a DPhil student as part of the Oxford Innovative Organic Synthesis for Cancer Research programme, under the supervision of Prof D. M. Hodgson. |
 David M. Hodgson | David M. Hodgson (BSc, University of Bath, and PhD, University of Southampton, with Prof P. J. Parsons) was a lecturer at the University of Reading for five years before moving in 1995 to the University of Oxford, where he is now a Professor of Chemistry, with research interests broadly in the development of synthetic methods and their application in natural product synthesis. |
Introduction
The potential of metal complex-catalysed alkene metathesis as useful synthetic methodology began to be widely assimilated by organic chemists in the early 1990s. This followed the development and demonstrated utility of easy to handle and functional group-tolerant Ru catalysts.1,2 The chemistry has proved especially convenient in carbo- and heterocyclic ring synthesis (Scheme 1). 5- and 6-membered oxacycles constitute important heterocyclic motifs, found in a variety of bioactive natural products.3 A significant number of total (or fragment) syntheses of such oxacycle-containing natural products have been reported using ring-closing alkene metathesis (RCM) as a key step, typically using Grubbs 1st or 2nd generation catalysts (GI, GII), or Hoveyda-Grubbs II catalyst (HGII).
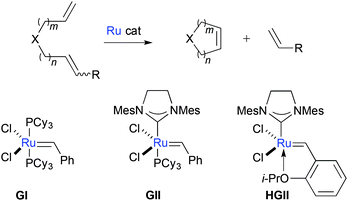 |
| | Scheme 1 Ring-closing alkene metathesis (RCM) and Ru catalysts commonly used. | |
This review focuses on total, formal and fragment syntheses of natural products that possess 5- or 6-membered oxacycles, where a dihydrofuran (DHF) or dihydropyran (DHP) is formed by RCM (Scheme 1, X = O, m = 0, 1; n = 1, 2, 3). A critical overview is given. The aim is to provide the reader with an appreciation of the various ways that such RCM chemistry has been, and could be, employed as a key strategic element to facilitate target synthesis. RCM substrate assembly and post-RCM manipulations are also analysed. Direct formations of furanones and pyranones by RCM of unsaturated esters have recently been nicely reviewed in the context of natural product syntheses,4 and are not further discussed here. In the current review, examples are grouped according to RCM product ring size and double bond position (2,5-DHF, 2,3-DHF, 3,6-DHP, 3,4-DHP sections); within the sections, similarly substituted systems, and the routes to them, are compared. Coverage is from mid-2003![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 to end-2015.6–8
5 to end-2015.6–8
2,5-Dihydrofurans
For 2-substituted-2,5-DHFs, RCM is a straightforward strategic disconnection, due to the ease of RCM substrate construction, typically by aldehyde C-vinylation–O-allylation. For example, the free-radical scavenger (−)-gloeosporiol (2) was accessed through RCM of an ether 1 available by O-allylation of the corresponding enzymatically-resolved benzylic alcohol (Scheme 2).9 Subsequent diastereoselective dihydroxylation and desilylation completed the synthesis.
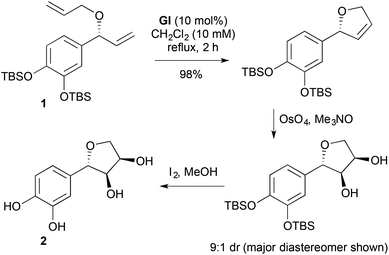 |
| | Scheme 2 Synthesis of (−)-gloeosporiol (2). | |
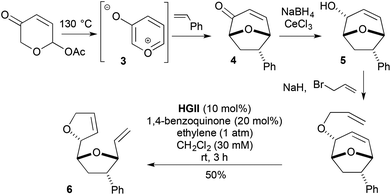
1,2-Reduction of an enone, then O-allylation delivers an alternative entry to metathesis substrates that lead to 2-substituted-2,5-DHFs. This strategy formed part of a stereochemically flexible approach to bis-THF containing acetogenins (Scheme 3),10 where the enones (e.g., 4) were derived from regio- and stereo-selective [5 + 2] cycloaddition of in situ generated 3-oxidopyrylium (3) with alkene dipolarophiles. In these cases, the 2-substituted-2,5-DHF 6 is produced through strain relief-driven ring rearrangement metathesis (RRM), and is carried out in the presence of 1,4-benzoquinone under an ethylene atmosphere. The quinone alleviates competitive allyl ether to 1-propenyl ether isomerisation, likely catalysed by Ru hydrides generated in the reaction. The ethylene promotes catalyst release following ring rearrangement. Mitsunobu inversion at the allylic alcohol 5 stage broadens the methodology to encompass stereochemically different acetogenin targets.
 |
| | Scheme 3 Ring rearrangement metathesis (RRM) towards acetogenins. | |
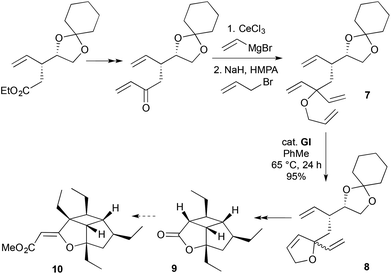
For 2,2-disubstituted-2,5-DHFs, ketone C-vinylation–O-allylation provides a convenient approach to RCM substrates. For example, the tricyclic core 9 of hippolachnin A (10) was recently synthesised from a D-mannitol-derived RCM substrate 7 made in this way (Scheme 4).11 Metathesis using GI likely initiated at the least hindered terminal olefin of the tetraene 7, with RCM occurring non-stereoselectively at the formally diastereotopic vinyl groups. Following acetonide manipulation, only one of the two diastereomeric 2,5-DHFs 8 subsequently underwent intramolecular [2 + 2] photocycloaddition. In principle, a chiral RCM catalyst12 could induce stereoselectivity in the RCM step.
 |
| | Scheme 4 Synthesis of the core of hippolachnin A (10). | |
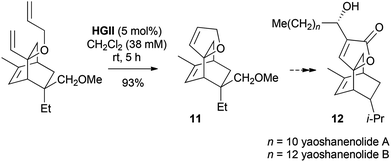
With cyclic ketones, C-vinylation–O-allylation followed by RCM leads to spiro-fused DHFs. In a model study, the tricyclic framework 11 of the cytotoxic yaoshanenolides 12 was completed in this fashion (Scheme 5).13,14
 |
| | Scheme 5 Model studies towards yaoshanenolides A and B. | |
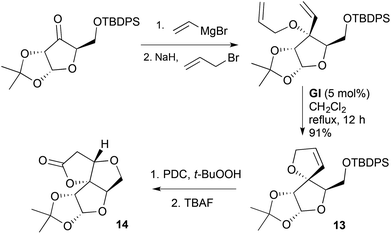
The strategy of C-vinylation–O-allylation followed by RCM has been applied in several instances to cyclic ketones bearing α-hydroxymethyl functionality.15,16 In such cases, subsequent allylic oxidation of the spiro-fused DHF 13 generates the corresponding furanone, which undergoes oxa-Michael addition; this provides a rapid entry to tricyclic systems containing the furo[3,2-b]furanone motif 14 (Scheme 6).17 The ABC ring systems 15 of the nortriterpenoid anti-HIV agents micrandilactone A (16) and lacnifodilactone G were prepared from D-mannitol using this strategy (Scheme 7).18
 |
| | Scheme 6 Synthesis of embedded furo[3,2-b]furanone in a trioxatriquinane. | |
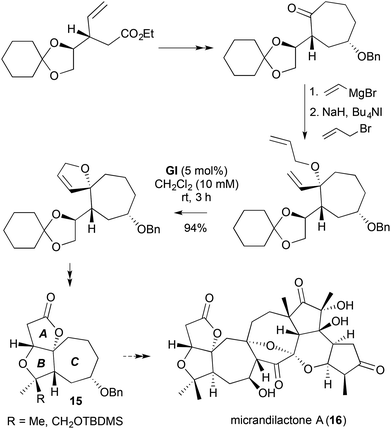 |
| | Scheme 7 Synthesis of the ABC ring systems of micrandilactone A and lacnifodilactone G. | |
Direct access to 2,3-disubstituted-2,5-DHFs by RCM requires a 2,2-disubstituted-1-alkene in the substrate. Syntheses of the isospongian diterpenoids (−)-marginatafuran, (−)-marginatone and (−)-20-acetoxymarginatone used such an approach (Scheme 8).19 Carvone-derived α,β-epoxy ketones 17 underwent stereoselective reduction, then O-allylation and regioselective epoxide to allylic alcohol isomerisation using diethyl aluminium-tetramethylpiperidide (TMP), to give the RCM precursors 18. Homodimerisation and allyloxy to enol ether isomerisation in the RCM step were suppressed by dilution and addition of 1,4-benzoquinone. Subsequent oxidation of the fused DHF 19 with DDQ gave the furan motif present in the natural products.
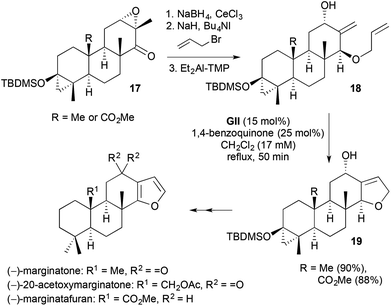 |
| | Scheme 8 Synthesis of isospongian diterpenoids. | |
A synthesis of the furan cembranolide (−)-(Z)-deoxypukalide (24) involved generation of a transient 2,3,5-trisubstituted DHF by RCM (Scheme 9).20 Selective ozonolysis of the trisubstituted alkene in the TIPS ether of (S)-perillyl alcohol 20, followed by aldehyde selective addition of a vinyl alane from methyl propiolate gave an allylic alcohol 21 (1:1 dr, mixture inconsequential). Acid-catalysed acetal exchange with acrolein diethyl acetal gave the RCM precursor 22. RCM, initiating at the less-substituted terminal alkene, gave an α-ethoxy-substituted DHF, which underwent acid-catalysed elimination of EtOH/aromatisation; the resulting 2,3-disubstituted furan 23 was taken forward to the target macrocycle 24.
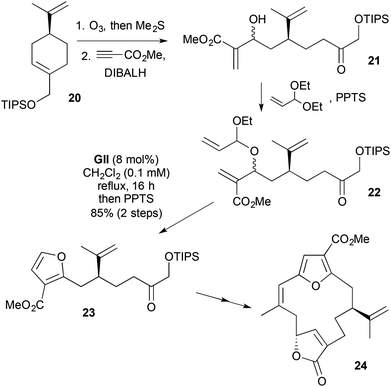 |
| | Scheme 9 Synthesis of (−)-(Z)-deoxypukalide (24). | |
A synthesis of the NBoc-protected α-amino acid antibiotic (+)-furanomycin 28 (Scheme 10),21 illustrates that the aldehyde C-vinylation–O-allylation RCM strategy can be extended to 2,5-disubstituted DHFs 27. The RCM substrate 26 synthesis involved syn-selective alkynylation of serine-derived Garner's aldehyde 25 and non-stereoselective O-allylation. After RCM, hydrolysis and oxidation gave NBoc-furanomycin 28, along with its methyl epimer.
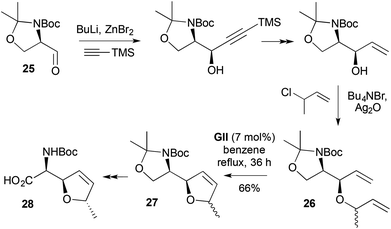 |
| | Scheme 10
C-vinylation–O-allylation RCM strategy to NBoc-furanomycin 28. | |
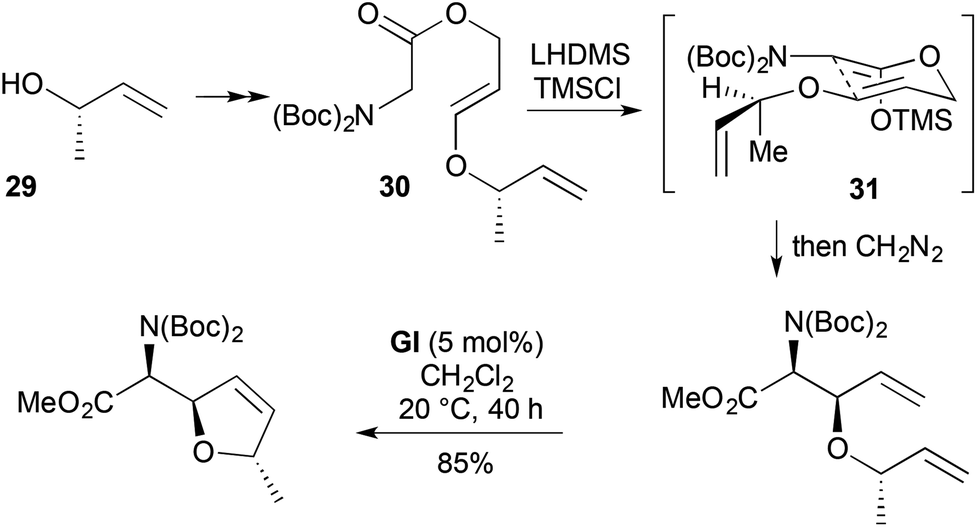
An alternative approach to a similar RCM substrate for furanomycin synthesis involved Ireland–Claisen rearrangement (Scheme 11).22 Base-catalysed conjugate addition of (S)-but-3-en-2-ol (29) to methyl propiolate led to an allylic E-enol ester. DIBALH reduction gave the corresponding alcohol, which coupled with NBoc2-protected glycine to give the rearrangement substrate 30. [3,3] Sigmatropic rearrangement of the Z-silyl ketene acetal 31 proceeded with complete relative stereocontrol for the two newly created stereocentres and modest (72:28) stereocontrol relative to the pre-existing stereocentre.
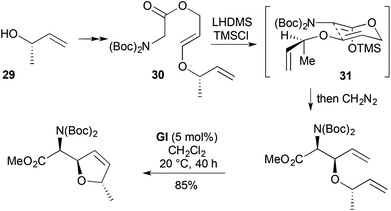 |
| | Scheme 11 Ireland–Claisen RCM strategy to furanomycin. | |
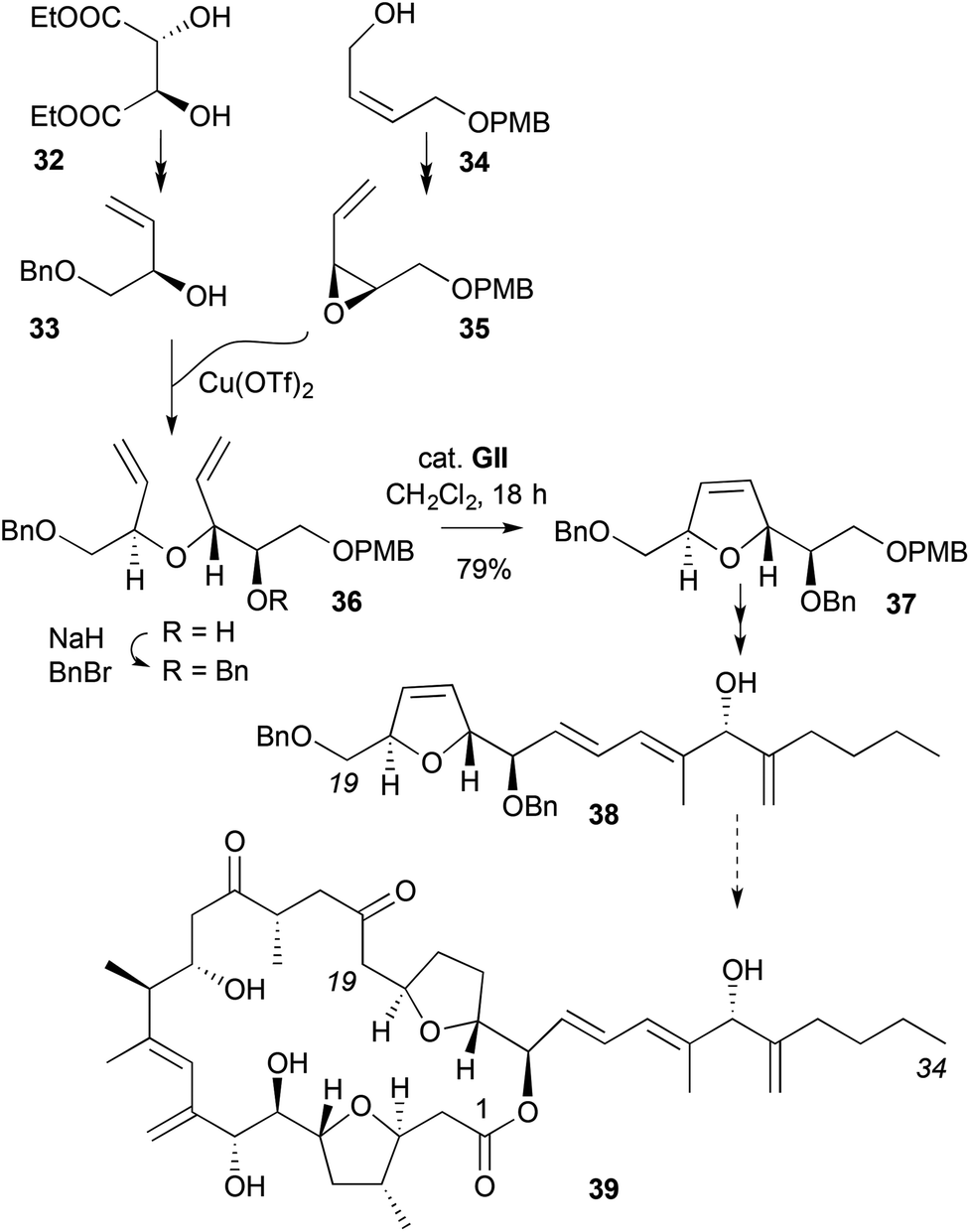
A synthesis of a 2,5-disubstituted DHF 37, where the α-,α′-stereochemistry in the acyclic ether RCM precursor 36 is assembled with high stereocontrol, is found in studies to the C19–C34 segment 38 of the cytotoxic marine natural product amphidinolide C (39) (Scheme 12).23,24 One of the stereocentres was developed from (+)-diethyl tartrate (32) as an enantiopure allylic alcohol 33, while the other was generated via Sharpless asymmetric epoxidation (SAE, 92:8 er) on the mono PMB ether of cis-2-butene-1,4-diol 34; the latter was inverted at the allylic position via Lewis acid-mediated opening of epoxide 35 by the allylic alcohol 33.
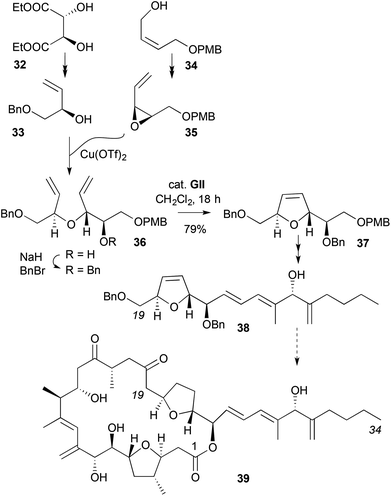 |
| | Scheme 12 Stereocontrolled synthesis of C19–C34 segment of amphidinolide C. | |
A synthesis of the anticancer agent (−)-mucocin (43) involved construction of a 2,5-disubstituted DHF 42 by RCM, where stereochemistry is introduced onto an acyclic ether 41 prior to RCM (Scheme 13).25,26 The first DHF stereocentre was generated via kinetic resolution using SAE. Subsequent O-alkylation with bromoacetic acid and introduction of Evans’ auxiliary gave an N-glycolyl oxazolidinone 40. A Lewis acid-catalysed syn-aldol reaction with acrolein generated the second (α′-) stereocentre, as well as the adjacent hydroxyl stereocentre (destined to be exocyclic). Auxiliary removal and homologation provided the RCM substrate 41, with an unsaturated tether that was found to be crucial in providing RCM selectivity. RCM without the tether led to a mixture of desired DHF 42 and 2,5-DHP, due to the similar affinity of either terminal alkene for the catalyst. With the engineered tether, relay RCM occurs: the catalyst first reacts with the less-hindered, electronically activated terminal alkene, then liberates a molecule of 2,5-DHF, resulting in the metallocarbene at the desired position for selective 2,5-DHF formation. Three-fragment assembly of the mucocin skeleton was then achieved via cross-metathesis to install the tetrahydropyran section, followed by Sonogashira coupling to add the 2,5-dihydrofuranone. The tetrahydropyran portion was also made by RCM, and is discussed later in this review (Scheme 32). Completion of the synthesis was achieved by mild hydrogenation of the connecting unsaturated double bond using diimide.
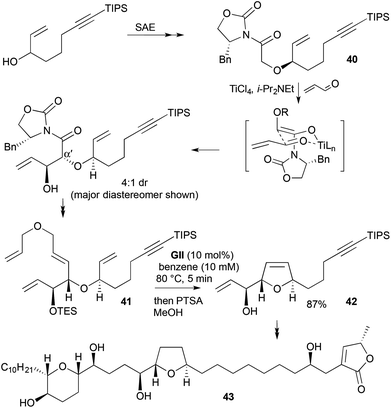 |
| | Scheme 13 Synthesis of (−)-mucocin (43). | |
RCM has been used to generate bridged/2,5-substituted DHFs from cyclic ethers bearing vinyl groups at the α- and α′-positions. A simplified analogue 47 of the anti-mitotic diterpenoid eleutherobin was constructed in this fashion (Scheme 14).27 Claisen rearrangement of a 2-deoxy-D-ribose derivative gave a 9-membered lactone 44. 3:1 dr At the α-,α′-positions was generated through chloroselenation–reductive dechlorination of the methylenated lactone 45. RCM using GI on the derived divinyl ether 46 of the major diastereomer gave a mixture of the desired bridged DHF 47 (69%) and a bis-cyclopentenyl ether 48 (22%) from RRM; the more active GII gave more of the RRM product 48. Formation of the latter was avoided by temporary epoxidation of the cis double bond in the 9-membered ring; deoxygenation was effected after RCM using WCl6 and BuLi. A related potential RCM substrate 46 (R = OMe) failed to undergo RCM, likely due to steric hindrance.
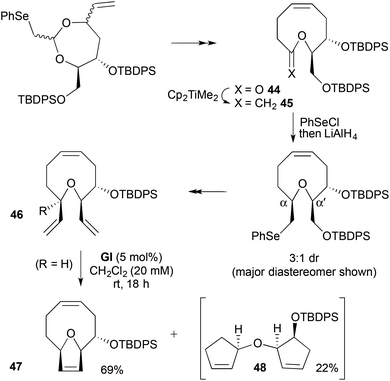 |
| | Scheme 14 Synthesis of an eleutherobin analogue. | |
RCM was used to make a DHF 52 present in the smaller bridged framework of bruguierol A (53) (Scheme 15).28O-Ethylation and homologation of 3-hydroxybenzaldehyde was followed by Friedel–Crafts acylation to give a ketoaldehyde 49. Double vinylation and acid-catalysed dehydrative cyclisation gave an inseparable 1:1 mixture of diastereomeric divinyl ethers 50 and 51, but only the cis-divinyl system 50 underwent reaction with GII, to give the bridged DHF 52. Hydrogenation of the alkene, followed by de-ethylation gave (±)-bruguierol A (53).
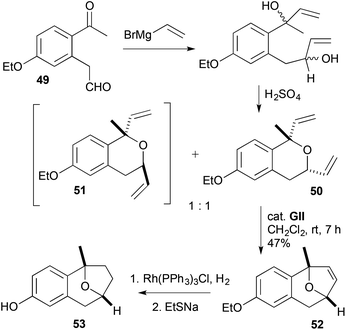 |
| | Scheme 15 Synthesis of (±)-bruguierol A (53). | |
2,3-Dihydrofurans
The enol ether character of 2,3-DHFs (and 3,4-DHPs) leads to application in spiroketal synthesis, by cyclisation of a hydroxyl-bearing tether at the 5-position. Direct access to 2,3-DHFs through RCM requires an acyclic enol ether of a homoallylic alcohol as the precursor. A convergent strategy involving this approach was described to the C15–C38 fragment 60 of the protein phosphatase (PP1 and PP2A) inhibitor okadaic acid (61) (Scheme 16).29 Regioselective hydroboration of a terminal alkene 54, followed by immediate Suzuki coupling with an unsaturated ester-derived enol phosphate 55 gave an enol ether 56 that was used directly in RCM; potential intramolecular Heck-type chemistry of the enol phosphate was not a complicating issue. DDQ-induced PMB deprotection on the resulting acid-sensitive crude DHF 57 led to spontaneous spirocyclisation, giving a ∼3:1 diastereomeric mixture of spiroketals 58 and 59. The undesired minor diastereomer 59 could be equilibrated under acidic conditions to provide more of desired spiroketal 58 (71%). DHF 57 was originally planned to come from Suzuki coupling of a lactone-derived enol phosphate, avoiding an RCM step; however, the cyclic enol phosphate was found to readily hydrolyse back to the lactone.
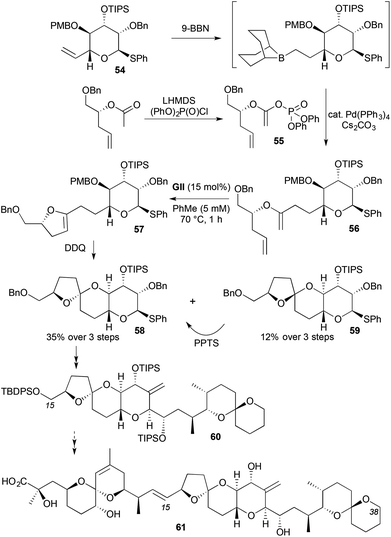 |
| | Scheme 16 RCM-spiroketalisation approach to okadaic acid (61). | |
3,6-Dihydro-2H-pyrans
Between 2003 and 2015, there were over 30 reported applications of RCM generating 3,6-DHPs in natural product synthesis. Allylation of homoallylic alcohols is a popular approach to the RCM substrates, with a variety of methods being used to access the homoallylic alcohols, depending on the required substitution pattern. For example, 2-methyl-3,6-DHP (63), prepared by RCM from allylated (R)-4-penten-2-ol (62), provided divergent access to ophiocerins A–C (Scheme 17).30
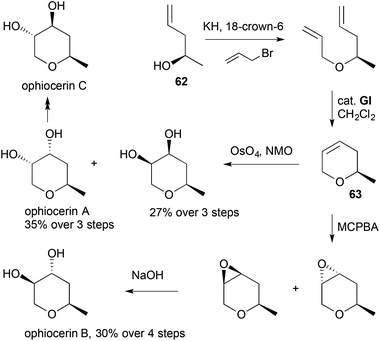 |
| | Scheme 17 Synthesis of ophiocerins A–C. | |
Anti C-allylation of D-mannitol-derived isopropylidene glyceraldehyde 64, followed by O-allylation, isopropylidene cleavage and RCM, gave protected 2-hydroxymethyl-3,6-DHPs (e.g., 65) that have been used in pyrano-fused pyrazine synthesis31 (e.g., Scheme 1831b). The corresponding epoxide of the RCM product underwent ring-opening with azide, then oxidation to give an azido ketone 66. Reduction of the azide 66 to the amine led to dehydrative dimerisation–aromatisation in the presence of air, to give (S,S)-palythazine (67).
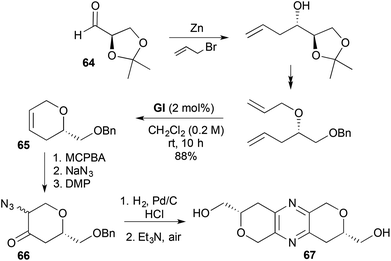 |
| | Scheme 18 Synthesis of (S,S)-palythazine (67). | |
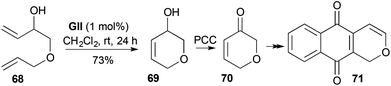
RCM of the allyl alcohol–butadiene monoepoxide addition product 68, to give 3-hydroxy-3,6-DHP (69), was involved in the preferred route to the corresponding pyranone 70 that was used in a synthesis of the pyranonaphthoquinone pentalongin (71) (Scheme 19).32 Prior syntheses of the pyranone 70 used vinyl stannane-unsaturated acid chloride Pd-catalysed coupling and subsequent RCM (17%), or a Hg(II)-mediated ring-closure from an alkyne (21%).
 |
| | Scheme 19 Synthesis of pentalongin (71). | |
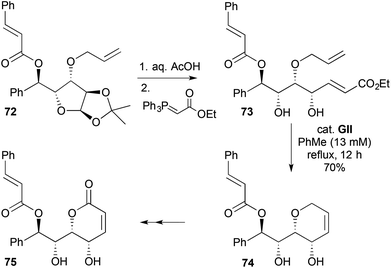
A 2,3-disubstituted-3,6-DHP 74 was made by RCM in a synthesis of the styryl-lactone (+)-howiionol (75) (Scheme 20).33 Deacetalisation of a glucose derivative 72 gave a hemiacetal that underwent olefination with a stabilised Wittig reagent, to give the RCM substrate 73. Following RCM, diol protection using 2,2-dimethoxypropane, then PDC-induced allylic oxidation and finally acetonide removal gave (+)-howiionol (75).
 |
| | Scheme 20 Synthesis of (+)-howiionol (75). | |
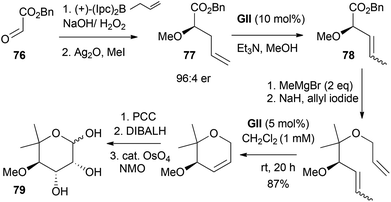
Two syntheses of L-(+)-noviose (79), the sugar component of the anticancer agent novobiocin, involve formation of 2,2,3-trisubstituted-3,6-DHPs by RCM (Schemes 21 and 22). In the first approach (Scheme 21),34 key steps to the RCM substrate are asymmetric Brown allylation of benzyl glyoxylate (76), and terminal alkene isomerisation to the 2-propenyl equivalent using GII (77 → 78); a Ru–H is the likely active catalytic species, formed in situ from heating GII in reagent grade methanol. After RCM, noviose (79) was obtained by allylic oxidation to the pyranone, 1,2-reduction and dihydroxylation. Compared with earlier carbohydrate-based syntheses, triol generation at the end avoided hydroxyl group protection/deprotection manipulations.
 |
| | Scheme 21 Synthesis of L-(+)-noviose (79) from benzyl glyoxylate. | |
 |
| | Scheme 22 Synthesis of L-(+)-noviose (79) from ethyl S-lactate. | |
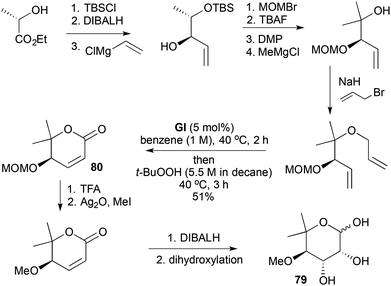
The second synthesis of L-(+)-noviose (79) uses diastereoselective (90:10 dr) vinylation of a lactic acid-derived aldehyde to introduce the methoxy-bearing stereocentre (Scheme 22).35 RCM with GI is followed by addition of t-BuOOH and conversion to a pyranone 80 in a one-pot procedure; the hydroperoxide converts GI into a catalyst for allylic oxidation. Subsequent MOM deprotection and methylation intersects with the earlier synthesis.
Two approaches to the microtubule-stabilising agent laulimalide (83) use RCM to make the 2,4-disubstituted-3,6-DHP that is attached to the macrocyclic lactone (Schemes 23 and 24). Both studies apply Julia–Kocieński olefination chemistry from the same DHP-containing sulfone 82 to extend the 2-substitution, where the sulfone-stabilised anion does not cleave the DHP by β-elimination. In the first synthesis (Scheme 23),36 access to the DHP sulfone 82 for aldehdye olefination begins with a Mitsunobu reaction on (R)-glycidol using a tetrazole thiol. This was followed by terminal epoxide opening with isopropenyl copper, Pd-catalysed allylation of the zinc alkoxide, oxidation to the sulfone 81 and RCM. In the second approach, the DHP stereocentre is installed by aldehyde methallylation under Keck conditions, followed by Williamson etherification (Scheme 24).37 The resulting diallyl ether was elaborated to the DHP sulfone 82 either by performing RCM prior to tetrazole formation (as shown; GI sufficed in this case), or by PMB ether conversion using Mitsunobu chemistry to the same RCM sulfone substrate 81 shown in Scheme 23.
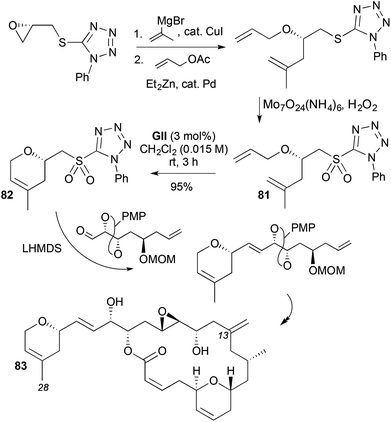 |
| | Scheme 23 Synthesis of laulimalide (83). | |
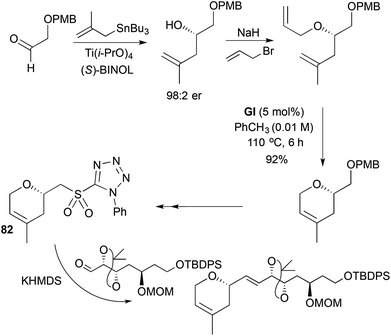 |
| | Scheme 24 Synthesis of C13–C28 portion of laulimalide. | |
The 2,5-disubstituted-3,6-DHP found in the marine sponge metabolites cacospongionolides B and E 87 has been prepared by RCM (Scheme 25).38 The unsaturated decalone portion 84 of the natural products, prepared by asymmetric Robinson annulation of 2-methylcyclohexane-1,3-dione and ethyl vinyl ketone, underwent reductive conjugate addition to an enone 85 (available from Brown asymmetric allylboration of 3-furfural), followed by double ketone Wittig methylenations to generate the RCM substrate 86. Exposure of this triene 86 to GII led to RCM, likely initiated at the less-hindered terminal olefin, and completed the carbon skeleton of the natural products.
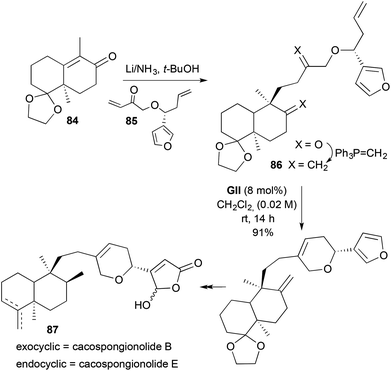 |
| | Scheme 25 Syntheses of cacospongionolides B and E. | |
A modestly diastereoselective DDQ-mediated oxidative allylation was used to make the allylic-homomoallylic ether RCM substrate 88 in a synthesis of the 2,6-cis-disubstituted tetrahydropyran core 89 of (±)-centrolobine (90) (Scheme 26).39
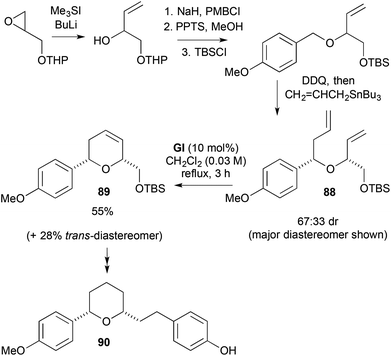 |
| | Scheme 26 Synthesis of centrolobine (90). | |
A cis-2,6-disubstituted-3,6-DHP was made by RCM during studies on the configuration of the antifungal goniodomin A (94) (Scheme 27).40 Two chiral alcohols were connected using bromoacetic acid. The resulting glycolate ether 91 gave the RCM substrate 93 following Ireland–Claisen rearrangement through the methyldichlorosilyl ketene acetal 92; use of less reactive TMSCl gave significant by-product due to [2,3] Wittig rearrangement.
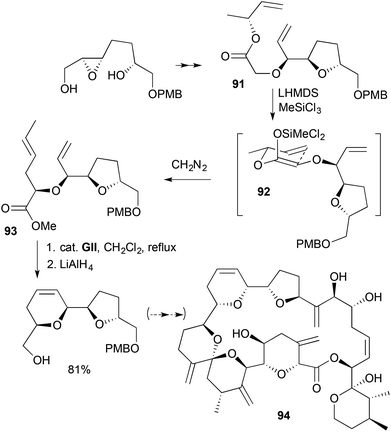 |
| | Scheme 27 Studies towards goniodomin A (94). | |
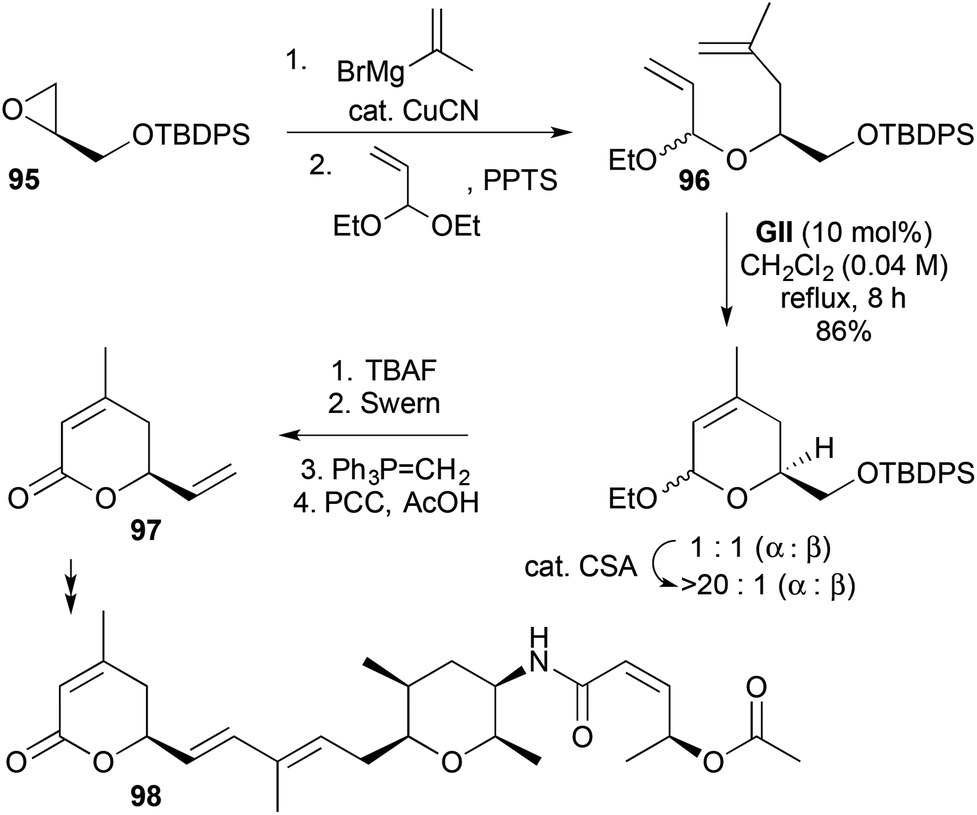
2,6-Disubstituted-3,6-DHPs bearing further substitution at the 4- or 5-positions have been accessed for natural product synthesis using RCM of more substituted alkenes (Schemes 28–30). In the synthesis of spliceostatin E (98), isopropenylcopper-induced regioselective ring-opening of an epoxide 95, followed by acid-catalysed acetal exchange with acrolein diethyl acetal gave the RCM substrate 96 (Scheme 28).41 Cross-metathesis was used to develop the pyranone 97 derived from RCM to the natural product 98.
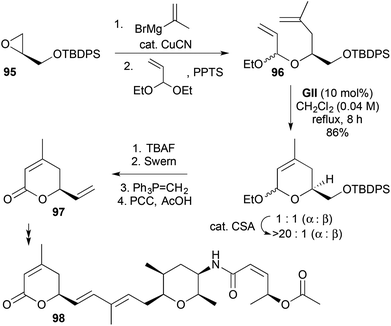 |
| | Scheme 28 Synthesis of spliceostatin E (98). | |
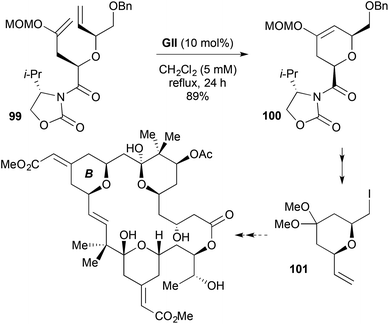 |
| | Scheme 29 Studies towards bryostatin 11. | |
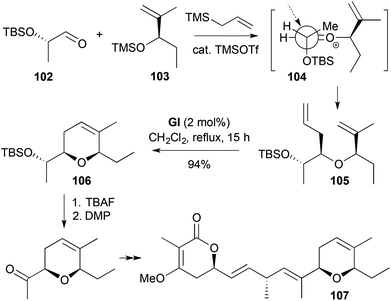 |
| | Scheme 30 Synthesis of jerangolid D (107). | |
In an approach to a bryostatin B-ring building block 101, RCM involving a MOM enol ether as one of the reacting alkenes was used to form a cis-2,6-disubstituted-3,6-DHP 100 (Scheme 29).42 The RCM substrate 99 was prepared by diastereoselective allylation of the enolate of an N-glycolyl oxazolidinone.
A 2,5,6-trisubstituted-3,6-DHP formed by RCM requires alkene substitution on the allylic part of the allylic–homoallylic ether RCM substrate (e.g., 105, Scheme 30).43 In this synthesis of jerangolid D (107), TMSOTf-catalysed three-component coupling gave an acyclic ether 105 for cis-2,6-disubstituted-3,6-DHP 106 formation by RCM. The required ether 105 was generated by completely diastereoselective allylation of the oxonium species from the aldehyde 102 and TMS ether 103, with the sense of asymmetric induction being rationalised through a Felkin-Anh transition state 104.
The use of RCM to produce 2,3,6-trisubstituted-3,6-DHPs requires assembly of a RCM substrate containing up to 3 stereocentres. In a synthesis of the protein phosphatase 2A inhibitor phoslactomycin B (111) (Scheme 31),44,45 the required 2,3-stereochemistry was set by a [2,3] Wittig rearrangement (dr > 96:4), where the rearrangement precursor 108 came from Noyori catalytic asymmetric reduction of an ynone (91:9 er). Subsequent transacetalisation with acrolein dimethyl acetal gave the RCM substrate 109. Relay RCM using GII gave a DHP 110 as an inconsequential 1:1 epimeric mixture. The corresponding acrylate was originally studied in the relay RCM step; however, this transformation was found to be difficult. Relay RCM was used in this synthesis to direct initiation, potentially avoiding enyne metathesis.
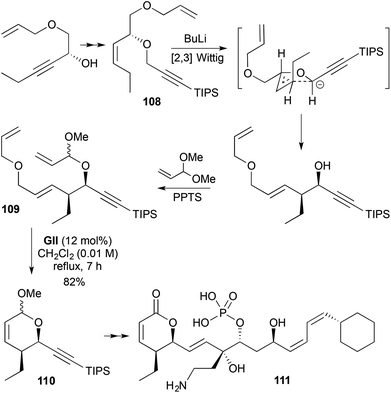 |
| | Scheme 31 Synthesis of phoslactomycin B (111). | |
As mentioned earlier, in a synthesis of (−)-mucocin (43) both DHF 42 (Scheme 13) and DHP 114 (Scheme 32) rings were generated using RCM steps.25 Similarly to the DHF RCM precursor synthesis (Scheme 13), the 2,3,6-trisubstituted-3,6-DHP precursor 113 was stereoselectively accessed through a titanium enolate-mediated syn-aldol reaction of a N-glycolyl oxazolidinone 112 with acrolein.
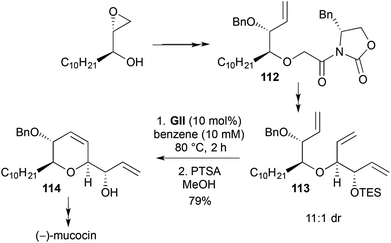 |
| | Scheme 32 Synthesis of a DHP in total synthesis of (−)-mucocin. | |
In the RCM step described above (Scheme 32), non-productive (reversible) metathesis might be considered to be occurring at the allylic silyl ether site, with 4- or 7-membered ring formation not being favoured. However, with a related substrate 115 in a synthesis of the C13–C34 fragment of (−)-mucocin (Scheme 33),46 an allylic silyl ether was also unaffected during RCM, but the corresponding allylic alcohol gave 1:1 mixture of dihydropyran 116 and a cyclooctene 117. These results show the lower propensity of allylic silyl ethers to engage in metathesis.
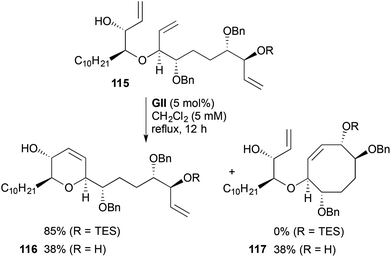 |
| | Scheme 33 Synthesis of THP fragment of (−)-mucocin. | |
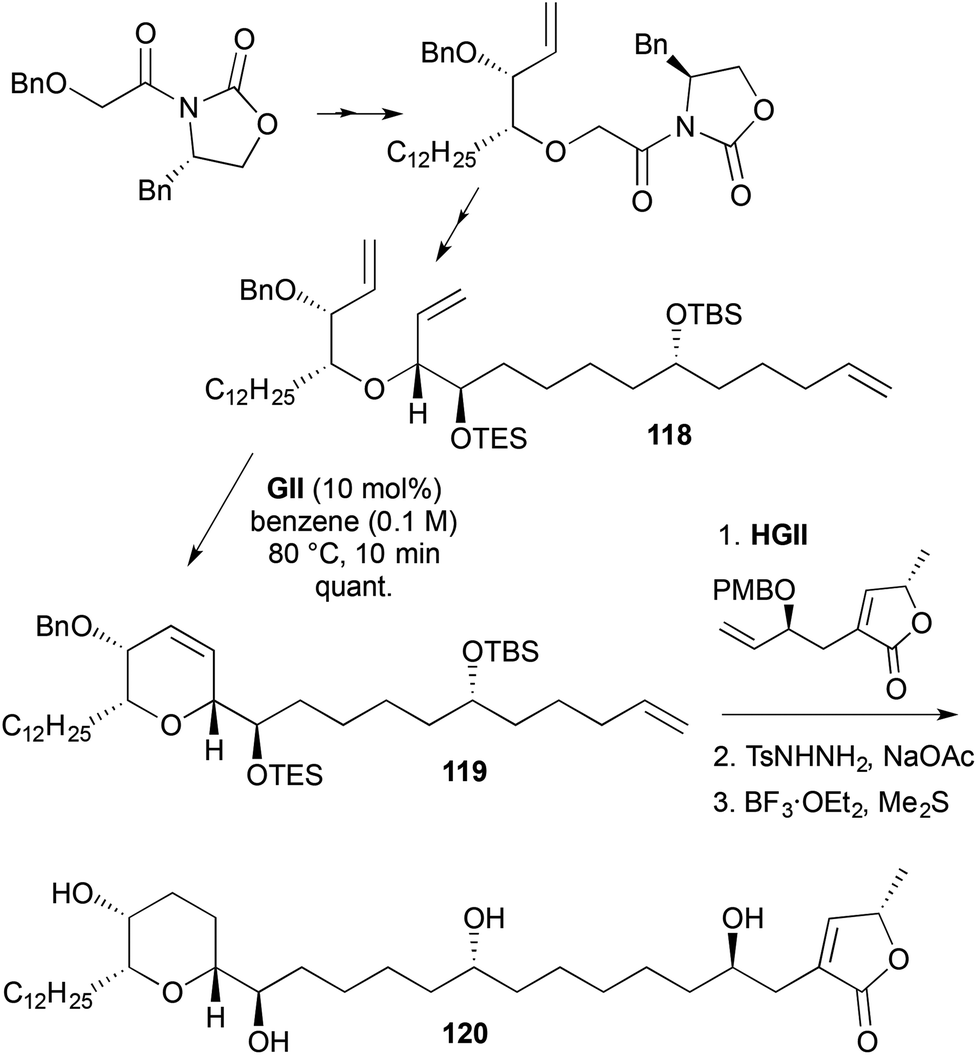
A synthesis of pyranicin (120) involved RCM to generate a 2,3,6-trisubstituted-3,6-DHP 119 (Scheme 34).47 The RCM substrate 118 was prepared via a double Evans chiral auxiliary approach. The RCM product 119 underwent cross-metathesis using HGII to give a triene. While RCM was used to form the DHP ring, the newly formed double bond was not required in the product and it, along with the exocyclic alkene from the cross-metathesis, were hydrogenated using diimide; subsequent alcohol deprotections gave pyranicin (120).
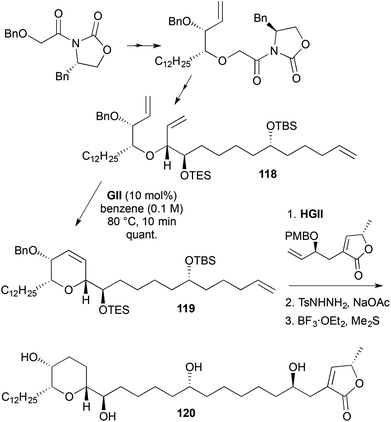 |
| | Scheme 34 Synthesis of pyranicin (120). | |
(−)-Brevisamide (123) contains a 2,3,5,6-tetrasubstituted tetrahydropyran that has been accessed in a stereocontrolled manner (Scheme 35).48 A SAE-derived allylic alcohol 121 was extended with bromoacetic acid to give an N-glycolyl oxazolidinone 122 that underwent a syn-aldol with acrolein, as described earlier (Scheme 13). The RCM step involves a 2,2-disubstituted-1-alkene, allowing the final ring stereocentre to be installed by stereocontrolled hydrogenation.
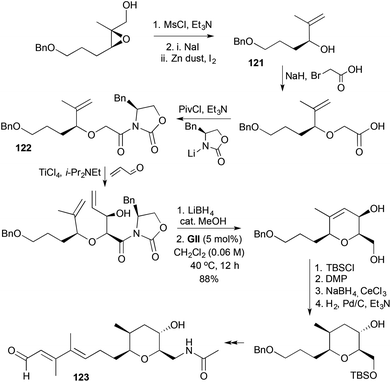 |
| | Scheme 35 Synthesis of (−)-brevisamide (123). | |
Spiro-, fused- and bridged-3,6-DHP-containing natural products have also been approached through RCM-based strategies (Schemes 36–44). Synthesis of the B ring of the protein phosphatase 2A inhibitor (+)-spirostrellolide A (126) is achieved by cyclic ketal-tethered RCM (Scheme 36).49 Triflimide-induced ketal formation under stereoelectronic control gives the RCM precursor 125, where the axially introduced homoallylic alcohol 124 is derived from an aldehyde anti-isocrotylation using (−)-(E)-crotyldiisopinocampheylborane.
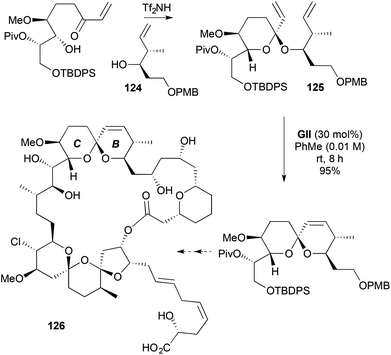 |
| | Scheme 36 B and C ring assembly of spirastrellolide A. | |
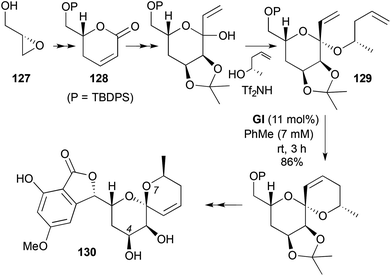 |
| | Scheme 37 Total synthesis of (+)-aigialospirol (130). | |
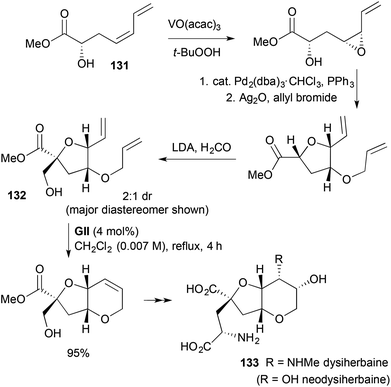 |
| | Scheme 38 Total synthesis of (−)-dysiherbaine (133). | |
 |
| | Scheme 39 Synthesis of the dysiherbaine core using RRM. | |
 |
| | Scheme 40 Domino metathesis step to dysiherbaine and neodysiherbaine. | |
 |
| | Scheme 41 Synthesis of norhalichondrin B (138) using RRM. | |
 |
| | Scheme 42 Synthesis of 3-O-benzyl-1,3,5-tri-epi-calystegine B2 (141). | |
 |
| | Scheme 43 Synthesis of thromboxane B2 (146). | |
 |
| | Scheme 44 Synthesis of didemniserinolipid B (148). | |
Another example of cyclic ketal-tethered RCM is found in the total synthesis of (+)-aigialospirol (130) (Scheme 37).50 (S)-Glycidol (127) was converted via a pyrone 128 into the RCM substrate 129. Although possessing the wrong spirocentre stereochemistry at the RCM step, acid-catalysed acetonide removal at the end of the synthesis resulted in epimerisation to the natural configuration, stabilised by an intramolecular H-bond between C4-OH and O7.
A fused tetrahydropyran system found in the naturally occurring amino acids dysiherbaine and neodysiherbaine has been made by three RCM-based approaches (Schemes 38–40).
In one synthesis of (−)-dysiherbane (133) (Scheme 38),51,52 the RCM substrate 132 was constructed by hydroxyl-directed epoxidation of a methyl glycidate-derived diene 131, followed by Pd-catalysed epoxide ring-opening with retention, allylation and hydroxymethylation.
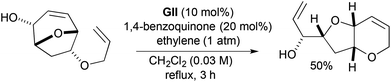
A second route to the 1,5-dioxaoctahydroindene core modifies the RRM chemistry outlined in Scheme 3, by using residual unsaturation originating from the dipolarophile component in the [5 + 2] cycloadduct (Scheme 39).10
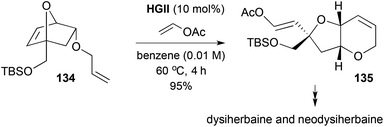
A metathesis-induced oxabicyclic rearrangement strategy related to that shown in Scheme 39 allowed a formal synthesis of dysiherbaine and neodysiherbaine (Scheme 40).53 The metathesis substrate 134 was accessed from Diels–Alder cycloaddition of TBS-protected furfuryl alcohol and an unsaturated sulfone, followed by resolution. RRM–cross-metathesis using HGII in the presence of a large excess of vinyl acetate gave a 1,5-dioxaoctahydroindene 135, with the olefins suitably differentiated to allow progress to dysiherbaine and neodysiherbaine.
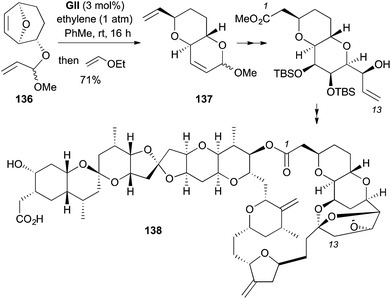
In a total synthesis of the marine polyether norhalichondrin B (138), a trans-fused pyranopyran 137 was formed by RRM of a furan-derived 8-oxabicyclo[3.2.1]oct-6-ene bearing a 2-exo-allylic ether 136 (Scheme 41).54 Addition of ethyl vinyl ether at the end of the RRM poisons the catalyst by formation of a stable Fisher carbene, preventing undesired post-RRM chemistry.
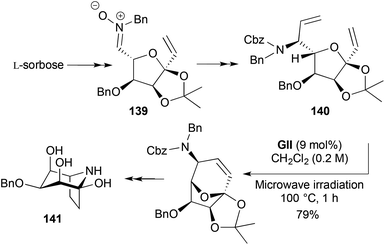
Syntheses of 1,3,5-tri-epi-calystegine B2 (as its 3-O-benzyl derivative 141), thromboxane B2 (TXB2) (146) (the stable hydrolysis product of the prostanoid signaling molecule TXA2), and didemniserinolipid B (148) all feature RCM steps leading to bridged DHPs (Schemes 42–44). The strategy to the calystegine RCM substrate 140 (Scheme 42),55 involved nitrone formation through a sorbose-derived aldehyde condensing with benzylhydroxylamine. The nitrone 139 underwent vinylation and chemoselective reduction, leaving the diene functionality intact. Following RCM, hydrogenation with concomitant N-deprotection and then acetonide hydrolysis led to the ring-closed carbinolamine target 141.
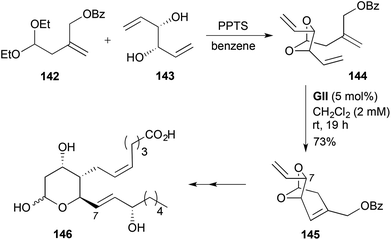
The route to TXB2 (146) (Scheme 43),56 used transacetalisation of a unsaturated acetal 142 with a tartaric acid-derived C2-symmetric dienediol 143 to give a pseudo-C2-symmetric RCM substrate 144, which on RCM with GII led to a bridged dihydropyran 145. Subsequent chemoselective cross-metathesis involving the terminal olefin allowed homologation for eventual conversion to the allylic alcohol side-chain, whereas reagent (SAE)-controlled epoxidation of the endocyclic alkene led to installation of the unsaturated acid side-chain.
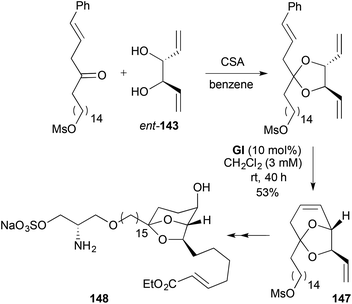
The strategy to didemniserinolipid B (148) (Scheme 44),57 is closely related to that for TXB2, involving ketalisation with the enantiomeric dienediol ent-143 followed by RCM (53%, 81% brsm). The use of GI, as opposed to the more active GII, may be to minimize cross-metathesis of the RCM product 147 with the styrene by-product; the presence of the aryl group was necessary to minimise double bond migration in the earlier ketalisation step.
3,4-Dihydro-2H-pyrans
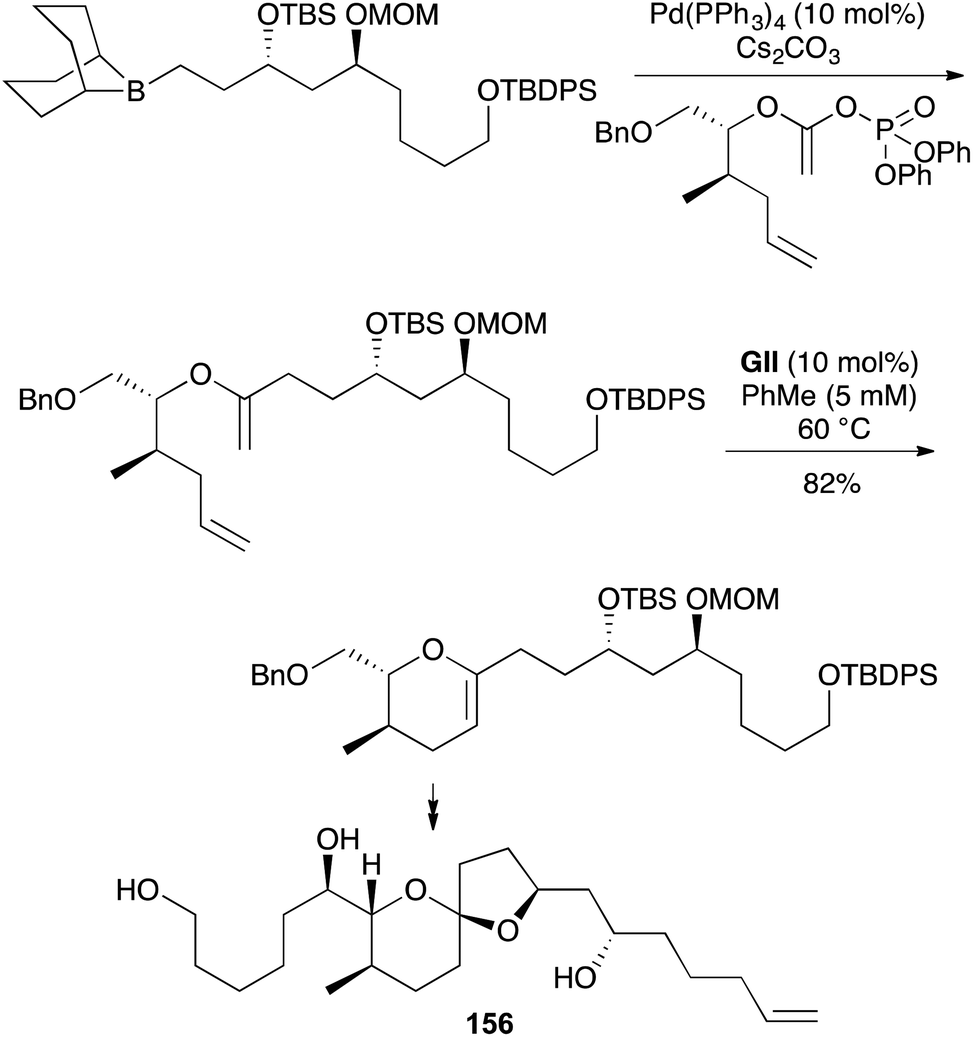
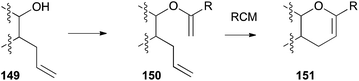
Direct formation of 3,4-DHPs 151 by RCM requires an enol ether 150 of a bishomoallylic alcohol 149 as the substrate (Scheme 45). This strategy, followed by elaboration of the enol ether functionality in the RCM product to build fused or spiro ethers, has found significant application in polyether natural product synthesis. Such cases often involve the creation of a 2,3,6-trisubstituted 3,4-DHP 151 by RCM, where the 2,3-substitution is connected as another oxacycle. Methods to make the enol ether RCM substrates from alcohols include: alkynylation–carbometallation (Scheme 46), cross-coupling the enol phosphate of a derived ester (Schemes 47 and 48), alkylidenation of a derived ester (Schemes 49–51), or Hg(II)-catalysed alkoxy exchange with an alkyl vinyl ether (Scheme 52).
 |
| | Scheme 45 RCM approach to 3,4-dihydro-2H-pyrans. | |
 |
| | Scheme 46 Two-directional approach to gambieric acid A. | |
 |
| | Scheme 47 RCM-spiroketalisation approach to (−)-attenol A (156). | |
 |
| | Scheme 48 DHP formation by RCM in a total synthesis of (+)-gambieric acid A (157). | |
 |
| | Scheme 49 Possible Ti-alkylidene pathways to 3,4-DHPs 151. | |
 |
| | Scheme 50 Total synthesis of (−)-gambierol (165). | |
 |
| | Scheme 51 Takai-Utimoto RCM towards ABCD core of azaspiracid-1. | |
 |
| | Scheme 52 OLEC and RCM chemistry towards adriatoxin (174). | |
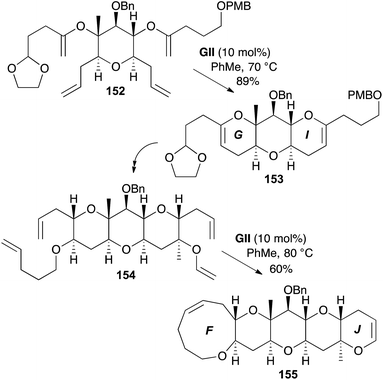
A two-directional approach to the F–J fragment 155 of the enantiomer of (+)-gambieric acid A involved RCM chemistry to form both the G and I rings in one step from the corresponding bis(enol ether) 152 (Scheme 46).58 The RCM substrate 152 was formed via successive carbocuprations from a bis(alkynyl ether). Double hydroboration of the RCM product 153 gave a diol for further functionalisation. The F and J rings were also formed in one step using RCM (154 → 155).
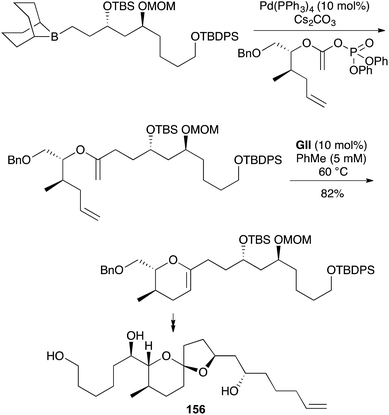
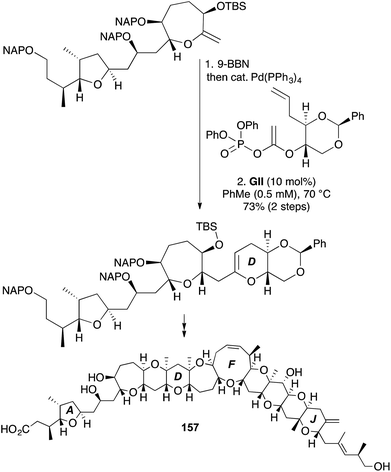
The strategy of convergent RCM substrate synthesis by Suzuki coupling with an unsaturated ester-derived enol phosphate, seen earlier in a 2,3-DHF approach towards okadaic acid (61) (Scheme 16), has also been applied in 3,4-DHP synthesis towards the attenol marine toxins (e.g., (−)-attenol A (156), Scheme 47),59 and the D-ring of (+)-gambieric acid A (157) in the latter's first total synthesis (Scheme 48, NAP = 2-napthylmethyl),60 and (+)-neopeltolide and analogues.61
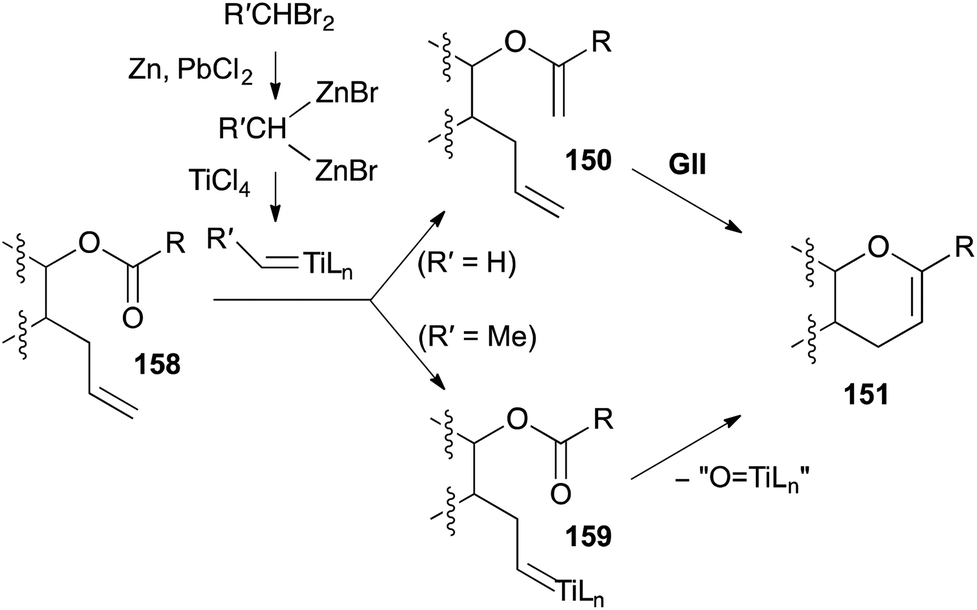
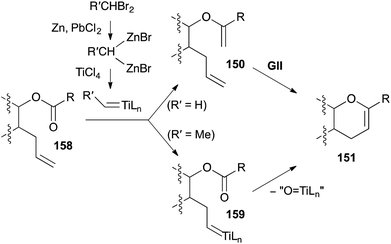
A popular route to 3,4-DHPs 151 from unsaturated esters 158 uses a reduced titanium alkylidene, derived from TiCl4 and a 1,1-dibromoalkane (Takai-Utimoto reagent, Scheme 49).62 Initial studies with a Ti methylidene generated under these conditions indicated that with unhindered esters, mainly ester methylenation occurs (giving 150) and efficient DHP formation required subsequent addition of a metathesis catalyst such as GII (Schemes 50 and 51). More hindered esters predominantly underwent direct DHP formation through alkylidene exchange (via159) and cyclisation on the ester with loss of “O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) TiLn”. Later studies revealed that Ti ethylidene facilitates direct DHP formation also from less-hindered unsaturated esters. While these latter olefinic ester cyclisations (OLECs) are strictly not RCM reactions and theoretically require stoichiometric reagents (in practice large excesses are employed), they are included here for comparison with the two-step procedures and because they offer the advantage of direct access to DHPs from unsaturated esters (Schemes 52–56).
TiLn”. Later studies revealed that Ti ethylidene facilitates direct DHP formation also from less-hindered unsaturated esters. While these latter olefinic ester cyclisations (OLECs) are strictly not RCM reactions and theoretically require stoichiometric reagents (in practice large excesses are employed), they are included here for comparison with the two-step procedures and because they offer the advantage of direct access to DHPs from unsaturated esters (Schemes 52–56).
 |
| | Scheme 53 OLEC towards maitotoxin (177). | |
 |
| | Scheme 54 Total synthesis of brevanal (180). | |
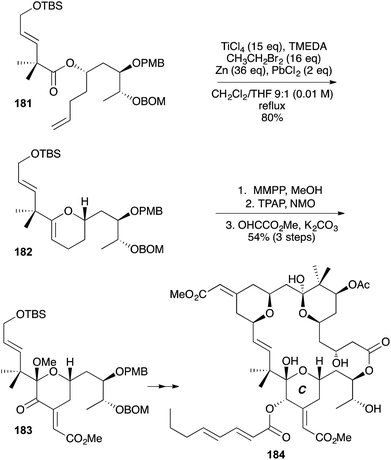 |
| | Scheme 55 Total synthesis of bryostatin 1 (184). | |
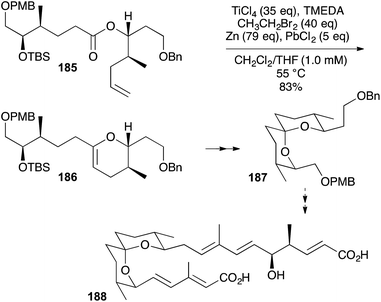 |
| | Scheme 56 OLEC towards spirofungin A. | |
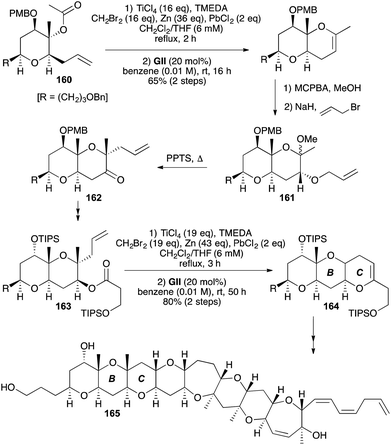
In a total synthesis of the polycyclic ether marine toxin (−)-gamberiol (165), both the B and C rings were made using DHPs generated from unsaturated esters (160 and 163, respectively) by reaction with a Ti methylidene, followed by GII (Scheme 50).63 For the B ring, RCM was carried out on a 1:1 mixture of acyclic and cyclic enol ethers. Subsequent epoxidation with concomitant methanolysis, and then O-allylation followed by Claisen rearrangement (161 → 162) induced through acid-catalysed elimination of methanol with PPTS on heating, were key steps leading to the unsaturated ester C-ring precursor 163. For this more hindered ester 163, the Ti methylidene provided a 8.5:1.5 mixture of acyclic and cyclic enol ethers that was fully converted to the DHP 164 using GII. The Ti methylidene–GII sequence has also been used in the synthesis of A–E fragment of gambieric acid A, forming the D ring as a 2,3,5,6-tetrasubstituted 3,4-DHP.64
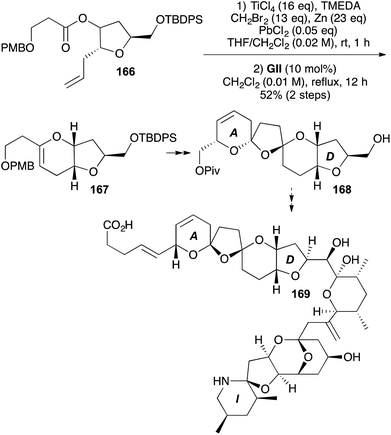
In the synthesis of the ABCD trioxadispiroketal subunit 168 of azaspiracid-1 (169), a ribose-derived tetrahydrofuran 166 underwent Takai-Utimoto methylenation and the resulting enol ether was purified on neutral alumina (74%) before undergoing RCM with GII to give the C ring 167 (70%, Scheme 51).65
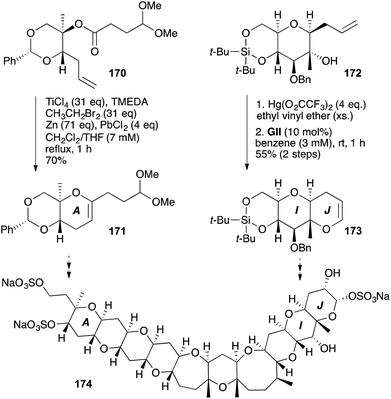
The direct access to DHPs from unsaturated esters by olefinic ester cyclisation (OLEC) using Ti ethylidene has been applied towards several natural products (Schemes 52–56). The A ring (as well as the 7-membered E ring) of the polycyclic ether marine toxin adriatoxin (174) was constructed by esterification, followed by OLEC (Scheme 52);66 70% yield for A ring formation by OLEC (170 → 171) compares with 50% yield obtained from a more conventional 2-step enol ether-olefin RCM sequence. In contrast, the J ring of adriatoxin was installed, as a C-6 unsubstituted DHP 173, using GII on a vinyl ether formed from an alcohol 172 using mercuric trifluoroacetate in ethyl vinyl ether.
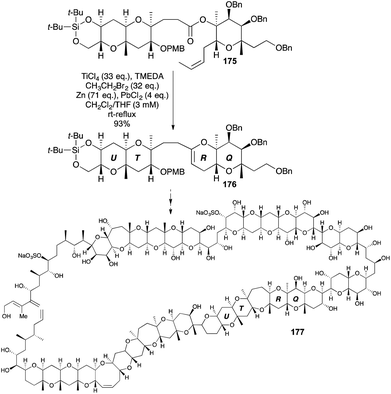
The R ring of maitotoxin (177), a polycyclic ether marine toxin with 32 rings and a molecular weight of 3422 Da, has been made by OLEC (Scheme 53).67 Despite the complexity of the OLEC substrate 175, assembled from acid chloride and alcohol precursors, formation of the DHP 176 occurred in 93% yield.
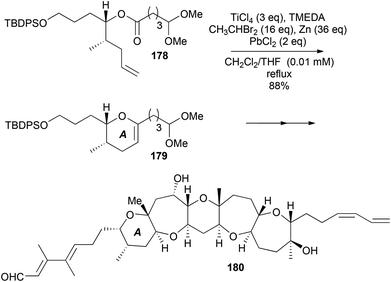
Three examples illustrate that a cyclic template is not required for OLECs (Schemes 54–56). The first example (178 → 179) forms the A ring of the marine toxin brevanal (180), in 88% yield (Scheme 54); 75% yield was obtained if Takai-Utimoto methylenation was followed by RCM using GII.68 The latter two examples show OLEC applications away from fused polycyclic ether structures containing pyrans (Schemes 55 and 56).
The macrolide lactone bryostatin 1 (184) is currently in phase 2b clinical trials as a treatment for Alzheimer's disease. As part of the total synthesis of bryostatin 1 (184), a C ring-containing enoate fragment 183 was constructed starting from (R)-isobutyl lactate (Scheme 55).69 The sequence involved OLEC (181 → 182), epoxidation (using magnesium monoperoxyphthalate, MMPP) – methanolytic ring-opening, oxidation to a methoxyketone, and an aldol condensation with methyl glyoxylate.
The spiroacetal core 187 of spirofungin A (188) was made by OLEC (185 → 186) followed, after desilylation, by NIS-induced trans-diaxial addition to the DHP and reductive deiodination (Scheme 56).70
3,4-DHPs can also be accessed in a one-flask operation by RCM of allyl homoallyl ethers to give 3,6-DHPs, followed by catalyst conversion to a Ru–H to induce alkene isomerisation (cf., Scheme 21) to the enol ether; the natural products centrolobine (90) (cf., Scheme 26) and 5,6-dehydro-de-O-methyl centrolobine (192) (Scheme 57) have been prepared using this strategy.71 The RCM substrate 190 used in these syntheses was generated through asymmetric addition of a chiral allylic silane to an aldehyde 189 (Scheme 57), with the post-RCM isomerisation being induced by addition of i-PrOH and NaOH. Completion of the synthesis of 5,6-dehydro-de-O-methyl centrolobine (192) was achieved from the 3,4-DHP 191 by a regio- and diastereoselective Heck reaction and desilylation.
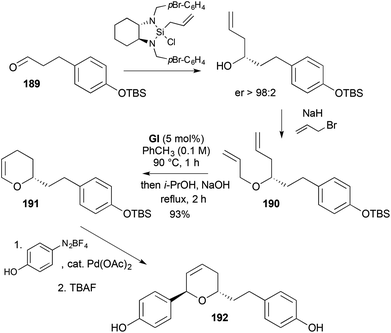 |
| | Scheme 57 Synthesis of 5,6-dehydro-de-O-methyl centrolobine (192) using RCM-isomerisation. | |
Conclusions
The structurally constraining demands of natural product synthesis provide a challenging environment for synthetic methodology applications. In this review, we have highlighted diverse and inventive applications from the last dozen years of ring-closing alkene metathesis (RCM) towards the commonest oxacycles (5 and 6-membered) for use towards such targets. Despite the many alternative ways available to construct such systems, the fact that RCM has found significant utility in this area and in several cases towards highly complex natural products, is a testament to the confidence that the synthetic community has in basing a strategy around this methodology. The attractiveness of this chemistry stems from a combination of the flexibility and convergent nature of RCM precursor construction, the range of functional group tolerant catalysts now (commercially) available that react with a variety of alkene substitution patterns in a predictable fashion, and the scope for post RCM manipulation of the newly formed unsaturation. We hope this review will serve to inspire further applications and developments of this chemistry in target-driven synthesis.
Acknowledgements
R. J., R. P., N. A. P., C. E. S. and P. W. S. are supported by the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1), and A. R. by the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme (FP7/2007–2013) under REA grant agreement no. 316955. R. P. also thanks the University of Oxford Clarendon Fund for a scholarship.
Notes and references
- G. C. Fu, S. T. Nguyen and R. H. Grubbs, J. Am. Chem. Soc., 1993, 115, 9856–9857 CrossRef CAS.
-
(a)
Olefin Metathesis: Theory and Practice, ed. K. Grela, John Wiley & Sons, Hoboken, NJ, USA, 2014 Search PubMed;
(b)
Handbook of Metathesis, ed. R. H. Grubbs and D. J. O'Leary, Wiley-VCH, Weinheim, Germany, 2015 Search PubMed.
-
(a)
B. A. Keay, J. M. Hopkins and P. W. Dibble, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, vol. 3, pp. 571–623 Search PubMed;
(b)
B. W. Fravel, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, vol. 7, pp. 701–726 Search PubMed.
- M. Bassetti and A. D'Annibale, Curr. Org. Chem., 2013, 17, 2654–2677 CrossRef CAS.
- For examples prior to mid-2003, see: A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199–2238 CrossRef CAS PubMed.
- For other recent reviews on RCM, see:
(a)
Metathesis in Natural Product Synthesis, ed. J. Cossy, S. Arseniyadis and C. Meyer, Wiley-VCH, Weinheim, 2010 Search PubMed;
(b) X. Lei and H. Li, Top. Curr. Chem., 2012, 327, 163–196 CrossRef CAS PubMed;
(c) B. Dassonneville, L. Delaude, A. Demonceau, I. Dragutan, V. Dragutan, K. S. Etsè and M. Hans, Curr. Org. Chem., 2013, 17, 2607–2651 CrossRef;
(d) K. C. Majumdar, R. K. Nandi and K. Ray, Adv. Org. Synth., 2013, 6, 355–435 CAS;
(e)
B. Schmidt, S. Hauke, S. Krehl and O. Kunz, in Comprehensive Organic Synthesis II, ed. P. Knochel and G. A. Molander, Elsevier, Amsterdam, 2014, vol. 5, pp. 1400–1482 Search PubMed;
(f)
K. M. Dawood and P. Metz, in Domino Reactions: Concepts for Efficient Organic Synthesis, ed. L. F. Tietze, Wiley-VCH, Weinheim, Germany, 2014, pp. 31–66 Search PubMed.
- For a recent review on strategies to natural product-related tetrahydropyrans, see: N. M. Nasir, K. Ermanis and P. A. Clarke, Org. Biomol. Chem., 2014, 12, 3323–3335 CAS.
- Where published, catalyst loading and substrate concentration for the RCM step are included in the schemes in this review.
-
(a) M. Femenía-Ríos, C. M. García-Pajón, R. Hernández-Galán, A. J. Macías-Sánchez and I. G. Collado, Bioorg. Med. Chem. Lett., 2006, 16, 5836–5839 CrossRef PubMed;
(b) G. Mancilla, M. Femenía-Ríos, M. Grande, R. Hernández-Galán, A. J. Macías-Sánchez and I. G. Collado, Tetrahedron, 2010, 66, 8068–8075 CrossRef CAS.
- P. Escavabaja, J. Viala, Y. Coquerel and J. Rodriguez, Adv. Synth. Catal., 2012, 354, 3200–3204 CrossRef CAS.
- R. Datta, R. J. Dixon and S. Ghosh, Tetrahedron Lett., 2016, 57, 29–31 CrossRef CAS.
- S. Kress and S. Blechert, Chem. Soc. Rev., 2012, 41, 4389–4408 RSC.
- B. Das, S. M. Mobin and V. Singh, Tetrahedron, 2014, 70, 4768–4777 CrossRef CAS.
- For another spiro-fused example, towards havellockate, see: R. L. Beingessner, J. A. Farand and L. Barriault, J. Org. Chem., 2010, 75, 6337–6346 CrossRef CAS PubMed.
- A. M. Lone, B. A. Bhat and G. Mehta, Tetrahedron Lett., 2013, 54, 5619–5623 CrossRef CAS.
- M. F. Hossain, K. Matcha and S. Ghosh, Tetrahedron Lett., 2011, 52, 6473–6476 CrossRef CAS.
- K. Ramakrishna and K. P. Kaliappan, Synlett, 2011, 2580–2584 CAS.
- S. Maity, K. Matcha and S. Ghosh, J. Org. Chem., 2010, 75, 4192–4200 CrossRef CAS PubMed.
- A. Gris, N. Cabedo, I. Navarro, I. de Alfonso, C. Agulló and A. Abad-Somovilla, J. Org. Chem., 2012, 77, 5664–5680 CrossRef CAS PubMed.
- T. J. Donohoe, A. Ironmonger and N. M. Kershaw, Angew. Chem., Int. Ed., 2008, 47, 7314–7316 CrossRef CAS PubMed.
- A. Bandyopadhyay, B. K. Pal and S. K. Chattopadhyay, Tetrahedron: Asymmetry, 2008, 19, 1875–1877 CrossRef CAS.
- J. P. Tellam and D. R. Carbery, Tetrahedron Lett., 2011, 52, 6027–6029 CrossRef CAS.
- D. K. Mohapatra, H. Rahaman, M. S. Chorghade and M. K. Gurjar, Synlett, 2007, 567–570 CrossRef CAS.
- For an earlier strategically related strategy, to solamin, see: G. Prestat, C. Baylon, M.-P. Heck, G. A. Grasa, S. P. Nolan and C. Mioskowski, J. Org. Chem., 2004, 69, 5770–5773 CrossRef CAS PubMed.
- M. T. Crimmins, Y. Zhang and F. A. Diaz, Org. Lett., 2006, 8, 2369–2372 CrossRef CAS PubMed.
- For a related asymmetric glycolate–RCM approach to a 2,5-DHF in the total synthesis of gigantecin, see: M. T. Crimmins and J. She, J. Am. Chem. Soc., 2004, 126, 12790–12791 CrossRef CAS PubMed.
-
(a) G. C. H. Chiang, A. D. Bond, A. Ayscough, G. Pain, S. Ducki and A. B. Holmes, Chem. Commun., 2005, 1860–1862 RSC;
(b) S. Y. F. Mak, G. C. H. Chiang, J. E. P. Davidson, J. E. Davies, A. Ayscough, G. Pain, J. W. Burton and A. B. Holmes, Tetrahedron: Asymmetry, 2009, 20, 921–944 CrossRef.
- D. Sarkar and R. V. Venkateswaran, Tetrahedron Lett., 2011, 52, 3232–3233 CrossRef CAS.
- H. Fuwa, K. Sakamoto, T. Muto and M. Sasaki, Tetrahedron, 2015, 71, 6369–6383 CrossRef CAS.
- H.-J. Hong, D.-M. Lee and H.-Y. Kang, Bull. Korean Chem. Soc., 2010, 31, 555–556 CrossRef CAS.
-
(a) S. Praveen Kumar, K. Nagaiah and M. S. Chorghade, Tetrahedron Lett., 2006, 47, 7149–7151 CrossRef;
(b) K. Nagaiah, K. Srinivasu, S. Praveen Kumar, J. Basha and J. S. Yadav, Tetrahedron: Asymmetry, 2010, 21, 885–889 CrossRef CAS.
- S. Claessens, D. Naidoo, D. Mulholland, L. Verschaeve, J. van Staden and N. De Kimpe, Synlett, 2006, 621–623 CAS.
- G. V. M. Sharma and S. Mallesham, Tetrahedron: Asymmetry, 2010, 21, 2646–2658 CrossRef CAS.
- S. Hanessian and L. Auzzas, Org. Lett., 2008, 10, 261–264 CrossRef CAS PubMed.
- B. Schmidt and S. Hauke, Eur. J. Org. Chem., 2014, 1951–1960 CrossRef CAS.
- B. M. Trost, W. M. Seganish, C. K. Chung and D. Amans, Chem. – Eur. J., 2012, 18, 2948–2960 CrossRef CAS PubMed.
- S. Raghavan and P. K. Samanta, Synlett, 2013, 1983–1987 CrossRef CAS.
- A. K. Cheung, R. Murelli and M. L. Snapper, J. Org. Chem., 2004, 69, 5712–5719 CrossRef CAS PubMed.
- H. Kim and D. Lee, Synlett, 2015, 2583–2587 Search PubMed . For other RCM approaches to centrolobine, see: V. Bohrsch and S. Blechert, Chem. Commun., 2006, 1968–1970 RSC; D. K. Mohapatra, R. Pal, H. Rahaman and M. K. Gurjar, Heterocycles, 2010, 80, 219–227 CrossRef CAS.
- T. Katagiri, K. Fujiwara, H. Kawai and T. Suzuki, Tetrahedron Lett., 2008, 49, 233–237 CrossRef CAS.
- A. K. Ghosh, A. M. Veitschegger, V. Reddy Sheri, K. A. Effenberger, B. E. Prichard and M. S. Jurica, Org. Lett., 2014, 16, 6200–6203 CrossRef CAS PubMed.
- K. Nakagawa-Goto and M. Crimmins, Synlett, 2011, 1413–1418 CrossRef CAS.
- J. Pospíšil and I. E. Markó, J. Am. Chem. Soc., 2007, 129, 3516–3517 CrossRef PubMed.
- V. Druais, M. J. Hall, C. Corsi, S. V. Wendeborn, C. Meyer and J. Cossy, Tetrahedron, 2010, 66, 6358–6375 CrossRef CAS.
- For a Brown allylation–transacetalisation–RCM approach to a related 2,3,6-trisubstituted-3,6-DHP for sorangicin synthesis, see: M. T. Crimmins, M. W. Haley and E. A. O'Bryan, Org. Lett., 2011, 13, 4712–4715 CrossRef CAS PubMed.
- S. Raghavan and S. G. Subramanian, Tetrahedron, 2011, 67, 7529–7529 CrossRef CAS.
- M. T. Crimmins and D. L. Jacobs, Org. Lett., 2009, 11, 2695–2698 CrossRef CAS PubMed.
- J. Lee, H.-S. Oh and H.-Y. Kang, Tetrahedron Lett., 2015, 56, 1099–1102 CrossRef CAS.
- Y. Tang, J.-H. Yang, J. Liu, C.-C. Wang, M.-C. Lv, Y.-B. Wu, X.-L. Yu, C. Ko and R. P. Hsung, Heterocycles, 2012, 86, 565–598 CrossRef CAS . For a related cyclic ketal-tethered RCM to the E ring of spirostrellolide A, see: Y.-B. Wu, Y. Tang, G.-Y. Luo, Y. Chen and R. P. Hsung, Org. Lett., 2014, 17, 4550–4553 CrossRef PubMed.
- R. Figueroa, R. P. Hsung and C. C. Guevarra, Org. Lett., 2007, 9, 4857–4859 CrossRef CAS PubMed.
- H. Do, C. W. Kang, J. H. Cho and S. R. Gilbertson, Org. Lett., 2015, 17, 3972–3974 CrossRef CAS PubMed.
- For a related RCM approach to the trans-fused system, found in malayamycin A, see: O. Loiseleur, H. Schneider, G. Huang, R. Machaalani, P. Sellès, P. Crowley and S. Hanessian, Org. Process Res. Dev., 2006, 10, 518–524 CrossRef CAS.
- H.-Y. Lee, S.-S. Lee, H. S. Kim and K. M. Lee, Eur. J. Org. Chem., 2012, 4192–4199 CrossRef CAS.
- K. L. Jackson, J. A. Henderson, H. Motoyoshi and A. J. Phillips, Angew. Chem., Int. Ed., 2009, 48, 2346–2350 CrossRef CAS PubMed.
- D. Lo Re, F. Franco, F. Sánchez-Cantalejo and J. A. Tamayo, Eur. J. Org. Chem., 2009, 1984–1993 CrossRef CAS.
- C. C. Marvin, A. J. L. Clemens and S. D. Burke, Org. Lett., 2007, 9, 5353–5356 CrossRef CAS PubMed.
- C. C. Marvin, E. A. Voight and S. D. Burke, Org. Lett., 2007, 9, 5357–5359 CrossRef CAS PubMed.
- J. S. Clark, M. C. Kimber, J. Robertson, C. S. P. McErlean and C. Wilson, Angew. Chem., Int. Ed., 2005, 44, 6157–6162 CrossRef CAS PubMed.
- H. Fuwa and M. Sasaki, Org. Lett., 2008, 10, 2549–2552 CrossRef CAS PubMed.
- K. Ishigai, H. Fuwa, K. Hashizume, R. Fukazawa, Y. Cho, M. Yotsu-Yamashita and M. Sasaki, Chem. – Eur. J., 2013, 19, 5276–5288 CrossRef CAS PubMed.
- H. Fuwa, A. Saito, S. Naito, K. Konoki, M. Yotsu-Yamashita and M. Sasaki, Chem. – Eur. J., 2009, 15, 12807–12818 CrossRef CAS PubMed.
- U. Majumder and J. D. Rainier, Tetrahedron Lett., 2005, 46, 7209–7211 CrossRef CAS; K. Iyer and J. D. Rainier, J. Am. Chem. Soc., 2007, 129, 12604–12605 CrossRef PubMed.
- U. Majumder, J. M. Cox, H. W. B. Johnson and J. D. Rainier, Chem. – Eur. J., 2006, 12, 1736–1746 CrossRef CAS PubMed.
- S. W. Roberts and J. D. Rainier, Org. Lett., 2007, 9, 2227–2230 CrossRef CAS PubMed.
- X. Li, J. Li and D. R. Mootoo, Org. Lett., 2007, 9, 4303–4306 CrossRef CAS PubMed.
- C. Osei Akoto and J. D. Rainier, Angew. Chem., Int. Ed., 2008, 47, 8055–8058 CrossRef PubMed.
- K. C. Nicolaou, C. F. Gelin, J. Hong Seo, Z. Huang and T. Umezawa, J. Am. Chem. Soc., 2010, 132, 9900–9907 CrossRef CAS PubMed.
- Y. Zhang, J. Rohanna, J. Zhou, K. Iyer and J. D. Rainier, J. Am. Chem. Soc., 2011, 133, 3208–3216 Search PubMed.
- G. E. Keck, Y. B. Poudel, T. J. Cummins, A. Rudra and J. A. Covel, J. Am. Chem. Soc., 2011, 133, 744–747 CrossRef CAS PubMed.
- J. Neumaier and M. E. Maier, Synlett, 2011, 187–190 CAS.
-
(a) B. Schmidt and H. Hölter, Chem. – Eur. J., 2009, 15, 11948–11953 CrossRef CAS PubMed;
(b) B. Schmidt, H. Hölter, A. Kelling and U. Schilde, J. Org. Chem., 2011, 76, 3357–3365 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2016 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a