
Synthesis and reactivities of monofluoro acylboronates in chemoselective amide bond forming ligation with hydroxylamines
Hidetoshi
Noda
a and
Jeffrey W.
Bode
*ab
aLaboratorium für Organische Chemie, Department of Chemistry and Applied Bioscience, ETH Zürich, 8093 Zürich, Switzerland. E-mail: bode@org.chem.ethz.ch; Web: http://www.bode.ethz.ch
bInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Chikusa, Nagoya 464-8602, Japan
First published on 4th November 2015
Abstract
The development of chemoselective reactions is a cornerstone of bioorganic chemistry and chemical ligation. Our contributions in this area include the development of two mechanistically distinct amide bond forming reactions, the α-ketoacid–hydroxylamine (KAHA) ligation for the chemical synthesis of proteins and the potassium acyltrifluoroborate (KAT) ligation for bioconjugation. The remarkable stability and unique reactivity of KATs with hydroxylamines prompted us to explore other acylborons as a ligation partner, which have been unappreciated in synthetic organic chemistry. A new convenient synthetic route was devised from KATs and suitable bidentate ligands, allowing for the preparation of a wide range of new acylboronates that retain one fluoride ligand on the boron. Our results established that a ligand on the boron is responsible for their reactivity and stability and provide a firm basis for further development of the acylboron–hydroxylamine amide bond forming ligation.
 Hidetoshi Noda | Hidetoshi Noda was born and raised in Tokyo (Japan). He conducted his bachelor's and master's research in the group of Professor Dr Masakatsu Shibasaki at the University of Tokyo. After three years of experience in industry, he moved to Switzerland in 2011 to pursue his doctorate with Professor Dr Jeffrey W. Bode at ETH-Zürich. In 2015, he obtained his Dr Sc. by the research on acylboronates. He is currently a JSPS fellow at the Institute of Microbial Chemistry (BIKAKEN), Tokyo. |
 Jeffrey W. Bode | Jeffrey W. Bode studied at Trinity University in San Antonio, TX (U.S.A.). Following doctoral studies at the California Institute of Technology and ETH-Zürich, and postdoctoral research at the Tokyo Institute of Technology (Japan), he began his independent academic career at UC Santa Barbara in 2003. In 2007, he accepted an appointment as an Associate Professor at the University of Pennsylvania, and, in 2010, as a Full Professor at ETH-Zürich. He has also been a Principal Investigator and Visiting Professor at the Institute of Transformative Biomolecules (WPI-ITbM) at Nagoya University (Japan) since 2013. |
Chemoselective reactions: what are we interested in?
Chemoselective reactions have revolutionized the way scientists synthesize and understand large, complex molecules. They are essential for studying biomolecules of interest by labeling or capturing them through a unique functional group in the presence of a diverse range of unprotected functional groups.1 They also make it possible to chemically synthesize large biomolecules (mw > 5000) through a specific covalent bond formation between large segments. The chemical synthesis of proteins is a flourishing research area in bioorganic chemistry,2 and currently synthetic chemists aim at preparing previously formidable, macromolecule constructs such as protein–protein conjugates in a structurally defined manner.3 The development of a new chemoselective reaction can greatly simplify synthesis and would offer a synthetic means to prepare such chemically engineered macromolecules otherwise difficult or impossible to obtain. As a research group of synthetic organic chemistry, we are particularly fascinated with this potential of chemoselective reactions.4As an initial step toward this dream, we developed the α-ketoacid–hydroxylamine (KAHA) ligation,5 which allows for the formation of a native amide bond between unprotected peptide fragments under aqueous conditions. The KAHA ligation is very suitable for the assembly of peptide fragments, and it has been routinely utilized in our group for the chemical syntheses of proteins.6 Its kinetics is, however, not sufficiently fast to work with large macromolecules; it requires relatively high concentration (5–20 mM) and relatively high temperatures (40–60 °C). We have been looking for an alternative, faster variant of the KAHA ligation.7
In 2012, we disclosed that potassium acyltrifluoroborates (KATs) possess much higher reactivity towards O-Bz hydroxylamines than α-ketoacids in water.8 KATs are categorized as acylborons, an underexplored class of entities in the literature.9 The unique reactivity of KATs with hydroxylamines under aqueous conditions led us to develop two convenient synthetic routes to KATs and a wide range of KATs can be prepared on a gram scale either from aldehydes10 or aryl halides.11 The O-Bz hydroxylamines employed in the initial disclosure were found to be somewhat unstable under various conditions and were not compatible with Fmoc-based solid phase peptide synthesis (SPPS). Further examinations on the hydroxylamine structures revealed that the use of O-diethylcarbamoylhydroxylamines reaches an optimum level between reactivity and stability, allowing for incorporations of the hydroxylamine moiety onto unprotected peptides via Fmoc-SPPS with its reactivity retained. The kinetic study of this ligation with an unprotected peptide substrate revealed that its second order rate constant is 20 M−1 s−1 in acidic aqueous media. Site specific introductions of PEGs, biotin, dye moiety and lipid chain onto an unprotected GLP-1 analogue were demonstrated with an equimolar ratio of reactants.12 One of the current drawbacks of the KAT ligation is its pH dependent kinetics; the ligation is relatively slow at pH 7.4, which is undesirable when it comes to working with folded proteins and in vivo ligations.13 Our strategy to tackle this problem was identifying a suitable ligand for the boron atom that enhances reactivity of the acylboron in amide formations. In this Perspective, we summarize our recent efforts on the chemistry of acylboronates,14 and illustrate a future direction along this research.
What was the initial problem in this research?
The main issue we had faced was only a limited number of acylborons were known in the literature (Fig. 1),15,16 and there was no general way to synthesize such a class of compounds. The existing literature on boron complexes did not readily translate into a useful guide for the design and synthesis of novel acylboronates, and the complexes did not seem stable under aqueous conditions that are essential for our applications. Given the established procedure for the synthesis of KATs from widely available starting materials, we envisioned that a series of acylboronates could be obtained from KATs by a simple ligand exchange on the boron. Based on the high stability of KATs and precedents to stabilize 2-pyridylboronic acid in the literature,17 we hypothesized that either anionic borates or complexes from a multidentate ligand would be stable under the ligation conditions. The report by Yudin15e,18 on N-methyliminodiacetyl (MIDA) acylboronates 3 prompted us to examine the synthesis of MIDA acylboronates 3 as a model target and evaluate their reactivity in the amide bond forming reactions.19 The combination of BF3·Et2O and the TMS-activated MIDA ligand (TMS2-2) in dry acetonitrile was found to be optimal, affording the desired acylboronate in good yield. In this transformation, BF3·Et2O first abstracts one of the fluorides from KAT 1 to generate the unstable acyldifluoroborane intermediate 4,20 followed by the ligand exchange leading to the formation of MIDA acylboronates 3 (Scheme 1a). It turned out, however, the optimized conditions only worked for the parent MIDA ligand; even slight changes in the structure of the MIDA ligand, led to the formation of unstable constructs that were difficult to obtain in pure form (Scheme 1b). These results emphasized the instability of many acylboron compounds in general and highlighted the special character of KATs and MIDA acylboronates. | ||
| Fig. 1 List of reported fully characterized acylborons. Dipp = 2,6-(iPr)2C6H3. R = alkyl, aryl. | ||
 | ||
| Scheme 1 (a) Synthesis of MIDA acylboronates from KAT. (b) List of MIDA analogues that did not form stable adducts. R = alkyl, aryl, and heteroaryl. | ||
What was the tipping point in this research?
Extensive ligand screening with a variety of tridentate ligands led to Schiff base 5 that formed the stable adduct. Surprisingly the product was neither the expected bicyclic boronate 7 nor 5-membered ring boronate 8, but rather a six-membered ring acylboronate 6 (Scheme 2).21 This finding encouraged us to investigate a series of bidentate ligand, and many ligands appeared to form stable acylboronates that retained one of the fluoride ligands on the boron atom (Scheme 3). The structures of all monofluoroacylboronates in Scheme 3 were established by single X-ray crystallography.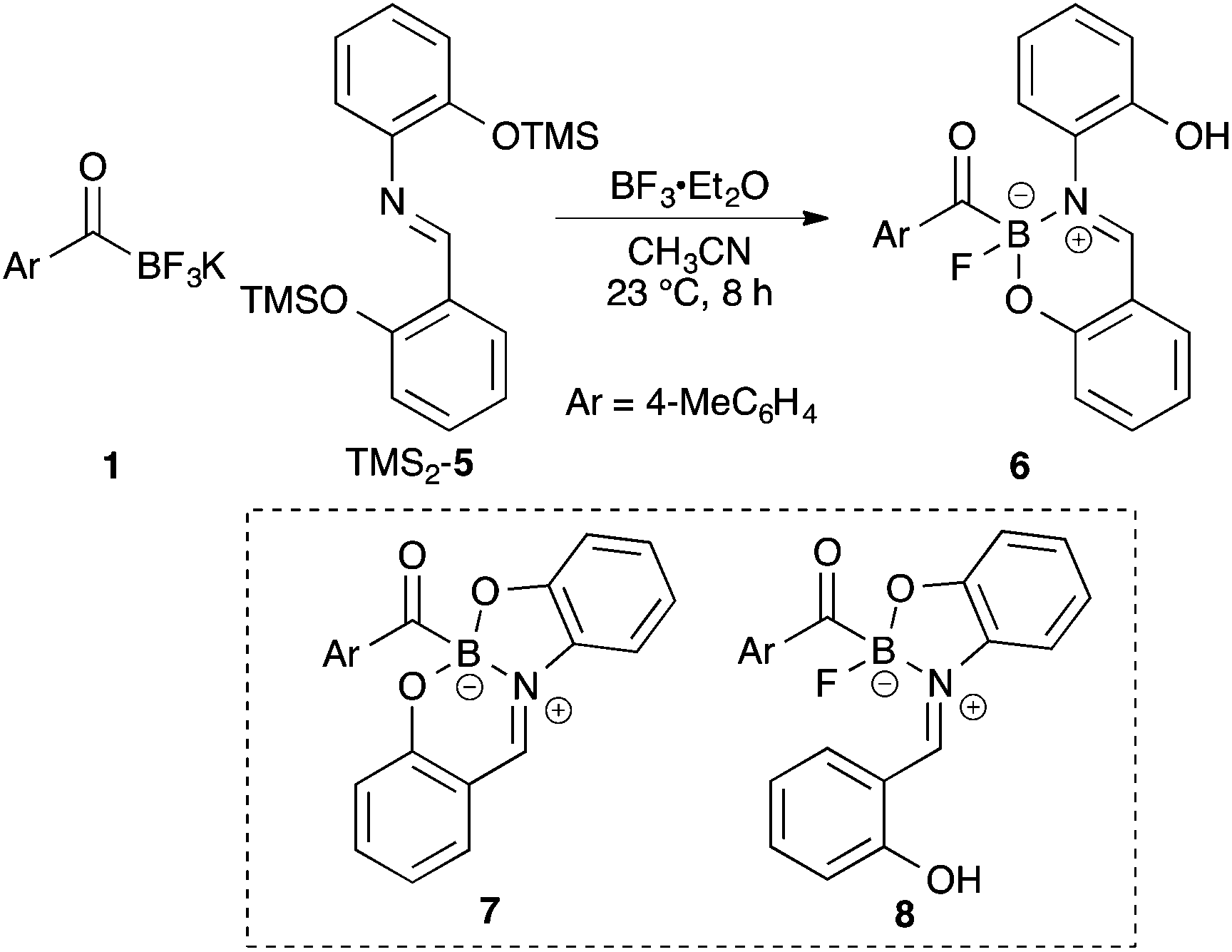 | ||
| Scheme 2 Formation of six-membered acylboronate 6 from the tridentate Schiff base ligand 5. | ||
 | ||
| Scheme 3 Synthesis of six-membered monofluoroacylboronates from various bidentate ligands. | ||
What was the most important result in this research?
Many precedents on a dramatic ligand effect on the reactivity of organometallic complexes led us to study the influences of ligands on acylboronates. Importantly, a modular nature of the bidentate ligands in Scheme 3 allowed us to easily prepare a set of structurally similar acylboronates by following the exactly same procedure; no substrate specific optimization was required. Identification of a bidentate ligand was crucial for this study; KATs have no derivatizable points and the preparation of suitable MIDA analogues was difficult (vide supra).Monofluoroacylboronates 9–19 derived from substituted 2-(2-pyridyl)phenol ligands were prepared for this study. The solid state structures of the obtained acylboronates revealed that a slight difference in the ligand structure could alter the structural parameters around the boron atom, such as dihedral angles and bond lengths. These structural changes were expected to correlate with their reactivity and stability. We evaluated the reactivity of acylboronates with hydroxylamines under neutral aqueous conditions by comparison with the parent acylboronate 9; KAT 1 and MIDA acylboronate 3 were also included as references. The half-lives of these acylboronates were also examined in aqueous DMSO. These data are presented together in Fig. 2.
 | ||
| Fig. 2 Evaluations of acylboronates on their reactivity and stability. (a) List of evaluated acylboronates. (b) Relative reactivity and stability to 9. | ||
Among monofluoroboronates, 13 and 19 were unique; these two differ in reactivity by more than 100-fold. MIDA acylboronate 3 also showed enhanced reactivity, albeit decomposing quickly under the conditions examined. On the other hand, KAT 1 was completely stable.22 These results established that the ligand can modulate the reactivity and stability of acylboronates, even though at the current stage of investigation it is not clear which factors in a ligand play a key role.
What are future perspectives from this research?
One of the direct extensions of this work is identifying a suitable ligand that forms a more reactive complex with hydroxylamines at aqueous neutral pH. Even though we cannot easily predict which kinds of ligands form stable adducts and which kinds do not, several guidelines were obtained through the ligand screening described here. Further mechanistic studies in the amide bond forming reaction, including computational approaches, will provide a rational design guideline to modulate the steric and electronic properties of ligands. Innovative ideas to utilize these novel acylboronates in organic chemistry are awaited.Our ultimate goal with the acylboron–hydroxylamine amide forming ligation is to offer a chemical means to synthesize macromolecule constructs including protein–protein conjugates. Towards this goal, the preparation of amino acid analogues containing an acylboron moiety is essential both on its side-chain and at the C-terminus (Fig. 3a). Unlike other bioorthogonal ligation reactions such as oxime formations and cycloadditions, this amide bond forming ligation generates side products derived from the boron moiety and hydroxylamines. For many biological applications, toxicity assessments of these side products are necessary. We recently obtained promising results on this issue; PEG-based hydrogels prepared by the KAT ligation were suitable to encapsulate bovine chondrocytes with high viability, demonstrating the biocompatibility of the KAT ligation.23 Further examinations in our group will support the innocuous nature of the ligation. Together with the development of methodologies to install acylboronate and hydroxylamine moieties onto the proteins of interest chemically or biologically (Fig. 3b), the acylboron–hydroxylamine amide bond forming ligation will become a truly powerful chemical tool to construct a protein–protein bond.
 | ||
| Fig. 3 (a) Fmoc-protected glutamic acid (Glu) and leucine (Leu) derivatives with an acylboron moiety for Fmoc-based SPPS. (b) Protein substrates for the acylboron–hydroxylamine amide bond forming ligation. Protein structures were adopted and modified from PDB 1V80. | ||
With a suitable technology, we envision a time when the synthesis of large, complex molecules is no longer the bottleneck of biological research and the focus can shift to the design and evaluation of such molecules. The acylboron–hydroxylamine amide forming ligation will play a pivotal role in the future.
Acknowledgements
This work was supported by ETH Zurich and ETH Research Grant (ETH 43 13-2).Notes and references
- (a) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed; (b) J. Kalia and R. T. Raines, Curr. Org. Chem., 2010, 14, 138–147 CrossRef CAS PubMed.
- (a) P. E. Dawson and S. Kent, Annu. Rev. Biochem., 2000, 69, 923–960 CrossRef CAS PubMed; (b) L. Raibaut, N. Ollivier and O. Melnyk, Chem. Soc. Rev., 2012, 41, 7001–7015 RSC; (c) H. P. Hemantha, N. Narendra and V. V. Sureshbabu, Tetrahedron, 2012, 68, 9491–9537 CrossRef CAS.
- (a) C. J. Hawker, Science, 2005, 309, 1200–1205 CrossRef CAS PubMed; (b) C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS; (c) E. M. Pelegri-O'Day, E.-W. Lin and H. D. Maynard, J. Am. Chem. Soc., 2014, 136, 14323–14332 CrossRef PubMed; (d) M. Haj-Yahya, B. Fauvet, Y. Herman-Bachinsky, M. Hejjaoui, S. N. Bavikar, S. V. Karthikeyan, A. Ciechanover, H. A. Lashuel and A. Brik, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17726–17731 CrossRef CAS PubMed.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- (a) J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248–1252 CrossRef CAS PubMed; (b) V. R. Pattabiraman, A. O. Ogunkoya and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 5114–5118 CrossRef CAS PubMed; (c) T. G. Wucherpfennig, F. Rohrbacher, V. R. Pattabiraman and J. W. Bode, Angew. Chem., Int. Ed., 2014, 53, 12244–12247 CrossRef CAS PubMed.
- (a) A. O. Ogunkoya, V. R. Pattabiraman and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 9693–9697 CrossRef CAS PubMed; (b) C. He, S. S. Kulkarni, F. Thuaud and J. W. Bode, Angew. Chem., Int. Ed., 2015, 54, 12996–13001 CrossRef CAS PubMed.
- I. Pusterla and J. W. Bode, Nat. Chem., 2015, 7, 668–672 CrossRef CAS PubMed.
- A. M. Dumas, G. A. Molander and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 5683–5686 CrossRef CAS PubMed.
- Stevenson mentioned that “no verified examples of acylboron derivatives had ever been isolated and theoretical calculations suggested that acylboranes were highly reactive species and prone to rearrangement”. P. J. Stevenson, in Comprehensive Organic Functional Group Transformations II, ed. A. R. Katritzky and R. J. Taylor, Elsevier, Oxford, 2005, pp. 375–398 Search PubMed.
- A. M. Dumas and J. W. Bode, Org. Lett., 2012, 14, 2138–2141 CrossRef CAS PubMed.
- G. Erős, Y. Kushida and J. W. Bode, Angew. Chem., Int. Ed., 2014, 53, 7604–7607 CrossRef PubMed.
- H. Noda, G. Erős and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 5611–5614 CrossRef CAS PubMed.
- F. Saito, H. Noda and J. W. Bode, ACS Chem. Biol., 2015, 10, 1026–1033 CrossRef CAS PubMed.
- H. Noda and J. W. Bode, J. Am. Chem. Soc., 2015, 137, 3958–3966 CrossRef CAS PubMed.
- (a) Y. Segawa, Y. Suzuki, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 16069–16079 CrossRef CAS PubMed; (b) J. Monot, A. Solovyev, H. Bonin-Dubarle, É. Derat, D. P. Curran, M. Robert, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2010, 49, 9166–9169 CrossRef CAS PubMed; (c) M. Sajid, G. Kehr, C. G. Daniliuc and G. Erker, Angew. Chem., Int. Ed., 2014, 53, 1118–1121 CrossRef CAS PubMed; (d) G. A. Molander, J. Raushel and N. M. Ellis, J. Org. Chem., 2010, 75, 4304–4306 CrossRef CAS PubMed; (e) Z. He, P. Trinchera, S. Adachi, J. D. St Denis and A. K. Yudin, Angew. Chem., Int. Ed., 2012, 51, 11092–11096 CrossRef CAS PubMed.
- A new synthetic route to acylborons using a boryl zinc reagent has been reported recently. J. Campos and S. Aldridge, Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201507627.
- For selected examples addressing this issue, see: (a) K. L. Billingsley and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 4695–4698 CrossRef CAS PubMed; (b) G. R. Dick, E. M. Woerly and M. D. Burke, Angew. Chem., Int. Ed., 2012, 51, 2667–2672 CrossRef CAS PubMed.
- (a) Z. He, A. Zajdlik and A. K. Yudin, Dalton Trans., 2014, 43, 11434–11451 RSC; (b) J. D. S. Denis, Z. He and A. K. Yudin, ACS Catal., 2015, 5, 5373–5379 CrossRef.
- H. Noda and J. W. Bode, Chem. Sci., 2014, 5, 4328–4332 RSC.
- E. Vedejs, R. W. Chapman, S. C. Fields and S. Lin, J. Org. Chem., 1995, 60, 3020–3027 CrossRef CAS.
- Ligand 5 formed a stable bicyclic boronate similar to 7 with phenylboronic acid or boron trifluoride. (a) A. Abreu, S. Jesùs Alas, H. I. Beltrán, R. Santillan and N. Farfán, J. Organomet. Chem., 2006, 691, 337–348 CrossRef CAS; (b) F. Umland, E. Hohaus and K. Brodte, Chem. Ber., 1973, 106, 2427–2437 CrossRef CAS.
- The high stability of KATs in pH 7.5 buffer was reported. Z. Liu, D. Chao, Y. Li, R. Ting, J. Oh and D. M. Perrin, Chem. – Eur. J., 2015, 21, 3924–3928 CrossRef CAS PubMed.
- D. Mazunin, N. Broguiere, M. Zenobi-Wong and J. W. Bode, ACS Biomater. Sci. Eng., 2015, 1, 456–462 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |