
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and functionalization of monodisperse near-ultraviolet and visible excitable multifunctional Eu3+, Bi3+:REVO4 nanophosphors for bioimaging and biosensing applications†
Alberto
Escudero
*ab,
Carolina
Carrillo-Carrión
ac,
Mikhail V.
Zyuzin
a,
Sumaira
Ashraf
a,
Raimo
Hartmann
a,
Nuria O.
Núñez
b,
Manuel
Ocaña
b and
Wolfgang J.
Parak
a
aAG Biophotonik, Fachbereich Physik, Philipps-Universität Marburg, Renthof 7. D-35037, Marburg, Germany
bInstituto de Ciencia de Materiales de Sevilla, CSIC – Universidad de Sevilla. C. Américo Vespucio 49. E-41092, Seville, Spain. E-mail: alberto.escudero@csic.es
cCIC biomaGUNE. Paseo Miramón 182. E-20009, San Sebastian, Spain
First published on 13th May 2016
Abstract
Near-ultraviolet and visible excitable Eu- and Bi-doped NPs based on rare earth vanadates (REVO4, RE = Y, Gd) have been synthesized by a facile route from appropriate RE precursors, europium and bismuth nitrate, and sodium orthovanadate, by homogeneous precipitation in an ethylene glycol/water mixture at 120 °C. The NPs can be functionalized either by a one-pot synthesis with polyacrylic acid (PAA) or by a Layer-by-Layer approach with poly(allylamine hydrochloride) (PAH) and PAA. In the first case, the particle size can also be tuned by adjusting the amount of PAA. The Eu- Bi-doped REVO4 based nanophosphors show the typical red luminescence of Eu(III), which can be excited through an energy transfer process from the vanadate anions, resulting in a much higher luminescence intensity in comparison to the direct excitation of the europium cations. The incorporation of Bi into the REVO4 structure shifts the original absorption band of the vanadate anions towards longer wavelengths, giving rise to nanophosphors with an excitation maximum at 342 nm, which can also be excited in the visible range. The suitability of such nanophosphors for bioimaging and biosensing applications, as well as their colloidal stability in different buffer media of biological interest, their cytotoxicity, their degradability at low pH, and their uptake by HeLa cells have been evaluated. Their suitability for bioimaging and biosensing applications is also demonstrated.
1. Introduction
Nanoparticles (NPs) are currently attracting wide research interest due to their potential use in medical1 or energy-conversion/storage applications. In the case of biomedically motivated research, luminescent NPs are interesting because of their optical properties, and can be used as optical biolabels for imaging of tissues or intracellular structures, as sensors to detect biological molecules, and as tracking devices.2–7For biomedical applications usually uniform NPs with controlled size, shape, composition, surface chemistry and other physicochemical properties are desired,8 given the strong effect that the morphology, size, and structure of the particles may have on many physicochemical properties of these materials.9–12
Among the different luminescent nanomaterials available in the literature,13 including quantum dots14 and nanostructures functionalized with organic dyes,6 Rare Earth (RE) based nanophosphors exhibit important advantages due to their lower toxicity, photostability, high thermal and chemical stabilities, high luminescence quantum yields, and sharp emission bands.15–17 These nanomaterials usually consist of an inorganic host matrix doped with luminescent lanthanide (Ln) cations. Red-emitting Eu3+ is frequently used as a luminescent cation, as the autofluorescence of tissues is minimized in this wavelength range.18 The main disadvantage of Ln-doped nanophosphors is their relatively low global intensity luminescence, caused by the low absorptions of the parity forbidden Ln3+ 4f–4f transitions, and constituting a serious limitation for their use for imaging applications. Different energy transfers schemes from the host materials to the Ln3+ cations are employed to enhance the global luminescence of lanthanide-doped nanophosphors, including the use of vanadate or oxyfluoride matrices.19–23 However, ultraviolet excitation radiations with wavelengths below 300 nm are normally required, resulting in limitations for the use of such materials for bioimaging. It has been reported that the Eu3+:REVO4 excitation peak can be shifted toward longer wavelengths upon doping with Bi3+, resulting in phosphors excitable by near-ultraviolet light.24–29
Eu3+:Bi3+ doped YVO4 micro- and NPs of different morphologies (sphere-, granular-, and rod-like) have been synthesized by various wet-chemical processes including coprecipitation,30 and hydrothermal31–33 methods, and thin films have been produced by sol–gel processes.34 Micrometric Eu3+:Bi3+:GdVO4 particles have been produced by a hydrolysed colloid reaction (HCR) technique35 and by a combustion process.36 Gd bearing NPs are also of special interest due to their use as a relaxation agent in magnetic resonance imaging (MRI), owing to the magnetic moment and nanosecond time scale electronic relaxation time of Gd3+.37
A functionalization process is normally required for the biomedical use of NPs. This process not only increases the colloidal stability of the NPs by introducing electrostatic and/or steric repulsions,38 but also provides anchors for adding functional ligands of biomedical interest such as antibodies, peptides, proteins, and some anticancer drugs.39 Functionalization of Eu3+:Bi3+:YVO4 nanophosphors with additives such as sodium citrate40 and branched polyethylenimine (BPEI)32,33 have been reported.
Alternatively, NPs can also be coated with polymers.41 Poly(acrylic acid) (PAA) is a frequently used polymeric ligand for the biofunctionalization of NPs, because of its negligible toxicity,42 its hydrophilic character,43 and its high affinity for cellular membranes.44 Usually, the PAA surface modification of the NPs is carried out through the adsorption or ionic interaction process.41 Another common functionalization strategy consists of the encapsulation of the NPs by multiple layers of polymers, leading to hybrid core/(polymer-)shell NPs.45 The Layer-by-Layer (LbL) approach, which is based on the electrostatic deposition of layers of polyelectrolytes with alternating charge on the surface of the particles,46 has been widely used to functionalize different kinds of nanostructured materials, such as gold NPs47 and, especially, for the synthesis of polyelectrolyte capsules.48 The application for RE-based NPs is however not common so far.41,49
We report here a facile and fast synthesis method of Eu3+:Bi3+:REVO4 (RE = Y and Gd) nanophosphors, based on a homogeneous precipitation process from solutions of appropriate RE and orthovanadate precursors in ethylene glycol (EG)/water mixtures at 120 °C. Both kinds of nanophosphors are functionalized with poly(acrylic acid) (PAA) by a one-pot synthesis strategy (i.e. PAA is added during the synthesis process), and also by a Layer-by-Layer approach with poly(allylamine hydrochloride) (PAH) and PAA. The effect of the level of Bi3+ doping on the luminescence properties of both kinds of nanophosphors has also been studied, as well as their colloidal stability in different physiological buffers, their degradability at low pH, and their cell viability. Finally, we show how the europium and bismuth doped rare earth orthovanadate NPs can be used to image human cervical carcinoma (HeLa) cells by using near-ultraviolet and visible radiation as excitation sources, and as pH sensors, after their conjugation with a pH-sensitive dye such as Nile Blue. The influence of the morphology and chemical composition of the NPs on the cellular uptake is also shown.
2. Results and discussion
2.1 Synthesis, PAA functionalization and physicochemical characterization of the NPs
A homogeneous process with constant reaction kinetics is normally required to obtain uniform NPs by precipitation. This can be achieved by a slow and controlled release of the precipitating anions or cations within the solution.50 In our case, a homogeneous precipitation is produced by using Y(acac)3 as the yttrium precursor for the synthesis of YVO4-based particles. This compound decomposes on heating, and gives rise to a slow release of the Y cations.51 It has also been described that the presence of additives during the synthesis process, such as PAA, can affect the final morphology of the particles.52 Moreover, a specific reaction kinetics is also demanded, which is established after an analysis of the effects of the experimental reaction parameters on the morphological features of the precipitated particles.50 The synthesis protocol herein reported for the synthesis of europium and bismuth doped YVO4 produces NPs with similar morphological characteristics, irrespective of the Bi content. A representative transmission electron microscopy (TEM) micrograph of the Bi20YPAA2 sample (see Table 1 for sample information) is shown in Fig. 1A. As it can be observed, NPs with a quasi-spherical morphology and a mean size of 93 nm ± 7 nm were obtained. The NPs are single crystals, as shown by the SAED pattern obtained from a single particle, which is compatible with the tetragonal symmetry of YVO4 (space group I41/amd) and could be indexed along the [001] zone axis (inset in Fig. 1A). The high-angle annular dark-field (HAADF) image of the sample Bi20YPAA2 indicates a homogeneous distribution of bismuth in the NPs (Fig. 1D). The mean hydrodynamic diameter of such nanoparticles obtained by dynamic light scattering (DLS) was 108 ± 10 nm, which is expected for well-dispersed particles free of aggregation. The analogous sample synthesized in the absence of PAA (sample Bi20Y) consisted of NPs with slightly sharper edges and a mean size of (107 ± 14) nm (Fig. S1A†). Also in this case, the mean hydrodynamic diameter of the Bi20Y NPs obtained by DLS (138 ± 7 nm) indicated that they were free of aggregation. It should be noted that the presence of bismuth in the europium-doped YVO4 NPs had almost no effect on the particle morphology, since similar NPs with a mean size of 88 ± 14 nm were observed in the absence of Bi (sample Bi0YPAA2, Fig. S1B†). By increasing the concentration of PAA from 2 to 8 mg mL−1 (sample Bi20YPAA8), a significant decrease in the mean particle size to 51 ± 5 nm was observed (Table 2) without alteration of the morphology (Fig. 1B). This difference in the particle size produced on the addition of PAA to the solutions containing the precursors may be attributed to the adsorption of these molecules on the NP surface, which could block the growth of the NPs.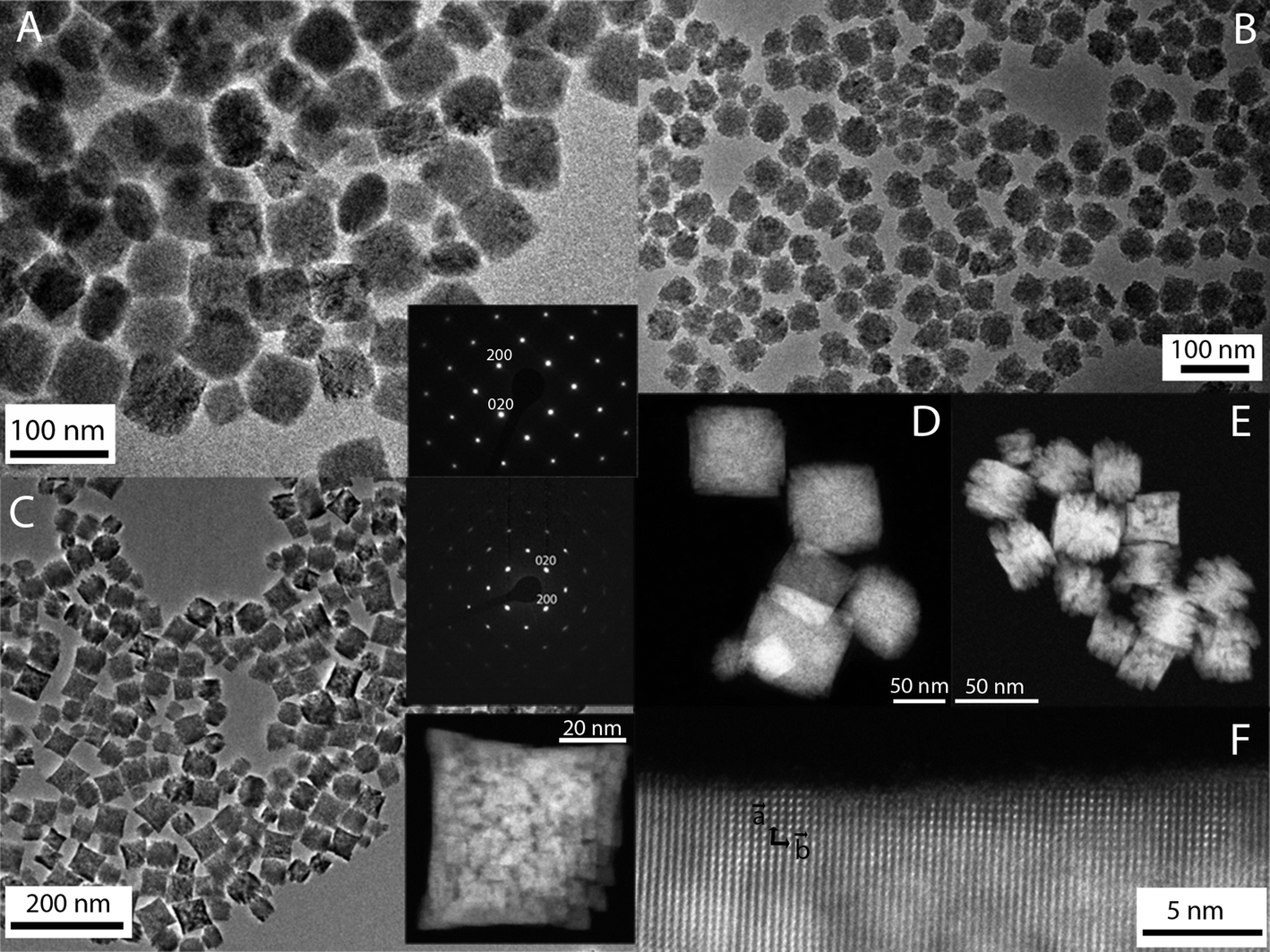 | ||
| Fig. 1 TEM micrographs of the Bi20YPAA2 (A, scale bar 100 nm), Bi20YPAA8 (B, scale bar 100 nm), and Bi20GdPAA (C, scale bar 200 nm) samples. HAADF images of the Bi20YPAA2 (D, scale bar 50 nm), and Bi20GdPAA (E, scale bar 50 nm) samples. (F, scale bar 5 nm) HRTEM image of a particle of the Bi20GdPAA2 sample. | ||
| Sample | [Eu3+] (M) | [Bi3+] (M) | [Y3+] (M) | [VO43−] (M) | PAA (mg mL−1) |
|---|---|---|---|---|---|
| Bi0YPAA | 0.0016 | 0 | 0.0184 | 0.1 | 2 |
| Bi20YPAA2 | 0.0016 | 0.004 | 0.0144 | 0.1 | 2 |
| Bi20YPAA8 | 0.0016 | 0.004 | 0.0144 | 0.1 | 8 |
| Bi20Y | 0.0016 | 0.004 | 0.0144 | 0.1 | 0 |
| [Eu3+] (M) | [Bi3+] (M) | [Gd3+] (M) | [VO43−] (M) | PAA (mg mL−1) | |
|---|---|---|---|---|---|
| Bi0GdPAA | 0.0016 | 0 | 0.0184 | 0.1 | 2 |
| Bi20GdPAA | 0.0016 | 0.004 | 0.0144 | 0.1 | 2 |
| Bi20Gd | 0.0016 | 0.004 | 0.0144 | 0.1 | 0 |
| Sample | Particle size (TEM) (nm) | Hydrodynamic diameter (nm) | Crystallite size (nm) | ζ-Potential (mV) | Unit cell volume (Å3) |
|---|---|---|---|---|---|
| Bi0YPAA | 88 ± 14 | 82 ± 3 | 62 | −37 ± 2 | 319.24 ± 0.03 |
| Bi20YPAA2 | 93 ± 7 | 108 ± 10 | 79 | −42 ± 3 | 325.58 ± 0.02 |
| Bi20YPAA8 | 51 ± 5 | 51 ± 2 | 57 | −39 ± 2 | 325.61 ± 0.03 |
| Bi20Y | 107 ± 14 | 138 ± 7 | N.r. | −19 ± 1 | N.r. |
| Bi0GdPAA | 39 ± 7 | 40 ± 2 | 53 | −45 ± 1 | 330.32 ± 0.04 |
| Bi20GdPAA | 40 ± 8 | 41 ± 2 | 65 | −41 ± 2 | 335.37 ± 0.02 |
| Bi20Gd | 51 ± 15 | 58 ± 4 | N.r. | −25 ± 1 | N.r. |
Similar results were observed for the Eu- Bi-doped GdVO4 samples. A representative TEM micrograph of the Bi20GdPAA sample is shown in Fig. 1C. As it can be seen, non-aggregated NPs with a smaller size and slightly sharper edges when compared with those based on YVO4 (TEM size 40 ± 8 nm, DLS 41 ± 2 nm) were produced after the synthesis in the presence of PAA. Such NPs were also single crystals, as inferred from the SAED pattern of a single particle, which is compatible with tetragonal GdVO4 (space group I41/amd), zone axis [001], and from the high resolution TEM images (insets of Fig. 1C and F). Analogous to the yttrium vanadate samples, a homogeneous distribution of Bi in the NPs was indicated by the HAADF-TEM images (Fig. 1E). Much more polydisperse and bigger NPs with a not so well defined shape (TEM size 51 ± 15 nm, DLS 58 ± 4 nm, Fig. S1C†) were obtained in the absence of PAA (sample Bi0Gd). Similar to the YVO4-based NPs, the presence of Bi did not affect the particle morphology, since NPs with a mean size of 39 ± 7 nm were obtained in the absence of Bi (sample Bi0GdPAA, Fig. S1D†). A summary of the data corresponding to all the synthesized samples is shown in Table S2.†
The presence of PAA on the surface of both Bi20YPAA2 and Bi20GdPAA NPs was confirmed by Fourier transform infrared spectroscopy (FTIR) analysis. The FTIR spectra of the Bi20Y, Bi20Gd, Bi20YPAA2, and Bi20GdPAA samples are shown in Fig. 2. In all the samples, the characteristic bands of vanadate anions (<1000 cm−1)53 and adsorbed water (broad bands at 3400 and 1600 cm−1) can be observed, as well as some much weaker features in the 1500–1300 cm−1 region, which can be attributed to the presence of carbonate anions from the adsorption of atmospheric CO2.54 Samples Bi20YPAA2 and Bi20GdPAA show extra signals around 1458 and 1550 cm−1 (marked with asterisks in Fig. 2), which can be ascribed to the symmetric and antisymmetric stretching frequencies of the carboxylate ion (COO−), respectively.52 Taking into account that the carbonyl stretching (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) vibration appears at 1717 cm−1 for pure PAA, the shift towards lower frequencies indicates that most of the carboxylate functional groups of the polymer are fully ionized55 and, therefore, that the PAA molecules are bonded to the NP surface though the carboxylate groups.
O) vibration appears at 1717 cm−1 for pure PAA, the shift towards lower frequencies indicates that most of the carboxylate functional groups of the polymer are fully ionized55 and, therefore, that the PAA molecules are bonded to the NP surface though the carboxylate groups.
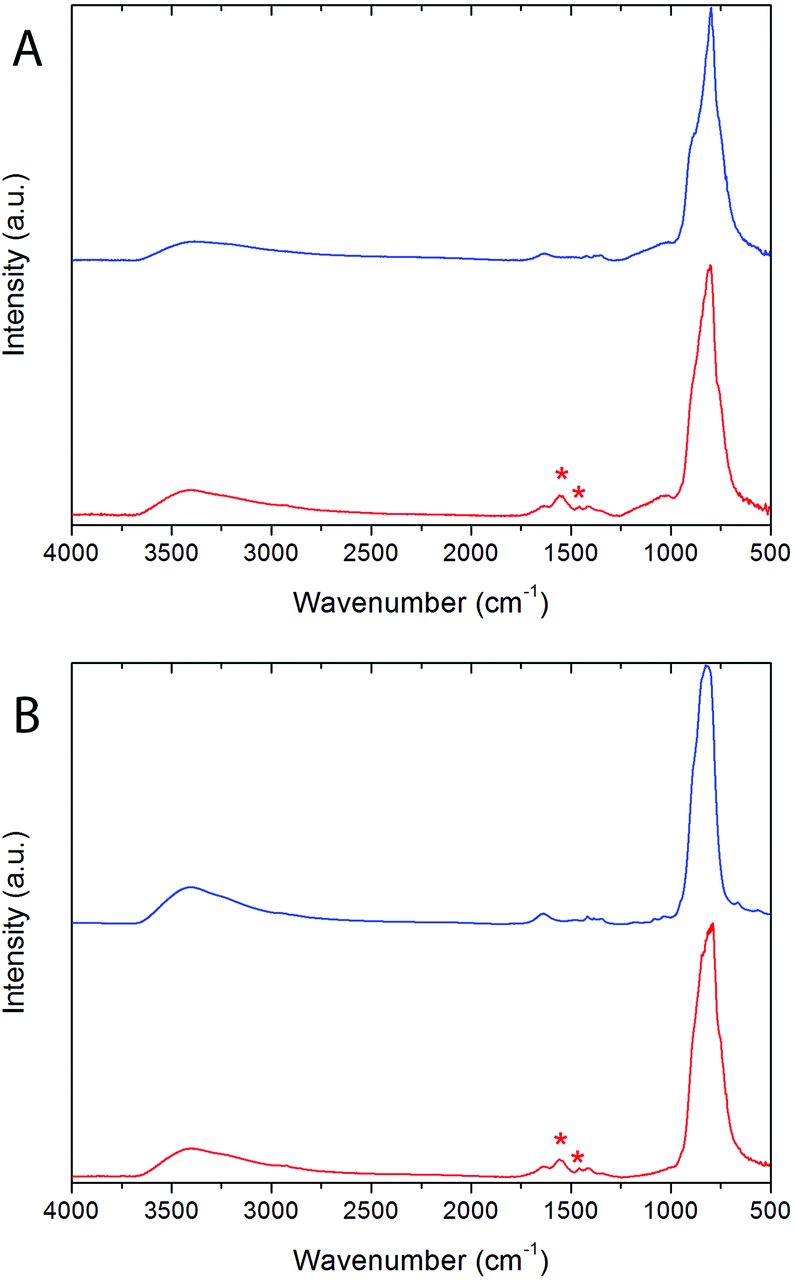 | ||
| Fig. 2 Fourier transform infrared spectroscopy (FTIR) spectra of (A) the Bi20Y (blue) and Bi20YPAA2 (red) samples; and (B) of the Bi20Gd (blue) and Bi20GdPAA (red) samples. | ||
Further evidence of the PAA functionalization of the particles was obtained by comparing the ζ-potential of aqueous suspensions of the non-functionalized and PAA-functionalized particles. Bi20Y particles showed a ζ-potential of −19 mV in water at pH 7, whereas the value of this magnitude for the NPs synthesized in the presence of PAA was −42 mV at the same pH. Analogously, the ζ-potential of the Bi20GdPAA NPs was −41 mV, to be compared to the one of −25 mV as observed for the non-functionalized Bi20Gd particles. Thus, NPs synthesized in the presence of PAA are already functionalized with carbonyl groups. An estimation of the amount of PAA on the surface of the PAA-functionalized NPs was obtained from thermogravimetry (TG) analysis (Fig. S2†). The weight loss observed at temperatures below 250 °C can be associated with the elimination of absorbed water. At higher temperatures (up to 600 °C), the loss most likely is connected with the decomposition of the adsorbed PAA molecules, which was higher for the sample prepared with more PAA (ca. 2% for sample Bi20YPAA2, ca. 15% for sample Bi20YPAA8 – including more adsorbed water-, and ca. 4% for sample Bi20GdPAA).
The successful incorporation of europium and bismuth into the NPs was evidenced from the energy dispersive X-ray spectra (EDX) obtained for single particles, which displayed europium and bismuth peaks, in addition to those of yttrium, vanadium, gadolinium and oxygen (Fig. S3†). Moreover, no extra particles with different morphologies or with a higher Bi or Eu content were observed. Induced coupled plasma atomic emission spectroscopy (ICP-AEP) measurements indicated that the experimental molar ratios corresponding to the doped particles were in good agreement with the nominal values (Table S3†).
The X-ray diffraction (XRD) diagrams obtained for the Bi0YPAA2 and Bi20YPAA2 samples (Fig. 3A) consisted of peaks that are very similar to the standard pattern of tetragonal YVO4 (PDF 01-070-1281). Similar results were observed for the Bi0GdPAA and Bi20GdPAA samples, whose XRD diagrams (Fig. 3B) were also very similar to the standard pattern of tetragonal GdVO4 (PDF 01-086-0996). Based on these data, the possible formation of slightly non-stoichiometric phases of rare earth orthovanadates can however not be fully discarded.56 It should also be pointed that for similar orthovanadate systems, an excess of [Na3VO4] in the starting solutions was used to adjust the precipitation kinetic, yielding NPs with smaller size.19,21,23 The slight differences in peak positions and intensities in both Y and Gd bearing samples when increasing the Bi content (see peaks under the vertical guide lines) indicate changes in the unit cell dimensions and composition. This was confirmed by the measurements of the unit cell parameters of all the doped samples synthesized in the presence of PAA, which showed in both cases a linear increase of the unit cell volume as the Bi doping level increased (Table 2, Fig. S4†). Such results are in agreement with the higher ionic radius of the Bi3+ cations (1.17 Å) when compared to those of Y3+ (1.019 Å) and Gd3+ (1.053 Å), all of them in eightfold coordination.57 The average crystal size (Table 2) estimated by using the Scherrer formula from several XRD peaks (101, 200, 112, 220, and 321) for all the particles was very similar to the size observed by TEM and DLS, also suggesting that the particles were single crystals.
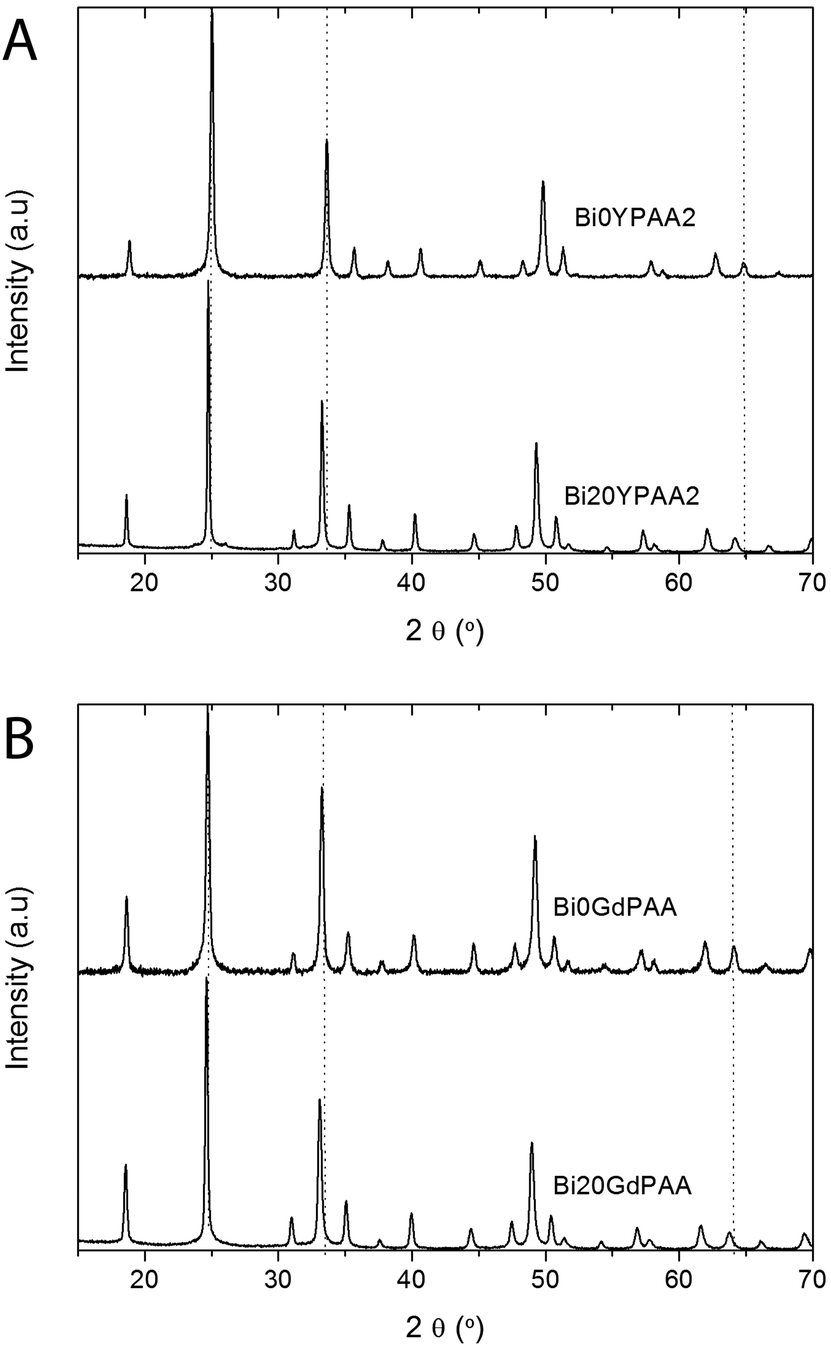 | ||
| Fig. 3 XRD diagrams of (A) the Bi0YPAA2 and Bi20YPAA2 samples; and (B) of the Bi0GdPAA and Bi20GdPAA samples. Vertical guides are only intended as a guide to the eye. | ||
2.2 Layer-by-Layer functionalization
The LbL functionalization of NPs usually requires an optimization of the experimental parameters in order to avoid particle agglomeration. Several variables are known to play an important role in this process, including the polyelectrolyte stoichiometric excess, the NP concentration, the size and degree of polymerization of the polyelectrolyte chains, and the salt concentration.58 In general terms, an excess of polyelectrolyte, low NP concentrations, and short polyelectrolyte chains help obtain NPs free of aggregation. Regarding the salt concentration, low concentrations prevent agglomeration, but high concentrations normally increase the polyelectrolyte amount that is deposited onto the NP surface.59Functionalized particles free of aggregation (as concluded from the fact that hydrodynamic diameters as determined by DLS were only slightly bigger than inorganic core diameters as determined by TEM) were obtained for both kinds of bare NPs (samples Bi20Y and Bi20Gd) by using 10 mg per mL polyelectrolyte solutions in 0.05 M NaCl aqueous solutions at pH 6.5, as described in the Experimental section. The success of functionalization and the absence of a significant particle agglomeration were confirmed by the surface charge reversal observed after the deposition of every polyelectrolyte layer, and by the evolution of the hydrodynamic diameter of the particles, respectively (Fig. 4). The selection of PAH and PAA as polymers for the Layer-by-Layer functionalization of the NPs also provides reactive –COOH and –NH2 groups, and increases the possibility of further conjugation with different molecules of biomedical interest.
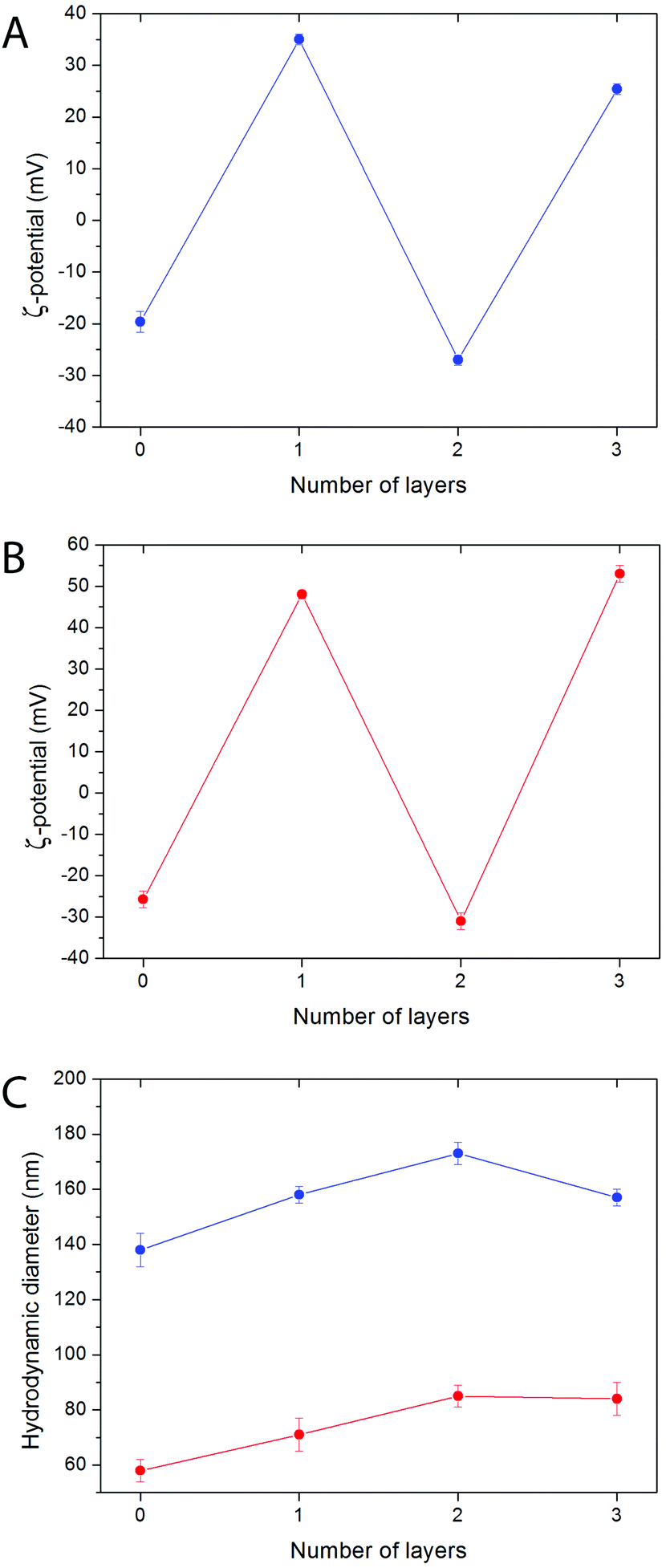 | ||
| Fig. 4 ζ-Potential of the Bi20Y (A) and Bi20Gd (B) nanoparticles functionalized with PAH and PAA by the LbL technique after the deposition of different polyelectrolyte layers, measured in water at pH 7. (C) Evolution of the hydrodynamic diameter of the Bi20Y (blue) and Bi20Gd (red) functionalized nanoparticles with the number of deposited polyelectrolyte layers. | ||
2.3 Colloidal stability
The colloidal stability of both, PAA- and LbL PAH/PAA/PAH functionalized NPs, was analyzed by studying the mean hydrodynamic diameter (dh) obtained by DLS in water and in different buffer media of biological interest, containing salt and proteins.60 The evolution of the hydrodynamic diameter with increasing time was also studied.The hydrodynamic diameters measured for freshly prepared suspensions of PAA-functionalized NPs in different buffers are shown in Fig. 5A and B, and in Table S4.† For both PAA-functionalized NPs similar hydrodynamic diameters were observed in water at pH 7 and in MES buffer at pH 6.5. Such values are very close to the size determined by TEM for both samples, indicating the absence of significant aggregation and, therefore, colloidal stability in both media. Bi20YPAA2 NPs also showed colloidal stability in PBS buffer at pH 7.4, whereas some aggregation was observed for the Bi20GdPAA NPs in this same buffer. A strong level of aggregation was observed for both PAA-functionalized NPs in cell medium, which was minimized in serum-supplemented cell medium. Proteins in solution are well-known to attach to the surface of the NPs, forming the so-called protein corona,61 and provide the NPs with additional colloidal stability. No changes were observed in the mean hydrodynamic diameter of the YVO4-based PAA-functionalized NPs (sample Bi20YPAA2) with increasing aging time in water at pH 7, MES buffer at pH 6.5, PBS at pH 7.4, and serum-supplemented cell medium (Fig. S5A–D†), and similar results were observed for the Bi20GdPAA sample with increasing aging time in water at pH 7, in MES at pH 6.5, and serum-containing cell medium, with only some aggregation when aging in PBS at pH 7.4 (Fig. S5F–J†), manifesting that no particle aggregation took place upon aging in these buffer media at the studied pHs.
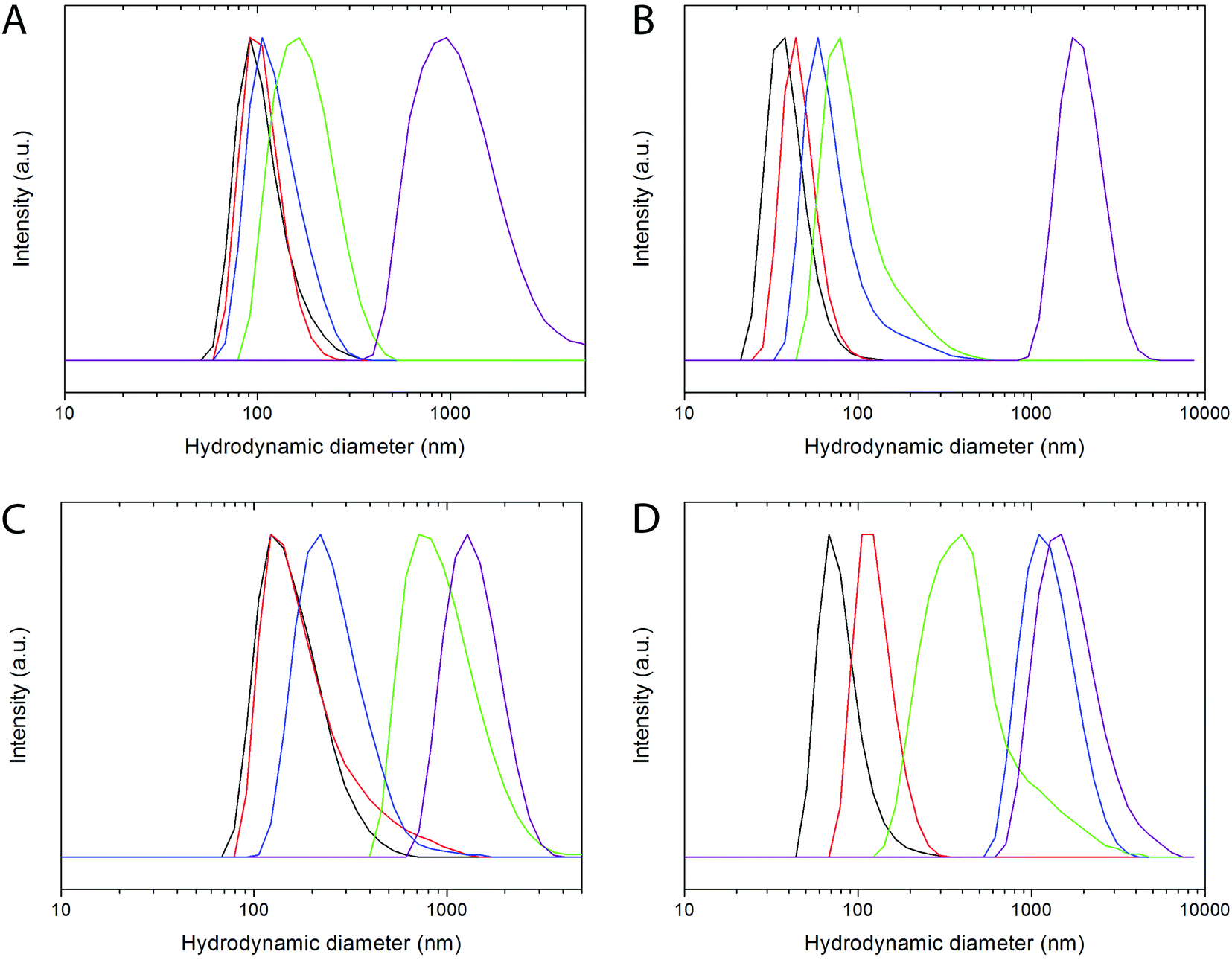 | ||
| Fig. 5 Hydrodynamic diameter of the Bi20YPAA2 (A), Bi20GdPAA (B), Bi20Y@PAH@PAA@PAH LbL-functionalized (C), and Bi20Gd@PAH@PAA@PAH LbL-functionalized (D) nanoparticles in water at pH 7 (black lines), MES 50 mM at pH 6.5 (red lines), PBS at pH 7.4 (blue lines), serum-supplemented cell medium (green lines), and cell medium (purple lines). | ||
Data corresponding to the PAH/PAA/PAH LbL functionalized YVO4 NPs are shown in Fig. 5C and Table S3.† No significant aggregation was observed in MES buffer at pH 6.5. It should be mentioned that at least two polyelectrolyte layers were required to achieve colloidal stability in MES buffer at pH 6.5 (Fig. S6†). A decrease of the colloidal stability was observed for the LbL functionalized particles in both PBS at pH 7.4 and in cell medium. Similar to the observation for the PAA-functionalized NPs, a considerable reduction in the agglomeration level was observed for the LbL functionalized particles in serum-supplemented cell medium, which was however not so strong as in the previous example. At this point, it worth pointing out that a similar level of aggregation (dh = 240 nm, Fig. S7A†) was observed for the non-functionalized bare particles (sample Bi20Y) in serum-supplemented cell medium, which indicates the strong effect of the protein absorption on the final colloidal stability of the NPs. Slightly worse colloidal stability was observed for the LbL functionalized GdVO4-based NPs (Fig. 5D, Table S4†). Very slight agglomeration was also observed for the bare Bi20Gd NPs in serum-supplemented cell medium (Fig. S7B†). The level of aggregation of both kinds of LbL functionalized NPs increased with aging in MES, PBS, and cell medium. No changes were observed with aging time in water at pH 7 and in serum-supplemented cell medium (Fig. S5†).
These results indicate that the colloidal stability of the one pot PAA synthesized and functionalized NPs is higher than the one shown by the NPs functionalized in a second step by the LbL approach. The incorporation of either functionalizing agents or adsorbed organic molecules onto the NP surface during the synthesis process seems to play a role in the final colloidal stability of the particles, at least when dealing with polymeric functionalizing agents. In particular, incorporation of multiple particles into one polymer shell may lead to aggregation. However, the LbL strategy offers a useful functionalization alternative for systems in which no functionalizing agents attach to the NP surface during the synthesis process. The colloidal stability of the NPs functionalized by the LbL strategy increases with the number of deposited layers.
2.4 Optical properties
Excitation spectra of both, 8% Eu- 20% Bi-doped YVO4 and GdVO4 PAA functionalized NPs (samples Bi20YPAA2 and Bi20GdPAA, Fig. 6A and B, respectively) were recorded by monitoring at a wavelength of 622 nm, which corresponds to the main emission line of Eu3+ cations in rare earth vanadate matrices. Both spectra show a broad band with maxima at 342 nm, associated with the indirect excitation of the Eu3+ cations. For both, Y- and Gd-bearing nanophosphors (i.e. Bi-free samples), a broad excitation band with a maximum centered at around 280 nm can be observed (Fig. S8†).19,21 This band has been ascribed to the optical excitation of the vanadate anions (originated from the O2− → V5+ charge transfer within the vanadate groups), followed by an energy transfer to the Eu3+ cations.62 The incorporation of Bi into the REVO4 lattices produces changes in both the shape and the position of the maximum of the broad absorption band. Increasing the Bi content produces wider absorption bands with the maxima shifting toward longer wavelengths. (Fig. S8†). The observed changes can be explained by the appearance of an extra charge transfer band from the 6s level of Bi3+ to the 3d levels of V5+, which is centered around 340 nm.24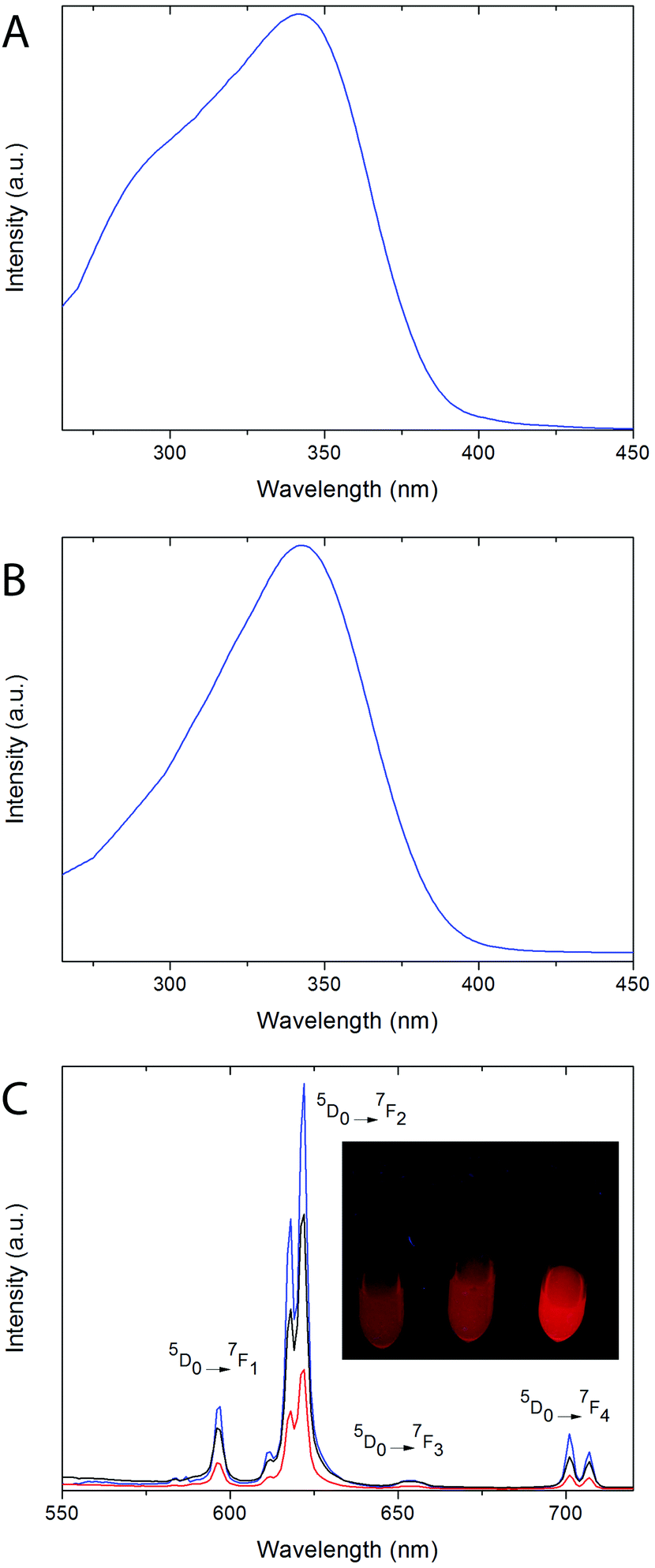 | ||
| Fig. 6 Excitation spectra (emission recorded at λem = 622 nm) of the Bi20YPAA2 (A) and Bi20GdPAA (B) samples. (C) Emission spectra of the Bi20GdPAA (blue line), Bi20YPAA2 (black line), and Bi20YPAA8 (red line) samples (excitation at λexc = 342 nm). The photograph shows the red luminescence of water suspensions (0.05 mg mL−1) of the Bi20YPAA8 (left), Bi20YPAA2 (center), and Bi20GdPAA (right) samples when irradiated with a UV lamp. | ||
Fig. 6C shows the emission spectra of the suspensions of the Bi20YPAA2 (black line), Bi20YPAA8 (red line), and Bi20GdPAA (blue line) samples, recorded under a UV excitation of 342 nm. The spectra display the typical emissions corresponding to the 5D0 → 7FJ (J = 1, 2, 3, and 4) electronic transitions expected for the Eu3+ cation.62 The most intense emission (5D0 → 7F2 transitions) is split into two bands at 618 and 622 nm owing to the crystal field.62 It can also be observed that the relative intensity of the 5D0 → 7F2 emission band is in all cases higher than that associated with the 5D0 → 7F1 transition, which is expected for Eu3+ cations located in crystallographic sites without an inversion center. This is compatible with Eu3+ cations replacing Y3+ or Gd3+ on the normal yttrium sites of the tetragonal REVO4 structure, in which the RE sites show a local symmetry of D2d.62
The different emission intensities observed for the different samples should be connected with their different luminescence quantum yields, since the spectra were recorded from NP suspensions owing to the same NP concentration. At least qualitatively, the GdVO4 host material seems to show a higher luminescence quantum yield than YVO4. The differences observed for the YVO4-based NPs of different sizes show that bigger particles are more efficient. This should be connected with their lower surface area per particle volume. As the size of the NPs decreases, there is a significant increase of the surface area per particle volume, as well as an increase in the number of surface defects and higher number of capping ligand molecules, causing a decrease in the luminescence efficiency.63,64
It should also be mentioned that it has been reported that Eu, Bi-doped YVO4 might exhibit photobleaching under continuous UV irradiation.65,66 In our case, Bi20YPAA2 NPs were continuously irradiated with UV light (Fig. S9†). After 10 minutes of irradiation, around 80% of the initial intensity was still observed, being reduced to about 70% after 25 minutes of irradiation.
2.5 Cellular uptake and imaging
Human cervical carcinoma cell line (HeLa) cells were used as a system to show the suitability of both Eu- Bi-doped YVO4 and GdVO4 NPs for bioimaging. Two different microscopes were used to image the NPs. On one hand, the broad absorption band of the NPs with a maximum at around 342 nm permits their excitation with near-ultraviolet light in a fluorescence microscope. On the other hand, the shift of the absorption band of the Bi-doped vanadate matrices toward longer wavelengths makes also possible their excitation with a visible radiation by using a laser as the excitation source in a confocal laser scanning microscope (cLSM). The study of the NP internalization by the cells was performed by observing the co-localization of the fluorescence of the NPs with those produced by different endocytic fluorescent markers that specifically attach to the cell membranes and organelles.67 A strong red luminescence in the interior of the cells could be observed after irradiation at (340 ± 24) nm after 24 h of incubation (illustrated in Fig. 7 for the Bi20YPAA2 sample). This fluorescence was clearly co-localized with the yellow one produced by LAMP1, a marker for the membrane of lysosomes, indicating that the NPs are eventually located in the cell's lysosomes after being internalized. Such red fluorescence was not observed under UV light excitation in the control experiments, in which HeLa cells were incubated without NPs (Fig. S10†). PAA-functionalized Eu- Bi-doped GdVO4 NPs, as well as both Eu- and Bi-doped YVO4 and GdVO4 NPs functionalized by LbL could also be observed in the cell interior (Fig. S11–S14†).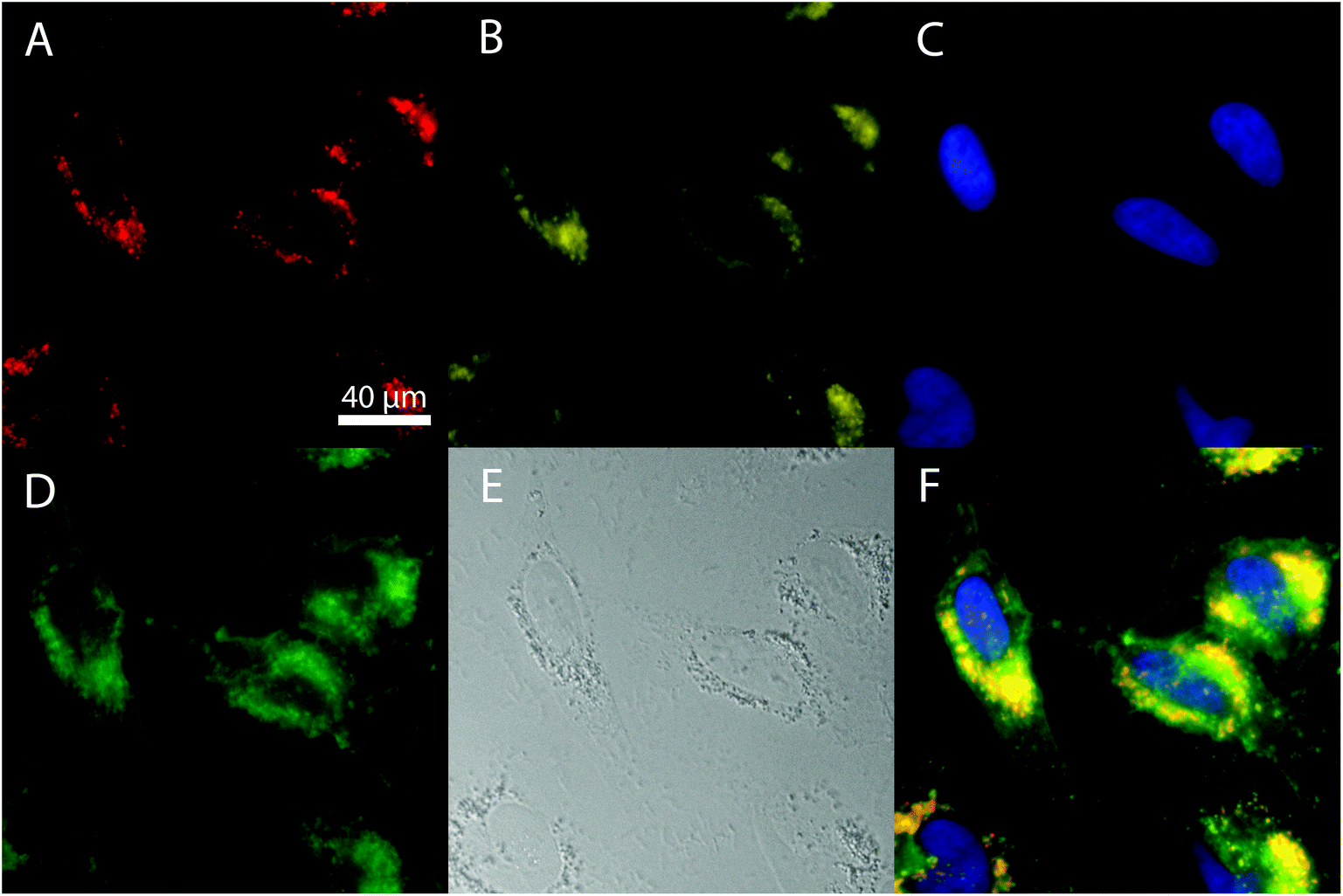 | ||
| Fig. 7 Fluorescence images of HeLa cells incubated with Bi20YPAA2 nanoparticles for 24 h. (A) Red channel, nanoparticles; (B) yellow channel, lysosomes; (C) blue channel, cell nuclei; (D) green channel, cell membranes; (E) transmission image; and (F) merged all channels. | ||
As it was previously mentioned, it is also possible to image both, YVO4- and GdVO4-based NPs with a confocal laser scanning microscope (cLSM) using visible light as the excitation source (wavelength of 405 nm, laser power 20%). The red luminescence of the NPs can clearly be observed to be associated with HeLa cells after 24 hours of incubation (Fig. 8, illustrated for sample Bi20YPAA2). Images of the control sample and of HeLa cells incubated with samples Bi20YPAA8 and Bi20GdPAA are shown in Fig. S15.†
 | ||
| Fig. 8 Confocal microscopy images of HeLa cells incubated with Bi20YPAA2 nanoparticles for 24 hours. (A) Transmission channel, (B) fluorescence channel, (C) merged image. | ||
2.6 Biosensing applications
The presence of free carboxyl groups on the NP surface not only provides colloidal stability but also facilitates the conjugation of the NPs with various biomolecules, which paves way for further bio-applications. In this work, Nile Blue (NB) was chosen as the fluorescent pH indicator, since it has an amino group that is capable of forming a covalent bond with the carboxyl groups of the PAA layer onto the NP surface. Samples Bi20YPAA2 and Bi20GdPAA were chosen for conjugation with Nile Blue.Changes of the fluorescence response of the NB-PAA-NPs towards pH were evaluated over a wide pH range (pH = 3–11) recording independently the emission of the Eu3+ ions from the NP cores (excitation wavelength 342 nm) and the emission of the NB from the NP shells (excitation wavelength 600 nm). The maximum emission of the Eu3+ ions at 622 nm was found to be insensitive to the pH value and therefore can be used as reference. The emission of the NB attached to the NPs was recorded at 675 nm, and showed a clear dependence with the pH (illustrated in Fig. 9A for the Bi20YPAA2 NPs conjugated with Nile Blue at different pH values). Such behaviour allowed the use of the ratiometric intensity at these two different emission wavelengths as analytical signal, which is advantageous in terms of accuracy and reliability, because errors associated with environmental variations, probe distribution, and instrumental performance are diminished. The sigmoidal response obtained from the calibration curve of the ratio between the pH and the emission intensities at 622 and 675 nm (Fig. 9B) demonstrates that such particles can be used as pH sensors at pH between 4 and 11. A similar behaviour was observed for the GdVO4 based nanophosphors (sample Bi20GdPAA) conjugated with NB (Fig. 9C).
 | ||
| Fig. 9 (A) Emission spectra of the Bi20YPAA2 nanoparticles functionalized with Nile Blue (NB) at pH 5 (red), 7 (green), and 10 (blue). Emission of Eu3+ (left part) was collected with excitation at λex = 342 nm, while λex for NB was 600 nm (emission on the right part). (B) Intensity ratio of the Eu3+ (λem = 622 nm, λex = 342 nm) and the NB (λem = 675 nm, λex = 600 nm) emission at different pH values for the Bi20YPAA2-NB nanoparticles, and Bi20GdPAA-NB particles (C). In both last cases, the error bars are smaller than the size of the symbols. | ||
2.7 Biocompatibility, cell viabilities and degradation of the particles
Biocompatibility studies of both, Eu- Bi-doped YVO4- and GdVO4-based nanophosphors were undertaken by evaluating the cell viability of HeLa cells on the basis of the metabolic activity of living cells using a Resazurin based assay.68 PAA-functionalized YVO4 based NPs (sample Bi20YPAA2, Fig. 10A) showed under the used exposure conditions negligible toxicity effects, with viability percentages around 75–80% for concentrations up 1 mg mL−1, and a LD50 value measured from the sigmoidal curve (logistic dose response fit) of 1.52 ± 0.61 mg mL−1. These toxicity data show low toxicity in comparison to similar particles,33 which is probably associated with the low toxicity of the PAA.42 However, an increase of the cytotoxicity as the particle size decreases is observed (sample Bi20YPAA8, Fig. 10A), with a measured LD50 value of 0.16 ± 0.09 mg mL−1. The PAA-functionalized GdVO4 based NPs (sample Bi20GdPAA, Fig. 10B) showed much less toxic effects than their YVO4-based counterparts of similar size, with a measured LD50 value of 1.10 ± 0.29 mg mL−1. Such results indicate that the incorporation of Bi into the GdVO4 particles had no considerable effects on the final cytotoxicity of the NPs, since similar values have been reported for the same Bi-free particles.21 The LbL functionalization of both kinds of nanophoshors with PAH and PAH layers reduced their cytotoxicity effects (Fig. 10A and B), as it has been observed in similar systems.69,70 These cytotoxicity results indicate the potential suitability of such nanophosphors for sensing and cell imaging applications, even more when taken into account that the NP concentrations used in cell imaging and FACS experiments were between 1 or 2 orders of magnitude lower than the measured LD50 values. However, it clearly needs to be stated that concerning future in vivo use extended toxicity studies would be required, involving, in particular, the possible effects of long-term exposure.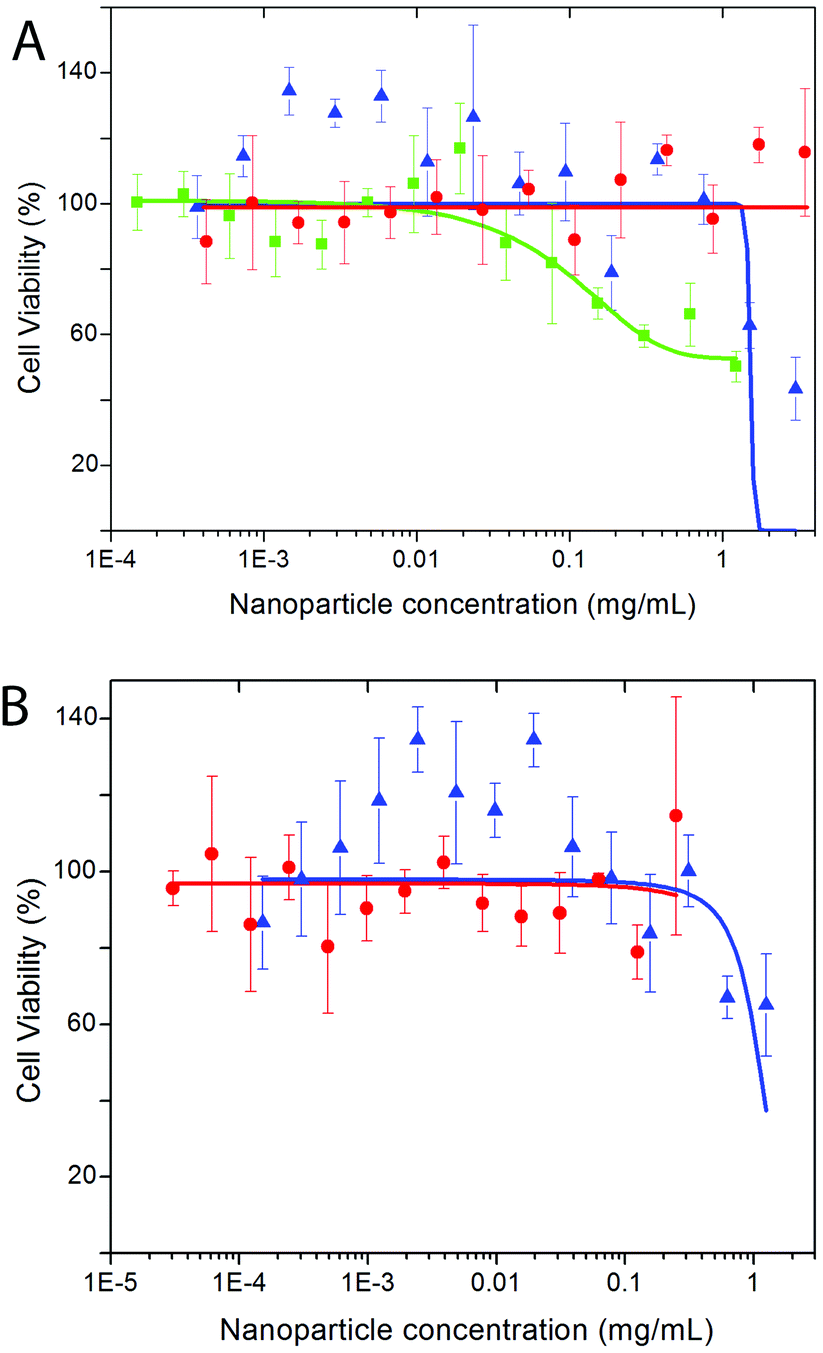 | ||
| Fig. 10 Cell viabilities for HeLa cells observed after 24 hours of incubation of (A) YVO4-based nanoparticles (Bi20YPAA2, blue triangles; Bi20YPAA8, green cubes; and Bi20Y sample LbL-functionalized with PAH and PAA, red circles); and (B) GdVO4-based nanoparticles (Bi20GdPAA, blue triangles; and Bi20Gd sample LbL functionalized with PAH and PAA, red circles). 100% viability corresponds to control cells which have not been exposed to NPs. The sigmoidal curves for the fits are also shown. | ||
The dissolution of the particles at low pH was carried out under conditions mimicking the lysosome, namely acidic pH, by analysing the evolution of the hydrodynamic diameter measured by DLS with increase in the aging time in MES 50 mM buffer at pH 3.5 (Fig. 11). Both, Bi20YPAA2 and Bi20GdPAA NPs aggregate in MES buffer at pH 3.5, forming agglomerates of around 1 μm. In both cases the size of the agglomerates decreased with increase in the aging time, and finally clear solutions were observed (Fig. S16†). This shows how both, YVO4- and GdVO4-based NPs can be degraded in the cell lysosomes, where eventually the NPs are located, resulting in an efficient method for the NP excretion.
 | ||
| Fig. 11 Evolution of the hydrodynamic diameter of Bi20YPAA2 (A) and Bi20GdPAA (B) NPs in MES 50 mM buffer at pH 3.5 at room temperature after different incubation times The lines represent a guide to the eye. | ||
2.8 Flow cytometry study
To reveal the influence of both, the chemical composition and the size of the NPs on the cellular uptake, flow cytometry studies with HeLa cells were performed for samples Bi20YPAA2, Bi20YPAA8, and Bi20GdPAA. Fig. 12A shows the mean red intensity fluorescence associated with every cell with increase in the incubation time. In general terms, this value increases with increase in the incubation time for the three analysed samples, indicating that the amount of NPs internalized by HeLa cells increases with time. After 24 hours of incubation, the highest red fluorescence was observed for the Bi20YPAA2 sample (black bar), followed by Bi20GdPAA (blue bar) and Bi20YPAA8 (red bar). The 2D density plots of the control measurements are shown in Fig. S17.† Such results, which are indeed consistent with the LSM images taken for HeLa cells after NP incubation (Fig. 8 and Fig. S14,† at least qualitatively), should however not be related to the amount of NPs associated with the cells, since the emission of the analyzed samples upon excitation at 355 nm depends on both chemical composition and the NP size (Fig. S10†). These different emission intensities were taken into account in Fig. 12B, whose results can then be related to the amount of NPs associated with the HeLa cells. A much higher amount of YVO4-based NPs is associated with HeLa cells at any time, when compared to GdVO4-based NPs. This is clearly observed when comparing the uptake of NPs of similar sizes but of different compositions, since the amount of particles of the Bi20YPAA8 sample associated with HeLa cells after 24 h of incubation is about 5 times higher than the observed for Bi20GdPAA NPs. Uptake of most NPs by cells takes place via endocytosis, whereby alterations in the characteristics of nanomaterials such as the radius of curvature, surface functionalization, size, geometry, NP volume, charge, and toxicity can drastically affect the uptake mechanisms of the NPs.60,71–73,74 The possible effect of the slightly different morphology shown by the GdVO4-based NPs, which consisted of particles with slightly sharper edges (Fig. 1C), should however not be ignored. The fact that particles with higher aspect ratios (i.e. rods) are normally taken up faster than spherical particles75,76 may have some effect on the NP uptake, as reported here. Moreover, there is also indication that there is an optimum NP size yielding the fastest NP uptake.76 In this study, 51 nm YVO4-based particles (sample Bi20YPAA8) were up taken 2–3 times faster than the bigger 93 nm particles (sample Bi20YPAA2). Such results also suggest the existence of a correlation between the observed cytotoxicity and the amount of NP internalized by the cells, as the toxicity of the particles increases with the amount of up taken NPs of the same chemical composition.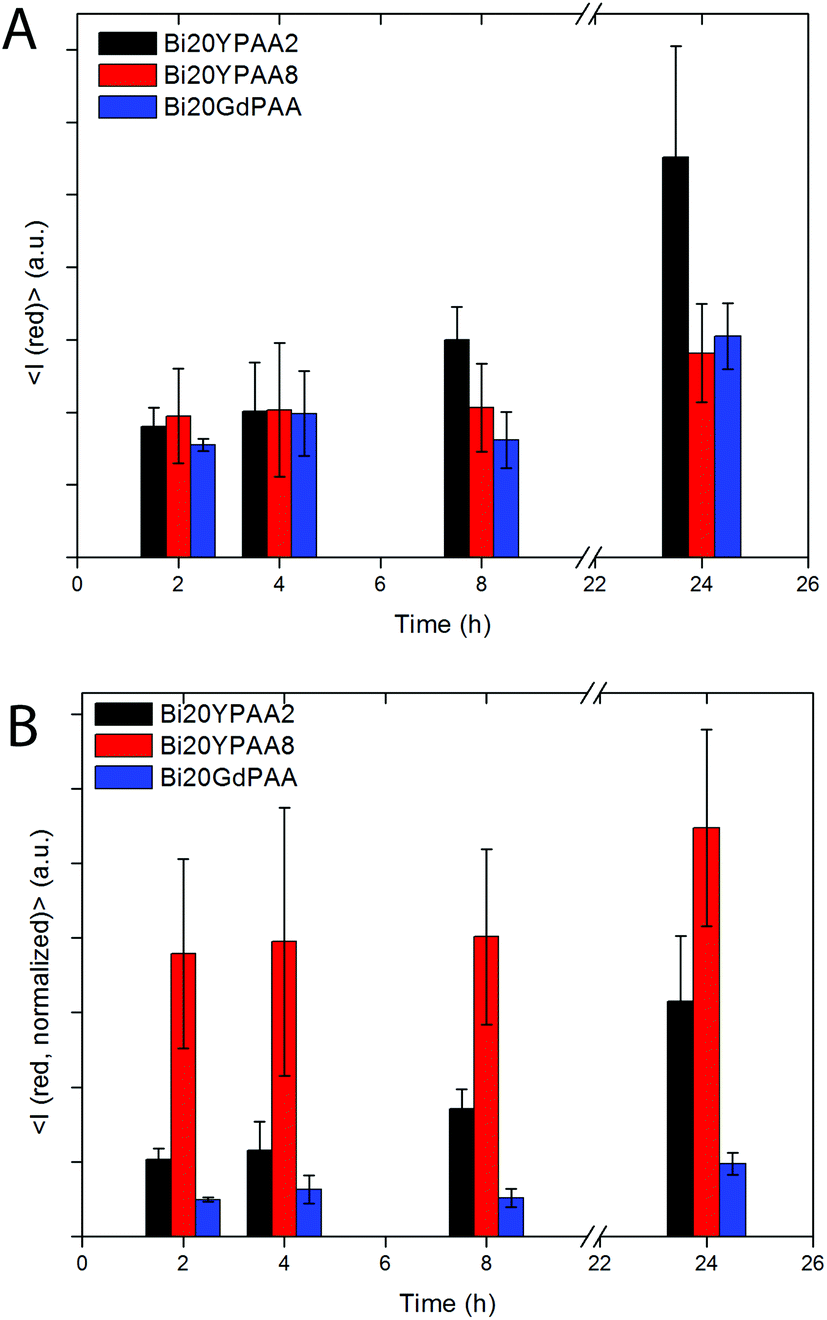 | ||
| Fig. 12 (A) Average red intensity emission associated with every cell after different incubation times as obtained by flow cytometry. (B) The normalized values are taking into account the different emission intensities of the analyzed samples. | ||
3. Conclusions
Monodisperse near-ultraviolet and visible excitable NPs with a quasi-spherical morphology based on YVO4 and GdVO4 have been produced after doping with europium and bismuth by precipitation at 120 °C in a mixture of ethylene glycol and water (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 by volume) and appropriate rare earth precursors. The particle size could be tuned by modifying the amount of added PAA. The NPs can efficiently be excited in the near-ultraviolet and visible range, with an excitation maximum up to 342 nm, and emit the characteristic red light of europium(III), whose intensity is higher for the GdVO4-based NPs than for the YVO4-based ones. PAA-functionalized particles are stable in water at pH 7, MES buffer at pH 6.5, and PBS at pH 7.4, and show a quite acceptable colloidal stability in cell medium supplemented with protein serum. The bare particles can also be functionalized by using a LbL approach with PAH and PAA, showing, however, less colloidal stability. Both YVO4- and GdVO4-based NPs show low cell toxicity, and can be used for biomedical applications, such as optical biolabels for imaging HeLa cells, and as biosensors, after an appropriate conjugation with analyte-sensitive dyes. Cell cytometry studies show that the uptake of YVO4-based NPs by HeLa cells is faster than for GdVO4-based ones. The influence of the particle size has also been evaluated, showing that smaller YVO4-based NPs (51 nm) are taken up faster than their bigger counterparts (93 nm). Both types of luminescent NPs are dissolved at acidic pHs, i.e. pH 3.5, which offers a way for the NP excretion.
:30 by volume) and appropriate rare earth precursors. The particle size could be tuned by modifying the amount of added PAA. The NPs can efficiently be excited in the near-ultraviolet and visible range, with an excitation maximum up to 342 nm, and emit the characteristic red light of europium(III), whose intensity is higher for the GdVO4-based NPs than for the YVO4-based ones. PAA-functionalized particles are stable in water at pH 7, MES buffer at pH 6.5, and PBS at pH 7.4, and show a quite acceptable colloidal stability in cell medium supplemented with protein serum. The bare particles can also be functionalized by using a LbL approach with PAH and PAA, showing, however, less colloidal stability. Both YVO4- and GdVO4-based NPs show low cell toxicity, and can be used for biomedical applications, such as optical biolabels for imaging HeLa cells, and as biosensors, after an appropriate conjugation with analyte-sensitive dyes. Cell cytometry studies show that the uptake of YVO4-based NPs by HeLa cells is faster than for GdVO4-based ones. The influence of the particle size has also been evaluated, showing that smaller YVO4-based NPs (51 nm) are taken up faster than their bigger counterparts (93 nm). Both types of luminescent NPs are dissolved at acidic pHs, i.e. pH 3.5, which offers a way for the NP excretion.
4. Experimental section
4.1 Materials
Yttrium(III) acetylacetonate hydrate (Y(C5H7O2)3·xH2O Aldrich, 99.95%, #438790), gadolinium(III) nitrate hexahydrate (Gd(NO3)3·6H2O, Aldrich, 99.99%, #451134) and europium(III) nitrate pentahydrate (Eu(NO3)3·5H2O, Aldrich, 99.9%, #207918) were selected as Ln precursors. Bismuth(III) nitrate pentahydrate (Bi(NO3)3·5H2O, Aldrich, 99.999%, #254150), and sodium orthovanadate (Na3VO4, Aldrich, 99.98%, #450243) were used as bismuth and vanadium sources, respectively. A mixture of ethylene glycol (EG, Fluka, >99.5%, #03750)–water was used as a solvent. Deionized water (ddH2O, MilliQ® Academic, Millipore, Billerica, USA) with a resistance greater than 18.2 MΩ cm−1 was used for all experiments. For functionalization, polyacrylic acid (PAA, average Mw ∼ 1800 Da, Aldrich, #323667), poly(allylamine hydrochloride) (PAH, average Mw ∼ 17500 Da, Aldrich, #283215), sodium chloride (NaCl, Roth, >99.9%, #HN00.2), Nile Blue A perchlorate (NB, Aldrich, 95%, #370088), and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC, Sigma-Aldrich, >98.0%, #03450) were employed. Different buffer media of biological interest were prepared from 2-(N-morpholino)ethanesulfonic acid hydrate (MES, Sigma, >99.5%, #M8250), Dulbecco's phosphate buffered saline without Ca2+ and Mg2+ (PBS, Biochrom, #L1825), fetal bovine serum superior (FBS, Biochrom, #S0615), Dulbecco's modified Eagle's medium–high glucose (DMEM, Sigma, #D1145), and Eagle's minimum essential medium (MEM, Sigma, #M5650). Hanks’ balanced salt solution (HBSS, Sigma-Aldrich, #H8264), paraformaldehyde solution (8%, Electron Microscopy Sciences, #157-8-100), WGA (wheat germ agglutinin)-Alexa Fluor 488 (WCA 488, Oregon green, Invitrogen, #W6748), Hoechst reagent (Hoeschst 33342, trihydrochloride, trihydrate, Thermo Fisher, #H1399), lysosomal-associated membrane protein 1 (LAMP 1, mouse anti-human IgG1; the Developmental Studies Hybridoma Bank, University of Iowa, Department of Biology, USA, #H4A3, Supernatant), Dylight 649 (donkey anti-mouse IgG (H + L), secondary antibody, Jackson ImmunoResearch Laboratories, #715-605-150), glycine (Roth, #3908.1), saponin (Sigma-Aldrich, #S7900), bovine serum albumin (BSA, Jackson ImmunoResearch Laboratories, #001-000-161), trypsin/EDTA 0.05% solution (Gibco, Germany, #25300-054), and 4′,6-diamidino-2-phenylindole (DAPI, ThermoFisher, #D3571) were used for the fixation, staining and imaging of the cells, and for flow cytometry studies. All chemicals were used as received.
4.2 One pot synthesis and PAA functionalization of the Eu- and Bi-doped REVO4 NPs
Y(acac)3 (i.e. (Y(C5H7O2)3·xH2O), Eu(NO3)3·5H2O, and Bi(NO3)3·5H2O were dissolved in EG in three different vials. To facilitate the dissolution of the reagents in EG, the solutions were mildly heated (∼80 °C) under magnetic stirring. At the same time, Na3VO4 and PAA were dissolved in a mixture of water and EG. After cooling down to room temperature, appropriate amounts of the precursor solutions were mixed while continuing the magnetic stirring. The Eu/(Eu + Bi + Y) molar ratio was fixed to 8% in all the cases, while the Bi/(Eu + Bi + Y) molar ratio was varied from 0 to 20%. In the final solutions, the total [Y + Bi + Eu], and Na3VO4 concentrations were 0.02 M, and 0.1 M, respectively, while the PAA concentration was varied from 2 to 8 mg mL−1. The final EG:H2O volumetric ratio was 3.5:1.5. The so prepared solutions (final volume = 5 mL) were then aged for 5 h in tightly closed test tubes using an oven preheated at 120 °C. After aging, the resulting dispersions containing the precipitated NPs were cooled down to room temperature, centrifuged to remove the supernatants and washed twice with ethanol and once with distilled water (i.e. adding around 10 mL of the corresponding liquid, centrifuge and remove supernatants). Samples without PAA were also synthesized for LbL functionalization. Most of the NPs were dispersed in water, but some portions were dried in order to carry out further characterization. Eu- and Bi-doped GdVO4 NPs were synthesized following the same protocol but using Gd(NO3)3·6H2O as the Gd precursor. For these samples, the final PAA concentration was 2 mg mL−1. A summary of the synthesized samples is shown in Tables 1 and S1.†
4.3 Layer-by-Layer functionalization with PAH and PAA
Different polyelectrolyte layers were deposited onto the surface of the non PAA-functionalized 8% Eu- and 20% Bi-doped REVO4 NPs. The following positive and negative polyelectrolytes were used: poly(allylamine hydrochloride) (PAH, average Mw ∼ 17500 Da), and polyacrylic acid (PAA, average Mw ∼ 1800 Da). The first positive PAH layer was deposited as follows: 500 μL of an aqueous PAH solution (10 mg mL−1, 0.05 M in NaCl, pH 6.5) were added to 150 μL of a water dispersion containing 0.5 mg of the NPs. The resulting dispersion was sonicated in cold water for 5 minutes and then shaken for 15 minutes. The so functionalized particles were washed three times with MilliQ water by centrifugation. For the deposition of the second layer, 500 μL of an aqueous PAA solution (10 mg mL−1, 0.05 M in NaCl, pH 6.5) were added, followed by 5 min of sonication and 15 minutes of shaking. Extra polyelectrolyte layers of alternate charge were deposited onto the NPs surface by following the same methodology. The final architecture was NPs@PAH@PAA@PAH.4.4 Characterization
The particle morphology was examined by transmission electron microscopy (TEM, JEOL Model JEM 3010) operating at an acceleration voltage of 300 keV. High-angle annular dark-field imaging (HAADF) was carried out in a JEOL JEM 2200FS. Particle size distributions were obtained by counting about 100 particles from the TEM micrographs as recorded of particles in the dried state. By dynamic light scattering (DLS, Malvern Zetasizer Nano-ZS90) the size distribution of the NP suspensions in water (0.5 mg mL−1) at pH 7 was recorded. The chemical composition of the NPs was studied by energy dispersive X-ray analysis (EDX) in a Bruker XFlash 5060 detector attached to the JEOL JEM 2200FS microscope. Bismuth, europium, yttrium, gadolinium, and vanadium contents were assessed by induced coupled plasma atomic emission spectroscopy (ICP-AES) using an ICP Horiba Jobin Yvon Ultima 2 spectrometer, for which the NPs were dissolved in concentrated HCl. The infrared spectra of the nanophosphors diluted in KBr pellets were recorded in a Jasco FT/IR-6200 Fourier transform infrared (FTIR) spectrophotometer. X-ray diffraction (XRD) studies were carried out using a Panalytical, X'Pert Pro diffractometer equipped with an X-Celerator detector. The crystallite size for several crystallographic directions (planes (101), (200), (112), (200), and (312)) of the tetragonal REVO4 structures was determined by using the Scherrer formula. A value of 0.9 for the K shape factor was employed. Zeta (ζ) potential measurements of NP suspensions in water (0.5 mg mL−1) at pH 7 were carried out in a Malvern Zetasizer Nano-ZS90 apparatus. The colloidal stability of the NP suspensions in different media (water at pH 7, 50 mM MES buffer at pH 6.5, PBS at pH 7.4, serum-supplemented (10% vol. fetal bovine serum) and serum free cell culture media) was monitored by analyzing the evolution of the hydrodynamic particle diameter with aging time, as measured by DLS. For these analyses the NP concentration was set to 0.5 mg mL−1. Each measurement was repeated three times. The fluorescence emission and excitation spectra of the NPs in water suspensions (0.05 mg mL−1) were measured on a PTI QuantaMaster™ spectrofluorometer (Photon Technology International) equipped with a 75 W xenon short arc lamp and the model 814 PTM detection system. Felix 32 software was used to collect and process the fluorescence data. The emission and excitation slit widths were both 2 nm. All optical measurements were made at room temperature, using a micro quartz cuvette of 10 mm light path. The photostability of the NPs was evaluated by analyzing the emission of immobilized NPs under continuous UV irradiation using a wide-field fluorescence microscope (Axiovert 200M, Zeiss, Germany).4.5 Cell culture
Human cervical cancer cells (HeLa) were seeded and grown in Eagle′s minimal essential medium supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine and 1% penicillin/streptomycin. The cells were incubated at 37 °C in an atmosphere of 95% air and 5% CO2.4.6 Cellular uptake and imaging
HeLa cells were seeded in 8 well μ-ibidi plates (Ibidi #80826, 1 cm2 per well) at a density of 15000 cells per well. Each well was always filled with 300 μL of cell medium. After incubation for 24 h, their medium was replaced with fresh growth media containing the luminescent particles at a concentration of 25 μg mL−1. After incubation for 24 h (37 °C and 5% CO2), the samples were washed with HBSS (Hanks’ Balanced Salt Solution), fixed with fixation solution (4% paraformaldehyde in PBS) for 15 minutes at room temperature, and washed three times with HBSS. Cell and nuclear membranes were stained with WGA (wheat germ agglutinin) conjugated with Alexa Fluor 488 (20 μg mL−1), and Hoechst reagent (5 μg mL−1) for 20 min at room temperature, respectively. In order to prove the intracellular location of the NPs, LAMP 1 (2 μg mL−1) and Dylight 649 (4 μg mL−1) were used as primary and secondary antibodies for immunostaining the lysosomal membranes. Samples were permeabilized with a permeabilization solution (5 mg glycine and 0.5 mg saponin/1 mL of PBS) for 5 minutes at room temperature, and then incubated at 37 °C with a blocking solution (20 mg BSA/1 mL of permeabilization solution) for 30 minutes. Cells were then incubated at 37 °C with a primary antibody (in blocking solution) for one hour, washed three times with the blocking solution and again incubated at 37 °C for one hour with a secondary antibody (in PBS; phosphate-buffered saline). Samples were washed three times with PBS before imaging. Imaging was performed with a wide-field fluorescence microscope (Axiovert 200M, Zeiss, Germany excitation filter for the NP visualization = 340 ± 24 nm, emission filter Long pass 600 nm).
For live imaging by using a visible laser radiation as the excitation source, HeLa cells were also seeded in 8 well μ-ibidi plates at a density of 10000 cells per well, and were incubated with the luminescent NPs at a concentration of 10 μg mL−1 for 24 h. Cells (neither fixed nor immunostained) were washed 3 times and observed with a confocal laser scanning microscope (CLSM Meta 510, Zeiss, excitation diode laser 405 nm, emission band pass filter 604–754 nm). Further information on the microscope set up and used filters and lasers can be found in the ESI.†
4.7 Conjugation with Nile Blue
PAA-functionalized NPs doped with 8% Eu and 20% Bi were chosen for further conjugation with Nile Blue (NB) through a 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) mediated reaction. In a typical experiment, PAA-NPs (0.5 mg) and EDC (1 mL at 50 μM) were mixed and stirred for 10 min. Afterwards, NB solution (1 mL at 5 μM) was added and the mixture was incubated at room temperature overnight under gentle shaking. The mixture was further purified by centrifugation (10000 rpm, 5 min) in order to separate the excess of reagents. The supernatant containing the not linked NB was discarded and the precipitate (functionalized NPs) was washed by adding 1 mL of water and stirring 10 min. Then, the NPs were again centrifuged (10000 rpm, 5 min) and the same washing procedure was repeated three times. In the last washing, the supernatant had no colour, indicating that the free NB had been completely removed. The NB-PAA-NPs were finally dispersed in water at a final concentration of 0.5 mg mL−1. A 20-fold dilution of the NB-PAA-NP solution (0.025 mg mL−1) in the different buffer solutions was performed. Aqueous universal buffer solutions pH = 3–11 were prepared by mixing an appropriate volume of acidic and alkaline buffer components. The acidic buffer component comprised 0.04 M phosphoric acid, 0.04 M boric acid, and 0.04 M acetic acid, whereas the alkaline buffer component was 0.2 M sodium hydroxide solution. The resulting pH of the mixtures was measured using a pH meter (Crison pH Meter Basic 20). The fluorescence emission of the NB-PAA-NPs was recorded at different pH values (from 3 to 11) under two excitation wavelengths (342 and 600 nm). The ratiometric analytical signal, i.e. the fluorescence intensity at 622 nm (under 342 nm excitation) divided by the fluorescence intensity at 675 nm (under 600 nm excitation), was calculated for studying the pH response of the NPs.
4.8 Cytotoxicity assays and degradability
Cell viability assays were performed using a fluorescence-based approach employing resazurin. Resazurin is a non-toxic non-fluorescent compound, which in living cells is converted into fluorescent resorufin.68 7500 HeLa cells were seeded per well in a 96-well plate (100 μL medium per well, 0.32 cm2 per well surface area, Corning) and incubated for 24 h. After 24 h, cells were washed with phosphate buffered saline (PBS), and the medium was replaced with fresh medium containing the PAA- and LbL-functionalized luminescent NPs in varying concentrations. After incubation again for 24 h, cells were washed with PBS and 10% of fresh resazurin solution in complete growth media was added. After 3.5 h of incubation with resazurin (37 °C and 5% CO2) the samples were analysed with a fluorometer for the presence of resorufin, which is the reduced product of resazurin (excitation 560 nm, emission between 572 and 650 nm). The background emission (640–650 nm) was subtracted from each value of fluorescence emission. For evaluation the maximum fluorescence emission intensity of resorufin (580–590 nm) was used, which is assumed to correlate with the viability of the cells. Each measurement was repeated three times to obtain the mean values and the standard deviation. The normalized fluorescence intensity values were plotted against the NP concentrations. When possible, the LD50 of the NPs (defined as the lethal dose toxic for 50% of the cells under the given experimental conditions) was calculated from a fit with a sigmoidal curve (logistic dose response fit). Control experiments were performed with cells that were not incubated with NPs.The dissolution of the NPs at acidic pH was studied by analyzing the evolution of the hydrodynamic particle diameter with aging time of NP suspensions (0.5 mg mL−1) in MES 50 mM at pH 3.5 at room temperature (around 20 °C). Prior to the DLS measurements, the suspensions were sonicated for 10 minutes. DLS measurements were repeated three times.
4.9 Flow cytometry study
HeLa cells were seeded into 24-well plates (TPP, Switzerland, 1.86 cm2 per well) with an amount of 40000 cells per well with 700 μL of cell medium, and left overnight in an incubator at 37 °C and 5% CO2. NPs were added to the cells (final concentration 10 μg mL−1). The cells were incubated with NPs for 2, 4, 8, and 24 h. After incubation, the cells were washed 3 times with phosphate buffered saline (PBS, Biochrom) and detached with typsin/EDTA 0.05% solution. Trypsin was then neutralized with 400 μL of cell culture medium. Detached cells were transferred to FACS tubes (Falcon, Germany) and centrifuged at 250g for 5 minutes. The supernatant was removed and 500 μL of PBS were added. In order to discard the dead cells, 1 μL (1.1 mM) of 4′,6-diamidino-2-phenylindole (DAPI) was added to each tube. Resuspended cells with internalized particles were then analyzed with a flow cytometer (LSRFortessa) equipped with a genesis CX 355 STM compact laser system (UV laser 355 nm). In addition, three control measurements (cells without NPs, cells with DAPI, and dead cells with DAPI) were performed to exclude cell duplets and dead cells. The red fluorescence from the cells with internalized particles was measured by using a band pass 610/20 nm filter. The blue (BP 450/50 nm) fluorescence was also collected to discard dead cells in the counts. 10000 events per sample were measured and every experiment was repeated 3 times. The mean red fluorescence intensity coming from the cells was calculated with FACSDiva software and plotted versus the time periods of incubation.
Acknowledgements
This work was supported by a Junta de Andalucía (Spain) Talentia Postdoc Fellowship, co-financed by the European Union's Seventh Framework Programme, grant agreement no 267226. Rocío Félix and Maximilian Schnabel are gratefully acknowledged for help with EDX and HAADF measurements, and sample preparation, respectively. CCC and SA acknowledge Alexander von Humboldt Foundation for a postdoctoral fellowship. Preliminary data corresponding to yttrium vanadate nanoparticles have been already shown in an extended abstract at the 2016 SPIE Photonic West conference.29 Parts of this work were supported by the European Commission (grant FutureNanoNeeds to WJP), and by the Spanish Ministerio de Economía y Competitividad (project MAT2014-54852-R to MO). We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).References
- S. K. Murthy, Int. J. Nanomed., 2007, 2, 129–141 CAS.
- S. Jiang, K. Y. Win, S. Liu, C. P. Teng, Y. Zheng and M.-Y. Han, Nanoscale, 2013, 5, 3127–3148 RSC.
- J. A. Barreto, W. O'Malley, M. Kubeil, B. Graham, H. Stephan and L. Spiccia, Adv. Mater., 2011, 23, H18–H40 CrossRef CAS PubMed.
- S. Ashraf, C. Carrillo-Carrion, Q. Zhang, M. G. Soliman, R. Hartmann, B. Pelaz, P. Del Pino and W. J. Parak, Curr. Opin. Pharmacol., 2014, 18, 98–103 CrossRef CAS PubMed.
- L. Y. Wang, R. X. Yan, Z. Y. Hao, L. Wang, J. H. Zeng, H. Bao, X. Wang, Q. Peng and Y. D. Li, Angew. Chem., Int. Ed., 2005, 44, 6054–6057 CrossRef CAS PubMed.
- F. Wang, W. B. Tan, Y. Zhang, X. P. Fan and M. Q. Wang, Nanotechnology, 2006, 17, R1–R13 CrossRef CAS.
- D. K. Chatterjee, M. K. Gnanasammandhan and Y. Zhang, Small, 2010, 6, 2781–2795 CrossRef CAS PubMed.
- Y. N. Xia, Nat. Mater., 2008, 7, 758–760 CrossRef CAS PubMed.
- S. Piana, M. Reyhani and J. D. Gale, Nature, 2005, 438, 70–73 CrossRef CAS PubMed.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CAS.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630–4660 CrossRef CAS PubMed.
- H.-X. Mai, Y.-W. Zhang, R. Si, Z.-G. Yan, L.-D. Sun, L.-P. You and C.-H. Yan, J. Am. Chem. Soc., 2006, 128, 6426–6436 CrossRef CAS PubMed.
- C. Feldmann, Nanoscale, 2011, 3, 1947–1948 RSC.
- X. H. Gao and S. M. Nie, Trends Biotechnol., 2003, 21, 371–373 CrossRef CAS PubMed.
- G. F. Wang, Q. Peng and Y. D. Li, Acc. Chem. Res., 2011, 44, 322–332 CrossRef CAS PubMed.
- H. Dong, S.-R. Du, X.-Y. Zheng, G.-M. Lyu, L.-D. Sun, L.-D. Li, P.-Z. Zhang, C. Zhang and C.-H. Yan, Chem. Rev., 2015, 115, 10725–10815 CrossRef CAS PubMed.
- C. Bouzigues, T. Gacoin and A. Alexandrou, ACS Nano, 2011, 5, 8488–8505 CrossRef CAS PubMed.
- J. Chang, Y. Liu, J. Li, S. Wu, W. Niu and S. Zhang, J. Mater. Chem. C, 2013, 1, 1168–1173 RSC.
- N. Núñez, J. Sabek, J. García-Sevillano, E. Cantelar, A. Escudero and M. Ocaña, Eur. J. Inorg. Chem., 2013, 2013, 1301–1309 CrossRef.
- A. Escudero, E. Moretti and M. Ocaña, CrystEngComm, 2014, 16, 3274–3283 RSC.
- N. O. Núñez, S. Rivera, D. Alcántara, J. M. De la Fuente, J. García-Sevillano and M. Ocaña, Dalton Trans., 2013, 42, 10725–10734 RSC.
- M. Abdesselem, M. Schoeffel, I. Maurin, R. Ramodiharilafy, G. Autret, O. Clément, P.-L. Tharaux, J.-P. Boilot, T. Gacoin, C. Bouzigues and A. Alexandrou, ACS Nano, 2014, 8, 11126–11137 CrossRef CAS PubMed.
- N. O. Núñez, P. Zambrano, J. García-Sevillano, E. Cantelar, S. Rivera-Fernández, J. M. De la Fuente and M. Ocaña, Eur. J. Inorg. Chem., 2015, 2015, 4546–4554 CrossRef.
- D. Chen, Y. Yu, P. Huang, H. Lin, Z. Shan, L. Zeng, A. Yang and Y. Wang, Phys. Chem. Chem. Phys., 2010, 12, 7775–7778 RSC.
- X. Y. Huang, J. X. Wang, D. C. Yu, S. Ye, Q. Y. Zhang and X. W. Sun, J. Appl. Phys., 2011, 109, 113526 CrossRef.
- W. Xu, H. Song, D. Yan, H. Zhu, Y. Wang, S. Xu, X. Bai, B. Dong and Y. Liu, J. Mater. Chem., 2011, 21, 12331–12336 RSC.
- S. Neeraj, N. Kijima and A. K. Cheetham, Solid State Commun., 2004, 131, 65–69 CrossRef CAS.
- U. Rambabu, N. R. Munirathnam, S. Chatterjee, B. S. Reddy and S. D. Han, Ceram. Int., 2013, 39, 4801–4811 CrossRef CAS.
- A. Escudero, C. Carrillo-Carrión, M. Zyuzin, R. Hartmann, S. Ashraf and W. J. Parak, Proc. SPIE, 2016, 9722, 972202 CrossRef.
- B. Yan and X. Q. Su, J. Non-Cryst. Solids, 2006, 352, 3275–3279 CrossRef CAS.
- H. D. Nguyen, S. I. Mho and I. H. Yeo, J. Lumin., 2009, 129, 1754–1758 CrossRef CAS.
- Y.-C. Chen, Y.-C. Wu, D.-Y. Wang and T.-M. Chen, J. Mater. Chem., 2012, 22, 7961–7969 RSC.
- Y. C. Chen, S. C. Huang, Y. K. Wang, Y. T. Liu, T. K. Wu and T. M. Chen, Chem. – Asian J., 2013, 8, 2652–2659 CrossRef CAS PubMed.
- S. Tanaka and S. Fujihara, Langmuir, 2011, 27, 2929–2935 CrossRef CAS PubMed.
- B. N. Mahalley, S. J. Dhoble, R. B. Pode and G. Alexander, Appl. Phys. A: Mater. Sci. Process., 2000, 70, 39–45 CrossRef CAS.
- B. N. Mahalley, R. B. Pode and P. K. Gupta, Appl. Phys. A: Solid Surf., 2000, 177, 293–302 CAS.
- I. Bertini and C. Luchinar, NMR of Paramagnetic Molecules in Biological Systems, Menlo Park, CA, 1991 Search PubMed.
- T. Pellegrino, S. Kudera, T. Liedl, A. Muñoz Javier, L. Manna and W. J. Parak, Small, 2005, 1, 48–63 CrossRef CAS PubMed.
- N. T. K. Thanh and L. A. W. Green, Nano Today, 2010, 5, 213–230 CrossRef CAS.
- S. Takeshita, T. Isobe, T. Sawayama and S. Niikura, J. Lumin., 2009, 129, 1067–1072 CrossRef CAS.
- S. Wilhelm, M. Kaiser, C. Wurth, J. Heiland, C. Carrillo-Carrion, V. Muhr, O. S. Wolfbeis, W. J. Parak, U. Resch-Genger and T. Hirsch, Nanoscale, 2015, 7, 1403–1410 RSC.
- Q. Wang, Y. Bao, X. Zhang, P. R. Coxon, U. A. Jayasooriya and Y. Chao, Adv. Healthcare Mater., 2012, 1, 189–198 CrossRef CAS PubMed.
- F. Wang, D. Banerjee, Y. S. Liu, X. Y. Chen and X. G. Liu, Analyst, 2010, 135, 1839–1854 RSC.
- Z. Wang, C. Liu, L. Chang and Z. Li, J. Mater. Chem., 2012, 22, 12186–12192 RSC.
- F. Zhang, E. Lees, F. Amin, P. Rivera_Gil, F. Yang, P. Mulvaney and W. J. Parak, Small, 2011, 7, 3113–3127 CrossRef CAS PubMed.
- G. B. Sukhorukov, E. Donath, S. Davis, H. Lichtenfeld, F. Caruso, V. I. Popov and H. Möhwald, Polym. Adv. Technol., 1998, 9, 759–767 CrossRef CAS.
- R. M. Fratila, S. G. Mitchell, P. Del Pino, V. Grazu and J. M. De la Fuente, Langmuir, 2014, 30, 15057–15071 CrossRef CAS PubMed.
- G. B. Sukhorukov, D. V. Volodkin, A. M. Gunther, A. I. Petrov, D. B. Shenoy and H. Mohwald, J. Mater. Chem., 2004, 14, 2073–2081 RSC.
- L. Wang, R. Yan, Z. Huo, L. Wang, J. Zeng, J. Bao, X. Wang, Q. Peng and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 6054–6057 CrossRef CAS PubMed.
- E. Matijevic, Chem. Mater., 1993, 5, 412–426 CrossRef CAS.
- S. Rodríguez-Liviano, F. J. Aparicio, T. C. Rojas, A. B. Hungría, L. E. Chinchilla and M. Ocaña, Cryst. Growth Des., 2012, 12, 635–645 Search PubMed.
- A. Escudero, M. E. Calvo, S. Rivera-Fernández, J. M. De la Fuente and M. Ocaña, Langmuir, 2013, 29, 1985–1994 CrossRef CAS PubMed.
- J. Wang, Y. Xu, M. Hojamberdiev, J. Peng and G. Zhu, J. Non-Cryst. Solids, 2009, 355, 903–907 CrossRef CAS.
- A. K. Levine and F. C. Palilla, Appl. Phys. Lett., 1964, 5, 118–120 CrossRef CAS.
- L. J. Kirwan, P. D. Fawell and W. van Bronswijk, Langmuir, 2003, 19, 5802–5807 CrossRef CAS.
- T. Katsumata, H. Takashima, T. Michino and Y. Nobe, Mater. Res. Bull., 1994, 29, 1247–1254 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751–767 CrossRef.
- G. Schneider and G. Decher, Langmuir, 2008, 24, 1778–1789 CrossRef CAS PubMed.
- Y. Liu, D. Tu, H. Zhu, E. Ma and X. Chen, Nanoscale, 2013, 5, 1369–1384 RSC.
- P. Rivera-Gil, D. Jimenez De Aberasturi, V. Wulf, B. Pelaz, P. Del Pino, Y. Zhao, J. M. De La Fuente, I. Ruiz De Larramendi, T. Rojo, X.-J. Liang and W. J. Parak, Acc. Chem. Res., 2013, 46, 743–749 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- K. Riwotzki and M. Haase, J. Phys. Chem. B, 1998, 102, 10129–10135 CrossRef CAS.
- D. C. Rodriguez Burbano, R. Naccache and J. A. Capobianco, in Handbook on the Physics and Chemistry of Rare Earths, ed. B. Jean-Claude and K. P. Vitalij, Elsevier, 2015, vol. 47, pp. 273–347 Search PubMed.
- F. Wang, J. A. Wang and X. G. Liu, Angew. Chem., Int. Ed., 2010, 49, 7456–7460 CrossRef CAS PubMed.
- S. Takeshita, H. Ogata, T. Isobe, T. Sawayama and S. Niikura, J. Electrochem. Soc., 2010, 157, J74–J80 CrossRef CAS.
- H. Hara, S. Takeshita, T. Isobe, T. Sawayama and S. Niikura, Mater. Sci. Eng., B, 2013, 178, 311–315 CrossRef CAS.
- L. Kastl, D. Sasse, V. Wulf, R. Hartmann, J. Mircheski, C. Ranke, S. Carregal-Romero, J. A. Martínez-López, R. Fernández-Chacón, W. J. Parak, H.-P. Elsasser and P. Rivera-Gil, ACS Nano, 2013, 7, 6605–6618 CrossRef CAS PubMed.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Eur. J. Biochem., 2000, 267, 5421–5426 CrossRef.
- L. Shen, J. Funct. Biomater., 2011, 2, 355–372 CrossRef CAS PubMed.
- M. Yu, S. H. Huang, K. J. Yu and A. M. Clyne, Int. J. Mol. Sci., 2012, 13, 5554–5570 CrossRef CAS PubMed.
- H. Herd, N. Daum, A. T. Jones, H. Huwer, H. Ghandehari and C.-M. Lehr, ACS Nano, 2013, 7, 1961–1973 CrossRef CAS PubMed.
- B. Parakhonskiy, M. V. Zyuzin, A. Yashchenok, S. Carregal-Romero, J. Rejman, H. Mohwald, W. J. Parak and A. G. Skirtach, J. Nanobiotechnol., 2015, 13, 13 CrossRef PubMed.
- I. Adjei, B. Sharma and V. Labhasetwar, in Nanomaterial, ed. D. G. Capco and Y. Chen, Springer, Netherlands, 2014, vol. 811, ch. 5, pp. 73–91 Search PubMed.
- M. Nazarenus, Q. Zhang, M. G. Soliman, P. Del Pino, B. Pelaz, S. Carregal-Romero, J. Rejman, B. Rothen-Rutishauser, M. J. D. Clift, R. Zellner, G. U. Nienhaus, J. B. Delehanty, I. L. Medintz and W. J. Parak, Beilstein J. Nanotechnol., 2014, 5, 1477–1490 CrossRef PubMed.
- H. Meng, S. Yang, Z. Li, T. Xia, J. Chen, Z. Ji, H. Zhang, X. Wang, S. Lin, C. Huang, Z. H. Zhou, J. I. Zink and A. E. Nel, ACS Nano, 2011, 5, 4434–4447 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional details of experiments and results (NP characterization, NPs’ uptake and imaging). See DOI: 10.1039/c6nr03369e |
| This journal is © The Royal Society of Chemistry 2016 |