
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electron beam controlled covalent attachment of small organic molecules to graphene†
Alexander
Markevich
a,
Simon
Kurasch
b,
Ossi
Lehtinen
b,
Oliver
Reimer
c,
Xinliang
Feng
d,
Klaus
Müllen
d,
Andrey
Turchanin
e,
Andrei N.
Khlobystov
a,
Ute
Kaiser
b and
Elena
Besley
*a
aSchool of Chemistry, University of Nottingham, NG7 2RD Nottingham, UK. E-mail: elena.besley@nottingham.ac.uk
bElectron Microscopy Department of Material Sciences, University of Ulm, 89081 Ulm, Germany
cFaculty of Physics, University of Bielefeld, 33615 Bielefeld, Germany
dMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany
eInstitute of Physical Chemistry, Friedrich Schiller University Jena, Lessingstraße 10, D-07743 Jena, Germany
First published on 4th January 2016
Abstract
The electron beam induced functionalization of graphene through the formation of covalent bonds between free radicals of polyaromatic molecules and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of pristine graphene surface has been explored using first principles calculations and high-resolution transmission electron microscopy. We show that the energetically strongest attachment of the radicals occurs along the armchair direction in graphene to carbon atoms residing in different graphene sub-lattices. The radicals tend to assume vertical position on graphene substrate irrespective of direction of the bonding and the initial configuration. The “standing up” molecules, covalently anchored to graphene, exhibit two types of oscillatory motion – bending and twisting – caused by the presence of acoustic phonons in graphene and dispersion attraction to the substrate. The theoretically derived mechanisms are confirmed by near atomic resolution imaging of individual perchlorocoronene (C24Cl12) molecules on graphene. Our results facilitate the understanding of controlled functionalization of graphene employing electron irradiation as well as mechanisms of attachment of impurities via the processing of graphene nanoelectronic devices by electron beam lithography.
C bonds of pristine graphene surface has been explored using first principles calculations and high-resolution transmission electron microscopy. We show that the energetically strongest attachment of the radicals occurs along the armchair direction in graphene to carbon atoms residing in different graphene sub-lattices. The radicals tend to assume vertical position on graphene substrate irrespective of direction of the bonding and the initial configuration. The “standing up” molecules, covalently anchored to graphene, exhibit two types of oscillatory motion – bending and twisting – caused by the presence of acoustic phonons in graphene and dispersion attraction to the substrate. The theoretically derived mechanisms are confirmed by near atomic resolution imaging of individual perchlorocoronene (C24Cl12) molecules on graphene. Our results facilitate the understanding of controlled functionalization of graphene employing electron irradiation as well as mechanisms of attachment of impurities via the processing of graphene nanoelectronic devices by electron beam lithography.
Introduction
Graphene, two-dimensional sp2-hybridizied carbon atoms arranged in a honeycomb pattern, is one of the most extensively studied materials due to its impressive range of superior mechanical, electronic, optical and transport properties. In its pristine form, graphene has been found to be a promising component in a great number of applications including touch screens, spintronic devices, fuel cells and flexible electronics.1–5 In spite of its great application potential, pristine graphene is believed to be rather inert to chemical reactions, which weakens its competitive strength in the areas of semiconductor devices and sensors. Recently, there has been a surge of interest in functionalization of graphene using chemical reactions with organic and inorganic molecules as well as chemical modification of large areas of its surface aimed at gaining control over the band gap opening of graphene. The manipulation of a band gap by chemical doping is a desirable and powerful feature for the use of graphene in functional nanoelectronic devices.6,7It is also worth noting that the purity of graphene is naturally, and very easily, affected by the environment. Organic residues, charged impurities, surface contaminants, even atmospheric oxygen, all contribute to local doping of graphene. Contaminants have an adverse effect on production and reliable synthesis of large-scale, high-quality pure graphene, which remains key to its successful applications. Adsorption of contaminants on graphene surface and unintentional doping may lead not only to its structural deformation but also to an inhomogeneous charge density and a shift in Dirac voltage.8–12 In general, contaminant molecules act as external scattering centers and modify the properties of graphene, with the covalently attached contaminants having the strongest effect.13–17 While smart choices of solvents and chemicals are required to limit contamination in graphene-based electronics, organic residues from fabrication are inherent in any graphene processing and in particular by electron beam lithography.
In this paper, we explore functionalization reactions of graphene through the formation of covalent bond(s) between free radicals and CC bonds of pristine graphene surface using first principles calculations and high-resolution transmission electron microscopy (HRTEM). Graphene displays particularly high affinity to the so-called hydrocarbon contamination18 dominated by π–π interactions to the flat surface19 thus making polycyclic aromatic hydrocarbons (PAHs) one of the most common organic contaminants of graphene. The hydrophobic properties and molecular sizes of PAHs affect their adsorption on graphene. The HRTEM images20 reveal that after loading with PAHs, the potential adsorption sites of graphene can be altered during the imaging process. However, the detailed HRTEM study of the adsorption behaviors of PAHs on graphene at a molecular level is hindered by electron irradiation damage during imaging. The imaging electron beam (e-beam) breaks readily the C–H bond and destroys individual small PAH molecules over time. The prime example is a coronene molecule, which has been observed to decompose very fast in the HRTEM at energies of 80–120 keV.21 A simple and elegant solution to increase stability of organic molecules under the e-beam has been proposed recently22 through substitution of protium isotope of hydrogen for deuterium. Isotopic substitution has no effect on the molecular structure but increases the atomic weight of the hydrogen atoms by 100%, which drastically improves the stability of organic molecules (or crystals) in the HRTEM, thus allowing imaging of individual species. Another effective approach to enhance the stability of organic molecules in HRTEM is halogenation, although substitution of hydrogen with halogens alters the original chemical properties of the molecules. It has been observed23,24 that halogenated substituents of benzene, benzoquinone and phthalocyanine, where all hydrogen atoms are replaced for much heavier atoms, exhibit greater durability against electron irradiation. A HRTEM study of perchlorocoronene, C24Cl12, suggests its high stability against the e-beam.25 The damaging mechanisms for disintegration of crystalline C24Cl12 during electron radiation have been previously studied using HRTEM, electron diffraction, and electron energy-loss spectroscopy.26
It is well known that hydrocarbons adsorbed on graphene diffuse along its surface towards the edge of the irradiated area or towards large structural defects (typically extended vacancy lines or holes) where they are immobilized by forming covalent bonds with carbon atoms at the edge of the defected area.18 An alternative example of chemical functionalization of the graphene surface is an addition of organic free radicals to sp2 carbon atoms of pristine graphene. The free radical addition method has been recently tested27 to achieve the reaction of benzoyl peroxide with graphene sheets, which was induced photochemically (using focused Ar-ion laser beam). The attachment of the phenyl groups was directly indicated by the appearance of a strong D-band at 1343 cm−1 in the Raman spectrum, which is due to the formation of sp3 carbon atoms in the basal plane of graphene by covalent attachment of phenyl groups. In this case, the covalent addition of phenyl groups to graphene led to a significant decrease in conductivity due to disruption of the aromatic system via transformation of carbon atoms from sp2 to sp3 hybridization.
In the present work we demonstrate the creation of free radicals of small organic molecules and their covalent chemical attachment to graphene during HRTEM imaging. The radicals strongly anchored to graphene are amenable to HRTEM imaging, unlike highly mobile physisorbed molecules. Since imaging of PAHs on graphene by HRTEM is not currently possible, we have deposited perchlorocoronene, C24Cl12, molecules on graphene and observed in HRTEM structural transformations induced by the e-beam in real time with near-atomic resolution. We find that perchlorocoronene radicals are preferably aligned along the armchair direction in graphene, which is consistent with our theoretical predictions for PAHs. Time series of HRTEM imaging clearly demonstrate switching between the vertical (“stand up”) and near horizontal (“lie down”) positions of perchlorocoronene radical. This can be understood by the interplay between the strain in covalent bonds formed between the radicals and graphene and the van der Waals attraction, as demonstrated by classical molecular dynamics (MD) simulations. The comparison between theoretically predicted behavior of PAH radicals anchored to graphene and stable halogenated PAH radicals on graphene observed in HRTEM reveals common dynamic behavior and chemical transformations of small radicals on graphene and enables capturing the atomistic mechanisms of the chemical bond formation.
Results and discussion
Aromatic hydrocarbons, ubiquitous impurities present during the growth and processing of graphene, undergo rapid damage under the e-beam of a HRTEM during the imaging process, which primarily starts at the C–H bond as the extremely light hydrogen atoms are rapidly removed at the energies of e-beam of 60–80 keV, which are typically used for imaging graphene in high resolution. This results in the formation of PAH free radicals. The structure and energetic properties of most commonly occurring PAH radicals, namely, benzene, C6H6, naphthalene, C10H8, anthracene, C14H10, phenanthracene, C14H10, and coronene, C24H12, adsorbed on graphene, both in chemisorption and physisorption, have been studied using density functional theory (DFT) with PBE form of the exchange–correlation functional28 and Grimme dispersion corrections.29 The counterpoise corrected30 values of the binding energy calculated as the difference in the total energy of isolated and interacting fragments are shown in Table 1 for the molecules and their radicals physisorbed on graphene in the AB stacking configuration. The experimental values of the binding energy determined with the use of the Redhead analysis31 of the thermal desorption spectra have been reported to be Eb = (0.50 ± 0.08) eV for benzene, Eb = (0.8 ± 0.1) eV for naphthalene, and Eb = (1.3 ± 0.2) eV for coronene molecule.32 The Falconer–Madix analysis33 of the same thermal desorption spectra produced the values of Eb = (0.90 ± 0.07) eV for naphthalene and Eb = (1.5 ± 0.1) eV for coronene molecule. These experimental values are slightly higher than the values reported in the present work. However, the calculated binding energies agree well with previously published theoretical data for physisorption energies, which vary in the range of 0.34 eV–0.66 eV for benzene molecule,19,34,35 0.59 eV–0.76 eV for naphthalene and 1.29 eV–1.73 eV for coronene19,35 depending on the method of treatment of van der Waals forces and basis set size used in the calculations. As shown in Table 1 the binding energies of physisorbed radicals were found to be only slightly smaller than the binding energies of the corresponding molecules suggesting that there is no significant direct interaction between the under-coordinated C atoms of a radical and graphene. This indicates that covalent bonds between the radicals and pristine graphene are not likely to form instantaneously.| Molecule | C6H6 | C10H8 | C14H10 | C14H10 | C24H12 |
|---|---|---|---|---|---|
| E b, eV | 0.38 | 0.60 | 0.81 | 0.82 | 1.33 |
| Radical | C6H5 | C10H6 | C14H6 | C14H7 | — |
|---|---|---|---|---|---|
| E b, eV | 0.36 | 0.56 | 0.74 | 0.76 | — |
Formation of covalent bonds between free radicals and graphene can be either thermodynamically driven or promoted by collisions with high-energy electrons of the imaging beam. The outcome of functionalization depends on the structure of a newly formed reactive carbon edge (armchair or zigzag) and the relative position of the radical edge with respect to the graphene substrate. For the smallest member of the studied family – benzene molecule – four possible chemisorption configurations on graphene are considered, as depicted in Fig. 1a–d, which are labeled to distinguish between different bonding sites within a hexagon in the graphene lattice. Mono-radical of benzene C6H5 (phenyl radical), shown in Fig. 1a, exhibits the binding energy of Eb = 0.99 eV and the magnetic moment of M = 1μB. In contrast, benzene bi-radical demonstrates different bonding modes on graphene, which are denoted as the 1,2-bonding (Fig. 1b), the 1,3-bonding (Fig. 1c), and the 1,4-bonding (Fig. 1d) positions. In the 1,2- and 1,4-bonding positions, the bi-radical is attached to graphene carbon atoms residing in different sub-lattices (shown in Fig. 1e in different color: yellow and blue), and it is aligned along the armchair direction in graphene. The binding energies of the 1,2- and 1,4-bonding positions are found to be very similar, namely Eb = 1.15 eV for the 1,2-bonding and Eb = 1.13 eV for the 1,4-bonding, and both configurations have zero magnetic moment. In a metastable 1,3-bonding position, the bi-radical of benzene is bound to carbon atoms in the same graphene sub-lattice, and it is aligned along the zigzag direction in graphene. The calculated binding energy for the 1,3-bonding position is only Eb = 0.29 eV, which is even smaller than a typical physisorption binding energy for benzene radicals. The magnetic moment M = 2μB of the 1,3-bonding position indicates that the lack of stability is related to the fact that the 1,3-bonding to graphene leads to a structure with open electronic shell (two topologically isolated unpaired electrons which cannot recombine). As a result the 1,2- and 1,4-bonding structures are both much more stable than the 1,3-bonding structure, even though they are more sterically strained. The lengths of the C–C bonds formed between the radicals and graphene vary between 1.54 Å for the case of 1,2-bonding of the C6H4 radical (Fig. 1b) and 1.59 Å for the C6H5 radical (Fig. 1a) indicating that in all cases chemical bonds between the radicals and graphene are formed. The table showing the calculated binding energies and magnetic moments for all studied radicals can be found in ESI (Table S1†).
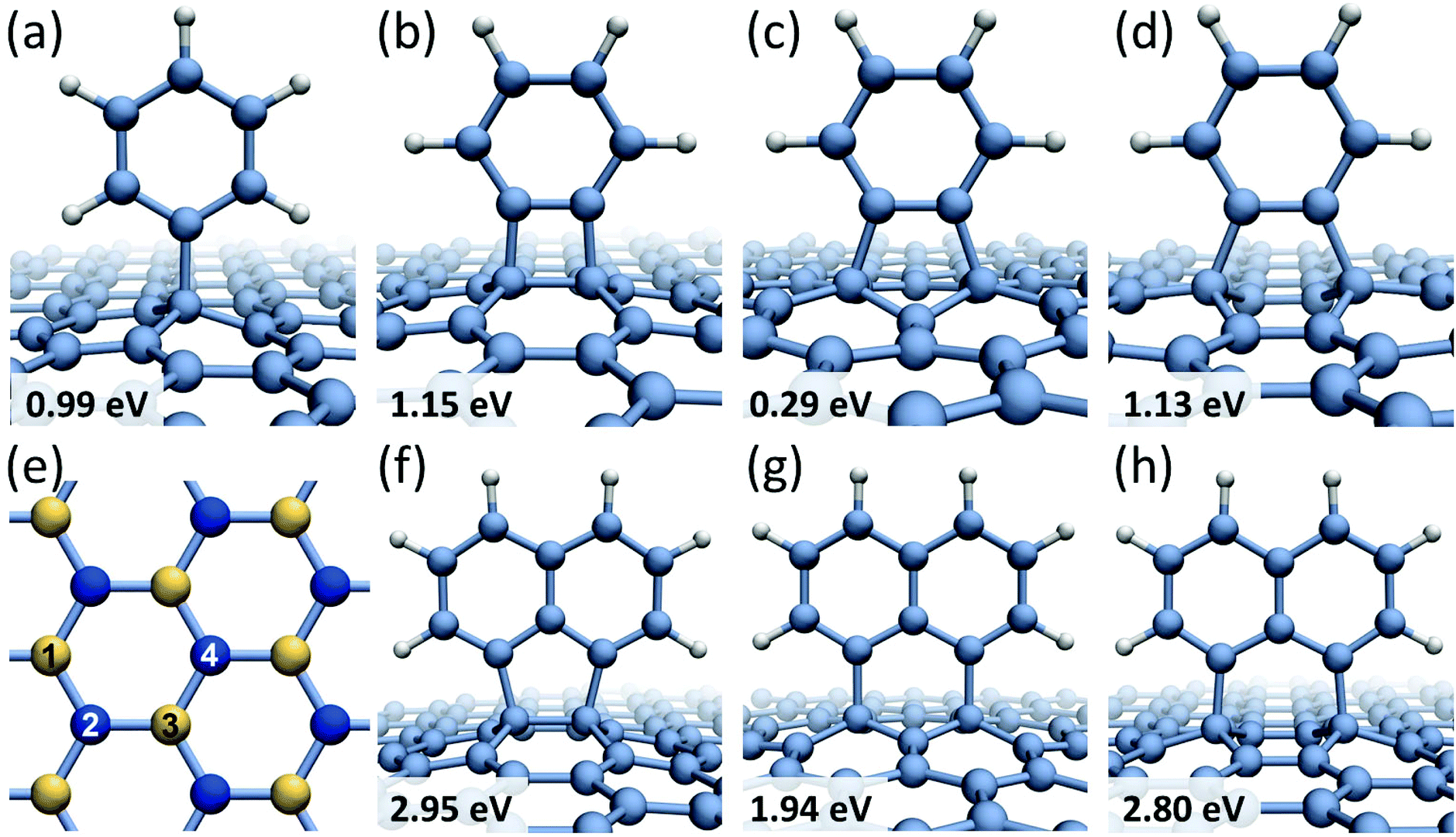 | ||
| Fig. 1 Chemisorption sites for benzene and naphthalene radicals on graphene with the calculated binding energies: (a) the bonding position of phenyl radical; (b) the 1,2-bonding; (c) the 1,3-bonding; (d) the 1,4-bonding positions of benzene bi-radical; (e) a diagram showing labeling of carbon atoms in graphene lattice; atoms corresponding to different graphene sub-lattices are shown in different color (yellow and blue); (f) the 1,2-bonding; (g) the 1,3-bonding; (h) the 1,4-bonding positions of the 1,8-C10H6 bi-radical. | ||
Similar chemisorption behavior has been predicted for the 1,8-C10H6 bi-radical of naphthalene molecule. When the 1,8-C10H6 radical is bound to graphene carbon atoms in different sub-lattices (shown in Fig. 1f and h), the corresponding binding energies are very similar: Eb = 2.95 eV for the 1,2-bonding position and Eb = 2.80 eV for the 1,4-bonding position. In the 1,3-bonding position (Fig. 1g), the bi-radical 1,8-C10H6 is bound to carbon atoms in the same graphene sub-lattice showing a significantly lower binding energy of Eb = 1.94 eV. A general trend has been observed where benzene and naphthalene bi-radicals chemisorbed on graphene along the armchair direction have the total magnetic moment of M = 0, while in the case of chemisorption along the zigzag direction M = 2μB. The same conclusions have been previously reached for chemisorption of atomic hydrogen on graphene layer36,37 showing that if a pair of hydrogen atoms are bound to different graphene sub-lattices the total magnetic moment of the system is zero. However, if all hydrogen atoms are bound to the same graphene sub-lattice the magnetic moment of the system is equal to the number of covalent bonds formed.
Larger molecules form more complicated chemisorption patterns on graphene but they follow similar structural and energetic trends. For example, anthracene tri-radical, 1,8,9-C14H7, tends to align along the 1,4-bonding armchair direction in graphene (Fig. 2a) with the binding energy of Eb = 4.46 eV, which is about 1.5 eV more favorable than chemisorption along the 1,3-bonding zigzag direction (Fig. 2b). Fig. 2c and d show phenanthrene tetra-radical, 3,4,5,6-C14H6, aligned along the 1,2- and 1,4-bonding armchair direction in graphene. In the alignment along the 1,4-bonding armchair direction a significant deformation of graphene substrate layer occurs, however, this configuration remains energetically more favorable than the 1,2-bonding case. Regardless of the radical structure, all chemisorbed molecules are preferably bound to graphene carbon atoms residing in different sub-lattices and aligned along the armchair direction in graphene. This result agrees with a general conclusion38 that chemisorption of atoms as well as functional groups on two different graphene sub-lattices is energetically more favorable than chemisorption on the same sub-lattice.
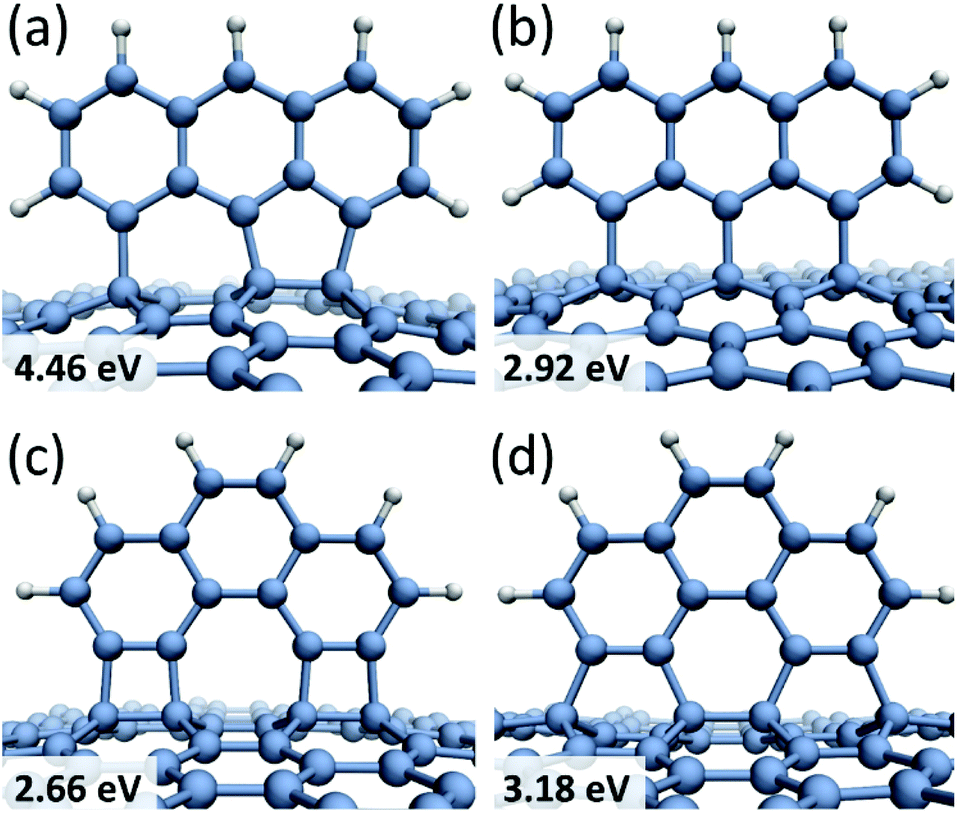 | ||
| Fig. 2 Chemisorption sites for anthracene and phenanthrene radicals on graphene with the calculated binding energies: 1,8,9-C14H7 tri-radical is aligned along (a) the 1,4-bonding armchair; (b) the 1,3-bonding zigzag direction; the 3,4,5,6-C14H6, C14H6 tetra-radical is aligned along (c) the 1,2-bonding armchair; (d) the 1,4-bonding armchair direction. | ||
The dynamical behaviour of chemisorbed PAH radicals on graphene substrate has been studied using classical molecular dynamics with the AIREBO39 potential. The C24H10 radical of coronene molecule has been modelled in the 1,2- and 1,4-bonding configurations. Two different initial geometries corresponding to the chemisorbed molecule either lying or standing on graphene substrate have been considered. The size of the considered molecule implies that the dispersion attraction to the substrate will make a significant contribution to the total binding energy. The physisorption energies calculated with the AIREBO potential were found to be Eb = 0.42 eV for C6H6, Eb = 1.41 eV for C24H12 and Eb = 2.97 eV for C54H18, in a good agreement with theoretical (DFT) and experimental results (see Table 1 and the accompanying text) thus suggesting that the AIREBO potential provides a realistic description of dispersion interactions. The MD simulations have shown that the C24H10 radical tends to assume vertical position on the graphene substrate irrespective of bonding direction and initial configuration. The “standing up” molecule exhibits two types of oscillating motion – bending (Fig. 3a) and twisting (Fig. 3b) – caused by the presence of acoustic phonons in graphene.
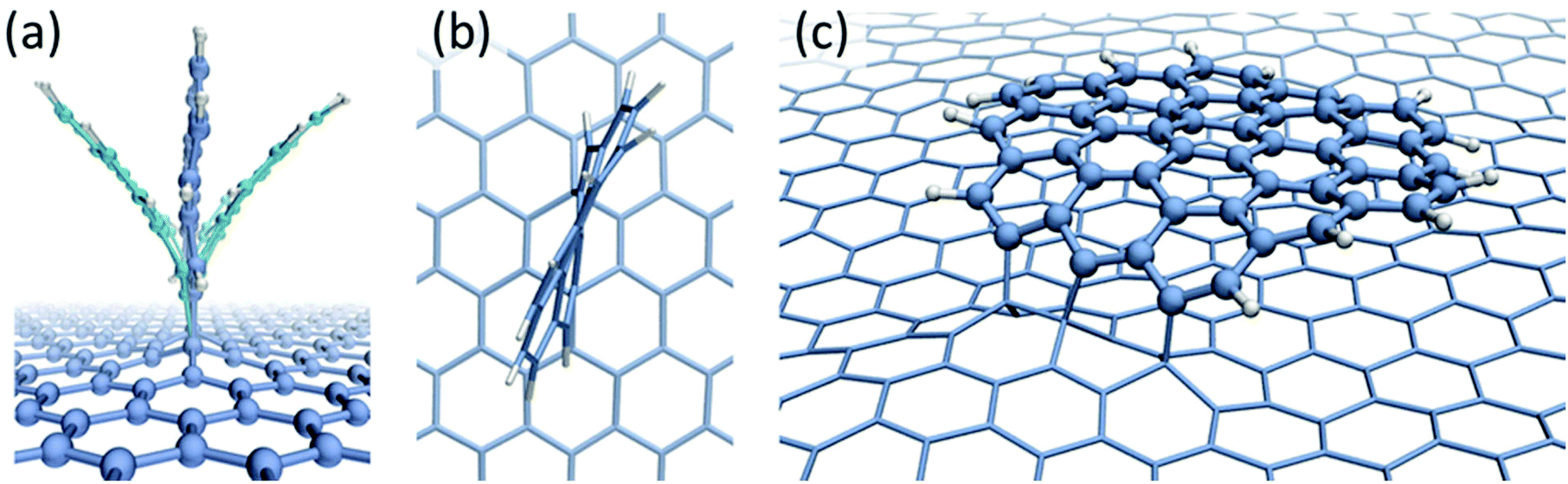 | ||
| Fig. 3 Snapshots of the classical MD simulations, using AIREBO potential, showing the bending oscillatory motion of a C24H10 radical chemisorbed on graphene (a) and twisting oscillations (b); (c) a snapshot taken after 5 ns of a larger C54H15 radical chemisorbed on graphene shows that, although three covalent bonds are formed with graphene, the dispersion attraction to graphene is higher than the strain energy caused by the bending of the sp2 carbon bonds, which causes the molecule to lie flat on graphene. | ||
A very different behaviour has been observed for the larger C54H15 radical in the 1,4-bonding configuration. If the chemisorbed radical is initially lying on the graphene surface it remains in the same configuration for the whole time of the MD simulation (Fig. 3c). In this case, the binding energy due to the dispersion interactions between the radical and graphene is higher than the strain energy caused by the bending of sp2 carbon bonds. However, if the initial configuration is a standing radical it does not lie down during simulation and the oscillations of the molecule are less pronounced than in the case of the C24H10 radical. Stabilization of the radical in this configuration can be explained by the fact that dispersion interactions are significantly reduced compared to the case of the lying molecule and contribute only 0.4 eV to the binding energy. Formation of three covalent bonds also makes the radical more stable with regards to the oscillations compared to the chemisorbed C24H10 radical in which only two covalent bonds are formed.
It is not possible to observe directly the dynamic behavior and chemical transformations of standard PAH molecules on graphene due to the rapid onset of knock-on damage caused by the fast electrons of the e-beam during HRTEM imaging.22 The structure of PAH molecule would need to be modified to prolong its lifetime under the e-beam exposure and to slow down the dynamics to the timescale of HRTEM imaging (e.g. few seconds). To achieve this, we have substituted all hydrogen atoms in coronene with chlorine atoms to form a perchlorocoronene molecule (C24Cl12) that exhibits a greater stability against electron irradiation. C24Cl12 molecules were then deposited onto graphene sheets suspended on HRTEM grids via physical vapor deposition at ultra-high vacuum conditions (see ESI† for details). The X-ray photoelectron spectroscopy (XPS) characterization of samples prepared at similar conditions on highly oriented pyrolytic graphite substrates demonstrates the adsorption of C24Cl12 molecules by appearance of the characteristic Cl 2s and Cl 2p3/2,1/2 peaks (see ESI†). The estimated coverage corresponds to ∼1% of a monolayer. If the mechanism of chemical bond formation to graphene is similar for small hydrogenated and halogenated aromatic molecules, it is expected that once two or more chemical bonds are formed between graphene and radicals (produced from C24Cl12 damaged by the e-beam), these molecules can also rotate into a vertical position and align along the preferential armchair direction, as theoretically predicted for PAHs.
The higher stability of C24Cl12 molecule in the e-beam enabled experimental investigation of the predicted mechanisms by aberration-corrected HRTEM imaging. The heavier chlorine atoms at the molecular edge produce a stronger contrast compared to lighter C atoms in the center, so the molecules are expected to have a ring-like appearance in HRTEM images as predicted by image simulation shown in Fig. 4b. Areas of graphene as large as tens to hundreds of nm in diameter were observed, which were free from contamination, similar to the case of imaging graphene with no deposition treatment but after pre-imaging cleaning.40
 | ||
| Fig. 4 C24Cl12 molecules deposited on graphene as observed by HRTEM: (a) schematic representation of the atomic structure of a C24Cl12 molecule; (b) a simulated TEM image of a C24Cl12 molecule on graphene; (c) an experimental high-magnification image of a C24Cl12 molecule on graphene (individual chlorine atoms cannot be resolved due to the high mobility of molecules on graphene); (d) overview of the sample, showing clean contamination-free areas of graphene with deposited C24Cl12 molecules observed as circles of dark contrast marked by the arrows; (e) series of HRTEM images of a single C24Cl12 molecule on graphene. | ||
Additionally, a large number of circles of dark contrast were observed on the otherwise clean area of graphene, as marked by the arrows in Fig. 4d. At higher magnification most of these features appeared to be ring-like structures of about 1 nm in diameter as shown in Fig. 4c and e. The shape and size of the observed structures and experimental conditions of the samples preparation indicate that the rings of dark contrast correspond to the 2D projection of the deposited C24Cl12 molecules. The molecules undergo a constant motion on the graphene surface (Fig. 4e) caused by thermal effects and electron irradiation so that the individual chlorine atoms cannot be resolved.
It has been observed that many of the adsorbed molecules occasionally undergo a reversible transformation between circular and linear projections. An example of such transformation is shown as a time series of HRTEM images in Fig. 5a. The time between the molecular transformations varies from few to hundred seconds at the dose rates used in experiment that range from 2 × 106 e nm−2 s−1 to 13 × 106 e nm−2 s−1. Often, just before the transformation to the linear projection, the molecule exhibits a crescent-like shape with diminishing contrast at one edge indicating the loss of Cl atoms and formation of a molecular radical. We suggest that the e-beam damages the initial structure of the molecule and promotes formation of covalent bonds between the radical and the graphene substrate, which induces the rotation of the radical by 90° with respect to the graphene plane, as theoretically predicted for PAHs. In such configuration the projection of the radical has nearly a linear shape. The reversible switching between two configurations suggests that under the e-beam irradiation conditions both formation and breaking of covalent bonds between the molecular radicals and graphene occurs.
 | ||
| Fig. 5 Time series of HRTEM images showing (a.1–a.10) a reversible transformation between circular and linear projections of the C24Cl12 molecule on graphene; (b.1–b.5) series shows liner projection of the standing up molecule switching its orientation to match different armchair directions of graphene. The dashed lines in (a) and (b) denote the armchair direction of graphene nearest to the C24Cl12 orientation. | ||
The molecules, when covalently attached to graphene, show remarkably consistent behaviour in terms of their orientation relative to the substrate. In full agreement with the computational predictions (both for the dynamics and stability), the molecules align along the armchair direction of graphene with no observed exceptions in our experiment. The sequences of images in Fig. 5 have the nearest armchair direction of graphene marked by the dashed lines whenever the molecule is in the ‘stand-up’ position. We note that the structure of the reactive edge and the binding configuration of the radicals on graphene cannot be determined from the TEM images. However, the fact “standing up” radicals are strictly aligned along the armchair direction of graphene and are stable in this configuration for extended period of time suggests that at least two covalent bonds have to be formed between the radical and graphene to promote rotation of the radical into the vertical position. Closer examination indicates that the size of the molecules tend to get smaller during imaging indicating gradual fragmentation and removal of atoms by the electron beam. Eventually the molecules are getting detached from the substrate, and either re-attach at, e.g., nearby contamination, or disappear completely.
The sharp line observed in TEM images for the “stand up” state of a radical of the C24Cl12 molecule suggests that no significant oscillations of the radical occur. This is in contrast with the strong oscillating motion predicted by MD simulations for the C24H10 radical chemisorbed on graphene (see Fig. 3). It should be noted, however, that in the case of the C24Cl12 molecule the chlorine atoms constitute about 75% of the molecular mass. Therefore, the heavy edge of the molecule will strongly suppress the oscillatory motion of the lighter C skeleton caused by the presence of phonons in graphene.
Conclusions
In this work we have presented the combined theoretical and experimental investigation of the chemisorption of small aromatic molecules on pristine graphene. The results of DFT calculations show that radicals of small polyaromatic hydrocarbons chemisorbed on graphene tend to assume vertical orientation with respect to the graphene plane, and they are preferably aligned along the armchair direction of graphene regardless of the structure of the reactive edge of the radical. The TEM images of individual perchlorocoronene (C24Cl12) molecules on graphene reveal that these molecules undergo reversible transformations between the ring- and line-like structures. It suggests that the electron beam of TEM promotes formation of free radicals of the C24Cl12 molecules and their covalent attachment to the graphene substrate. The chemisorbed radicals also assume the vertical position with respect to graphene, which in TEM images appear as a line-like structure. In agreement with the computational predictions, the molecules in their ‘line’ state are always aligned along the armchair direction of graphene. Experimental TEM observations of dynamics of PCC molecules on graphene represent a striking example of a real-time imaging of molecular transformations with near-atomic resolution. Our results facilitate the deeper understanding of controlled functionalization of graphene employing electron irradiation and effects of impurities in graphene-based devices produced by electron beam lithography.Methods
Simulations
PAHs adsorbed on graphene (both chemisorption and physisorption) have been studied using density functional theory (DFT) with PBE form of the exchange–correlation functional28 and Grimme dispersion corrections,29 as implemented in the AIMPRO code.41 The core electron levels were treated within the Hartwigsen–Goedecker–Hutter (HGH) pseudopotentials scheme.42 Kohn–Sham spin-polarised valence orbitals were represented by a set of atom centered s-, p-, and d-like Gaussian functions. Matrix elements of the Hamiltonian are determined using a plane wave expansion of the density and Kohn–Sham potential with a cut-off of 300 a.u. All structures were studied with periodic boundary conditions. Graphene substrate was represented by the hexagonal unit cell containing 128 carbon atoms. The Brillouin zone was sampled within the Monkhorst–Pack scheme43 using a (6 × 6 × 1) k-points grid. Optimization of the atomic positions was performed using a conjugate-gradient algorithm until the maximum force on any atom is less than 0.01 eV Å−1.The dynamic behaviour of the C24H10 and C54H15 radicals covalently bonded to graphene substrate has been investigated using classical MD calculations with the AIREBO potential.39 The calculations have been carried out using LAMMPS molecular dynamics simulator.44 The graphene layer has been represented with 680 carbon atoms and periodic boundary conditions have been employed. All structures have been initially optimized using the conjugate gradient energy minimization scheme and then equilibrated at temperature 300 K over 10 ns. The dynamic behavior of the radicals was studied in the subsequent NVE simulation for another 10 ns.
The image simulations were conducted with the QSTEM software45 using a spherical aberration value of 20 μm, focal spread of 6 nm and Scherzer focus. The value of the electron dose was taken to be 107 e nm−2. The experimentally measured modulation transfer function (MTF) of a CCD camera46 at an 80 kV accelerating voltage was then applied to obtain an accurate signal to noise ratio.
Experiment
Graphene was grown on 25 μm thin copper foils (99.8%, Alfa Aesar GmbH & Co KG, Karlsruhe) using low-pressure chemical vapour deposition.47,48 C24Cl12 powder was introduced in a Knudsen cell (Kentax Evaporator TCE-BSC), put into ultra-high vacuum and was baked out over night at 473 K. The temperature was then further increased to 523 K and after 30 min of thermal equilibration the compound was evaporated for 30 minutes onto graphene. A detailed description of the preparation of graphene samples and C24Cl12 powder can be found in ESI.†The samples were characterized by aberration-corrected high-resolution transmission electron microscopy using an FEI Titan 80–300 microscope, equipped with an image-side spherical aberration corrector. The microscope was operated at 80 kV, and the extraction voltage of the field emission gun was set to 2 kV to reduce the energy spread of the electron beam. The spherical aberration coefficient was tuned to <20 μm and the imaging was conducted at under focus resulting in atoms appearing as dark spots in the images. The electron dose rate was (2–13) × 106 e nm−2 s−1.
Author contributions
AM and EB produced static and dynamic chemical calculations elucidating the mechanisms of the electron beam assisted attachment of molecules to graphene; SK and OL performed AC-HRTEM imaging on graphene and image analysis; OR prepared the samples of perchlorocoronene deposited on graphene and characterized them by XPS; XF and KM synthesised perchlorocoronene used for graphene samples; AT conceived experiments on the AC-HRTEM imaging of perchlorocoronene on graphene, supervised preparation and XPS characterization of the samples and contributed to interpretation of the AC-HRTEM data; UK conceived experiments on the AC-HRTEM imaging of perchlorocoronene on graphene, supervised the AC-HRTEM work and contributed to interpretation of the results; ANK carried out initial HRTEM imaging and contributed to interpretation of the results; AM, AT, UK, ANK and EB co-wrote the manuscript.Acknowledgements
EB and ANK acknowledge ERC Consolidator Grants and EPSRC for financial support. AM and EB are grateful to the High Performance Computing (HPC) Facility at the University of Nottingham for providing computational time. AT, OL, UK and XF acknowledge support via “Graphene Flagship” and DFG SPP “Graphene”. UK, SK, OL acknowledge the DFG and the Ministry of Science, Research and the Arts (MWK) of Baden-Wuerttemberg in the frame of the SALVE (Sub Angstrom Low-Voltage Electron microscopy) project. AT and OR acknowledge the Heisenberg Programme and the EMRP project “GraphOhm”.References
- A. Reina, X. Jia, J. Ho, D. Nezich, H. Son, V. Bulovic, M. S. Dresselhaus and K. Jing, Nano Lett., 2009, 9, 30–35 CrossRef CAS PubMed.
- B. R. Van Noorden, Nature, 2011, 469, 14–16 CrossRef PubMed.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I. C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS PubMed.
- S. Huh, J. Park, K. S. Kim, B. H. Hong and S. Bin Kim, ACS Nano, 2011, 5, 3639–3644 CrossRef CAS PubMed.
- A. C. Ferrari, F. Bonaccorso, V. Fal'ko, K. S. Novoselov, S. Roche, P. Boggild, S. Borini, F. H. L. Koppens, V. Palermo, N. Pugno, J. A. Garrido, R. Sordan, A. Bianco, L. Ballerini, M. Prato, E. Lidorikis, J. Kivioja, C. Marinelli, T. Ryhanen, A. Morpurgo, J. N. Coleman, V. Nicolosi, L. Colombo, A. Fert, M. Garcia-Hernandez, A. Bachtold, G. F. Schneider, F. Guinea, C. Dekker, M. Barbone, Z. Sun, C. Galiotis, A. N. Grigorenko, G. Konstantatos, A. Kis, M. Katsnelson, L. Vandersypen, A. Loiseau, V. Morandi, D. Neumaier, E. Treossi, V. Pellegrini, M. Polini, A. Tredicucci, G. M. Williams, B. Hee Hong, J.-H. Ahn, J. Min Kim, H. Zirath, B. J. van Wees, H. van der Zant, L. Occhipinti, A. Di Matteo, I. A. Kinloch, T. Seyller, E. Quesnel, X. Feng, K. Teo, N. Rupesinghe, P. Hakonen, S. R. T. Neil, Q. Tannock, T. Lofwander and J. Kinaret, Nanoscale, 2015, 7, 4598–4810 RSC.
- J. Park, W. H. Lee, S. Huh, S. H. Sim, S. Bin Kim, K. Cho, B. H. Hong and K. S. Kim, J. Phys. Chem. Lett., 2011, 2, 841–845 CrossRef CAS PubMed.
- J. Park, S. B. Jo, Y. J. Yu, Y. Kim, J. W. Yang, W. H. Lee, H. H. Kim, B. H. Hong, P. Kim, K. Cho and K. S. Kim, Adv. Mater., 2012, 24, 407–411 CrossRef CAS PubMed.
- S. Adam, E. H. Hwang, V. M. Galitski and S. Das Sarma, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18392–18397 CrossRef CAS PubMed.
- J. Martin, N. Akerman, G. Ulbricht, T. Lohmann, J. H. Smet, K. von Klitzing and A. Yacoby, Nat. Phys., 2008, 4, 144–148 CrossRef CAS.
- E.-A. Kim and A. H. Castro Neto, EPL, 2008, 84, 57007 CrossRef.
- J.-H. Chen, C. Jang, S. Adam, M. S. Fuhrer, E. D. Williams and M. Ishigami, Nat. Phys., 2008, 4, 377–381 CrossRef CAS.
- Y. Zhang, V. W. Brar, C. Girit, A. Zettl and M. F. Crommie, Nat. Phys., 2009, 5, 722–726 CrossRef CAS.
- X. Du, I. Skachko, A. Barker and E. Y. Andrei, Nat. Nanotechnol., 2008, 3, 491–495 CrossRef CAS PubMed.
- Y. Dan, Y. Lu, N. J. Kybert, Z. Luo and A. T. C. Johnson, Nano Lett., 2009, 9, 1472–1475 CrossRef PubMed.
- F. Chen, J. Xia, D. K. Ferry and N. Tao, Nano Lett., 2009, 9, 2571–2574 CrossRef CAS PubMed.
- M. Ishigami, J. H. Chen, W. G. Cullen, M. S. Fuhrer and E. D. Williams, Nano Lett., 2007, 7, 1643–1648 CrossRef CAS PubMed.
- Z. Cheng, Q. Zhou, C. Wang, Q. Li, C. Wang and Y. Fang, Nano Lett., 2011, 11, 767–771 CrossRef CAS PubMed.
- R. F. Egerton, P. Li and M. Malac, Micron, 2004, 35, 399–409 CrossRef CAS PubMed.
- O. V. Ershova, T. C. Lillestolen and E. Bichoutskaia, Phys. Chem. Chem. Phys., 2010, 12, 6483 RSC.
- J. Wang, Z. Chen and B. Chen, Environ. Sci. Technol., 2014, 48, 4817–4825 CrossRef CAS PubMed.
- B. Botka, M. E. Füstös, H. M. Tõháti, K. Németh, G. Klupp, Z. Szekrényes, D. Kocsis, M. Utczás, E. Székely, T. Váczi, G. Tarczay, R. Hackl, T. W. Chamberlain, A. N. Khlobystov and K. Kamarás, Small, 2014, 10, 1369–1378 CrossRef CAS PubMed.
- T. W. Chamberlain, J. Biskupek, S. T. Skowron, P. A. Bayliss, E. Bichoutskaia, U. Kaiser and A. N. Khlobystov, Small, 2015, 11, 622–629 CrossRef CAS PubMed.
- M. Koshino, H. Kurata and S. Isoda, Microsc. Microanal., 2007, 13, 96–107 CrossRef CAS PubMed.
- T. Kobayashi and K. Reimwe, Bull. Inst. Chem. Res., Kyoto Univ., 1975, 53, 105–116 CAS.
- W. Dong, T. Baird, J. R. Fryer, C. J. Gilmore, D. D. MacNicol, G. Bricogne, D. J. Smith, M. A. O'Keefe and S. Hövmoller, Nature, 1992, 355, 605–609 CrossRef CAS.
- M. Koshino, H. Kurata and S. Isoda, Ultramicroscopy, 2010, 110, 1465–1474 CrossRef CAS PubMed.
- H. Liu, S. Ryu, Z. Chen, M. L. Steigerwald, C. Nuckolls and L. E. Brus, J. Am. Chem. Soc., 2009, 131, 17099–17101 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396–1396 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- P. A. Redhead, Vacuum, 1962, 12, 203–211 CrossRef CAS.
- R. Zacharia, H. Ulbricht and T. Hertel, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 155406 CrossRef.
- J. L. Falconer and R. J. Madix, J. Catal., 1977, 48, 262–268 CrossRef CAS.
- K. Berland and P. Hyldgaard, Phys. Rev. B: Condens. Matter, 2013, 87, 205421 CrossRef.
- J. Björk, F. Hanke, C. A. Palma, P. Samori, M. Cecchini and M. Persson, J. Phys. Chem. Lett., 2010, 1, 3407–3412 CrossRef.
- D. W. Boukhvalov, M. I. Katsnelson and A. I. Lichtenstein, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 035427 CrossRef.
- Y. Ferro, D. Teillet-Billy, N. Rougeau, V. Sidis, S. Morisset and A. Allouche, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 085417 CrossRef.
- D. W. Boukhvalov and M. I. Katsnelson, J. Phys.: Condens. Matter, 2009, 21, 344205 CrossRef CAS PubMed.
- S. J. Stuart, A. B. Tutein and J. A. Harrison, J. Chem. Phys., 2000, 112, 6472–6486 CrossRef CAS.
- G. Algara-Siller, O. Lehtinen, A. Turchanin and U. Kaiser, Appl. Phys. Lett., 2014, 104, 153115 CrossRef.
- R. Jones and P. R. Briddon, Semicond. Semimetals, 1998, 51, 287 Search PubMed.
- C. Hartwigsen, S. Goedecker and J. Hutter, Phys. Rev. B: Condens. Matter, 1998, 58, 3641 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef.
- C. Koch, Arizona State University, 2002.
- A. Lubk, F. Röder, T. Niermann, C. Gatel, S. Joulie, F. Houdellier, C. Magén and M. J. Hÿtch, Ultramicroscopy, 2012, 115, 78–87 CrossRef PubMed.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312–1314 CrossRef CAS PubMed.
- M. Woszczyna, A. Winter, M. Grothe, A. Willunat, S. Wundrack, R. Stosch, T. Weimann, F. Ahlers and A. Turchanin, Adv. Mater., 2014, 26, 4831–4837 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A table showing the calculated binding energies and magnetic moments for all studied molecular radicals; details of samples preparation and characterization; time series of TEM images showing transformations of a C24Cl12 molecule on graphene under electron irradiation. See DOI: 10.1039/c5nr07539d |
| This journal is © The Royal Society of Chemistry 2016 |