
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metabolic engineering with systems biology tools to optimize production of prokaryotic secondary metabolites†
Hyun Uk
Kim
*ab,
Pep
Charusanti
b,
Sang Yup
Lee
ab and
Tilmann
Weber
*b
aBioInformatics Research Center, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, Republic of Korea. E-mail: ehukim@kaist.ac.kr
bThe Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Hørsholm, Denmark. E-mail: tiwe@biosustain.dtu.dk
First published on 13th April 2016
Abstract
Covering: 2012 to 2016
Metabolic engineering using systems biology tools is increasingly applied to overproduce secondary metabolites for their potential industrial production. In this Highlight, recent relevant metabolic engineering studies are analyzed with emphasis on host selection and engineering approaches for the optimal production of various prokaryotic secondary metabolites: native versus heterologous hosts (e.g., Escherichia coli) and rational versus random approaches. This comparative analysis is followed by discussions on systems biology tools deployed in optimizing the production of secondary metabolites. The potential contributions of additional systems biology tools are also discussed in the context of current challenges encountered during optimization of secondary metabolite production.
 Hyun Uk Kim | Dr Hyun Uk Kim is currently Research Fellow at Korea Advanced Institute of Science and Technology (KAIST), South Korea, and visiting Senior Researcher at the Novo Nordisk Foundation Center for Biosustainability at Technical University of Denmark (DTU). His research field lies in systems biology, biochemical and metabolic engineering, and drug targeting and discovery. His current studies are focused on development of systems approaches to produce and characterize natural products. He earned his Ph.D. at KAIST under the supervision of Prof. Sang Yup Lee. |
 Pep Charusanti | Dr Pep Charusanti has dual affiliations with the University of California, San Diego, and the Novo Nordisk Foundation Center for Biosustainability at the Technical University of Denmark. His primary research interest is natural products discovery, especially novel secondary metabolites from actinomycetes that show antibacterial properties. A secondary interest is the application of mathematical models from systems biology to different topics, for example to guide the discovery of new enzyme targets for antibiotic development. He is co-inventor on a U.S. patent and has a strong interest in entrepreneurship. |
 Sang Yup Lee | Prof. Sang Yup Lee is Distinguished Professor at KAIST, and also Scientific Director at the Novo Nordisk Foundation Center for Biosustainability, DTU. He has published more than 500 journal papers, 60 books/book chapters, and 500 patents, largely in the field of systems metabolic engineering. He has received numerous awards: recently, Order of Service Merit-Red Stripes from President of Korea and The Ho-Am Prize. He is Fellow of several academic societies (e.g., Association for the Advancement of Science), Foreign Associate of National Academy of Engineering USA, Editor-in-Chief of Biotechnology Journal, and Associate Editor and board member of numerous journals. |
 Tilmann Weber | Dr Tilmann Weber is Co-Principal Investigator of the New Bioactive Compound section at the Novo Nordisk Foundation Center for Biosustainability of the Technical University of Denmark. He is interested in integrating bioinformatics, genome mining, and systems biology approaches into Natural Products discovery and characterization, and thus bridging the in silico and in vivo world. He received his Ph.D. (supervisor Prof. Dr Wolfgang Wohlleben) and his habilitation from the Eberhard Karls University Tübingen, Germany. |
1 Introduction
Microorganisms serve as an important source of secondary metabolites that have various medicinal and industrial uses.1 According to Newman and Cragg,2 who investigated the source of new drugs within a 30 year period from 1981–2010, 69% and 75% of newly introduced anti-infective and anti-cancer drugs, respectively, were natural products or their derivatives; this indicates the importance of natural products and the huge potential for the discovery of novel drug leads.3,4 Bacteria of the family Actinomycetaceae, in particular, are prolific producers of secondary metabolites. These bacteria are the biological source of many drugs that, for example, are used in the treatment of infections (e.g., streptomycin and erythromycin), as immunosuppressants (e.g., rapamycin), and as anthelmintics (e.g., ivermectins). In addition, many other genera, such as myxobacteria, bacilli, and many marine bacteria, have the capability to produce complex bioactive secondary metabolites. Bacteria, mainly of the Streptomyces genus, appear to be the fourth largest source of FDA-approved drugs at 16% overall.5 This amount is greater than the number of drugs derived from fungi (12% overall).In most described cases, biosynthetic pathways for secondary metabolites are organized as biosynthetic gene clusters (BGCs), which means that genes encoding core biosynthetic enzymes (e.g., polyketide synthase and non-ribosomal peptide synthetase), genes encoding tailoring enzymes, genes encoding for specific precursor biosynthesis pathways, cluster-situated regulators and often genes encoding transporters or resistance factors are physically clustered on the chromosome. Thus, all required genes for the production of a secondary metabolite are encoded within such BGCs (Fig. 1).6
 | ||
| Fig. 1 Three major stages that lead to the optimized production of secondary metabolites from secondary metabolite-producing microorganism. In the context of rational engineering, each indicated component (e.g., metabolite precursors, regulations, medium design, etc.) can be engineering targets. Five colored circles indicate different systems biology tools discussed, and are positioned near each component name where most applicable. Be noted, however, that applications of the shown systems biology tools are not necessarily confined to the indicated components. The word “Signals” in the figure can be any environmental conditions (e.g., aeration, co-culturing with another microorganism and temperature) or chemical elicitors (e.g., antibiotics at sub-lethal concentration and quorum sensing-dependent signaling molecules) that can influence the expression of secondary metabolite biosynthetic gene clusters.75 | ||
Recently, the discipline of metabolic engineering has increasingly been applied to the secondary metabolite studies to help boost commercial production of target molecules.6,7 The general objective of metabolic engineering is to overproduce chemicals that are valuable to mankind from microbial or mammalian cells, and was first coined in the field of biochemical engineering.8 By its definition, this discipline attempts to systematically understand and engineer a cell's metabolic network at a systems level.9–11 Although conventional metabolic engineering takes a rational approach, a random approach such as adaptive laboratory evolution is also considered to be a part of metabolic engineering in recent years.12 Moreover, due to advances in systems and synthetic biology, a suite of high-throughput molecular and computational tools are increasingly deployed in the practice of metabolic engineering such that the field is now often called ‘systems metabolic engineering’.11 Systems metabolic engineering has been rigorously applied to platform production strains such as Escherichia coli and Saccharomyces cerevisiae for the production of various chemicals, biofuels and biopolymers.13 In this Highlight, both ‘metabolic engineering’ and ‘systems metabolic engineering’ are used interchangeably.
Traditionally, the metabolic engineering and secondary metabolite communities have focused on different goals. In conventional metabolic engineering, quantitative values such as titer, yield and productivity for a target biochemical from a production host are more heavily emphasized.11,14 These quantitative values constitute a production host's performance metrics in strain development. In contrast, the secondary metabolite studies seem to have focused more on the discovery of novel and diverse chemical structures and any possible bioactivity they might have. This focus stems partly from the unique biochemical logic of secondary metabolite BGCs, whose manipulation can give rise to diverse chemical structures as output. Manipulation of BGCs, including inactivation or exchange of domains in polyketide synthase and non-ribosomal peptide synthetase, engineering of a domain active site and tailoring enzymes, and shuffling of modules, can all lead to the production of secondary metabolites with novel structures, and has been a classical topic in the secondary metabolite community.6,15 With this background in mind, it has become important to view the production of secondary metabolites from the metabolic engineer's perspective. A motivation is that once a structurally novel molecule is sufficiently determined to have commercial value, its production titer and yield need to be enhanced in order to implement larger-scale experiments, including (pre)clinical trials, and ultimately industrial production (Fig. 1).7 Such production optimization has recently become even more important because some secondary metabolites and their precursors have been identified by the metabolic engineering community to be important sources of industrial chemicals and biofuels that have conventionally been produced from petrochemical processes.16–18
To this end, this Highlight discusses recent studies on the metabolic optimization of native producers and other heterologous hosts for enhanced production of secondary metabolites. In particular, we focus on prokaryotic secondary metabolites because of their high contribution to currently marketed drugs,5 and their potential as a source of industrial chemicals and fuels.16–18 First, we analyze recent trends in metabolic engineering conducted to enhance the production of various secondary metabolites with emphasis on host selection and different types of engineering approaches used (i.e., rational, random or combined). This analysis is based on a survey of relevant studies reported since 2012. Second, we review various systems biology tools that have been applied to microbial hosts for the enhanced production of secondary metabolites. These tools are discussed in the context of current challenges encountered during the production of secondary metabolites.
2 Recent trends in metabolic engineering for production of secondary metabolites
2.1 Different considerations for host selection when producing primary and secondary metabolites
Selection of a production host determines the suite of tools to be deployed for strain optimization, and therefore is a very important starting point of metabolic engineering.11 While variables considered for selecting microbial host to produce primary metabolites are also applicable to secondary metabolite production studies, different priorities appear to exist when optimizing the production of primary and secondary metabolites as target bioproducts (Fig. 2). Representative primary metabolites considered for bioproducts include those biosynthesized in central carbon metabolism (e.g., ethanol, lactic acid and succinic acid), amino acids and recently fatty acids. Different priorities are largely caused by stark differences in the biochemistry of primary and secondary metabolites. Primary metabolites, in particular fermentation products and their derivatives, are often produced in high titers at the scale of grams per liter, whereas secondary metabolites are secreted at much lower levels, typically at the scale of micrograms or milligrams per liter. However, production of secondary metabolites can also reach the scale of grams per liter upon metabolic engineering.19,20 For this reason, units of titer, yield and productivity for production of primary metabolites are usually defined to be ‘g L−1’, ‘g g−1’ and ‘g L−1 h−1’, respectively, but can be varied for secondary metabolites.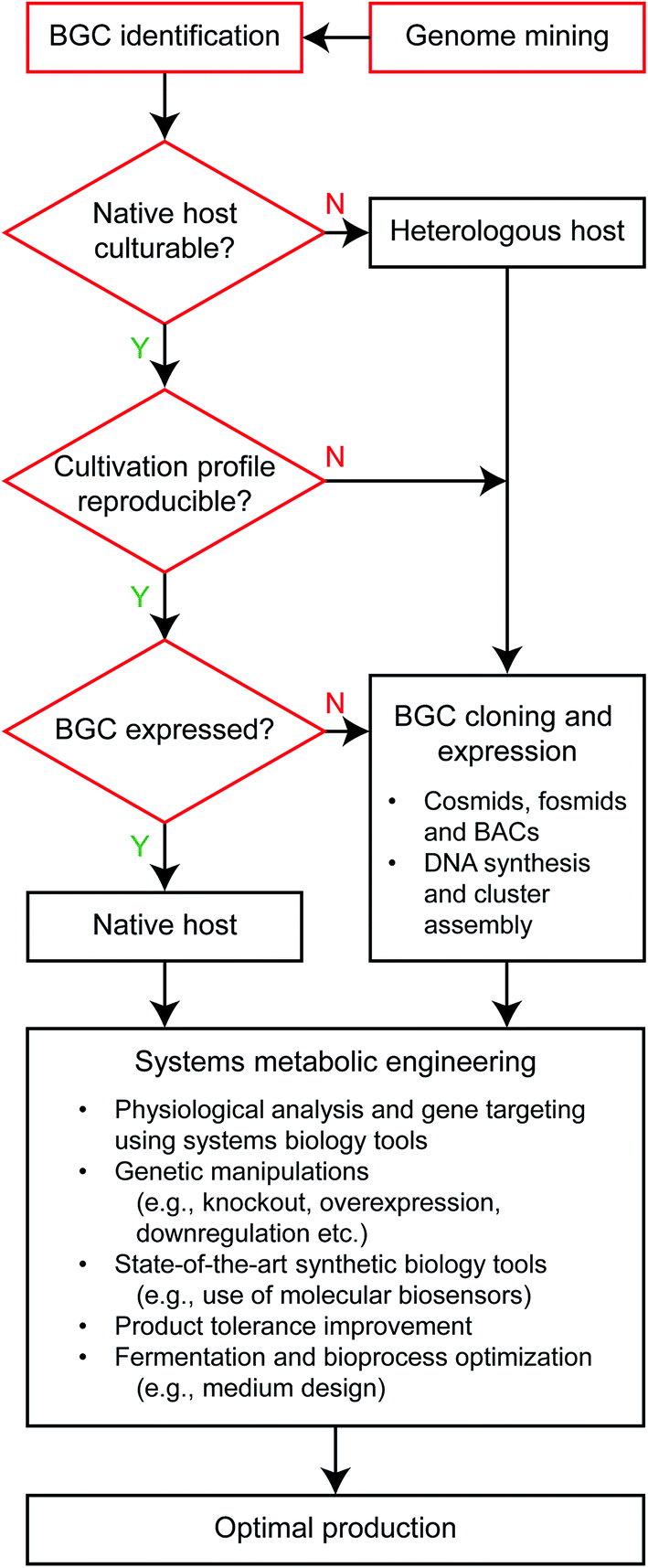 | ||
| Fig. 2 Flowchart of systems metabolic engineering considered for secondary metabolite production. Steps in red are specific to the secondary metabolite production, and can be considered altogether as examining the candidate host strain's native production capacity for a target bioproduct in the case of primary metabolite production. BAC and BGC stand for bacterial artificial chromosome and biosynthetic gene cluster, respectively. The step “Systems metabolic engineering” is also applicable to the primary metabolite production, and further details can be found elsewhere.11 | ||
A key consideration for host selection in conventional metabolic engineering (e.g., overproducing primary metabolites using model organisms) has been whether it is possible to maintain both high growth and production rates using defined minimal or industrial media, which are directly linked to the operation cost of a microbial bioprocess;21 cheaper nutrient utilization is always more favored from a bioprocess perspective. The availability of biosynthetic reactions for the production of a target biochemical is also an important criterion for host selection, but it has been somewhat overcome with state-of-the-art synthetic biology tools for model organisms.22,23 Industrially or medicinally valuable compounds, such as 1,4-butanediol and opioid compounds, have been successfully produced from engineered heterologous production hosts E. coli24 and S. cerevisiae,25 respectively, by constructing heterologous biosynthetic pathways.
Meanwhile, studies on the production of secondary metabolites using native hosts (e.g., actinomycetes) or heterologous hosts have additional considerations in addition to those already considered for the production of primary metabolites often using model organisms (Fig. 2). Culturability of a native production host can be a major issue because some secondary metabolite-producing microorganisms are unculturable, and do not grow fast enough or with sufficient reproducibility despite possession of secondary metabolite BGCs of interest.26 If a target BGC never gets expressed in a native host, a heterologous host should be considered. Doing so introduces the challenge of capturing or cloning the BGC, which can be 100 kb in length or more, and optimally expressing genes within the BGC. Advanced synthetic biological tools greatly aid this process.6,27 Finally, because general gene manipulation tools, such as knockout and overexpression of genes, have actively been applied to secondary metabolite-producing microorganisms as manifested in next section (Table S1†), they no longer appear to be a limiting factor for host selection when producing secondary metabolites. In the same line, state-of-the-art synthetic biology tools mostly developed with model organisms (e.g., E. coli) are increasingly available to secondary metabolite producers.28,29
2.2 Recent trends in host selection and engineering approaches: native versus heterologous hosts and rational versus random approaches
In order to gain insights on recent trends in host selection and engineering approaches deployed to optimize the production of secondary metabolites, we conducted a literature survey on relevant studies reported since 2012 (Table S1†). Our survey reveals that production studies using native producers outnumber those using heterologous hosts (Fig. 3A). For native producers, diverse hosts such as Saccharopolyspora spinosa, Streptomyces bingchenggensis, Streptomyces chattanoogensis and Streptomyces roseosporus, have actively been subjected to production optimization of their native secondary metabolites. These hosts are not necessarily model actinomycetal species. Meanwhile, heterologous production hosts were not confined to actinomycetes such as S. coelicolor30 and Streptomyces venezuelae31 because biologically distant and/or model organisms such as E. coli,32Bacillus subtilis33 and Myxococcus xanthus34 were also considered (Table S1†). Production performances of these heterologous hosts often appeared to be worse or at least not better than native producers, and the secondary metabolite production studies currently seem to favor the use of native producers.35 However, a potential advantage of using a model organism as a heterologous production host (e.g., E. coli) would be better access to the state-of-the-art synthetic biology approach in comparison with native secondary metabolite producers. In a recent study using E. coli for the deoxyviolacein production,36 deoxyviolacein biosynthetic pathway was divided into two modules with L-tryptophan as a key intermediate, and heterologous expression of each module was independently optimized. In this optimization procedure, a molecular biosensor was newly developed and used to detect the intracellular pool of L-tryptophan along with fluorescence-activated cell sorting. This approach led to a titer of 1.92 g L−1. | ||
| Fig. 3 Number of reported studies aimed at enhancing the production of prokaryotic secondary metabolites using (A) different types of production hosts and (B) engineering approaches (i.e., rational, random or combined). Summaries of each study are available in Table S1.† Search words in PubMed were: production[tiab] AND engineering[tiab] AND (secondary metabolites OR polyketides OR nonribosomal peptides OR lactam OR glycopeptide OR macrolide) NOT plant AND 2012:2016[dp]; (secondary metabolite[tiab] OR natural product[tiab] OR actinomycetes[tiab] OR streptomyces[tiab]) AND (design[tiab] OR engineering[tiab]) AND 2012:2016[dp]. | ||
Also, among the metabolic engineering studies examined (Table S1†), rational approaches were more frequently deployed than conventionally-used random (e.g., ribosome engineering using antibiotics at sub-lethal concentrations) and combined approaches (Fig. 3B).37,38 Examples of the rational approaches used to improve the production of secondary metabolites include the enhanced supply of intracellular precursors39,40 and the overexpression of positive regulators41 and/or removal of competing pathways leading to other byproducts.42 These rational approaches also employed systems biology tools such as statistical medium optimization43 and genome-scale metabolic modeling44,45 (see next section). Rational approaches are expected to remain dominant due to recent releases of precise gene manipulation tools specifically developed for actinomycetes, for instance CRISPR-Cas9.28,29 In one recent metabolic engineering study, Streptomyces pristinaespiralis was systematically engineered to improve its production of pristinamycin II.19 The pristinamycin II biosynthetic gene cluster was duplicated using a modified Gibson assembly method for its overexpression. Also, the combined effects of knocking out repressor genes and overexpressing activator genes were examined. Finally, macroreticular resin was added to the medium in order to facilitate separation of pristinamycin from the medium, thereby reducing feedback inhibition by pristinamycin. Final engineered strain produced 1.16 g L−1 of pristinamycin II from 5 L bioreactor, corresponding to 5.26-fold increase in titer, compared to the parental strain.
Although it is extremely difficult to predict changes in the relative frequency of using native and heterologous hosts for the optimal production of secondary metabolites in coming years, lines of evidence collected herein suggest that native producers can serve as competitive hosts. This conclusion is also supported by large fold increases in production titer (or yield) of secondary metabolites from native producers upon their engineering (Table S1†). Technical aspects of molecular technologies used for the enhanced production of secondary metabolites are extensively discussed elsewhere.6,15,35
3 Systems biology tools to optimize production of secondary metabolites
As just mentioned, many metabolic engineering efforts to boost the production of secondary metabolite are centered on engineering native producers. In this case, systems biology tools tend to be more important than synthetic biology tools initially because one needs to gain insights into the fundamental biochemistry of native producers before actually engineering them. Therefore, we next discuss systems biology tools that have been used in the study on optimization of secondary metabolite production (Table S1†). We also discuss additional systems tools that can be further considered to overcome current challenges associated with secondary metabolite production.3.1 Genome-scale metabolic models
Genome-scale metabolic modeling has become a popular rational approach to enhance the production of secondary metabolites.46,47 Genome-scale metabolic models continue to be an important tool in systems biology by predicting global metabolic flux distributions under given genetic and environmental conditions. A genome-scale metabolic model is a large-scale stoichiometric model that describes all the metabolic pathways experimentally and/or theoretically characterized through stoichiometric coefficients and mass balances of participating metabolites, and is simulated using numerical optimization.9 This modeling approach takes an assumption of pseudo-steady state, which can be best applied to simulating primary metabolism in an exponential growth phase.9 Metabolic questions that can be best addressed with genome-scale metabolic models include, but are not limited to, prediction of the most efficient pathway that leads to the maximal production yield of a target bioproduct, and optimization of precursor supply and intracellular redox balances, typically through prediction of the effects of gene knockouts and overexpressions.48,49 Due to its ease of implementation and relatively high predictive power, this modeling approach has contributed to a diverse array of applications,11,50 for example prediction of gene manipulation targets in metabolic engineering for enhanced biochemical production,21 and prediction of drug targets in microbial pathogens51 and cancers (e.g., hepatocellular carcinoma).52 Genome-scale metabolic models can be relatively easily created using the genome sequence of an organism.53,54 Several software programs have been introduced to automate a large part of the metabolic modeling procedure, which enable the rapid reconstruction of draft genome-scale metabolic models of multiple species.55,56In recent years, genome-scale metabolic models have been manually constructed for Amycolatopsis balhimycina,57S. coelicolor,45Saccharopolyspora erythraea,44S. spinosa,58Streptomyces lividans59 and Streptomyces tsukubaensis60 (Table S1†). Genome-scale metabolic models of S. erythraea and S. spinosa were used to identify the effects of supplementing amino acids in media on production yield,44,58 while those of A. balhimycina, S. coelicolor and S. tsukubaensis were used to identify gene manipulation targets to enhance target production.45,60,61 In these metabolic models, only experimentally known secondary metabolite biosynthetic pathways were considered. For example, separate biosynthetic pathways for actinorhodin, undecylprodigiosin, calcium-dependent antibiotic, ectoine, and germicidin were included in the latest version of the S. coelicolor metabolic model,45 while the S. erythraea metabolic model describes biosynthetic pathways for erythromycin, 2-methylisoborneol, rhamnosylflaviolin, and erythrochelin.44
Now that many BGCs can be effectively detected using software programs (e.g., antiSMASH53), incorporating their corresponding biosynthetic reactions into metabolic models becomes an important task. The biosynthetic reactions for several clusters have been characterized, but the majority have not. More complete information on secondary metabolite biosynthetic reactions would help to evaluate systematically the production capacity of secondary metabolite producers of interest using metabolic models. Zakrzewski et al. demonstrated a proof-of-concept study relevant to this issue by automatically generating genome-scale metabolic models of 38 actinobacteria, and predicting theoretical production capacity of each strain for 15 heterologously expressed secondary metabolites. The prediction outcomes showed that large genomic sizes do not necessarily lead to optimal production.56
Interestingly, in addition to the studies summarized in Table S1,† D'Huys et al. investigated the effects of growing S. lividans under complex media using its genome-scale metabolic model. The metabolic studies revealed that nutritionally rich media do not necessarily lead to maximal biomass formation.59 Although this study was aimed at heterologous production of proteins, it is noteworthy because actinomycetes are almost always cultivated in complex media, and elucidating the effects of complex media on the production of secondary metabolites will be an invaluable resource in the context of bioprocess optimization.
3.2 Medium design using statistical optimization
Although genome-scale metabolic modeling can partly address the problem of media optimization as mentioned above, this area often requires more thorough independent analyses. Media components heavily influence the production performance of microbial hosts because the cells use different metabolic pathways depending on the availability of different nutrients in the media. A challenge here is that the design of optimal media for the best possible production performance is often complicated by a large possible number of combinations of nutrients. For this reason, a ‘design-of-experiments (DOE)’ approach has often been applied to media optimization in bioprocess engineering for the enhanced production of various bioproducts, including secondary metabolites.62 Frequently deployed methods have been statistical optimization involving Plackett–Burman design and response surface methodology, leading to identification of key medium components along with their optimal concentrations. Recently, this statistical optimization approach has been applied to the production of daptomycin from a S. roseosporus mutant strain,63 neomycin from Streptomyces fradiae,64 nosiheptide from Streptomyces actuosus,65 and pikromycin from S. venezuelae.43 In all these studies, minerals and/or carbon sources that most affected the secondary metabolite production were selected, and their optimal concentrations were determined using the Plackett–Burman design and response surface methodology. The DOE approach will continue to play an important role in optimizing multiple bioprocess variables, including microbial cultivation media.3.3 13C-Metabolic flux analysis
A similar approach to the aforementioned genome-scale metabolic modeling is 13C-metabolic flux analysis, which also uses information on mass balance of metabolites with their stoichiometric coefficients.9 However, the two differ in that 13C-metabolic flux analysis uses 13C-labelling data from isotopic labelling experiments in addition to a stoichiometric metabolic network model in order to estimate in vivo metabolic fluxes under a given condition. Despite its precise measurement of in vivo flux values, 13C-metabolic flux analysis has not been frequently deployed to analyze metabolism of secondary metabolite-producing microorganisms in the past when compared to model organisms.46,66 Recently, Coze et al. investigated differences in metabolic flux distributions of an actinorhodin-producing S. coelicolor wild-type and its mutant, in which its native four BGCs were removed.6713C-Metabolic flux analysis revealed a few insights, including a more active pentose phosphate pathway in the mutant, and a competition for common precursors such as acetyl-CoA between primary and secondary metabolism. Although this study was not intended for production optimization, the 13C-metabolic flux analysis used in this study could be useful in analyzing the metabolic status of engineered strains towards enhanced production. Finally, use of dynamic 13C-metabolic flux analysis should be useful in analyzing metabolic status during the secondary metabolite production phase that is in non-steady state.68 Because many complex regulations take place during the secondary metabolite production, conventional 13C-metabolic flux analysis based on the pseudo-steady state assumption, in the strict sense, is not an ideal approach to analyze this specific metabolic status. Also, dynamic 13C-metabolic flux analysis can be more advantageous for the analysis of microbial metabolism in fed-batch fermentations because this fermentation mode has very dynamic cultivation conditions. Fed-batch fermentations are predominantly conducted in industry.3.4 A challenge of identifying metabolic and regulatory gene manipulation targets and further systems approaches to be considered
Identifying metabolic and/or regulatory genes responsible for the enhanced production of secondary metabolites stands as an important challenge. We expect that additional systems biology tools available in the metabolic engineering community, which have not been applied to the secondary metabolite production yet, will help to meet this challenge.First, the aforementioned genome-scale metabolic model contains information about the reactions that lead to secondary metabolite biosynthesis, but it does not contain any regulatory information. Probabilistic modeling approach is likely to be a good option to model regulation associated with secondary metabolite biosynthesis. As a relevant recent example, a regulatory network describing relationships between genes encoding transcription factors and their target metabolic genes was modeled by calculating conditional probabilities for Mycobacterium tuberculosis.69 In this modeling approach, conditional probabilities are used to describe relationship between transcription factors and their target metabolic genes; they reveal the probability that a metabolic gene gets expressed or inactivated, depending on the expression status of a transcription factor. This probabilistic information is used to calculate more accurate metabolic flux values using the genome-scale metabolic model of M. tuberculosis. A similar integrative modeling approach can be considered to identify regulatory genes that can boost the expression of metabolic genes related to the biosynthesis of a target secondary metabolite.
The above probabilistic regulatory modeling approach in turn requires a large transcriptome dataset to bestow greater reliability on the calculated conditional probabilities for metabolic genes and transcription factors. Of particular importance is that the large transcriptome (or other omics) dataset needs to be obtained from as many different environmental and/or genetic conditions as possible in order to accurately determine the conditional probabilities that describe relationships between genes. Massive transcriptome analyses have been conducted for S. coelicolor under multiple conditions in the past, for example, elucidating: nutritional stress response of an antibiotic regulator AfsS,70 a genome-wide regulatory network,71 genome-wide gene expression changes during a metabolic switch from exponential to stationary antibiotic production phase,72 and sigma factor-regulated genes in germination.73 Despite several massive transcriptome analyses conducted on actinomycetes (or heterologous model hosts), they have barely been deployed to enhance the production of secondary metabolites. Generation of massive omics data in the context of secondary metabolite production should be very important resources for optimizing the whole bioprocess.
3.5 A starting point of systems biology and metabolic engineering studies for secondary metabolites: the Secondary Metabolite Bioinformatics Portal
BGCs of secondary metabolites are a very complex system, and therefore optimization of secondary metabolite production requires relevant insights before actual metabolic engineering for production optimization begins. This step can be particularly challenging for metabolic engineers who are not familiar with the biology of actinomycetes. To this end, we recently released the Secondary Metabolite Bioinformatics Portal (SMBP) available at http://secondarymetabolites.org/, which provides a full list of databases and tools dedicated to secondary metabolite bioinformatics, along with their descriptions and URLs.54 This portal provides a concise gateway to various bioinformatic resources and tools that can facilitate metabolic engineering of actinomycetes and heterologous model hosts, including databases of secondary metabolites and BGC mining tools (e.g., antiSMASH). There are also other software tools with more generic applications, for example CRISPy-web, an application that supports the design of guideRNAs (sgRNAs) for CRISPR-Cas9 mediated genetic manipulation of microorganisms.743.6 Perspectives on the direction of secondary metabolite production studies
It should be noted that systems biology tools used in metabolic engineering are complementary to tools and strategies that have been uniquely developed and used for secondary metabolite studies. An obvious reason is that the production of secondary metabolites needs unique considerations that are distinct from those of the primary metabolite production (Fig. 2). For the optimal production and potential commercialization of secondary metabolites, following considerations can be useful. First, processes of systems metabolic engineering need to be taken into account at an early phase of the secondary metabolite production study (e.g., genome mining and host selection). This will enable decision-makings not only from a pure biochemistry perspective, but also from an engineering perspective, leading to an integral pipeline from novel secondary metabolite identification to its optimal production. Second, metabolic engineers also need to have a better understanding of the working mechanism of secondary metabolite BGCs. This will facilitate introduction of state-of-the-art tools to secondary metabolite producers, which were initially developed for model organisms. With recent efforts, more relevant achievements are awaiting to be realized.17,28,29,364 Conclusions
As the discipline of metabolic engineering has expanded into the realm of prokaryotic secondary metabolites, this Highlight aimed to review the current status of metabolic engineering for secondary metabolite production and the relevant systems biology tools used. While our analysis manifested the progress made for the optimal production of secondary metabolites, it also clearly pinpointed room for further development. In particular, state-of-the-art systems biology tools established for the metabolic engineering community, including integrative metabolic and regulatory modeling, (dynamic) 13C-metabolic flux analysis and omics data generation, have not been fully deployed in the production optimization of secondary metabolites. Upon successful implementation of such tools, more diverse secondary metabolites will be considered for industrial production and commercialization in both medicinal and chemical industries.5 Acknowledgements
This work was supported by the Novo Nordisk Foundation. H.U.K. and S.Y.L. are also supported by the Technology Development Program to Solve Climate Changes on Systems Metabolic Engineering for Biorefineries from the Ministry of Science, ICT and Future Planning (MSIP) through the National Research Foundation (NRF) of Korea (NRF-2012M1A2A2026556 and NRF-2012M1A2A2026557).6 Notes and references
- A. L. Demain, J. Ind. Microbiol. Biotechnol., 2014, 41, 185–201 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS PubMed.
- G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, 2013, 1830, 3670–3695 CrossRef CAS PubMed.
- L. Katz and R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2016, 43, 155–176 CrossRef CAS PubMed.
- E. Patridge, P. Gareiss, M. S. Kinch and D. Hoyer, Drug Discov. Today, 2016, 21, 204–207 CrossRef CAS PubMed.
- T. Weber, P. Charusanti, E. M. Musiol-Kroll, X. Jiang, Y. Tong, H. U. Kim and S. Y. Lee, Trends Biotechnol., 2015, 33, 15–26 CrossRef CAS PubMed.
- S. Y. Lee, H. U. Kim, J. H. Park, J. M. Park and T. Y. Kim, Drug Discov. Today, 2009, 14, 78–88 CrossRef CAS PubMed.
- J. E. Bailey, Science, 1991, 252, 1668–1675 CrossRef CAS PubMed.
- H. U. Kim, T. Y. Kim and S. Y. Lee, Mol. BioSyst., 2008, 4, 113–120 RSC.
- G. Stephanopoulos, Metab. Eng., 1999, 1, 1–11 CrossRef CAS PubMed.
- S. Y. Lee and H. U. Kim, Nat. Biotechnol., 2015, 33, 1061–1072 CrossRef CAS PubMed.
- M. Dragosits and D. Mattanovich, Microb. Cell Fact., 2013, 12, 64 CrossRef PubMed.
- C. Cho, S. Y. Choi, Z. W. Luo and S. Y. Lee, Biotechnol. Adv., 2015, 33, 1455–1466 CrossRef CAS PubMed.
- S. Van Dien, Curr. Opin. Biotechnol., 2013, 24, 1061–1068 CrossRef CAS PubMed.
- K. J. Weissman, Nat. Prod. Rep., 2016, 33, 203–230 RSC.
- A. Hagen, S. Poust, T. Rond, J. L. Fortman, L. Katz, C. J. Petzold and J. D. Keasling, ACS Synth. Biol., 2016, 5, 21–27 CrossRef CAS PubMed.
- R. M. Phelan, O. N. Sekurova, J. D. Keasling and S. B. Zotchev, ACS Synth. Biol., 2015, 4, 393–399 CrossRef CAS PubMed.
- S. Menendez-Bravo, S. Comba, M. Sabatini, A. Arabolaza and H. Gramajo, Metab. Eng., 2014, 24, 97–106 CrossRef CAS PubMed.
- L. Li, Y. Zhao, L. Ruan, S. Yang, M. Ge, W. Jiang and Y. Lu, Metab. Eng., 2015, 29, 12–25 CrossRef CAS PubMed.
- A. S. Eustaquio, L. P. Chang, G. L. Steele, C. J. O’Donnell and F. E. Koehn, Metab. Eng., 2016, 33, 67–75 CrossRef CAS PubMed.
- T. Y. Kim, J. M. Park, H. U. Kim, K. M. Cho and S. Y. Lee, Metab. Eng., 2015, 28, 63–73 CrossRef CAS PubMed.
- K. M. Esvelt and H. H. Wang, Mol. Syst. Biol., 2013, 9, 641 CrossRef PubMed.
- C. W. Song, J. Lee and S. Y. Lee, Biotechnol. J., 2015, 10, 56–68 CrossRef CAS PubMed.
- H. Yim, R. Haselbeck, W. Niu, C. Pujol-Baxley, A. Burgard, J. Boldt, J. Khandurina, J. D. Trawick, R. E. Osterhout, R. Stephen, J. Estadilla, S. Teisan, H. B. Schreyer, S. Andrae, T. H. Yang, S. Y. Lee, M. J. Burk and S. Van Dien, Nat. Chem. Biol., 2011, 7, 445–452 CrossRef CAS PubMed.
- S. Galanie, K. Thodey, I. J. Trenchard, M. Filsinger Interrante and C. D. Smolke, Science, 2015, 349, 1095–1100 CrossRef CAS PubMed.
- S. E. Ongley, X. Bian, Y. Zhang, R. Chau, W. H. Gerwick, R. Muller and B. A. Neilan, ACS Chem. Biol., 2013, 8, 1888–1893 CrossRef CAS PubMed.
- Y. Luo, B. Enghiad and H. Zhao, Nat. Prod. Rep., 2016, 33, 174–182 RSC.
- Y. Tong, P. Charusanti, L. Zhang, T. Weber and S. Y. Lee, ACS Synth. Biol., 2015, 4, 1020–1029 CrossRef CAS PubMed.
- R. E. Cobb, Y. Wang and H. Zhao, ACS Synth. Biol., 2015, 4, 723–728 CrossRef CAS PubMed.
- L. Huo, S. Rachid, M. Stadler, S. C. Wenzel and R. Muller, Chem. Biol., 2012, 19, 1278–1287 CrossRef CAS PubMed.
- W. S. Jung, E. Kim, Y. J. Yoo, Y. H. Ban, E. J. Kim and Y. J. Yoon, Appl. Microbiol. Biotechnol., 2014, 98, 3701–3713 CrossRef CAS PubMed.
- J. Jaitzig, J. Li, R. D. Sussmuth and P. Neubauer, ACS Synth. Biol., 2014, 3, 432–438 CrossRef CAS PubMed.
- J. Kumpfmuller, K. Methling, L. Fang, B. A. Pfeifer, M. Lalk and T. Schweder, Appl. Microbiol. Biotechnol., 2016, 100, 1209–1220 CrossRef PubMed.
- Y. Chai, S. Shan, K. J. Weissman, S. Hu, Y. Zhang and R. Muller, Chem. Biol., 2012, 19, 361–371 CrossRef CAS PubMed.
- S. E. Ongley, X. Bian, B. A. Neilan and R. Muller, Nat. Prod. Rep., 2013, 30, 1121–1138 RSC.
- M. Fang, T. Wang, C. Zhang, J. Bai, X. Zheng, X. Zhao, C. Lou and X. H. Xing, Metab. Eng., 2016, 33, 41–51 CrossRef CAS PubMed.
- L. Li, T. Ma, Q. Liu, Y. Huang, C. Hu and G. Liao, BioMed Res. Int., 2013, 2013, 479742 Search PubMed.
- J. Zhao, Y. Li, C. Zhang, Z. Yao, L. Zhang, X. Bie, F. Lu and Z. Lu, J. Ind. Microbiol. Biotechnol., 2012, 39, 889–896 CrossRef CAS PubMed.
- D. Chen, Q. Zhang, Q. Zhang, P. Cen, Z. Xu and W. Liu, Appl. Environ. Microbiol., 2012, 78, 5093–5103 CrossRef CAS PubMed.
- W. Wohlleben, Y. Mast, G. Muth, M. Rottgen, E. Stegmann and T. Weber, FEBS Lett., 2012, 586, 2171–2176 CrossRef CAS PubMed.
- S. R. Li, G. S. Zhao, M. W. Sun, H. G. He, H. X. Wang, Y. Y. Li, C. H. Lu and Y. M. Shen, Gene, 2014, 544, 93–99 CrossRef CAS PubMed.
- J. Zhang, J. An, J. J. Wang, Y. J. Yan, H. R. He, X. J. Wang and W. S. Xiang, Appl. Microbiol. Biotechnol., 2013, 97, 10091–10101 CrossRef CAS PubMed.
- J. S. Yi, M. S. Kim, S. J. Kim and B. G. Kim, J. Microbiol. Biotechnol., 2015, 25, 496–502 CrossRef CAS PubMed.
- C. Licona-Cassani, E. Marcellin, L. E. Quek, S. Jacob and L. K. Nielsen, Antonie van Leeuwenhoek, 2012, 102, 493–502 CrossRef CAS PubMed.
- M. Kim, J. Sang Yi, J. Kim, J. N. Kim, M. W. Kim and B. G. Kim, Biotechnol. J., 2014, 9, 1185–1194 CrossRef CAS PubMed.
- K. S. Hwang, H. U. Kim, P. Charusanti, B. O. Palsson and S. Y. Lee, Biotechnol. Adv., 2014, 32, 255–268 CrossRef CAS PubMed.
- R. Breitling, F. Achcar and E. Takano, ACS Synth. Biol., 2013, 2, 373–378 CrossRef CAS PubMed.
- B. Kim, W. J. Kim, D. I. Kim and S. Y. Lee, J. Ind. Microbiol. Biotechnol., 2015, 42, 339–348 CrossRef CAS PubMed.
- N. E. Lewis, H. Nagarajan and B. O. Palsson, Nat. Rev. Microbiol., 2012, 10, 291–305 CAS.
- H. U. Kim, S. B. Sohn and S. Y. Lee, Biotechnol. J., 2012, 7, 330–342 CrossRef CAS PubMed.
- H. U. Kim, S. Y. Kim, H. Jeong, T. Y. Kim, J. J. Kim, H. E. Choy, K. Y. Yi, J. H. Rhee and S. Y. Lee, Mol. Syst. Biol., 2011, 7, 460 CrossRef PubMed.
- R. Agren, A. Mardinoglu, A. Asplund, C. Kampf, M. Uhlen and J. Nielsen, Mol. Syst. Biol., 2014, 10, 721 CrossRef PubMed.
- T. Weber, K. Blin, S. Duddela, D. Krug, H. U. Kim, R. Bruccoleri, S. Y. Lee, M. A. Fischbach, R. Muller, W. Wohlleben, R. Breitling, E. Takano and M. H. Medema, Nucleic Acids Res., 2015, 43, W237–W243 CrossRef PubMed.
- T. Weber and H. U. Kim, Synth. Syst. Biotechnol., 2016 DOI:10.1016/j.synbio.2015.12.002.
- J. J. Hamilton and J. L. Reed, Environ. Microbiol., 2014, 16, 49–59 CrossRef PubMed.
- P. Zakrzewski, M. H. Medema, A. Gevorgyan, A. M. Kierzek, R. Breitling and E. Takano, PLoS One, 2012, 7, e51511 CAS.
- W. Vongsangnak, L. F. Figueiredo, J. Forster, T. Weber, J. Thykaer, E. Stegmann, W. Wohlleben and J. Nielsen, Biotechnol. Bioeng., 2012, 109, 1798–1807 CrossRef CAS PubMed.
- X. Wang, C. Zhang, M. Wang and W. Lu, Microb. Cell Fact., 2014, 13, 41 CrossRef PubMed.
- P. J. D'Huys, I. Lule, D. Vercammen, J. Anne, J. F. Van Impe and K. Bernaerts, J. Biotechnol., 2012, 161, 1–13 CrossRef PubMed.
- D. Huang, S. Li, M. Xia, J. Wen and X. Jia, Microb. Cell Fact., 2013, 12, 52 CrossRef CAS PubMed.
- M. Kim, J. S. Yi, M. Lakshmanan, D. Y. Lee and B. G. Kim, Biotechnol. Bioeng., 2016, 113, 651–660 CrossRef CAS PubMed.
- C. F. Mandenius and A. Brundin, Biotechnol. Prog., 2008, 24, 1191–1203 CrossRef CAS PubMed.
- G. Yu and G. Wang, Interdiscip. Sci.: Comput. Life Sci., 2015 DOI:10.1007/s12539-015-0133-8.
- B. M. Vastrad and S. E. Neelagund, Biotechnol. Res. Int., 2014, 2014, 674286 CAS.
- W. Zhou, X. Liu, P. Zhang, P. Zhou and X. Shi, Molecules, 2014, 19, 15507–15520 CrossRef CAS PubMed.
- I. Borodina, J. Siebring, J. Zhang, C. P. Smith, G. van Keulen, L. Dijkhuizen and J. Nielsen, J. Biol. Chem., 2008, 283, 25186–25199 CrossRef CAS PubMed.
- F. Coze, F. Gilard, G. Tcherkez, M. J. Virolle and A. Guyonvarch, PLoS One, 2013, 8, e84151 Search PubMed.
- R. W. Leighty and M. R. Antoniewicz, Metab. Eng., 2011, 13, 745–755 CrossRef CAS PubMed.
- S. Ma, K. J. Minch, T. R. Rustad, S. Hobbs, S. L. Zhou, D. R. Sherman and N. D. Price, PLoS Comput. Biol., 2015, 11, e1004543 Search PubMed.
- W. Lian, K. P. Jayapal, S. Charaniya, S. Mehra, F. Glod, Y. S. Kyung, D. H. Sherman and W. S. Hu, BMC Genomics, 2008, 9, 56 CrossRef PubMed.
- M. Castro-Melchor, S. Charaniya, G. Karypis, E. Takano and W. S. Hu, BMC Genomics, 2010, 11, 578 CrossRef PubMed.
- K. Nieselt, F. Battke, A. Herbig, P. Bruheim, A. Wentzel, O. M. Jakobsen, H. Sletta, M. T. Alam, M. E. Merlo, J. Moore, W. A. Omara, E. R. Morrissey, M. A. Juarez-Hermosillo, A. Rodriguez-Garcia, M. Nentwich, L. Thomas, M. Iqbal, R. Legaie, W. H. Gaze, G. L. Challis, R. C. Jansen, L. Dijkhuizen, D. A. Rand, D. L. Wild, M. Bonin, J. Reuther, W. Wohlleben, M. C. Smith, N. J. Burroughs, J. F. Martin, D. A. Hodgson, E. Takano, R. Breitling, T. E. Ellingsen and E. M. Wellington, BMC Genomics, 2010, 11, 10 CrossRef PubMed.
- E. Strakova, A. Zikova and J. Vohradsky, Nucleic Acids Res., 2014, 42, 748–763 CrossRef CAS PubMed.
- K. Blin, L. E. Pedersen, T. Weber and S. Y. Lee, Synth. Syst. Biotechnol., 2016 DOI:10.1016/j.synbio.2016.01.003.
- J. S. Zarins-Tutt, T. T. Barberi, H. Gao, A. Mearns-Spragg, L. Zhang, D. J. Newman and R. J. Goss, Nat. Prod. Rep., 2015, 33, 54–72 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1. See DOI: 10.1039/c6np00019c |
| This journal is © The Royal Society of Chemistry 2016 |