
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Calyculin: Nature's way of making the sponge-derived cytotoxin
Toshiyuki
Wakimoto
*a,
Yoko
Egami
a and
Ikuro
Abe
*b
aFaculty of Pharmaceutical Sciences, Hokkaido University, Kita 12, Nishi 6, Kita-ku, Sapporo 060-0812, Japan. E-mail: wakimoto@pharm.hokudai.ac.jp
bGraduate School of Pharmaceutical Sciences, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: abei@mol.f.u-tokyo.ac.jp
First published on 29th February 2016
Abstract
Covering: up to 2015.
Calyculin A is a major cytotoxic compound isolated from the Japanese marine sponge Discodermia calyx. Its potent cytotoxicity is attributable to the specific inhibition of protein phosphatases 1 and 2A, as in the case of okadaic acid and the microcystins. Its chemical structure is well-designed not only for enzyme inhibition but also for higher membrane permeability in order to impart its potent cytotoxicity. The biosynthetic gene cluster of this densely functionalized polyketide and nonribosomal peptide hybrid molecule was recently identified from the sponge–microbe association. The producer organism and the dynamic bioconversion process were also revealed. In this highlight, we focus on the recent studies addressing nature's design and biogenesis of the sponge-derived cytotoxin, calyculin A.
 Toshiyuki Wakimoto | Toshiyuki Wakimoto received his Ph.D. at the Graduate School of Agricultural and Life Sciences, University of Tokyo under the guidance of Professor Nobuhiro Fusetani in 2001, and went on to Professor Peter Wipf's lab at University of Pittsburgh as a postdoctoral researcher for 2 years. In 2003, he joined the faculty of the University of Shizuoka as an Assistant Professor, after which he moved to the Graduate School of Pharmaceutical Sciences, University of Tokyo as an Associate Professor. In 2015, he was appointed Professor of the Laboratory of Natural Products Chemistry at the Faculty of Pharmaceutical Sciences, Hokkaido University, where he continues his research involving the isolation, characterization, and biosynthesis of biologically active natural products from marine sponges, foods and traditional medicines. |
 Yoko Egami | Yoko Egami received her M.Sc. in 2010 from the University of Shizuoka under the direction of Professor Kunihiko Itoh. She moved to the University of Tokyo as a technical assistant (2010–2011) and a project researcher (2011–2015). Meanwhile, she obtained her Ph.D. in Pharmaceutical Sciences from the University of Tokyo under the supervision of Professor Ikuro Abe in 2015. She is now an Assistant Professor at Hokkaido University, working with Professor Toshiyuki Wakimoto. Her research focuses on the biosynthesis of marine natural products. |
 Ikuro Abe | Ikuro Abe is a Fellow of the Royal Society of Chemistry. He received his Ph.D. in 1989 from the University of Tokyo under the direction of Professor Yutaka Ebizuka, where he studied chemistry and biochemistry of natural product biosynthesis. After two years of postdoctoral research with Professor Guy Ourisson at the CNRS Institut de Chimie des Substances Naturelles, and mostly with Professor Michel Rohmer at the Ecole Nationale Supérieure de Chimie de Mulhouse (1989–1991), he moved to the USA to work with Professor Glenn D. Prestwich at the State University of New York at Stony Brook (1991–1996) and then at the University of Utah (1996–1998), as a Research Assistant Professor. In 1998, he returned to Japan to join the faculty at the School of Pharmaceutical Sciences, University of Shizuoka (1998–2009). In 2009, he was appointed Professor of Natural Products Chemistry at the Graduate School of Pharmaceutical Sciences, University of Tokyo. His research interests mostly focus on exploring and engineering natural product biosynthesis. |
1. Introduction
The marine sponge Discodermia calyx inhabits relatively shallow water in the central to southern areas of Japan. Originally, this marine sponge was part of the vast collection of Japanese marine organisms amassed by the German biologist Ludwig Döderlein, who was a visiting Professor at the University of Tokyo from 1879–1881, and it was identified based on a specimen collected in the Gulf of Sagami, Japan.1 Almost one hundred years later, Fusetani and co-workers reported that the marine sponge contains a major secondary metabolite, calyculin A (Fig. 1, 1, 0.15% wet weight), showing potent activity in the starfish egg assay.2 Subsequently, 1 was revealed to have pM range IC50 values against several cancer cell lines and tumor promotion activity, ascribed to the specific and potent inhibition of protein phosphatases 1 and 2A (PP1 and 2A),3 thereby rendering it a valuable tool to evaluate intracellular signal transduction. Its densely functionalized structure is naturally well-designed for the specific inhibition of PPs as well as cytotoxicity, and it has also attracted considerable attention from synthetic chemists, resulting in six total syntheses.4 | ||
| Fig. 1 Structures of calyculin derivatives and the crystal structure of ent-calyculin A. | ||
The natural source of the calyculins is not limited to D. calyx, as other sponge species also contain calyculin derivatives, such as the calyculinamides from the New Zealand sponge Lamellomorpha strongylata,5 the clavosines (10) from the Palauan sponge Myriastra clavosa,6 geometricin (11) from the Australian sponge Luffariella geometrica,7 and swinhoeiamide (12) from the Papua New Guinean sponge Theonella swinhoei (Fig. 2).8 However, no other organisms except for marine sponges have been reported to contain calyculin analogs. The cross-genera distribution of calyculin-related compounds suggests that they are produced by common or similar symbionts primarily associated with the marine sponges. Recently, the biosynthetic gene cluster and the producer organism were identified in the Japanese marine sponge D. calyx.9 This highlight focuses on the biological activity, the biosynthesis, and the producer organism of the calyculins.
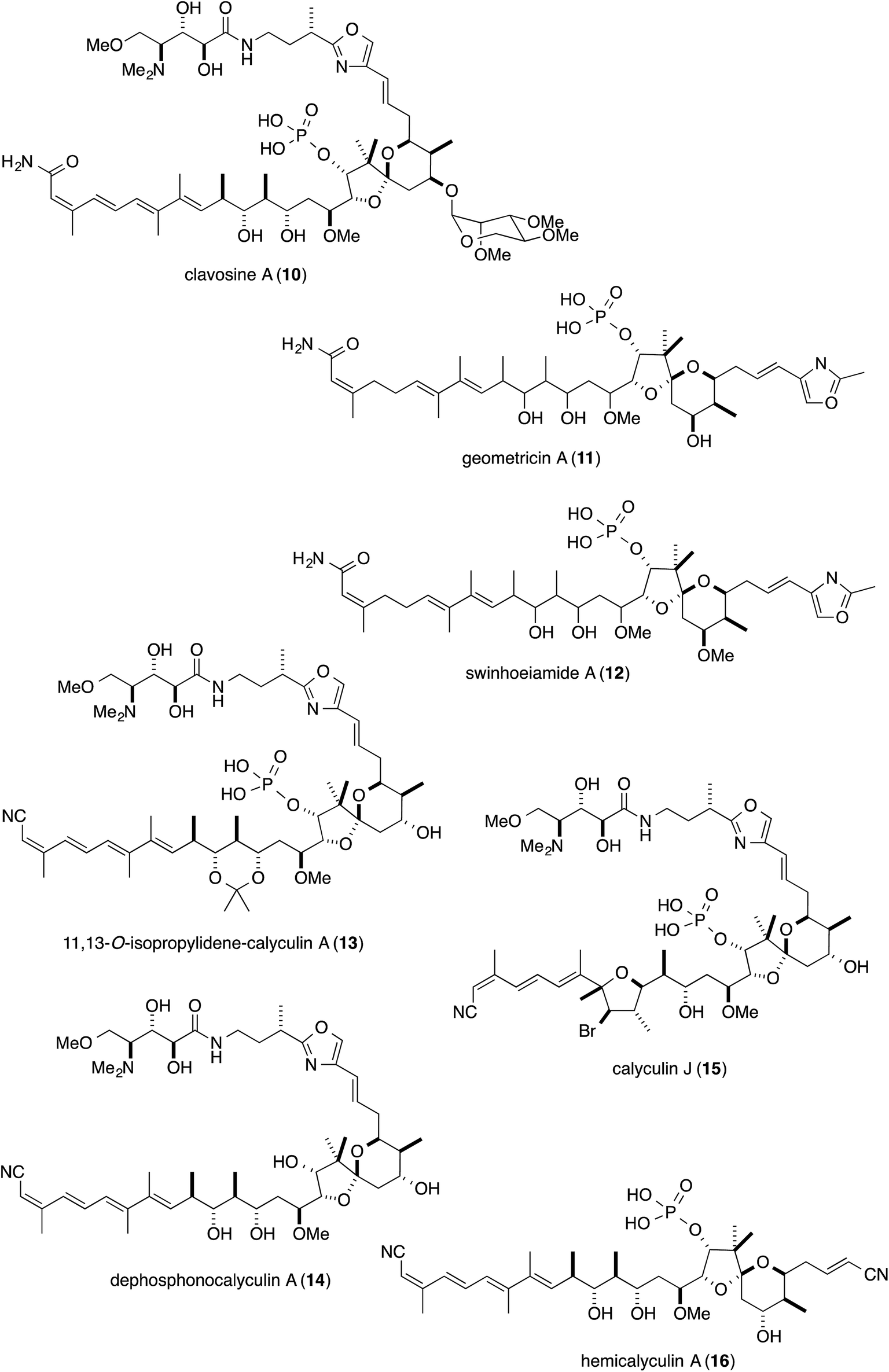 | ||
| Fig. 2 Structures of calyculin derivatives. | ||
2. Structure–activity relationship of calyculin A
Phosphatase inhibitors, in addition to 1, have been isolated from different biological sources, including cantharidin (17) from blister beetles,10 microcystins (18) from cyanobacteria,11 and okadaic acid (19) from marine sponges12 and dinoflagellates (Fig. 3).13 Actinomycetes are also important sources, producing tautomycin (20),14 fostriecin (21)15 and phoslactomycin (22) (Fig. 3).16 Notably, in addition to the distinct origins, the structural features of this class of molecules are also diverse, and include terpenoids such as cantharidin, microcystins with a peptidic nature, and polyketides including okadaic acid, tautomycin, fostriecin and phoslactomycin. In spite of the stark differences in their chemical structures, these natural molecules can inhibit the same enzyme in a competitive manner, although some inhibitors exhibited varied selectivities on PP1 and PP2A, cf. moderate selectivity against PP2A by okadaic acid17 and fostriecin,15 and the higher specificity of tautomycin14 towards PP1 than PP2A. This fact invokes the question of which common structural motifs are essential for the specific inhibition of protein phosphatases. Since all the molecules possess a hydrophobic moiety, in addition to an acidic functionality as the phosphate mimic, these common structural features had been suggested to be indispensable for the enzyme inhibition.18–20 In fact, the X-ray crystal structure of the complex between the natural inhibitors and the PPs has been found to firmly support the molecular basis for this proposal. Based on the crystal structure, the acidic functional groups of the PP inhibitors interact with the phosphate recognizing residue, Arg, of the enzyme. Furthermore, one of the Y-shaped grooves on the surface of the enzyme, which is occupied by hydrophobic amino acid residues (hydrophobic groove), indeed, interacts with the hydrophobic tails of the natural inhibitors.21–24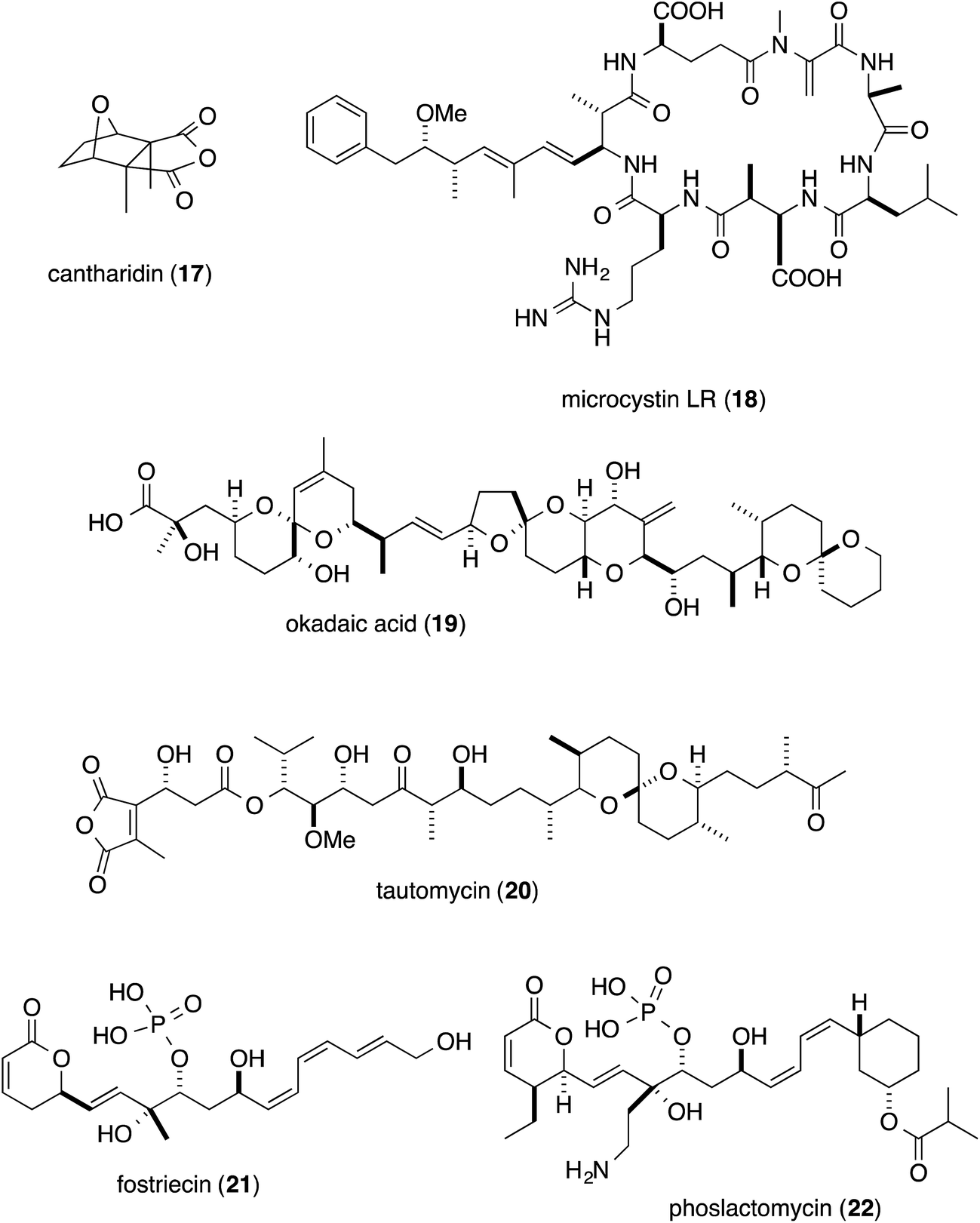 | ||
| Fig. 3 Structures of natural product inhibitors of PP1 and PP2A. | ||
Detailed structure–activity relationship studies have also been conducted on calyculins, with the natural variants isolated from the extract of the sponge D. calyx as well as semisynthetic analogs.25 The tetraene moiety of 1 is photoisomerized into geometrical isomers, calyculins B (2), E (6), and F (7) (Fig. 1).26,27 Calyculin C (3) has an additional methyl group on C32 of 1, and the geometrical isomers of this series are calyculins D (4), G (8), and H (9) (Fig. 1).26,27 The tetraene group corresponds to the hydrophobic motif found in other phosphatase inhibitors, and therefore the IC50 values of the geometrical isomers decrease by up to 3 orders of magnitude. The semisynthetic 11,13-O-isopropylidene-calyculin A25 (13) and the natural derivative, dephosphonocalyculin A28 (14) (Fig. 2), completely lacked both the enzyme inhibition and cytotoxic activities, demonstrating that the phosphate and the 1,3-diol are essential for inhibitory activity against protein phosphatases. The considerable retention of activity by calyculin J29 (Fig. 2, 15) indicated the importance of the 13-OH, rather than the 11-OH, for enzyme inhibition. On the other hand, hemicalyculin A25 (Fig. 2, 16), which is terminated by a nitrile in place of the oxazole ring, but retains the phosphate group, 13-OH, and tetraene, showed potent enzyme inhibition activities against both PP1 and PP2A comparable to those of 1. Therefore, the peptide portion composed of two γ-amino acids does not seem to be involved in the binding to PPs. In fact, the corresponding portion could not be detected in the X-ray crystal structure of the complex between 1 and PP1γ due to the indistinct electron densities, strongly supporting the idea that this motif does not function in the interaction.23
These findings then engendered the next question about the role of the peptide portion. Remarkably, the cytotoxicity of 16 was significantly diminished, and was more than four orders of magnitude weaker than that of 1,25 which clearly indicated that the peptide portion is essential for the cytotoxicity of 1. Again, the X-ray structure provided insight into the proposed role of this moiety. The free state of 1 forms the pseudocyclic conformation through an intramolecular salt bridge network between the phosphate group and the dimethylamino group, oxazole nitrogen, and 13-OH (Fig. 1).2 This conformation masks the charges of the phosphate group and decreases the polarity of this molecule, and thus enhances the membrane permeability. However, upon binding to PP1γ, the pseudocyclic conformation is drastically changed into the extended conformation, to expose the phosphate group to the amino acid residues in the phosphate recognition site.23 Thus, the sophisticated natural design for the preservation of the cytotoxicity is embedded in the entire skeleton of 1.
3. Biosynthetic gene cluster of calyculin A
Structurally, 1 has an oxazole ring that connects two biosynthetically dissimilar motifs: a peptide portion composed of two γ-amino acids and a polyketide portion containing a 5,6-spiroacetal, phosphate, tetraene, and a terminal nitrile. Thus, these structural features suggest a non-ribosomal peptide and polyketide hybrid biosynthetic pathway, with some noncanonical modifications. One of these modifications is the β-branched methyl groups on C3 and C7, implying a trans-AT PKS origin.30 PCR analysis with degenerate primers for KS was performed on the D. calyx metagenome, to obtain the trans-AT type ketosynthase DNA fragments. The screening of a fosmid library with the DNA fragment encoding the trans-AT KS sequences successfully yielded the PKS–NRPS hybrid gene cluster, ranging over 150 kb. The PKS–NRPS modular assembly line is composed of nine ORFs encoding thirty-four modules aligned until the TE domain (Fig. 4), whereas the downstream ORFs, calH and calI, encode some extra modules, whose putative products are missing in the calyculin structure. The five A domains encoded in the cal gene are supposed to accept Ser, Gly, Ser, Ala, and Ser, according to the NRPS codes, and among them the latter two, the A4 and A5 domains, are encoded in the extra module (Fig. 4).31 The first two Ser residues recruited by the A1 and A3 domains are converted into trimethylserine and oxazole, respectively, which agrees with the domain organization of the corresponding module (Fig. 4). The NRPS code of the A2 domain matched that accepting Gly, except for the replacement of Leu or Met by Thr, which presumably confers the promiscuity to this A domain and thus allows it to accept Ala as an additional substrate, to generate 3.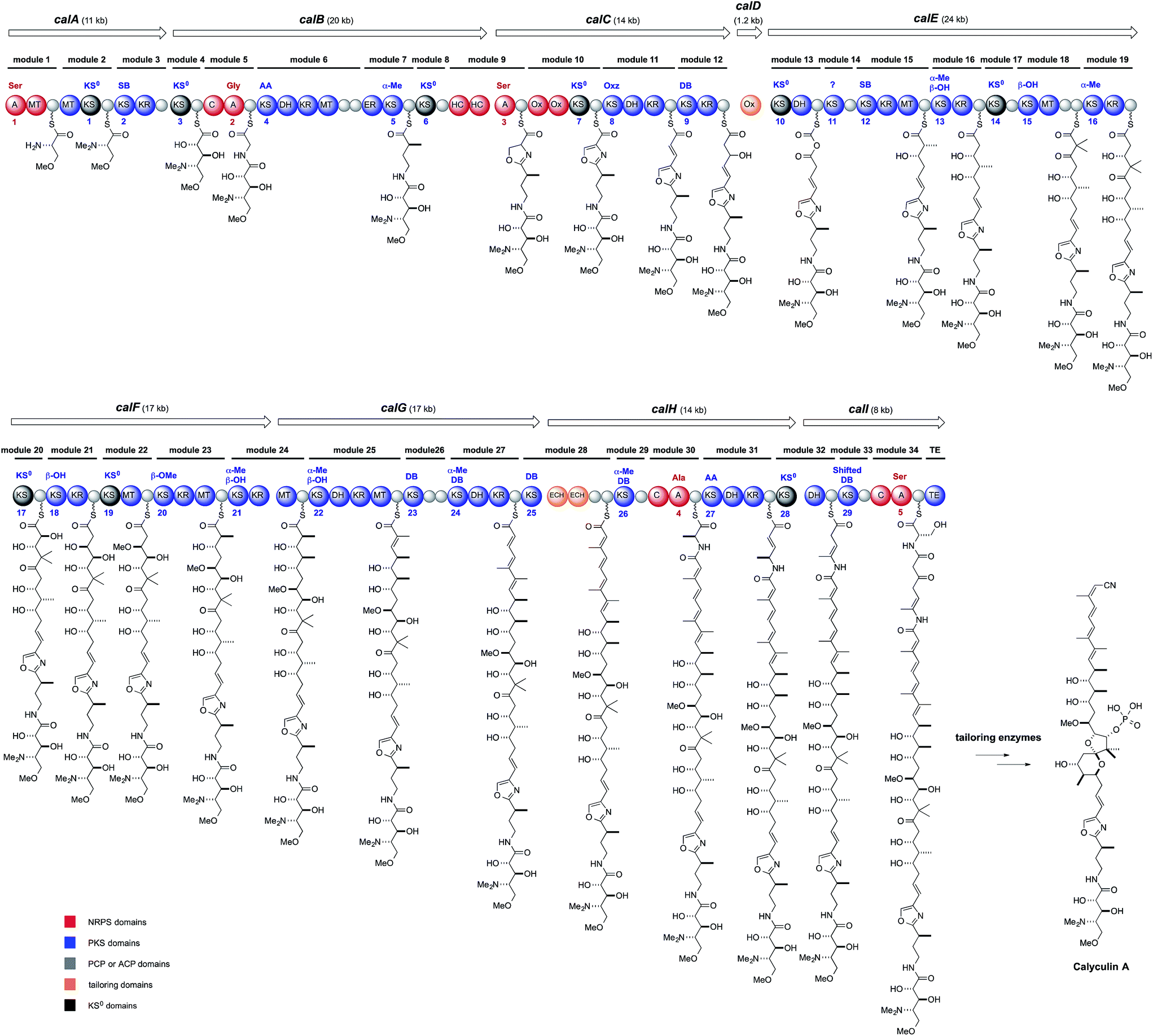 | ||
| Fig. 4 Proposed biosynthetic pathway of calyculin A. | ||
Notably, the cal gene encodes 29 KS domains in total, and among them, nine are non-elongating KS0 domains (Fig. 4, KS1, 3, 6, 7, 10, 14, 17, 19, and 28). The fact that nearly one-third of the total KS domains have non-elongating functions provides significant insights into the domain architecture exerting non-canonical modifications in the trans-AT PKS pathway. The N- and O-methylation proceeds on the module containing KS1 and KS19, respectively. The domain organization with KS28 and the succeeding DH catalyzes the β, γ-shift of the double bond to generate an enamide.32,33 The roles of KS6 and 7 are unknown, but they flank the NRPS module responsible for the oxazole formation. Aside from KS14, all of the remaining KS0 (Fig. 4, KS3, 10, and 17) domain-containing modules (Fig. 4, modules 4, 13, and 20) are involved in the α-hydroxylation. Modules 4 and 20 both have KS0 domains, and ACP could implement the α-hydroxylation by an unidentified oxygenase. Indeed, two monooxygenases are encoded upstream of the cal gene. While module 4 introduces the 34-OH, module 20 generates the 16-OH required for the 5,6-spiroacetal ring formation. Once the 16-OH is introduced, the tetrahydrofuran ring can be formed with the carbonyl group located at C19. The resulting lactol can generate an oxonium ion, which is captured by the 23-OH to afford a 5,6-spiroacetal. Since the natural C-19 S-isomer is thermodynamically more stable than the C-19 R-isomer,34 the 5,6-spiroacetal could be formed in a non-enzymatic manner.
Above all, module 13, along with the stand-alone oxygenase (CalD), functions in the important process of constructing the polyketide portion with an odd numbered carbon chain from C1 to C25. The exact mechanism of the chain shortening modification is still unknown, but a mechanism has been proposed, based on the domain organization. The architecture of Ox-KS0-DH suggests that α-hydroxylation by the Ox, followed by dehydration by the DH, generates the α-ketothioester intermediate (Fig. 4),35 which in turn is taken up again by the Ox to undergo a Baeyer–Villiger type oxidation. If the next elongation reaction occurs on the ester carbonyl group of the mixed anhydride, then a carbon unit is eliminated as carbon dioxide. The other KS0 is KS14, and its role is still unknown. Thus, the cal gene is an intricate showcase of the module architectures potentiating the non-canonical modifications in the trans-AT PKS pathway.
In contrast, there are two major discrepancies between the cal gene organization and the calyculin structure. One is the stereochemistry of C17, which was predicted by the KR sequence (module 19) to be antipodal to that found in 1. The stereochemical inversion may occur during the phosphorylation of 17-OH at the post-PKS modification stage. The other discrepancy is the existence of the extra module encoded by calH and calI. The nitrogen atom of Ala introduced by the A domain (A4) is obviously required for nitrile formation at the terminus. The succeeding domain architectures of modules 31 and 32 indicate the enamide formation at the corresponding position, as mentioned above. Considering that some natural calyculin derivatives have a terminal amide in place of the nitrile (Fig. 1 and 2), we envisioned that calyculin biosynthesis is terminated at the enamide functionality, to generate a terminal amide as the nascent product. A phosphate-based reaction was proposed36 as the mechanism for the transformation of the amide into a nitrile, since three ORFs within the upstream region are annotated as phosphotransferases.
4. Phosphocalyculin A as a protoxin
The three phosphotransferases encoded in the upstream region of the gene cluster were subjected to heterologous expression and functional analysis. Unexpectedly, one of them, CalQ, appended a phosphate group onto 1 as well as calyculinamide A (5). Previously, 1 had been considered to be the final product of the biosynthesis, since it was the major metabolite in the sponge extract prepared in the conventional manner. However, the alcoholic extract of the sponge tissue, which was freeze-dried after flash-freezing in liquid nitrogen, afforded phosphocalyculin A (30) as the major metabolite, in place of 1. This result suggested that an as yet unidentified phosphatase coexists along with 30, and they are compartmentalized in the sponge tissue. The conventional extraction procedure induces decompartmentalization, which immediately converts 30 into 1. Thus, 30 had been overlooked due to enzymatic dephosphorylation during the previous extraction procedure. Indeed, the induction of the “activating” process was observed immediately after wounding the sponge tissue.25,37 As mentioned in the structure–activity relationships study, since the phosphate group is essential for phosphatase inhibition, 30 is 1000 times less toxic than 1. These findings provide not only functional evidence of the gene cluster, but also the underlying mechanism of calyculin biogenesis.Wound-activated chemical defense systems are prevalent among terrestrial plants (Fig. 5). Cyanogen glycoside is a representative example, in which tissue wounding induces a quick increase in hydrogen cyanide via the hydrolytic liberation of the aglycone.38 The precursor molecule and the activating enzyme are compartmentalized in the plant tissue, which is readily disrupted by wounding to accomplish chemical defense. The activated defense strategy has also been found in a few marine sponges (Fig. 5).39 The quick enzymatic transformation of the protoxin to the toxin often prevents the prevalence of this system from being recognized, since the homogenization of sponge tissue during the extraction of secondary metabolites results in decompartmentalization, thus giving rise to the same reaction as in the case of wound activation. The first example was found in the marine sponge Aplysina aerophoba, which accumulates brominated isoxazoline alkaloids. The isoxazoline alkaloids (23) are stored in spherulous cells and are enzymatically converted to the more active toxin, dienone amide (25), in response to tissue disruption.40,41 This bioconversion is a multistep process comprising the liberation of the isoxazoline moiety with a terminal nitrile, followed by demethylation, dehydration, and hydration of the nitrile. The enzymes catalyzing these steps are membrane-bound or membrane-associated enzymes except for that catalyzing the final step, nitrile hydration.42 Proksch and coworkers recently reported the isolation of an aeroplysinin-1 (24)-specific nitrile hydratase, which lacks sequence homology to known nitrile hydratases and has narrow substrate specificity.42 Similar bioconversion events have been found in the marine sponges Luffariella variabilis and Aplysinella rhax, and are relatively simpler processes involving deacetylation and desulfonation, respectively (Fig. 5).43,44 However, the enzymes responsible for the bioconversions have not been identified yet. If the biosynthetic genes corresponding to the syntheses of these terpenoids and alkaloids are identified, it would be interesting to determine whether the enzymes involved in the deactivating or activating processes are clustered.
 | ||
| Fig. 5 Enzymatic wound-activated bioconversion in marine sponges. | ||
5. Calyculin A production in the sponge–microbe association
Faulkner and co-workers originally reported that the filamentous bacterium associated with Palauan Theonella swinhoei contained theopalauamide, by cell separation methodology.45 This bacterium was subsequently identified as “Candidatus Entotheonella palauensis”.46 Although the chemical localization study successfully illuminated the cellular distributions of the sponge-derived bioactive molecules, the site of biosynthesis might not always be the same as the site of storage. To determine the chemical localization in line with the production site, biosynthetic evidence is desirable. A few successful examples encompassing biosynthetic gene clusters have been reported. Several polychlorinated peptides isolated from the marine sponge Dysidea herbacea are closely related to barbamide, from the free-standing cyanobacterium Lyngbya majuscula.47 Gerwick's group applied the known sequence of the barbamide biosynthetic gene cluster to search for the biosynthetic gene responsible for the production of the polychlorinated peptides, which yielded the bar homolog from the sponge. A CARD-FISH analysis with the probe specific to the bar homolog revealed that the biosynthetic genes are housed within the cyanobacterial symbiont Oscillatoria spongeliae.48 In 2014, Piel and co-workers reported that Entotheonella belongs to the candidate phylum Tectomicrobia, and the bacterium of the phylotype “Candidatus Entotheonella factor” is the producer of almost all the metabolites isolated from the Japanese marine sponge T. swinhoei (yellow chemotype).49 Quite recently, the white chemotype T. swinhoei was also found to harbor the similar filamentous bacterium “Candidatus Entotheonella serta”, as the producer of misakinolide. The overlapped locations of the 16S rDNA and misakinolide were elucidated by CARD-FISH and MALDI imaging mass spectrometry, respectively, confirming that the biosynthesis and storage sites were identical.50A CARD-FISH analysis was also conducted with the calyculin biosynthetic gene cluster, in the sponge D. calyx.9 The gene cluster was localized within the filamentous bacterium “Candidatus Entotheonella” sp., as confirmed by single cell analysis using laser microdissection.9 The data provided the first clues about the biosynthetic gene and the symbiotic producer involved in the wound-activated process to generate a cytotoxic compound derived from a marine sponge. Considering that calyculins have almost no antibacterial activity, the self-toxicity to Entotheonella is considered to be low. However, the potent cytotoxicity of the calyculins would be harmful to the host sponge. The manner in which 1 is phosphorylated and deactivated by the bacterial symbiont suggests that the crosstalk between the sponge host and the symbiont plays an important role in not only the dynamic process for the on-demand generation of toxic 1, but also the self-resistance systems of the host.
6. Conclusions
The overall picture of the biosynthetic mechanisms of sponge-derived bioactive molecules is still largely unclear. However, recent comprehensive studies have illuminated some intriguing pieces of the complicated processes, such as chemical localizations, real producers, and dynamic bioconversions. The discovery of the association of Entotheonella with the genera Theonella and Discodermia has given rise to more questions about the ubiquity of secondary metabolite production by Entotheonella in other sponges, and whether other bacterial taxa function as symbiotic producers. Future studies will provide answers to these questions.Wound-activated bioconversion processes have been reported for brominated isoxazoline alkaloids and calyculins, which are both predominant metabolites in the sponges A. aerophoba and D. calyx, respectively. The spatial organization and the activation process might allow the sponges to store large amounts of highly potent toxins. Recent advances in mass spectrometry-based imaging technology will clarify not only the detailed localization but also the dynamic transformations occurring in response to tissue disruption.
As potential applications, the enzymatic activation and deactivation processes of bioactive molecules have historically been utilized as biochemical tools, including certain antibiotics as expression markers. Thus, understanding nature's way of producing bioactive metabolites in the sponge–microbe association will provide significant opportunities, not only for addressing supply problems but also for further applications of these metabolites in biochemical research. The sponge factory may have many more treasures than the medicinally important bioactive molecules themselves.
7. Acknowledgments
Our research on calyculin biosynthesis was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan. Support was also provided by the Mitsubishi Foundation, the Nagase Science Technology Foundation, Astellas Foundation for Research on Metabolic Disorders, the Sumitomo Foundation, the Takeda Science Foundation, and the Naito Foundation.8. References
- L. Döderlein, Studien an japanischen Lithistiden, Z. Wiss. Zool., Abt. A, 1883, 40, 62–104 Search PubMed.
- Y. Kato, N. Fusetani, S. Matsuanga, K. Hashimoto, S. Fujita and T. Furuya, Calyculin A, a novel antitumor metabolites from the marine sponge Discodermia calyx, J. Am. Chem. Soc., 1986, 108, 2780–2781 CrossRef CAS.
- M. Suganuma, H. Fujiki, H. Furuya-Suguri, S. Yoshizawa, S. Yamamoto, Y. Kato, N. Fusetani and T. Sugimura, Calyculin A, and inhibitor of protein phosphatases, a potent tumor promoter on CD-1 mouse skin, Cancer Res., 1990, 50, 3521–3525 CAS.
- A. E. Fagerholm, D. Habrant and A. M. P. Koskinen, Calyculins and related marine natural products as serine-threonine protein phosphatase PP1 and PP2A inhibitors and total syntheses of calyculin A, B, and C, Mar. Drugs, 2010, 8, 122–172 CrossRef CAS PubMed.
- E. J. Dumdei, J. W. Blunt, M. H. G. Munro and L. K. Pannell, Isolation of calyculins, calyculinamides, and swinholide H from the New Zealand deep-water marine sponge Lamellomorpha strongylata, J. Org. Chem., 1997, 62, 2636–2639 CrossRef CAS PubMed.
- X. Fu, F. J. Schmitz, M. Kelly-Borges, T. L. McCready and C. F. B. Holmes, Clavosines A-C from the marine sponge Myriastra clavosa: Potent cytotoxins and inhibitors of protein phosphatases 1 and 2A, J. Org. Chem., 1998, 63, 7957–7963 CrossRef CAS.
- S. Kehraus, G. M. König and A. D. Wright, A new cytotoxic calyculinamide derivative, geometricin A, from the Australian sponge Luffariella geometrica, J. Nat. Prod., 2002, 65, 1056–1058 CrossRef CAS.
- R. A. Edrada, R. Ebel, A. Supriyono, V. Wray, P. Schupp, K. Steube, R. van Soest and P. Proksch, Swinhoeiamide A, a new highly active calyculin derivative from the marine sponge Theonella swinhoei, J. Nat. Prod., 2002, 65, 1168–1172 CrossRef CAS.
- T. Wakimoto, Y. Egami, Y. Nakashima, Y. Wakimoto, T. Mori, T. Awakawa, T. Ito, H. Kenmoku, Y. Asakawa, J. Piel and I. Abe, Calyculin biogenesis from a pyrophosphate protoxin produced by a sponge symbiont, Nat. Chem. Biol., 2014, 10, 648–655 CrossRef CAS PubMed.
- R. E. Honkanen, Cantharidin, another natural toxin that inhibits the activity of serine/threonine protein phosphatases types 1 and 2A, FEBS Lett., 1993, 330, 283–286 CrossRef CAS PubMed.
- C. MacKintosh, K. A. Beattie, S. Klumpp, P. Cohen and G. A. Codd, Cyanobacterial microcystin-LR is a potent and specific inhibitor of protein phosphatases 1 and 2A from both mammals and higher plants, FEBS Lett., 1990, 264, 187–192 CrossRef CAS PubMed.
- K. Tachibana, P. J. Scheuer, Y. Tsukitani, H. Kikuchi, D. V. Engen, J. Clardy, Y. Gopichand and F. J. Schmitz, Okadaic acid, a cytotoxic polyether from two marine sponges of the genus Halichondria, J. Am. Chem. Soc., 1981, 103, 2469–2471 CrossRef CAS.
- Y. Murakami, Y. Osjima and T. Yasumoto, Identification of okadaic acid as a toxic component of a marine dinoflagellate Prorocentrum lima, Nihon Suisan Gakkaishi, 2006, 72, 4907–4916 Search PubMed.
- S. Mitsuhashi, N. Matsuura, M. Ubukata, H. Oikawa, H. Shima and K. Kikuchi, Tautomycetin is a novel and specific inhibitor of serine/threonine protein phosphatase type 1, PP1, Biochem. Biophys. Res. Commun., 2001, 287, 328–331 CrossRef CAS PubMed.
- M. Roberge, C. Tudan, S. M. F. Hung, K. W. Harder, F. R. Jirik and H. Anderson, Antitumor drug fostriecin inhibits the mitotic entry checkpoint and protein phosphatases 1 and 2A, Cancer Res., 1994, 54, 6115–6121 CAS.
- T. Usui, G. Marriott, M. Inagaki, G. Swarup and H. Osada, Protein phosphatase 2A inhibitors, phoslactomycins. Effects on the cytoskeleton in NIH/3T3 cells, J. Biochem., 1999, 125, 960–965 CrossRef CAS PubMed.
- A. Takai and G. Miekes, Inhibitory effect of okadaic acid on the p-nitrophenyl phosphate phosphatase activity of protein phosphatases, Biochem. J., 1991, 275, 233–239 CrossRef CAS PubMed.
- J. R. Bagu, B. D. Sykes, M. M. Craig and C. F. B. Holmes, A molecular basis for different interactions of marine toxins with protein phosphatase-1, J. Biol. Chem., 1997, 272, 5087–5097 CrossRef CAS PubMed.
- V. Gupta, A. K. Ogawa, X. Du, K. N. Houk and R. W. Armstrong, A model for binding of structurally diverse natural product inhibitors of protein phosphatases PP1 and PP2A, J. Med. Chem., 1997, 40, 3199–3206 CrossRef CAS PubMed.
- C.-M. Gauss, J. E. Sheppeck II, A. C. Nairn and R. Chamberlin, A molecular modeling analysis of the binding interactions between the okadaic acid class of natural product inhibitors and the Ser-Thr phosphatases, PP1 and PP2A, Bioorg. Med. Chem., 1997, 5, 1751–1773 CrossRef CAS PubMed.
- J. Goldberg, H. B. Huang, Y. G. Kwon, P. Greengard, A. C. Narin and J. Kuriyan, Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1, Nature, 1995, 376, 745–753 CrossRef CAS PubMed.
- J. T. Maynes, K. S. Bateman, M. M. Cherney, A. K. Das, H. A. Luu, C. F. B. Holmes and M. N. G. James, Crystal structure of the tumor-promoter okadaic acid bound to protein phosphatase-1, J. Biol. Chem., 2001, 276, 44078–44082 CrossRef CAS PubMed.
- A. Kita, S. Matsunaga, A. Takai, H. Kataiwa, T. Wakimoto, N. Fusetani, M. Isobe and K. Miki, Crystal structure of the complex between calyculin A and the catalytic subunit of protein phosphatase 1, Structure, 2002, 10, 715–724 CrossRef CAS PubMed.
- M. S. Keller, R. Page and W. Peti, Crystal structures of protein phosphatase-1 bound to nodularin-R and tautomycin: A novel scaffold for structure-based drug design of serine/threonine phosphatase inhibitors, J. Mol. Biol., 2009, 385, 11–21 CrossRef PubMed.
- T. Wakimoto, S. Matsunaga, A. Takai and N. Fusetani, Insight into binding of calyculin A to protein phosphatase 1: isolation of hemicalyculin A and chemical transformation of calyculin A, Chem. Biol., 2002, 9, 309–319 CrossRef CAS PubMed.
- Y. Kato, N. Fusetani, S. Matsunaga, K. Hashimoto and K. Koseki, Isolation and structure elucidation of calyculins B, C, and D, novel antitumor metabolites, from the marine sponge Discodermia calyx, J. Org. Chem., 1998, 53, 3930–3932 CrossRef.
- S. Matsunaga, H. Fujiki, D. Sakata and N. Fusetani, Calyculins E, F, G, and H, additional inhibitors of protein phosphatases 1 and 2A, from the marine sponge Discodermia calyx, Tetrahedron, 1991, 47, 2999–3006 CrossRef CAS.
- S. Matsunaga, T. Wakimoto, N. Fusetani and M. Suganuma, Isolation of dephosphonocalyculin A from the marine sponge Discodermia calyx, Tetrahedron Lett., 1997, 38, 3763–3764 CrossRef CAS.
- S. Matsunaga, T. Wakimoto and N. Fusetani, Isolation of four new calyculins from the marine sponge Discodermia calyx, J. Org. Chem., 1997, 62, 2640–2642 CrossRef CAS PubMed.
- E. J. N. Helfrich and J. Piel, Biosynthesis of polyketides by trans-AT polyketide synthases, Nat. Prod. Rep., 2016, 33, 231–316 RSC.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases, Chem. Biol., 1999, 6, 493–505 CrossRef CAS PubMed.
- B. Kusebauch, B. Busch, K. Scherlach, M. Roth and C. Hertweck, Functionally distinct modules operate two consecutive α,β→β,γ double-bond shifts in the rhizoxin polyketide assembly line, Angew. Chem., Int. Ed., 2010, 49, 1460–1464 CrossRef CAS PubMed.
- J. Moldenhauer, D. C. G. Götz, C. R. Albert, S. K. Bischof, K. Schneider, R. D. Süssmuth, M. Engeser, H. Gross, G. Bringmann and J. Piel, The final steps of bacillaene biosynthesis in Bacillus amyloliquefaciens FZB42: direct evidence for β,γ dehydration by a trans-acyltransferase polyketide synthase, Angew. Chem., Int. Ed., 2010, 49, 1465–1467 CrossRef CAS PubMed.
- A. B. Smith III, G. K. Friestad, J. Barbosa, E. Bertounesque, K. G. Hull, M. Iwashima, Y. Qiu, B. A. Salvatore, P. G. Spoors and J. J.-W. Duan, Total synthesis of (+)-calyculin A and (−)-calyculin B: asymmetric synthesis of the C(9-25) spiroketal dipropionate subunit, J. Am. Chem. Soc., 1999, 121, 10468–10477 CrossRef.
- M.-C. Tang, H.-Y. He, F. Zhang and G.-L. Tang, Baeyer–Villiger oxidation of acyl carrier protein-tethered thioester to acyl carrier protein-linked thiocarbonate catalyzed by a monooxygenase domain in FR901464 biosynthesis, ACS Catal., 2013, 3, 444–447 CrossRef CAS.
- P. B. Gomes, M. Nett, H.-M. Sattler, K. Martin and C. Hertweck, Bezerramycins A-C, antiproferative phenoxazinones from Streptomyces griseus featuring carboxy, carboxamide or nitrile substituents, Eur. J. Org. Chem., 2010, 2, 231–235 CrossRef.
- Y. Egami, T. Wakimoto and I. Abe, Phophocalyculin C as apyrophosphate protoxin of calyculin C in the marine sponge Discodermia calyx, Bioorg. Med. Chem. Lett., 2014, 24, 5150–5153 CrossRef CAS PubMed.
- A. Mithöfer and W. Boland, Plant defense against herbivores: chemical aspects, Annu. Rev. Plant Biol., 2012, 63, 431–450 CrossRef PubMed.
- S. Rohde, S. Nietzer and P. J. Schupp, Prevalence and mechanisms of dynamic chemical defenses in tropical sponges, PLoS One, 2015, 10, e0132236 Search PubMed.
- R. Ebel, M. Brenzinger, A. Kunze, H. J. Gross and P. Proksch, Wound activation of protoxins in marine sponge Aplysina aerophoba, J. Chem. Ecol., 1997, 23, 1451–1462 CrossRef CAS.
- C. Thoms, R. Ebel and P. Proksch, Activated chemical defense in Aplysina sponge revisited, J. Chem. Ecol., 2006, 32, 97–123 CrossRef CAS PubMed.
- B. Lipowicz, N. Hanekop, L. Schmitt and P. Proksch, An aeroplysinin-1 nitrile hydratase isolated from the marine sponge Aplysina cavernicola, Mar. Drugs, 2013, 11, 3046–3067 CrossRef PubMed.
- P. Ettinger-Epstein, C. A. Motti, R. de Nys, A. D. Wright, C. N. Battershill and D. M. Tapiolas, Acetylated sesterterpenes from the great barrier reef sponge Luffariella variabilis, J. Nat. Prod., 2007, 70, 648–651 CrossRef CAS PubMed.
- C. Thoms and P. J. Schupp, Activated chemical defense in marine sponges – a case study on Aplysinella rhax, J. Chem. Ecol., 2008, 34, 1242–1252 CrossRef CAS PubMed.
- C. A. Bewley, N. D. Holland and D. J. Faulkner, Two classes of metabolites from Theonella swinhoei are localized in distinct populations of bacterial symbionts, Experientia, 1992, 52, 716–722 CrossRef.
- E. W. Schmidt, A. Y. Obraztsova, S. K. Davidson, D. J. Faulkner and M. G. Haygood, Identification of the antifungal peptide-containing symbiont of the marine sponge Theonella swinhoei as a novel δ-proteobacterium, “Candidatus Entotheonella palauensis”, Mar. Biol., 2000, 136, 969–977 CrossRef CAS.
- J. Orjala and W. H. Gerwick, Barbamide, a chlorinated metabolite with molluscicidal activity from the Caribbean cyanobacterium Lyngbya majuscula, J. Nat. Prod., 1996, 59, 427–430 CrossRef CAS PubMed.
- P. M. Flatt, J. T. Gautschi, R. W. Thacker, M. Musafija-Girt, P. Crews and W. H. Gerwick, Identification of the cellular site of polychlorinated peptide biosynthesis in the marine sponge Dysidea (Lamellodysidea) herbacea and symbiotic cyanobacterium Oscillatoria spongeliae by CARD-FISH analysis, Mar. Biol., 2005, 147, 761–774 CrossRef CAS.
- M. C. Wilson, T. Mori, C. Rückert, A. R. Uria, M. J. Helf, K. Takada, C. Gernert, U. Steffens, N. Heycke, S. Schmitt, C. Rinke, E. J. N. Helfrich, A. O. Brachmann, C. Gurgui, T. Wakimoto, M. Kracht, M. Crüsemann, U. Hentschel, I. Abe, S. Matsunaga, J. Kalinowski, H. Takeyama and J. Piel, An environmental bacterial taxon with a large and distinct metabolic repertoire, Nature, 2014, 506, 58–62 CrossRef CAS PubMed.
- R. Ueoka, A. R. Uria, S. Reiter, T. Mori, P. Karbaum, E. E. Peters, E. J. N. Helfrich, B. I. Morinaka, M. Gugger, H. Takeyama, S. Matsunaga and J. Piel, Metabolic and evolutionary origins of actin-binding polyketides from diverse organisms, Nat. Chem. Biol., 2015, 11, 705–712 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |