DOI:
10.1039/C5NJ01741F
(Paper)
New J. Chem., 2016,
40, 512-520
Organosilane sulfonated graphene oxide in the Biginelli and Biginelli-like reactions
Received
(in Montpellier, France)
7th July 2015
, Accepted 3rd November 2015
First published on 17th November 2015
Abstract
Organosilane sulfonated graphene oxides (SSi-GO) have been synthesized by a two-step procedure involving the grafting of graphene oxide (GO) using 3-chloropropyltriethoxysilane (CCPTES) and subsequent oxidation using sulfanilic acid. It has been shown that organosilane sulfonated graphene oxide (SSi-GO) exhibits a superior catalytic performance to produce pyrimidines in the Biginelli and Biginelli-like reactions. This stronger acidity corresponds to the cooperative effects of the aryl sulfonic acid groups and other kinds of acid sites (carboxylic acids). However, the acidic functionalities bonded to the SSi-GO surface are stable under the catalytic reaction conditions resulting in its efficient reuse.
Introduction
Graphite oxide (GtO) is a carbonaceous layered material consisting of hydrophilic oxygenated graphene sheets (graphene oxide sheets). Generally, bulk graphite oxide can be prepared by the oxidative treatment of purified natural graphite powder using the modified Hummers method. This involved the use of strong concentrated oxidizing acids (HNO3 and H2SO4) and strong oxidizing salts (potassium permanganate).1 Despite the retention of the layered structure, graphite oxide has much lighter color than graphite powder owing to the loss of electronic conjugation afforded by the oxidation. Graphite oxides contain covalently attached oxygen-containing groups such as sp3-hybridized carbons containing hydroxyl and epoxide functional groups on above and below each sheet (the basal planes) as major components, and sp2-hybridized carbons containing carbonyl and carboxyl groups located at the edges of the basal planes and hole defects as minor components.2–5 These oxygen functionalities make GtO sheets strongly hydrophilic, therefore the mild sonication of graphite oxide (GtO) in both aprotic polar solvents and water results monolayer exfoliation to form homogeneous and stable aqueous dispersions containing sheets with atomic thickness.6 Indeed, GtO consists of graphene oxide sheets with both covalently bound oxygen and non-covalently bound water between the carbon layers. Therefore, graphene oxide – the oxygenated form of a monolayer graphene platelet – should be noticed as an amphiphile with a largely hydrophobic basal plane (polyaromatic islands of unoxidized benzene rings) and hydrophilic edges (–COOH groups).7–10 GO sheets are composed of planar and graphene-like aromatic domains with a hexagonal ring based carbon network in the chair configuration bearing oxygen functional groups.11 GO is a single-layer of graphite oxide with a two-dimensional network of sp2- and sp3-hybridized carbon atoms, while an ideal graphene sheet consists of 100% sp2-bonded carbon atoms.12 The unique electronic structure of graphene oxide (GO) is heterogeneous owing to the presence of mixed sp2 and sp3 hybridizations.13 The availability of oxygen-containing functional groups on the GO sheets allows them to be functionalized with a wide range of organic and inorganic materials in covalent or non-covalent and/or ionic approaches.14 Thus, GO is a significant building block to prepare various functional materials.15 These oxygen functional groups afford mild acidic and oxidative properties for GO,16 and further functionalization can make stronger acid sites on these carbons.17–19 Unique properties of graphene oxide (GO) including the 2D structure, high stability and high surface areas make it a novel type of promising carbocatalysts which their catalytic performance can be promoted with functionalities to both sides of the carbon sheets.20 The functionalized GO has great potential for applications in biosensing,21 drug delivery,22–24 bio-analysis,25 gene delivery and bioimaging,26 photothermal therapy,27 hydrogen storage,28 transparent film,29 high efficiency catalysis,30–32 electronics and optoelectronics,33 and chemical and biochemical sensors.34 It is because of its exclusive characteristics such as high mechanical strength,35 good water dispersibility,36 facile surface modification,37 and photoluminescence.38
A heterocyclic moiety is a key structure in many bioactive natural and therapeutic products. Nitrogen heterocycles are important branches of pharmacologically active substances and they have been utilized as significant precursors in the synthesis of novel drugs. Out of the five major bases in nucleic acids, three, i.e., cytosine, uracil and thymine are pyrimidine derivatives which are found in DNA and RNA.39,40 Therefore, pyrimidines have become very important in the world of synthetic organic chemistry.41 3,4-Dihydropyrimidinones as biologically active compounds have been extensively used as drug-like scaffolds due to their pharmacological and therapeutic properties.42 Their derivatives such as monastrol, enastron, piperastrol,43 amlodipine and nicardipine44 have been developed as drugs. Biginelli reaction is one of the most efficient and straightforward procedures to obtain the DHPMs including the acid-catalyzed three components condensation in one-pot. The efficiency of this process is greatly limited owing to strong acidic and harsh reaction conditions.45 Thus, new modified routes have been developed to improve the efficiency of the Biginelli reaction in the presence of highly active, stable, and friendly environmental catalysts.
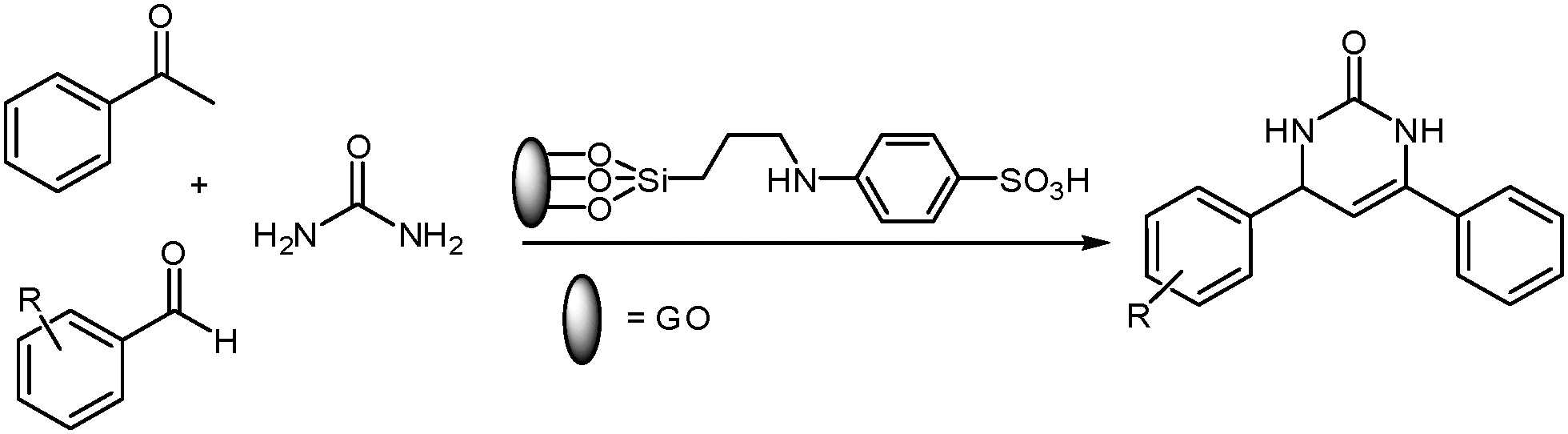
Herein, we report the highly efficient activity of organosilane sulfonated graphene oxide (SSi-GO) as an effective catalyst to prepare pyrimidinones (Schemes 1 and 2). First, the sulfonated graphene oxide nanosheets are prepared through facile covalent functionalization with sulfanilic acid. The functionalized GO is then used as a highly active, selective, reusable and stable catalyst to produce pyrimidinones. The carboxylic acid and sulfonic acid groups present in SSi-GO are potentially active sites for its superior catalytic performance. However, the organosilane sulfonated graphene oxide has not yet been used as a heterogeneous catalyst in the Biginelli reaction.
 |
| | Scheme 1 SSi-GO-catalyzed synthesis of dihydropyrimidinones. | |
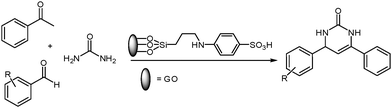 |
| | Scheme 2 SSi-GO-catalyzed synthesis of diarylpyrimidinones. | |
Experimental
Materials and methods
Chemical reagents were purchased from Merck and Aldrich with high purity and used as received without further purification. The completion of reactions was checked by the TLC technique on silica gel plates in the solvent system petroleum ether–EtOAc (V/V = 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). Melting points (°C) were determined in an open-glass capillary on an Electrothermal MK3 apparatus and are uncorrected. A Nicolet Magna FT-IR 550 spectrophotometer was used for recording the IR spectra using potassium bromide pellets in the range of 400–4000 cm−1. The proton and carbon NMR spectra were recorded on a Bruker DRX-400 spectrometer operated at 400 MHz using CDCl3 as a solvent and TMS as an interior standard. X-ray diffraction patterns were performed on a Holland Philips Xpert X-ray diffractometer with CuK radiation, (λ = 0.154056 nm) at a scanning speed of 2° min−1 from 10° to 100° (2θ). Scanning electron microscopy (SEM) was performed on a Quanta 200 SEM operated at 20 kV accelerating voltage. The samples were prepared for SEM by spreading a small drop containing GO onto a silicon wafer and by drying it almost completely in air at room temperature for 2 h; it was then transferred onto a SEM conductive tape. The transferred sample was coated with a thin layer of gold before measurement. Sonication was performed in a Shanghai Branson-BUG40-06 ultrasonic cleaner (with a frequency of 35 kHz and a nominal power of 200 W). A circulating water bath (DC2006, Shanghai Hengping Apparatus Factory) was adopted with an accuracy of 0.1 K to keep constant the reaction temperature.
:3). Melting points (°C) were determined in an open-glass capillary on an Electrothermal MK3 apparatus and are uncorrected. A Nicolet Magna FT-IR 550 spectrophotometer was used for recording the IR spectra using potassium bromide pellets in the range of 400–4000 cm−1. The proton and carbon NMR spectra were recorded on a Bruker DRX-400 spectrometer operated at 400 MHz using CDCl3 as a solvent and TMS as an interior standard. X-ray diffraction patterns were performed on a Holland Philips Xpert X-ray diffractometer with CuK radiation, (λ = 0.154056 nm) at a scanning speed of 2° min−1 from 10° to 100° (2θ). Scanning electron microscopy (SEM) was performed on a Quanta 200 SEM operated at 20 kV accelerating voltage. The samples were prepared for SEM by spreading a small drop containing GO onto a silicon wafer and by drying it almost completely in air at room temperature for 2 h; it was then transferred onto a SEM conductive tape. The transferred sample was coated with a thin layer of gold before measurement. Sonication was performed in a Shanghai Branson-BUG40-06 ultrasonic cleaner (with a frequency of 35 kHz and a nominal power of 200 W). A circulating water bath (DC2006, Shanghai Hengping Apparatus Factory) was adopted with an accuracy of 0.1 K to keep constant the reaction temperature.
General procedure for the preparation of graphene oxide (GO)
The graphene oxide (GO) nanosheets were prepared from natural graphite powder using the modified Hummer's method.1 In a typical synthesis process, graphite powder (3 g) and sodium nitrate (NaNO3, 1 g) were slowly added to a solution of 98% H2SO4 (46 mL) while stirring in an ice bath at 0–5 °C (15 min). Under vigorous stirring, potassium permanganate (KMnO4, 6 g) was slowly added to the suspension at 10–15 °C for 2 h. The resulting mixture was stirred continuously at 35 °C for another 30 min. Subsequently, distilled water (138 mL) was slowly added to the reaction vessel under vigorous stirring, which led to a color change to yellow and kept the temperature in the range of 95–98 °C for 1 h. Then, the reaction mixture was further diluted with warm distilled water (200 mL, 40 °C), followed by treated with 30% hydrogen peroxide (H2O2, 18 mL). The resultant brown solution was washed using 10% hydrochloric acid (HCl) solution, after that centrifuged for several times. The brown graphite oxide was collected by filtration and dried under vacuum at 60 °C for 24 h. To prepare graphene oxide (GO), 100 mg graphite oxide was dispersed in distilled water (100 mL) and sonicated in an ultrasonic bath cleaner (100 W) for hours to exfoliate graphitic oxide until the solution became clear. Indeed, graphene oxide (GO) in aqueous solution was generated by the oxidization and subsequently exfoliation of graphite. Afterwards, the GO solution was centrifuged for 10 min to remove any unexfoliated graphitic oxide. The GO powder was obtained after drying in a vacuum oven at 80 °C for 24 h. Scheme 3 briefly illustrates the used procedure to synthesize GO.
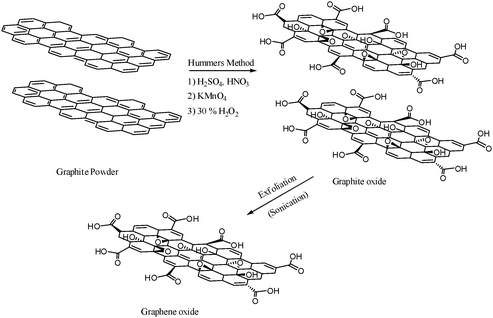 |
| | Scheme 3 Schematic model of the used procedure to synthesize graphene oxide. | |
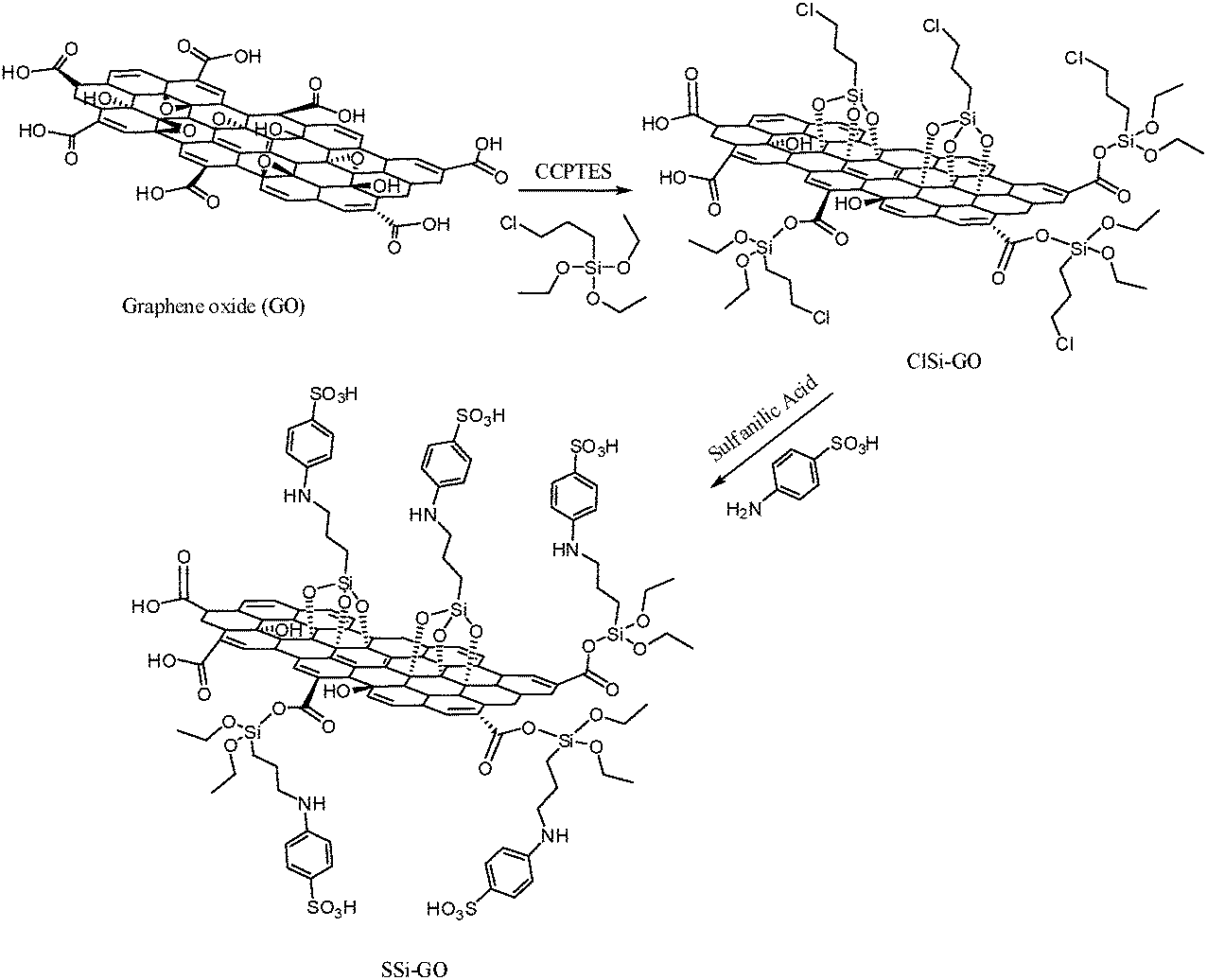
General procedure for the preparation of organosilane sulfonated graphene oxide (SSi-GO)
The process for the synthesis of organosilane sulfonated graphene oxide from graphene oxide was carried in the following manner. The functionalized, chemically converted graphene nanosheets (SSi-GO) can be synthesized via the covalent interaction between GO and sulfanilic acid. In a typical procedure, the functionalization of GO nanosheets was performed using 3-chloropropyltriethoxysilane (CPTES) as the sulfonic acid functional group precursor. The reaction was carried out in toluene at temperature of 110 °C under reflux conditions for 24 h, with ratios of 3:1:2 of toluene, GO and CPTES, respectively. Afterwards, the chloro groups were grafted onto the GO nanosheets to achieve chlorofunctionalized GO (ClSi-GO). The prepared samples were filtered and washed three times with ethanol to remove the precursor residue. Samples were then dried at temperature of 80 °C for 8 h. Finally, sulfanilic acid (1.5 g) was added to a mixture of ClSi-GO (1 g) and triethylamine (1.2 mL, 8.6 mmol) in 30 mL of toluene. The solution was continuously stirred at 110 °C under reflux conditions for 48 h to produce SSi-GO. After the reaction, the mixture was washed with toluene and chloroform for several times and then collected by a centrifuge and dried in a vacuum oven at 80 °C for 24 h. The synthesis of organosilane sulfonated graphene oxide was described in several steps in Scheme 4.
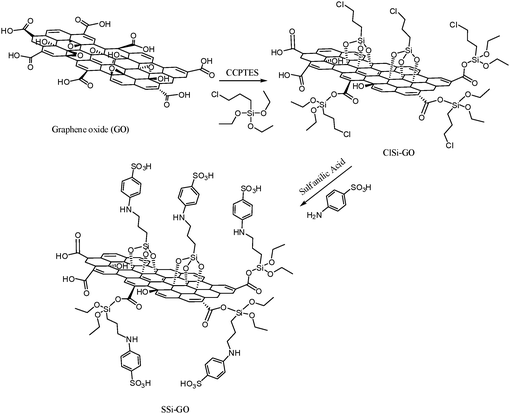 |
| | Scheme 4 Schematic illustration to prepare organosilane sulfonated graphene oxide. | |
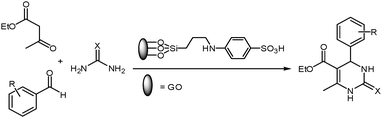
General procedure for the preparation of pyrimidinone derivatives
The solution of aromatic aldehyde (1 mmol), ethyl acetoacetate or acetophenone (1 mmol) and urea or thiourea (1.5 mmol) in 5 ml of ethanol was stirred and refluxed at 80 °C in the presence of SSi-GO (0.1 g). The completion of the reaction was determined on TLC plates using ethyl acetate/petroleum ether mixture as a mobile phase. After completion, the mixture was cooled to room temperature, then crushed ice was added to precipitate the product. The yellow solid precipitate was separated by filtration through a Buckner funnel and washed with cold water to remove excess of urea and dried under vacuum. In addition, it was further purified by recrystallization from hot ethanol to afford pure pyrimidinones. Then, the filtrate obtained was concentrated under reduced pressure to recover the catalyst. The recovered catalyst was washed with water, dried under a vacuum oven at 90 °C for about 3 h and reused for subsequent reaction.
Results and discussion
The preparation of graphene oxide nanosheets from graphite powder via the Hummer's method affords hydrophilic oxygen-containing functional groups such as carboxyl, epoxy, and hydroxyl on the surfaces of GO nanosheets, which can stabilize the dispersion of these sheets in aqueous media.46 Graphene oxide, which is introduced as oxidized graphene, was modified by treatment with sulfanilic acid to afford sulfur-containing acid groups onto the carbon surface.
Screening of reaction conditions
The catalytic activity of organosilane sulfonated graphene oxide was investigated to produce dihydropyrimidinones. Initially, to evaluate the effect of the catalyst, the reaction of benzaldehyde, ethyl acetoacetate, and urea in the presence and absence of organosilane sulfonated graphene oxide nanocatalyst under reflux conditions was selected as a model reaction. The results are summarized in Table 1. It could be seen that without any catalysts no desirable DHPMs product was obtained even after prolonging the reaction time (Table 1, entry 1). It indicated that the catalyst is necessary for this reaction. The yield increased linearly with increase in the amount of the catalyst. Hence, 0.1 g of the catalyst was used in the reaction. It should be noted that the high catalytic activity of solid acid of SSi-GO was attributed to the quantity and type of acidic groups and high surface area. In other experiments, the same reaction was carried out with different amounts of urea in the presence of the same quantity of the catalyst (Table 2). Based on these results, the yield increased with the addition of urea and the optimal amount of urea was 1.5 mmol. Thus, the best result was obtained with 1.0:1.0:1.5 molar ratios of aldehyde, 1,3-dicarbonyl compounds, urea or thiourea, and 0.1 g of SSi-GO.
Table 1 The effect of various amounts of the catalyst for Biginelli reaction
| Entry |
Catalyst (g) |
Time (min) |
Yield (%) |
| The yield was calculated according to the limiting factor (aldehyde). |
| 1 |
— |
50 |
— |
| 2 |
0.05 |
40 |
83 |
| 3 |
0.1 |
20 |
94 |
| 4 |
0.2 |
25 |
92 |
| 5 |
0.3 |
20 |
94 |
Table 2 Optimization of amount of urea for the synthesis of DHPMs
| Entry |
Urea (mmol) |
Yield (%) |
| 1 |
1 |
60 |
| 2 |
1.5 |
94 |
| 3 |
2 |
92 |
| 4 |
2.5 |
90 |
The potency of the reaction was influenced by temperature. It was indicated that the reaction did not proceed at room temperature (Table 3, entry 1). Furthermore, it was observed that the yield of the reaction increased with the increasing of the reaction temperature ranging from 60 to 80 °C (entries 2–4), but there was not much change when above 80 °C.
Table 3 Optimization of reaction temperature for the synthesis of DHPMs
| Entry |
Temperature (°C) |
Yield (%) |
| 1 |
r.t. |
— |
| 2 |
60 |
65 |
| 3 |
70 |
79 |
| 4 |
80 |
94 |
| 5 |
100 |
89 |
Next, the effect of various solvents on the improvement of reaction was tested. Typically, solvents such as H2O, CH3CN, DMF, EtOH and CH2Cl2 were selected for comparison. As shown, the polar solvents such as acetonitrile, H2O and EtOH resulted good yields, while low yield of the product was obtained using DMF and CH2Cl2 (Table 4, entries 5 and 6). This may be attributed to the better solubility of the starting materials in the polar solvents. In comparison with aprotic solvents, a significant increase in the yield of the product was obvioused in a shorter time period when the model reaction was occurred under solvent-free conditions. The highest yield of the product was obtained in EtOH as a protic and polar solvent within 20 min, but the reaction in other solvents afforded low yields in longer reaction times.
Table 4 Effect of various solvents on the Biginelli reaction
| Entry |
Solvent |
Time (min) |
Yield (%) |
| 1 |
Solvent-free |
35 |
80 |
| 2 |
Water |
40 |
84 |
| 3 |
EtOH |
20 |
94 |
| 4 |
CH3CN |
30 |
89 |
| 5 |
DMF |
35 |
65 |
| 6 |
CH2Cl2 |
40 |
60 |
The most important feature of the present reaction is the recyclability of the catalyst. Subsequently, the recycling and reusability performance of the SSi-GO nanocatalyst were investigated using the model reaction under the optimal conditions. After the catalytic reaction, water was added and the recovered catalyst was removed from the reaction mixture by filtration and washed with ethanol, and dried under vacuum at 90 °C for 3 h. The recovered catalyst was reused five times without significant loss of catalytic activity. These results show that the catalyst is very stable. This observation strongly confirms the high recycling efficiency of the nanocatalyst which is a significant property from the economical and environmental points of view (Fig. 1).
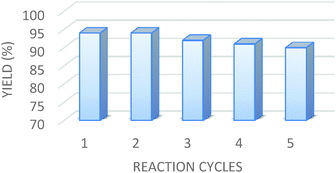 |
| | Fig. 1 Reusability of the catalyst for the synthesis of 4a. | |
The results were tabulated to compare the efficiency of the present catalyst with some of the reported catalysts for the promotion of the synthesis of DHPMs. The present method was more efficient according to Table 5.
Table 5 Comparative study using published methods
| Entry |
Catalyst |
Condition |
Time (min) |
Yield (%) |
Ref. |
| 1 |
PTA@MIL-101 |
Solvent free, 100 °C |
60 |
90 |
47
|
| 2 |
Cu@PMO-IL |
Solvent free, 70 °C |
50 |
96 |
48
|
| 3 |
[TEBSA]HSO4 |
Solvent free, reflux |
75 |
88 |
49
|
| 4 |
PPA |
Solvent free, grinding |
25 |
84 |
50
|
| 5 |
Nano-γ-Fe2O3-SO3H |
Solvent-free, 60 °C |
180 |
91 |
51
|
| 6 |
SiO2-BaCl2/SF |
Solvent-free, 85 °C |
45 |
93 |
52
|
| 7 |
Mn@PMO-IL |
Solvent free, 70 °C |
45 |
97 |
53
|
| 8 |
SiO2-H2PO3 |
Solvent-free, 60 °C |
150 |
92 |
54
|
| 9 |
ErCl3 |
Solvent-free, 120 °C |
30 |
92 |
55
|
| 10 |
SSi-GO |
Reflux, 80 °C |
20 |
94 |
This study |
To investigate the substrates scope, the reactions were carried out using various aldehydes, 1,3-dicarbonyl compounds (acetophenone), and urea (or thiourea) catalyzed by SSi-GO under the optimal conditions. The results are shown in Tables 6 and 7. The reactions proceeded very efficiently within relatively short reaction time. The results illustrate that the type and position of the substituent have no significant effect on the activity of the catalyst and the reaction yield. These observations confirm the high efficiency of the nanocatalyst to convert an extensive range of aldehyde substrates to a series of structurally diverse pyrimidinones in high purity. Additionally, thiourea was applied instead of urea to successfully provide the corresponding 3,4-dihydropyrimidin-2(1H)-thiones in good yields (Table 6, entries 10 and 11). It is essential to note that the methodology was also successfully used for acetophenone substrates and corresponding adducts obtained in good yields. However, ketones required a longer reaction time (Table 7). The products were confirmed by comparing their melting points with authentic samples, FTIR and 1H NMR spectroscopies.
Table 6 Synthesis of 3,4-dihydropyrimidin-2(1H)-one/thiones in the presence of the SSi-GO catalyst under reflux conditions
| Entry |
Aldehyde |
X |
Product |
Mp (°C) |
Time (min) |
Yield (%) |
| Obs. |
Lit. |
| 1 |
H |
O |
4a
|
200–202 |
200–20156 |
20 |
94 |
| 2 |
4-Cl |
O |
4b
|
211–215 |
209–21157 |
20 |
96 |
| 3 |
4-Me |
O |
4c
|
213–216 |
212–21458 |
25 |
90 |
| 4 |
2-OH |
O |
4d
|
201–203 |
200–20259 |
25 |
89 |
| 5 |
2-Cl |
O |
4e
|
215–218 |
216–21859 |
27 |
91 |
| 6 |
2-F |
O |
4f
|
239–240 |
233–23560 |
30 |
98 |
| 7 |
Thiophene |
O |
4g
|
215–217 |
215–21761 |
20 |
92 |
| 8 |
3-NO2 |
O |
4h
|
229–230 |
229–23162 |
23 |
94 |
| 9 |
4-NMe2 |
O |
4i
|
230–232 |
230–23263 |
35 |
95 |
| 10 |
Thiophene |
S |
4j
|
216–217 |
215–21662 |
30 |
90 |
| 11 |
H |
S |
4k
|
197–199 |
199–20064 |
25 |
93 |
Table 7 Synthesis of diarylpyrimidin-2(1H)-ones in the presence of the SSi-GO catalyst under reflux conditions
| Entry |
Aldehyde |
Ketone |
X |
Product |
Mp (°C) |
Yield (%) |
| Obs. |
Lit. |
| 1 |
H |
H |
O |
6a
|
229–231 |
228–23065 |
89 |
| 2 |
3,4-(OMe)2 |
H |
O |
6b
|
245–246 |
243–24565 |
90 |
| 3 |
3-OMe |
H |
O |
6c
|
258–259 |
257–25865 |
93 |
| 4 |
4-Cl |
H |
O |
6e
|
268–269 |
267–26966 |
98 |
| 5 |
4-Me |
H |
O |
6f
|
249–251 |
248–25065 |
87 |
| 6 |
4-OMe |
H |
O |
6g
|
260–261 |
259–26165 |
92 |
| 7 |
2-OMe |
H |
O |
6h
|
265–267 |
266–26765 |
89 |
| 8 |
4-OH |
H |
O |
6i
|
256–258 |
257–25866 |
77 |
| 9 |
2,4-Cl2 |
H |
O |
6j
|
270–272 |
271–27465 |
83 |
| 10 |
2-Cl |
H |
O |
6k
|
263–264 |
264–26565 |
91 |
Structural characterization of the catalyst
The crystalline phases of graphite and graphite oxide samples prepared were investigated by the X-ray diffraction (XRD) patterns, as shown in Fig. 2. It can be seen in Fig. 2 that the natural graphite presented the very strong diffraction peak at 2θ = 26.0° with the (002) plane of graphite. The diffraction peak at around 2θ = 43.1° is related to the (100) plane of the hexagonal structure of carbon.67 The spectrum of graphite oxide after oxidation for 30 min exhibited the same peak but a little bit weaker than raw graphite and another peak at 2θ = 11.7° appeared. However, after oxidation for 45 min, the peak becomes even weaker. It can be observed that after complete oxidation, the sharp diffraction peak disappeared in graphite nanosheets (2θ = 26.0°), and a new diffraction peak (2θ = 11.7° with 0.8 nm d-spacing corresponds to the (001) reflection) appeared in graphite oxide, indicating the damage of the regular crystalline pattern of graphite during the oxidation. The characteristic diffraction peak (001) of graphite oxide nanosheets and its increased d-spacing is associated to introduce oxygenated functional groups attached on both sides and edges of carbon sheets as well as water molecules trapped in the interlayer galleries of hydrophilic graphite oxide.
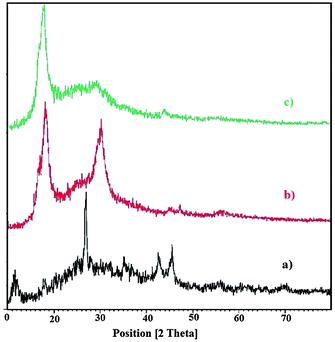 |
| | Fig. 2 X-ray diffraction patterns of (a) graphite, (b) graphite oxide after oxidation for 30 min and (c) graphite oxide after oxidation for 45 min. | |
Fig. 3 shows typical scanning electron microscopy (SEM) images of the GtO and GO. Indeed, SEM images show the structures and morphology of graphite oxide before (Fig. 3a and b) and after (Fig. 3c and d) exfoliation. Compared with graphite oxide which is a laminar compound with layers stucking together, graphene oxide exhibits a relatively rough surface and crumpling features with clear layers. The atomic scale roughness arises from structural defects (sp3 bonding) generated on the originally atomically flat graphite oxide sheet. It can be identified that GO nanosheets are relatively exfoliated and wrinkled and afforded an increase in the distance between adjacent sheets and reduction in interaction between sheets. This increased spacing is considerably different depending on the amount of water intercalated within the stacked-sheet structure.68 These factors have potential advantages as the active sites, which can be easily produced on both sides of the two dimensional graphene oxide sheets.
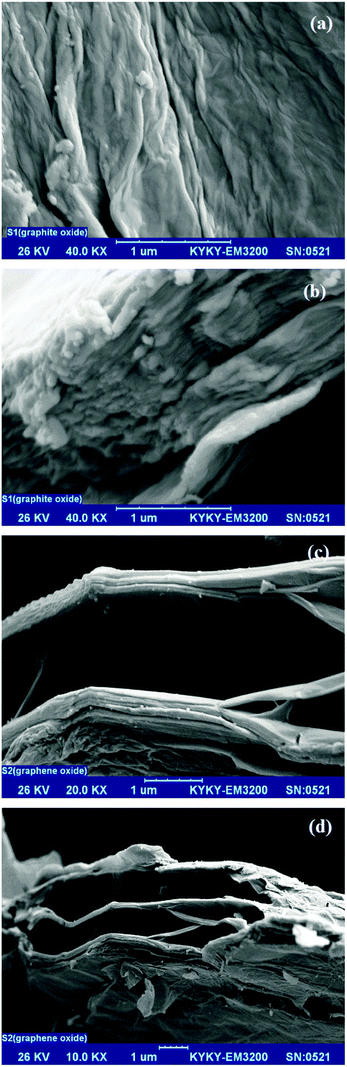 |
| | Fig. 3 SEM images (a) and (b) graphite oxide, (c) and (d) graphene oxide (GO). | |
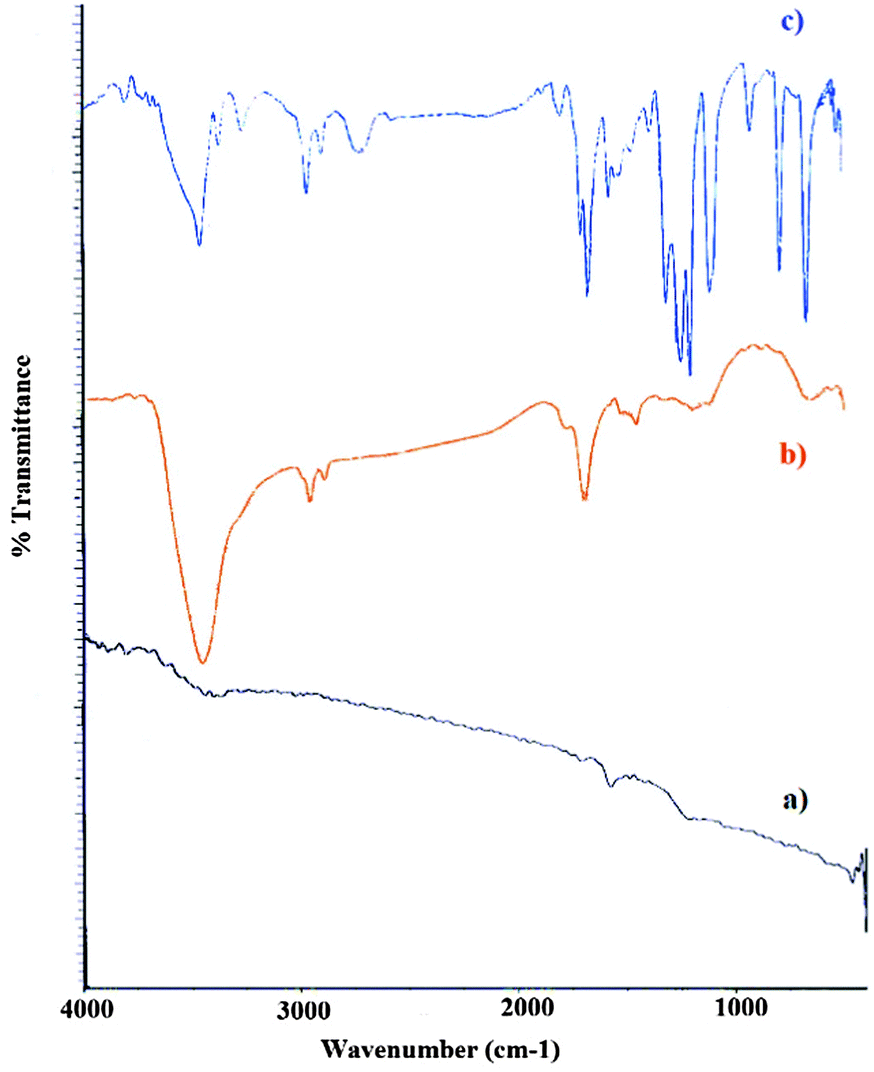
The FTIR spectra of graphite, graphite oxide and organosilane sulfonated graphene oxide were obtained to confirm the presence of different functional groups on the graphene nanosheets (Fig. 4). Graphite has two peaks at 3430 cm−1 (O–H stretching vibrations due to adsorbed water) and 1610 cm−1 (aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, skeletal vibrations of graphitic domains). Large amounts of oxygen-containing functional groups are found in the FTIR spectrum of graphite oxide nanosheets. The strong absorption bands at 3428, 2923, 1729, 1628, 1385, and 1121 cm−1 for graphite oxide confirm the existence of –OH, C–H, CO in COOH, unoxidized graphitic skeletal domains (CC aromatic) and the adsorbed water molecules,69 carboxylic C–OH stretching and C–O stretching vibration functional groups, respectively.70 These results are in good agreement with the structure and morphology of graphite oxide and confirm the successful oxidation of graphite. Functionalization with aryl SO3H does not change the structure of graphite oxide, as shown in Fig. 4c. The FTIR analysis in Fig. 4c indicates that aryl sulfonic acid groups were successfully introduced into GO sheets. The strong absorption peaks at 1159 and 1230 cm−1 are assigned to the symmetric and asymmetric stretching vibrations of SO in the –SO3H group, respectively.71 SSi-GO also exhibits additional bands at 1023 and 692 cm−1 which is indicated by S–phenyl and S–O groups, confirming the presence of aryl SO3H groups covalently bonded to the graphene sheet. A broad peak at 3228 cm−1 corresponded to –OH groups on the surface and also –SO3H. Additionally, the band stretching from 2888 to 2924 cm−1 attributed to the presence of aromatic CH groups and aliphatic C–H groups of the (CH2)3 chains in the CCPTES sulfanilic acid precursor.72 The broad bands were also seen in the regions 3426 and 832 cm−1 which are related to aromatic NH and OOP NH groups. The strong absorption peak at 1117 cm−1 attributed to the Si–O stretching vibrations.
C, skeletal vibrations of graphitic domains). Large amounts of oxygen-containing functional groups are found in the FTIR spectrum of graphite oxide nanosheets. The strong absorption bands at 3428, 2923, 1729, 1628, 1385, and 1121 cm−1 for graphite oxide confirm the existence of –OH, C–H, CO in COOH, unoxidized graphitic skeletal domains (CC aromatic) and the adsorbed water molecules,69 carboxylic C–OH stretching and C–O stretching vibration functional groups, respectively.70 These results are in good agreement with the structure and morphology of graphite oxide and confirm the successful oxidation of graphite. Functionalization with aryl SO3H does not change the structure of graphite oxide, as shown in Fig. 4c. The FTIR analysis in Fig. 4c indicates that aryl sulfonic acid groups were successfully introduced into GO sheets. The strong absorption peaks at 1159 and 1230 cm−1 are assigned to the symmetric and asymmetric stretching vibrations of SO in the –SO3H group, respectively.71 SSi-GO also exhibits additional bands at 1023 and 692 cm−1 which is indicated by S–phenyl and S–O groups, confirming the presence of aryl SO3H groups covalently bonded to the graphene sheet. A broad peak at 3228 cm−1 corresponded to –OH groups on the surface and also –SO3H. Additionally, the band stretching from 2888 to 2924 cm−1 attributed to the presence of aromatic CH groups and aliphatic C–H groups of the (CH2)3 chains in the CCPTES sulfanilic acid precursor.72 The broad bands were also seen in the regions 3426 and 832 cm−1 which are related to aromatic NH and OOP NH groups. The strong absorption peak at 1117 cm−1 attributed to the Si–O stretching vibrations.
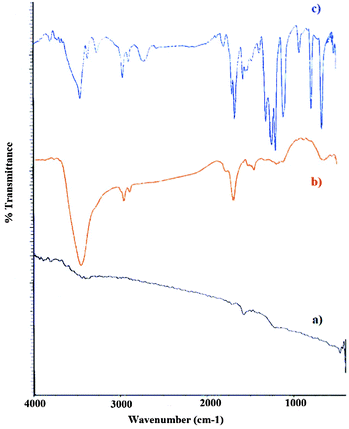 |
| | Fig. 4 FT-IR spectra for (a) graphite, (b) graphite oxide, and (c) SSi-GO. | |
Energy dispersive spectroscopy (EDS) reveals the presence of –OSO3H functional groups on SSi-GO. EDX elemental analysis shows that the element mass ratios of carbon, nitrogen, oxygen, silicon and sulfur in organosilane sulfonated graphene oxide are 58.32%, 17.42%, 20.79%, 0.39% and 3.09 wt%, respectively. The elemental analysis of SSi-GO presented that the calculated density of the sulfonic acid group on sulfonated graphene oxide is 0.96 mmol g−1 of –SO3H based on the sulfur percentage (3.09 wt%). The O/S atom ratio (6.7:1) is higher than 3:1 owing to the presence of residual epoxide, carboxyl and hydroxyl groups on the sulfonated graphene oxide sheets (Fig. 5).
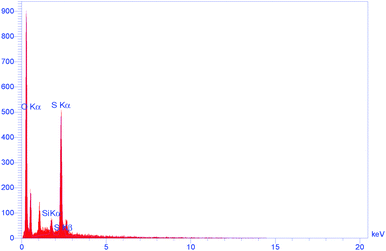 |
| | Fig. 5 The EDS spectrum of SSi-GO. | |
Furthermore, the carbon, nitrogen, oxygen, silicon and sulfur distribution maps (Fig. 6) show that the SSi-GO sheets were densely and homogeneously functionalized with sulfur, reflecting the uniformity of the modification treatment.
 |
| | Fig. 6 Elemental distribution maps in the SSi-GO region shown by EDX. | |
Conclusions
In summary, organosilane functionalized graphene oxides (SSi-GO) were successfully synthesized and well characterized. The functionalized chemically modified organosilane graphene oxide was simply prepared via covalent functionalization with a reactive surfactant, sulfanilic acid. Hence, this study highlights that SSi-GO can be a stable, active and efficient carbocatalyst to improve the yield of pyrimidinones through one-pot multi-component reaction of aromatic aldehydes, ethyl acetoacetate (or aromatic ketones) and urea or thiourea under reflux conditions. The strongly acidic aryl SO3H groups are responsible for the catalytic activity and high stability of this solid acid catalyst. Moreover, the usage of SSi-GO as a Brønsted acid and a reusable carbocatalyst can be extended to other organic reactions.
Acknowledgements
We thanks financial support for this work by the university of Kashan Research Council (Grant no. 363022/21).
References
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- N. I. Kovtyukhova, P. I. Olliver, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771 CrossRef CAS.
- A. B. Bourlinos, D. Gournis, D. Petridis, T. Szabó, A. Szeri and I. Dékány, Langmuir, 2003, 19, 6050 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. M. Kohlhass, A. Kleinhammes, Y. Y. Jia, Y. Wu, S. B. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS.
- H. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53 CrossRef CAS.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3(2), 101 CrossRef CAS PubMed.
- A. Lerf, H. Y. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477 CrossRef CAS.
- J. Kim, L. J. Cote, F. Kim, W. Yuan, K. R. Shull and J. X. Huang, J. Am. Chem. Soc., 2010, 132, 8180 CrossRef CAS PubMed.
- L. J. Cote, J. Kim, V. C. Tung, J. Y. Luo, F. Kim and J. X. Huang, Pure Appl. Chem., 2011, 83(1), 95 CAS.
- F. Kim, L. J. Cote and J. X. Huang, Adv. Mater., 2010, 22(17), 1954 CrossRef CAS PubMed.
- T. Szabo, O. Berkesi, P. Forgo, K. Josepovits, Y. Sanakis, D. Petridis and I. Dekany, Chem. Mater., 2006, 18, 2740 CrossRef CAS.
- K. A. Mkhoyan, A. W. Contryman, J. Silcox, D. A. Stewart, G. Eda, C. Mattevi, S. Miller and M. Chhowalla, Nano Lett., 2009, 9(3), 1058 CrossRef CAS PubMed.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577 CrossRef CAS.
-
(a) S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217 CrossRef CAS PubMed;
(b) H. Bai, Y. X. Xu, L. Zhao, C. Li and G. Q. Shi, Chem. Commun., 2009, 1667 RSC.
-
(a) S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS PubMed;
(b) Q. Su, S. Pang, V. Alijani, C. Li, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 3191 CrossRef CAS.
- D. R. Dreyer, H. P. Jia and C. W. Bielawski, Angew. Chem., 2010, 122, 6965 CrossRef.
- J. Li, G. Zhang, H. Chen, S. Wang, G. Zhang, F. Zhang and X. Fan, Chem. Sci., 2011, 2(3), 484 RSC.
- E. Lam, J. H. Chong, E. Majid, Y. liu, S. Hrapovic, A. C. W. Leung and J. H. T. Luong, Carbon, 2012, 50, 1033 CrossRef CAS.
- F. Liu, J. Sun, L. Zhu, X. Meng, C. Qi and F.-S. Xiao, J. Mater. Chem., 2012, 22, 5495 RSC.
- C. Su and K. P. Loh, Acc. Chem. Res., 2012, 46(10), 2275 CrossRef PubMed.
-
(a) C. H. Lu, H. H. Yang, C. L. Zhu, X. Chen and G. N. Chen, Angew. Chem., 2009, 121(26), 4879 CrossRef;
(b) S. J. He, B. Song, D. Li, C. F. Zhu, W. P. Qi, Y. Q. Wen, L. H. Wang, S. P. Song, H. P. Fang and C. H Fan, Adv. Funct. Mater., 2010, 20, 453 CrossRef CAS.
- Z. Liu, J. Robinson, X. M. Sun and H. J. Dai, J. Am. Chem. Soc., 2008, 130, 10876 CrossRef CAS PubMed.
- L. Zhang, J. Xia, Q. Zhao, L. Liu and Z. Zhang, Small, 2010, 6, 537 CrossRef CAS PubMed.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203 CrossRef CAS PubMed.
- L. Cui, Y. Song, G. Ke, Z. Guan, H. Zhang, Y. Lin, Y. Huang, Z. Zhu and C. J. Yang, Chem. – Eur. J., 2013, 19(32), 10442 CrossRef CAS PubMed.
- H. Kim, R. Namgung, K. Singha, I. K. Oh and W. J. Kim, Bioconjugate Chem., 2011, 22(12), 2558 CrossRef CAS PubMed.
- J. T. Robinson, S. M. Tabakman, Y. Liang, H. Wang, H. S. Casalongue, D. Vinh and H. Dai, J. Am. Chem. Soc., 2011, 133, 6825 CrossRef CAS PubMed.
- L. Wang, K. Lee, Y. Y. Sun, M. Lucking, Z. Chen, J. J. Zhao and S. B. Zhang, ACS Nano, 2009, 3, 2995 CrossRef CAS PubMed.
- J. Zhao, S. Pei, W. Ren, L. Gao and H. M. Cheng, ACS Nano, 2010, 4(9), 5245 CrossRef CAS PubMed.
- C. Chen, W. Cai, M. Long, B. Zhou, Y. Wu, D. Wu and Y. Feng, ACS Nano, 2010, 4, 6425 CrossRef CAS PubMed.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380 CrossRef CAS PubMed.
- J. Liu, H. Bai, Y. Wang, Z. Liu, X. Zhang and D. D. Sun, Adv. Funct. Mater., 2010, 20, 4175 CrossRef CAS.
- G. Eda and M. Chhowalla, Adv. Mater., 2010, 22(22), 2392 CrossRef CAS PubMed.
- J. Balapanuru, J. X. Yang, S. Xiao, Q. Bao, M. Jahan, L. Polavarapu, J. Wei, Q. H. Xu and K. P. Loh, Angew. Chem., Int. Ed., 2010, 49, 6549 CrossRef CAS PubMed.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457 CrossRef CAS PubMed.
- L. Fan, C. Luo, M. Sun, X. Li and H. Qiu, Colloids Surf., B, 2013, 103, 523 CrossRef CAS PubMed.
- S. M. Kang, S. Park, D. Kim, S. Y. Park, R. S. Ruoff and H. Lee, Adv. Funct. Mater., 2011, 21(1), 108 CrossRef CAS.
-
(a) Z. Luo, P. M. Vora, E. J. Mele, A. C. Johnson and J. M. Kikkawa, Appl. Phys. Lett., 2009, 94(11), 111909 CrossRef;
(b) Q. Mei, K. Zhang, G. Guan, B. Liu, S. Wang and Z. Zhang, Chem. Commun., 2010, 46(39), 7319 RSC.
- M. C. Etienne, S. Cheradame, J. L. Fischel, P. Formento, O. Dassonville, N. Renee, M. Schneider, A. Thyss, F. Demard and G. Milano, J. Clin. Oncol., 1995, 13, 1663 CAS.
- V. I. Saloutin, Y. V. Burgat, O. G. Kuzueva, C. O. Kappe and O. N. Chupakhin, J. Fluorine Chem., 2000, 103, 17 CrossRef CAS.
- A. I. McDonald and L. E. Overman, J. Org. Chem., 1999, 64, 1520 CrossRef CAS PubMed.
- C. O. Kappe, Eur. J. Med. Chem., 2000, 35, 1043 CrossRef CAS PubMed.
- H. L. Luo, W. Yang, Y. Li and S. F. Yin, Chem. Nat. Compd., 2010, 46(3), 412 CrossRef.
- G. C. Rovnyak, S. D. Kimball, B. Beyer, G. Cucinotta, J. D. Dimarco, J. ougoutas, A. Hedberg, M. Malley, J. P. McCarthy, R. Zhang and S. Moreland, J. Med. Chem., 1995, 38(1), 119 CrossRef CAS PubMed.
- P. Biginelli, Gazz. Chim. Ital., 1893, 23, 360 Search PubMed.
- R. Bissessur and S. F. Scully, Solid State Ionics, 2007, 178, 877 CrossRef CAS.
- M. Saikia, D. Bhuyan and L. Saikia, Appl. Catal., A, 2015, 505, 501 CrossRef.
- D. Elhamifar, F. Hosseinpoor, B. Karimi and S. Hajati, Microporous Mesoporous Mater., 2015, 204, 269 CrossRef CAS.
- M. S. SushilkumarDhanmane, Chem. Mater. Res., 2015, 7(3), 27 Search PubMed.
- Y. Zhao, Y. Zhao, J. Zhang, H. Liu and L. Liang, Indian J. Chem., 2015, 45B, 139 Search PubMed.
- E. Kolvari, N. Koukabi and O. Armandpour, Tetrahedron, 2014, 70, 1383 CrossRef CAS.
- F. Hatamjafari, J. Appl. Chem. Res., 2015, 9(1), 95 Search PubMed.
- D. Elhamifar, M. Nasr-Esfahani, B. Karimi, R. Moshkelgosha and A. Shabani, ChemCatChem, 2014, 6, 2593 CrossRef CAS.
- M. Pramanik and A. Bhaumik, ACS Appl. Mater. Interfaces, 2014, 6, 933 CAS.
- M. liverio, P. Costanzo, M. Nardi, I. Rivalta and A. Procopio, ACS Sustainable Chem. Eng., 2014, 2, 1228 CrossRef.
- A. Shaabani and A. Bazgir, Tetrahedron Lett., 2004, 45, 2575 CrossRef CAS.
- R. V. Yarapathi, S. Kurva and S. Tammishetti, Catal. Commun., 2004, 5(9), 511 CrossRef CAS.
- R. Tayebee, B. Maleki and M. Ghadamgahi, Chin. J. Catal., 2012, 33(4), 659 CrossRef CAS.
- A. Paraskar, G. Dewkar and A. Sudalai, Tetrahedron Lett., 2003, 44(16), 3305 CrossRef CAS.
- A. R. Gholap, K. Venkatesan, T. Daniel, R. Lahoti and K. Srinivasan, Green Chem., 2004, 6(3), 147 RSC.
- M. Nasr-Esfahani, S. J. Hoseini and F. Mohammadi, Chin. J. Catal., 2011, 32, 1484 CrossRef CAS.
- N. Y. Fu, Y. F. Yuan, Z. Cao, S. W. Wang, J. T. Wang and C. Peppe, Tetrahedron, 2002, 58, 4801 CrossRef CAS.
- C. V. Reddy, M. Mahesh, P. Raju, T. R. Babu and V. Reddy, Tetrahedron Lett., 2002, 43, 2657 CrossRef CAS.
- A. Rajack, K. Yuvaraju, C. Praveen and Y. Murthy, J. Mol. Catal. A: Chem., 2013, 370, 197 CrossRef CAS.
- B. Liang, X. Wang, J. Wang and Z. Du, Tetrahedron, 2007, 63, 1981 CrossRef CAS.
- Y. M. Ren and C. Cai, Monatsh. Chem., 2009, 140(1), 49 CrossRef CAS.
- J. J. Niu and J. N. Wang, Electrochim. Acta, 2008, 53, 8058 CrossRef CAS.
- W. Scholz and H. P. Boehm, Z. Anorg. Allg. Chem., 1969, 369(3–6), 327 CrossRef CAS.
- T. Szabo, O. Berkesi and I. Dekany, Carbon, 2005, 43(15), 3186 CrossRef CAS.
- G. I. Titelman, V. Gelman, S. Bron, R. L. Khalfin, Y. Cohen and H. B. Peled, Carbon, 2005, 43(3), 641 CrossRef CAS.
- L. Zhang, J. Xia, Q. Zhao, L. Liu and Z. Zhang, Small, 2010, 6, 537 CrossRef CAS PubMed.
- Y. Si and E. Samulski, Nano Lett., 2008, 8, 1679 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |