
The S2 Cu(I) site in CupA from Streptococcus pneumoniae is required for cellular copper resistance†
Yue
Fu‡
ab,
Kevin E.
Bruce
c,
Hongwei
Wu
a and
David P.
Giedroc
*a
aDepartment of Chemistry, Indiana University, Bloomington, Indiana 47405-7102, USA. E-mail: giedroc@indiana.edu
bGraduate Program in Biochemistry, Indiana University, Bloomington, Indiana 47405-7102, USA
cDepartment of Biology, Indiana University, Bloomington, Indiana 47405, USA
First published on 1st September 2015
Abstract
Pathogenic bacteria have evolved copper homeostasis and resistance systems for fighting copper toxicity imposed by the human immune system. Streptococcus pneumoniae is a respiratory pathogen that encodes an obligatorily membrane-anchored Cu(I) binding protein, CupA, and a P1B-type ATPase efflux transporter, CopA. The soluble, cytoplasmic domain of CupA (sCupA) contains a binuclear Cu(I) cluster consisting of S1 and S2 Cu(I) ions. The NMR solution structure of apo-sCupA reveals the same cupredoxin fold of Cu2–sCupA, except that the Cu(I) binding loop (residues 112–116, harboring S2 Cu ligands M113 and M115) is highly dynamic as documented by both backbone and side chain methionine methyl order parameters. In contrast to the more solvent exposed, lower affinity S2 Cu site, the high affinity S1 Cu-coordinating cysteines (C74, C111) are pre-organized in the apo-sCupA structure. Biological experiments reveal that the S1 site is largely dispensable for cellular Cu resistance and may be involved in buffering low cytoplasmic Cu(I). In contrast, the S2 site is essential for Cu resistance. Expression of a chimeric CopZ chaperone fused to the CupA transmembrane helix does not protect S. pneumoniae from copper toxicity and substitution of a predicted cytoplasm-facing Cu(I) entry metal-binding site (MBS) on CopA also gives rise to a Cu-sensitivity phenotype. These findings suggest that CupA and CopA may interact and filling of the CupA S2 site with Cu(I) results in stimulation of cellular copper efflux by CopA.
Introduction
All bacteria are capable of tightly regulating the concentrations of bioavailable late d-block transition metals from Mn to Zn.1,2 Although all cells accumulate transition metals to varying degrees, they have evolved to limit the availability of highly competitive, strongly binding, metals, e.g., Cu(I) and Zn(II), by minimizing the “free” or rapidly exchanging pool of these metals relative to more weakly bound metals, including Mn(II) and Fe(II).3,4 This is perhaps best understood for cuprous ion Cu(I), which will predominate under the reducing conditions of the cell cytoplasm.4 A number of reports reveal that elevated cytoplasmic Cu(I) leads to disassembly of solvent-exposed Fe–S clusters, which can have many downstream metabolic effects.5–7 Since there is accumulating evidence that Cu is concentrated in phagolysosomes in which facultative intracellular pathogens, e.g., Mycobacterium tuberculosis, are able to survive,8,9 there are increasing efforts to understand the molecular details of Cu resistance in bacterial pathogens.Many bacteria encode a small cytoplasmic high affinity Cu(I) binding protein, termed a copper chaperone, that plays a role in limiting the collateral damage of copper toxicity.10,11 The first copper chaperone to be functionally and structurally characterized was CopZ from Enterococcus hirae.12 Subsequent NMR and X-ray crystallographic studies showed that CopZ was indeed representative of a major class of copper chaperones in prokaryotes, designated CopZ or Atx1-like copper chaperones, and adopt a ferredoxin-like fold, harboring a CXXC Cu(I) binding motif.11,13 Although the early copper trafficking hypothesis proposed that the chaperone was responsible for delivering Cu(I) to a cellular target(s) without dissociation into solvent, thereby minimizing copper-mediated cellular damage,14 more recent studies suggest that Cu chaperones, alongside glutathione, buffer Cu(I) to very low “free” levels, thereby preventing Cu(I) from gaining access to other non-cognate metal sites.15,16
A new class of prokaryotic copper chaperone has been identified and characterized, exemplified by CupA from Streptococcus pneumoniae.17 The crystallographic structure of the soluble, cytoplasmic domain of Cu(I)-bound CupA, sCupA, reveals a binuclear Cu(I) center (Fig. 1) on what is essentially an eight-stranded β-barrel, cupredoxin-like fold, reminiscent of the CuA-binding subunit of bacterial ba3-type cytochrome c oxidase.18 Cys74 is a bridging ligand between the S1 and S2 Cu(I) ions (Fig. 1). The S1 Cu(I) ion is digonally coordinated by Cys74 and Cys111, while the S2 adopts a trigonal pyramidal structure, coordinated by Cys74, Met113 and Met115 with a fourth axial ligand derived from a solvent anion (Fig. 1).17 Cu(I) binds 1000-fold more tightly to the S1 site (KCu1 = 1017.9 M−1), relative to the S2 site (KCu1 = 1014.8 M−1).17 The structure of the N-terminal metal binding domain (MBD) of CopA was determined in the same study and found to adopt the same fold, although of opposite electrostatic surface charge. Although Cu(I) bound to the S2 site on sCupA could be facilely transferred to the S1 site on the MBD of CopA in vitro, loss of Cu(I)-binding by the CopA MBD in a C49S mutant strain (C49, like C74 in CupA, is the bridging ligand in the CopAMBD; see Fig. 1) in an otherwise wild-type CopA had no detectable effect on cellular copper resistance.17 Since Cu(I) binding by CupA was shown to be essential for Cu(I) resistance, how CupA functions in this process as well as the functional role played by each of the two Cu(I) sites is not yet established. Further, the structure of apo-sCupA is not yet available.
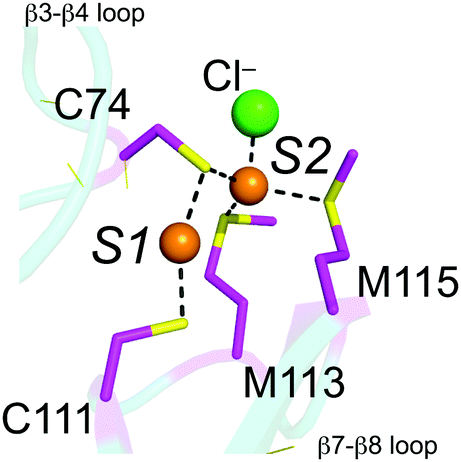 | ||
| Fig. 1 Close-up of the binuclear Cu(I) cluster in Cu2-bound sCupA.17 The S1 site is formed by C74 and C111, while the S2 site is formed by C74, M113, M115 and a solvent anion (Cl−). | ||
In this work, we present the NMR solution structure of apo-sCupA and evaluate the functionality of each Cu(I) site in CupA in vivo. Our findings reveal that apo-sCupA adopts the same cupredoxin fold as Cu2–CupA, but harbors a highly dynamic Cu(I) binding loop, as demonstrated by both backbone and side chain measurements. The high affinity S1 Cu(I) site is dispensable for cellular Cu(I) resistance, while the low affinity S2 Cu(I) binding site is essential for bacterial growth under copper stress. A triple substitution of the proposed metal entry site (MBS) in CopA19 abrogates Cu resistance at high Cu, and a non-cognate CopZ13 tethered to the plasma membrane via the CupA transmembrane (TM) helix also fails to protect the pneumococcus from Cu toxicity. Potential functions of CupA in cellular Cu resistance are discussed.
Results
Solution structure of apo-CupA
Uniformly 13C, 15N labeled apo-sCupA (residues 26–123) was subjected to multidimensional NMR spectroscopy using standard methods to obtain 91.6% of all possible proton assignments in the ordered regions and 87.9% overall. Four residues, 113–116, are characterized by missing amide resonances in the 1H, 15N-heteronuclear single quantum coherence (HSQC) spectrum of apo-sCupA;17 these include the two Met residues, M113 and M115, that coordinate the S2 Cu(I) ion in the β7–β8 loop harboring three of the four Cu(I) binding residues (Fig. 1). A lack of line-broadening in the immediate adjacent residues suggests that the resonances in this loop are broadened beyond detection due to flexibility in the μs–ms timescale. The three-dimensional structure of apo-sCupA§ is well defined by the NMR data with 2201 NOE-based distance constraints, including 1067 long-range constraints, and 130 dihedral angle constraints (Table 1). The final bundle of 18 structures reveals a pair-wise atomic rmsd values of 0.1 Å and 0.6 Å for the backbone and all heavy atoms, respectively (Fig. 2A), excluding the N-terminus and the disordered β7–β8 loop from residues 112–116 and including M113 and M115 (Fig. 2B). The ordered regions of apo-sCupA superimpose extremely well on the structure of Cu2–sCupA17 fully consistent with previous chemical shift perturbation experiments (Fig. 2B).17 In contrast, both the backbone and side chain donor atoms of the high-affinity S1 Cu(I) ion, C74 in the β3–β4 loop and C111 in the β7–β8 loop, appear pre-organized for Cu(I) binding (Fig. 2C).| Conformationally-restricting experimental constraintsa | |
|---|---|
| a Analysed for residues 26 to 123. b There are 92 residues with conformationally restricting constraints. c Calculated for all constraints for the given residues, using sum over r−6 (generated using PSVS 1.515). d Largest constraint violation among all the reported structures. e Ordered residues range 30–111, 117–122 [S(φ) + S(ψ) ≥ 1.8]. f With respect to mean and standard deviation for a set of 252 X-ray structures of <500 residues with resolution ≤1.80 Å, R factor ≤0.25 and Rfree ≤0.28; a positive value indicates a ‘better’ score (generated using PSVS 1.551). | |
| NMR distance and dihedral constraints | |
| NOE-based distance constraints | |
| Total NOE restraints | 2201 |
| Intra-residue (|i − j| = 0) | 347 |
| Sequential (|i − j| = 1) | 539 |
| Medium range (1 < |i − j| < 5) | 248 |
| Long range (|i − j| ≥ 5) | 1067 |
| NOE constraints/restrained resa | 23.9 |
| Dihedral-angle constraints | |
| φ(°) | 65 |
| ψ(°) | 65 |
| Total no. of restricting constraintsb | 2331 |
| Total no. of restricting constraints per restrained residueb | 25.3 |
| Restricting long-range constraints per restrained residueb | 11.6 |
| Residual constraint violationsa,c | |
| Average no. of distance violations per structure | |
| 0.1–0.2 Å | 5.5 |
| 0.2–0.5 Å | 0.44 |
| >0.5 Å | 0 |
| Maximum distance violationd (Å) | 0.35 |
| Average no. of dihedral angle violations per structure | |
| 1–10° | 0.83 |
| >10° | 0 |
| Maximum dihedral angle violationd (°) | 1.70 |
| RMSD Values | |
| Backbone atomse (Å) | 0.1 |
| Heavy atomse (Å) | 0.6 |
| Ramachandran plot statisticse | |
| Most favored regions (%) | 90.7 |
| Allowed regions (%) | 9.3 |
| Disallowed regions (%) | 0 |
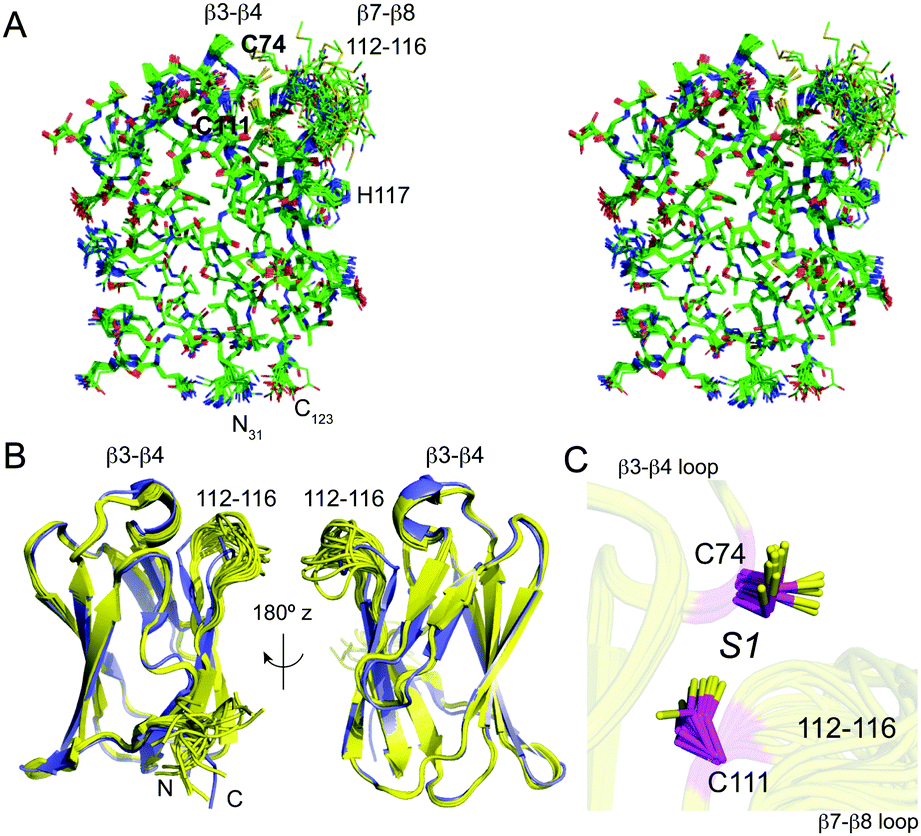 | ||
| Fig. 2 NMR solution structure of apo sCupA (PDB 2MRY). (A) Stereo view of an all heavy-atom ensemble representation of 18 structures of apo-sCupA (residues 31–123), with the side chains of the S1 Cu(I) site highlighted in bold. (B) Two views of a ribbon representation of the apo-sCupA ensemble (yellow) superimposed on a ribbon representation of Cu2 sCupA (slate). Cu(I) ions are not shown in this view. (C) Close-up view of the S1 Cu(I) site coordinating residues, C74 and C111 in the apo-sCupA ensemble. | ||
Dynamics of the Cu(I) binding β7–β8 loop as a function of Cu occupancy
Missing cross-peaks in the 1H, 15N-HSQC spectrum for residues 113–116 likely result from chemical exchange broadening between two or more distinct conformations on an intermediate timescale. Fast timescale (ps–ns) internal dynamical fluctuations can be approximated by the magnitude of 15N–(1H} NOE (hNOE), where a hNOE ≥ 0.8 is considered rigid, with correspondingly smaller hNOE values indicative of significant dynamics on this fast timescale. This experiment provides insights to changes in backbone flexibility upon sequential Cu(I) loading of the high affinity S1 and low affinity S2 Cu(I) sites (Fig. 3). Inspection of these data reveals that Cu(I) binding to the S1 site completely quenches backbone disorder from residue 112 to 116 in the β7–β8 loop in the S2 site, while loading of the second Cu(I) into the S2 site has little effect on backbone rigidity.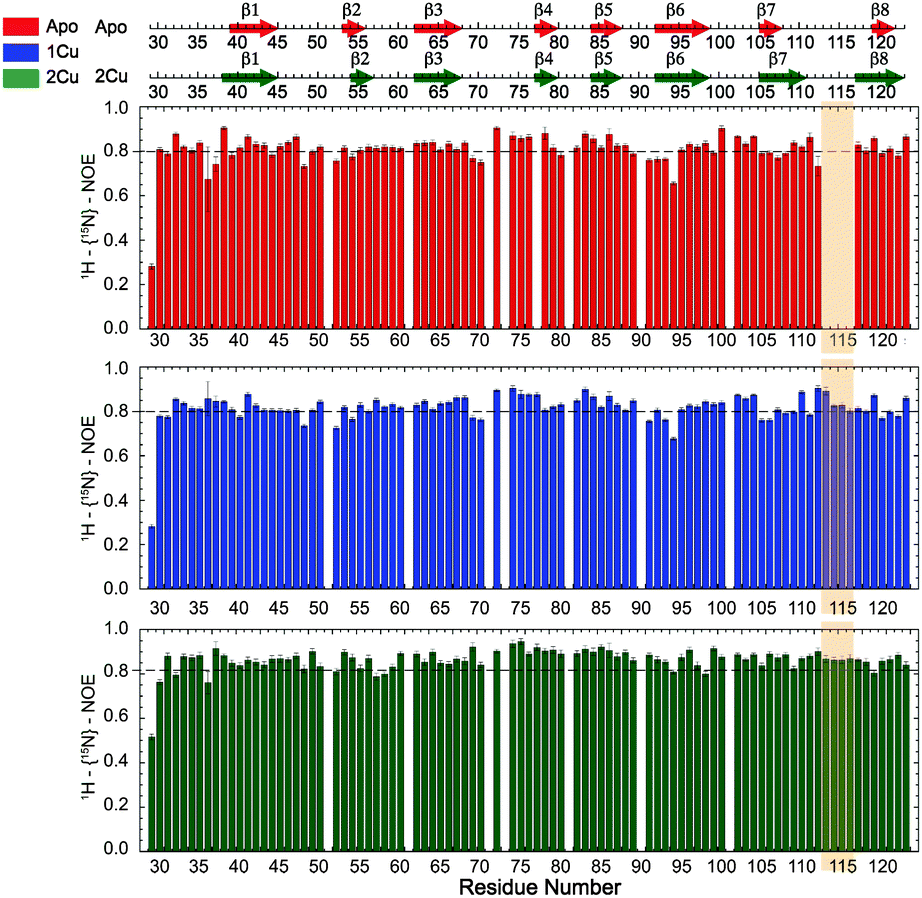 | ||
| Fig. 3 Backbone fast (ps–ns) timescale dynamics of sCupA in various Cu(I) ligation states with the secondary structural regions in the apo- and Cu2 states shown for reference. Residue-specific heteronuclear 15N–(1H} NOE (hNOE) values for apo-sCupA (top panel), Cu1 sCupA with 0.9 Cu(I) loaded (middle panel) and Cu2 sCupA with 1.8 Cu(I) loaded (bottom panel). The region that is stabilized by the first Cu(I) binding event is highlighted. Missing bars correspond to Pro in all other cases. | ||
Methyl groups are excellent reporters of side chain disorder on the ps–ns and μs–ms timescale.20 Given the relatively high abundance and distribution of methionine residues (7 total) in sCupA, particularly in the Cu(I) binding loop (M113, M115), we used the Met εCH3 groups to compare the flexibility of the Cu(I) binding loop with the core of the protein. Intra-methyl 1H–1H dipole–dipole cross-correlated relaxation rates (η) of the Met εCH3 groups (see Fig. S1, ESI†) report on the local dynamics on the ps–ns timescale from which one can calculate the axial order parameters of each side chain εCH3 group (S2axis) and are generally considered to report on side chain entropy.21 For side chain methyls that are highly dynamic in the ps–ns timescale, the S2axis value is close to 0, while for residues that are ordered the value is close to 1. We measured S2axis values of the Met εCH3 groups in all three states, apo, Cu1 (S1-bound), and Cu2 states, and compared them with one another (Fig. 4).22,23 M91, whose side chain is stabilized by interaction with neighboring residues in both the apo- and Cu2-states, shows consistently larger S2axis values of ≈0.4 while M28, near the N-terminus, has S2axis values of less than 0.05 for all three states. M46, solvent exposed and just C-terminal to the β1 strand generally gives low S2axis values as well. The two S2 ligands, M113 and M115, as well as M116 and M120 exhibit similar mobilities in the apo-state, S2axis ≈ 0.15, with the binding of the S1 and S2 Cu(I) ions inducing opposite effects. Filling of the S1 Cu(I) site detectably decreases motional disorder in M116 and M120, while enhancing disorder in the two S2–Cu ligands, M113 and M115. This disorder in M113 and M115 methyl groups is quenched in the Cu2 state as a result of formation of Cu–S coordination bonds, rising to an S2axis value comparable to that of M91. In contrast, S2axis for M116, closest to the S1 Cu, and M120 are characterized by increased flexibility in the fully Cu-loaded state. However, only M91, M113 and M115 in the Cu2 state show restricted mobility that could be classified as occurring within a single rotameric state (α-class), with all other residues classified as J-class, where there is significant sub-nanosecond rotameric interconversion.20 Met residues in proteins, in general, may well be over-represented in the J-class relative to other methyl-bearing side chains, due to the long side chain.24 These structural and dynamics studies reveal that the S1 and S2 Cu sites are characterized by distinct conformational dynamics, with the high affinity S1 site pre-organized to bind Cu, while the low affinity, more solvent exposed Met-rich S2 site is highly flexible in the apo- and Cu1 states. Previous NMR studies show that the Cu bound to the S2 site is preferentially transferred to the S1 site of the CopAMBD.17 A pre-organized S1 site would lower the reorganizational energy likely resulting in a fast on-rate while minimizing Cu(I) release. In contrast, Cu(I) binding to the S2 site would be entropically disfavored, perhaps contributing to the significantly lower affinity; on the other hand, these side chain dynamics of M113 and M115 are not entirely quenched and these residual dynamics may well facilitate Cu(I) transfer to a partner protein (see Discussion).
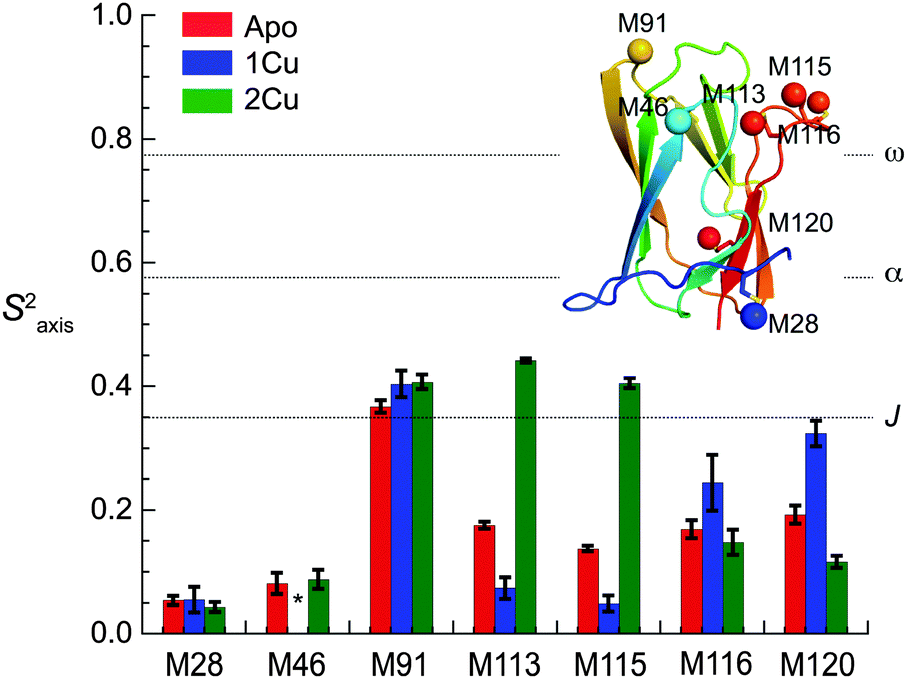 | ||
| Fig. 4 Side chain methionine ε-methyl group dynamics (S2axis) for each of the seven Met residues in sCupA as a function of Cu(I)-ligation state. Dashed horizontal lines indicate approximate maxima of three overlapping methyl-group motional regimes defined by S2axis values of 0.35 (J-class), 0.58 (α-class) and 0.78 (ω-class).20 Insert, ribbon representation of Cu2 sCupA highlighting the positions of the seven methionine methyl groups (Cε shown as spheres), colored sequentially blue to red, from N- to C-terminus. | ||
Differential biological functions for each Cu binding site in CupA
These physical studies make the prediction that the S1 site may be principally involved in chelating Cu(I) at low bioavailable [Cu], while S2 is largely responsible for transferring Cu(I) to CopA for export. To test this hypothesis, each individual Cu site was disrupted by site directed mutagenesis, and mutant bacterial strains expressing each allele were tested for biological copper resistance. A CupAC111S substitution is postulated to abrogate Cu(I) binding to the S1 site, while maintaining a functional S2 site. In contrast, the double mutant CupAM113A/M115A (CupA2MA) was designed to block Cu binding to the S2 site, without disruption of the S1 site. Anaerobic Cu(I) binding chelator competition experiments were carried out with each mutant to determine Cu(I) stoichiometry and affinity (Fig. S2, ESI†) in vitro. Both sCupA mutants bind a single mol equivalent of Cu(I) with the results consistent with retention of the remaining site in each case (Fig. S2, ESI;†Table 2). For CupA2MA, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KCu is 17.2 (±0.1), or within a factor of five of the S1 site in wild-type CupA. In contrast, for C111S CupA, logKCu is 12.5 (±0.3) or ≈200-fold weaker than for the S2 site in wild-type sCupA (Table 2). Thus, filling of the S1 site with Cu(I) enhances the stability of the S2–Cu(I) complex. Interestingly, 1H, 13C HSQC spectra in Cu(I)-loaded C111S sCupA exhibit features of chemical exchange broadening in the β7–β8 loop region and beyond, and thus do not provide clear evidence for Cu(I) ligation by M113 and M115 in the C111S mutant (Fig. S3A and B, ESI†). Substitution of the immediately adjacent residues, M116 and H117, with Ala does little to quench these dynamics, suggesting that non-native S2 Cu(I) coordination by these side chains is not the origin of this exchange broadening (Fig. S3C, ESI†).
KCu is 17.2 (±0.1), or within a factor of five of the S1 site in wild-type CupA. In contrast, for C111S CupA, logKCu is 12.5 (±0.3) or ≈200-fold weaker than for the S2 site in wild-type sCupA (Table 2). Thus, filling of the S1 site with Cu(I) enhances the stability of the S2–Cu(I) complex. Interestingly, 1H, 13C HSQC spectra in Cu(I)-loaded C111S sCupA exhibit features of chemical exchange broadening in the β7–β8 loop region and beyond, and thus do not provide clear evidence for Cu(I) ligation by M113 and M115 in the C111S mutant (Fig. S3A and B, ESI†). Substitution of the immediately adjacent residues, M116 and H117, with Ala does little to quench these dynamics, suggesting that non-native S2 Cu(I) coordination by these side chains is not the origin of this exchange broadening (Fig. S3C, ESI†).
| Protein |
|
|
|---|---|---|
| a Measured using a global fit to a series of anaerobic chelator competition assays (see Methods) as described previously.17 Conditions: 25 mM Hepes, pH 7.0, 0.2 M NaCl, ambient temperature. Conditions: 25 mM Hepes, pH 7.0, 200 mM NaCl, ambient temperature. | ||
| sCupA | 17.9 (±0.3) | 14.8 (±0.2) |
| C111S sCupA | — | 12.5 (±0.3) |
| M113A/M115A sCupA (2MA) | 17.2 (±0.1) | — |
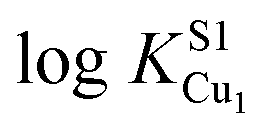
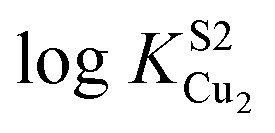
We next determined the Cu-sensitivity of the cupAC111S and cupA2MA mutant S. pneumoniae D39 strains relative to the wild-type and parent ΔcupA strains (Fig. 5A). Previous studies showed that 0.2 mM Cu added to the BHI growth medium was sufficient to completely inhibit growth of the ΔcupA mutant (Fig. 5A).17 Remarkably, the cupAC111S strain is indistinguishable from the wild-type strain under these conditions as well as at higher Cu (0.5 mM Cu; Fig. S4, ESI†); this reveals that the S1 site is not required for resistance of chronic Cu toxicity. In contrast, the cupA2MA strain exhibits a severe growth phenotype at both 0.2 and 0.5 mM Cu, but grows slightly better than the ΔcupA strain under low copper stress (0.1 mM) (Fig. 5A and Fig. S4, ESI†). The reduced growth rate of the cupA2MA strain is the result of copper toxicity as shown by elevated cell-associated copper, in contrast to the cupAC111S strain which has total cell associated Cu similar to the wild-type D39 strain under conditions of 0.2 mM Cu-shock (Fig. 5B).
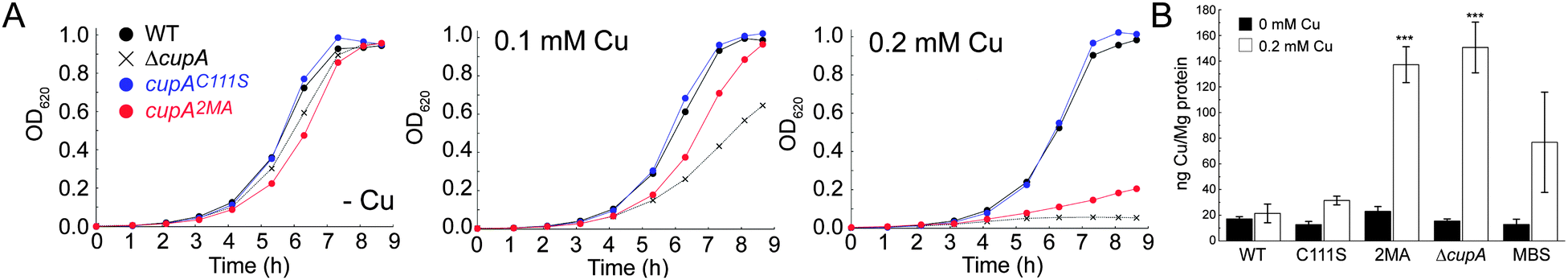 | ||
| Fig. 5 A cupA2MA mutant exhibits a growth phenotype and accumulates Cu under 0.2 mM Cu added to BHI, while a cupAC111S mutant grows like WT and does not accumulate Cu. (A) Microaerophilic growth rate analysis of cupAC111S (blue symbols) and cupA2MA (red symbols) strains compared to parent isogenic ΔcupA (X) and wild-type (WT) (black filled circles) strains grown in 0 mM Cu (–Cu, left panel), 0.1 mM Cu (middle panel) and 0.2 mM Cu (right panel) added to the BHI growth medium at t = 0. (B) Total [Cu] (ng Cu/mg protein) in the indicated strains measured by ICP-MS in the absence (filled bars) or presence (open bars) of 0.2 mM Cu stress added to cultures at OD620 ≈ 0.03. These data represent mean ± S.E. from three biological replicates. Statistical significance was determined using one-way ANOVA with Tukey post-test where ***p < 0.0001. WT, wild-type strain; C111S, cupAC111S strain; 2MA, cupA2MA strain; ΔcupA strain; MBS, copAMBS strain. | ||
These data reveal that the S2 Met-rich Cu site is necessary and sufficient for cellular Cu resistance. In addition, they reveal that although the S2 site in the cupAC111S strain has substantially lower Cu affinity and greater conformational dynamics, it is capable of functioning like a wild-type S2 chelate in the cell. In contrast, the high affinity S1 site may only involved in chelating cellular Cu(I) and may function to buffer low levels of free Cu, as previously described for other bacterial chaperones, thus effectively tuning the sensitivity of the cellular Cu response.16
A heterologous copper chaperone expressed and localized to the plasma membrane does not provide cellular copper resistance
While the CopAMBD is capable of stripping Cu from the S2 site of CupA, this transfer reaction is apparently not required for cellular Cu resistance since the copAC49S strain exhibits wild-type Cu resistance.17 The fact that the MBD is not required for cellular Cu resistance is consistent with many previous studies on Zn and Cu P1B-type ATPases (cf.ref. 25). These results therefore suggest that the CupA S2 site may transiently dock with some other region of CopA, and via intermolecular transfer mediated by ligand exchange, facilitates Cu efflux from the cell. This protein–protein interaction model makes the prediction that a non-cognate Cu chaperone may not support robust cellular Cu resistance.26To test this, we prepared a mutant strain designed to express a chimeric Bacillus subtilis CopZ (CopZBsu), which harbors the N-terminal transmembrane helix of CupA and full-length CopZBsu. We note that CopZBsu is characterized by a high Cu(I) binding affinity comparable to S1 site of CupA.27 Growth curves of the chimeric strain reveal a severe growth phenotype under 0.2 mM Cu stress (Fig. 6A), despite the fact that the CopZ chimera is expressed at high levels, is fully reduced in cells (Fig. 6B) and localizes to the plasma membrane (Fig. 6C), and thus would be capable of binding intracellular Cu. This mutant is functionally indistinguishable from the ΔcupA and cupA2MA strains, consistent with the hypothesis that specificity in a CupA–CopA interface in S. pneumoniae is crucial for Cu trafficking.26,28
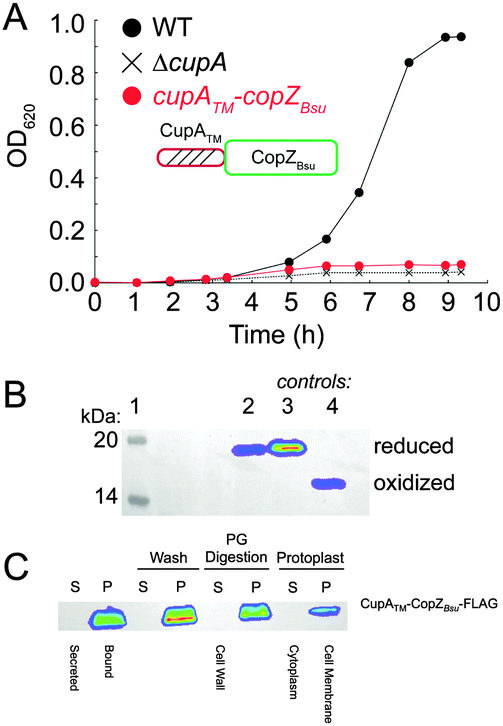 | ||
| Fig. 6 An S. pneumoniae D39 strain expressing a C-terminally FLAG-tagged chimeric, membrane-bound CopZ from B. subtilis (cupATM–copZBsu; see cartoon) can not restore the growth defect of ΔcupA strain grown in the presence of 0.2 mM Cu added to the BHI growth medium at t = 0. (A) Growth rate analysis of the cupATM–copZBsu strain (red symbols) compared to isogenic ΔcupA (X) and wild-type (WT) (black filled circles) strains. (B) Anti-FLAG Western blot of C-terminally FLAG-tagged CupATM–CopZBsu from cells (lane 2), with reduced (lane 3) and oxidized (lane 4) standards of CupATM–CopZBsu shown for reference (see Methods for details). (C) Subcellular localization of C-terminally FLAG-tagged CupATM–CopZBsu to the cell membrane of Cu-stressed S. pneumoniae cells, as probed by anti-FLAG Western blotting.17 S, supernatant; P, pellet fractions from cells first pelleted (first two lanes), then washed with PBS (Wash), digested with peptidoglycan hydrolases (PG Digestion), resulting in the formation of S and P Protoplast fractions (Protoplasts). | ||
The metal binding “entry” site (MBS) on CopA is required for Cu resistance at high Cu
Previous crystallographic studies of Legionella pneumophila CopA in the apo-state revealed a cluster of invariant, cytoplasmic-facing residues designated the entry metal binding site (MBS) and corresponding to M172, E216 and D347 in S. pneumoniae CopA.19 More recent X-ray absorption studies posit a key role for the residue corresponding to M172 in Cu(I) capture, and direct delivery to the transmembrane CPC site.29 The MBS is positioned just above the inner leaflet of the plasma membrane and the “platform (P) helix”, corresponding to the C-terminal half of the kinked MB helix unique to Cu/Zn P1B ATPase transporters (MB′; Fig. 7A).19 In an archeal CopA, this entry MBS was shown to be required for cognate Cu(I)–CopZ-stimulated CopA ATPase activity and Cu(I) transfer, but was not required for free Cu(I)-stimulated ATPase activity. This is consistent with the interpretation that the MBS provides a major site for copper chaperone-mediated Cu delivery.30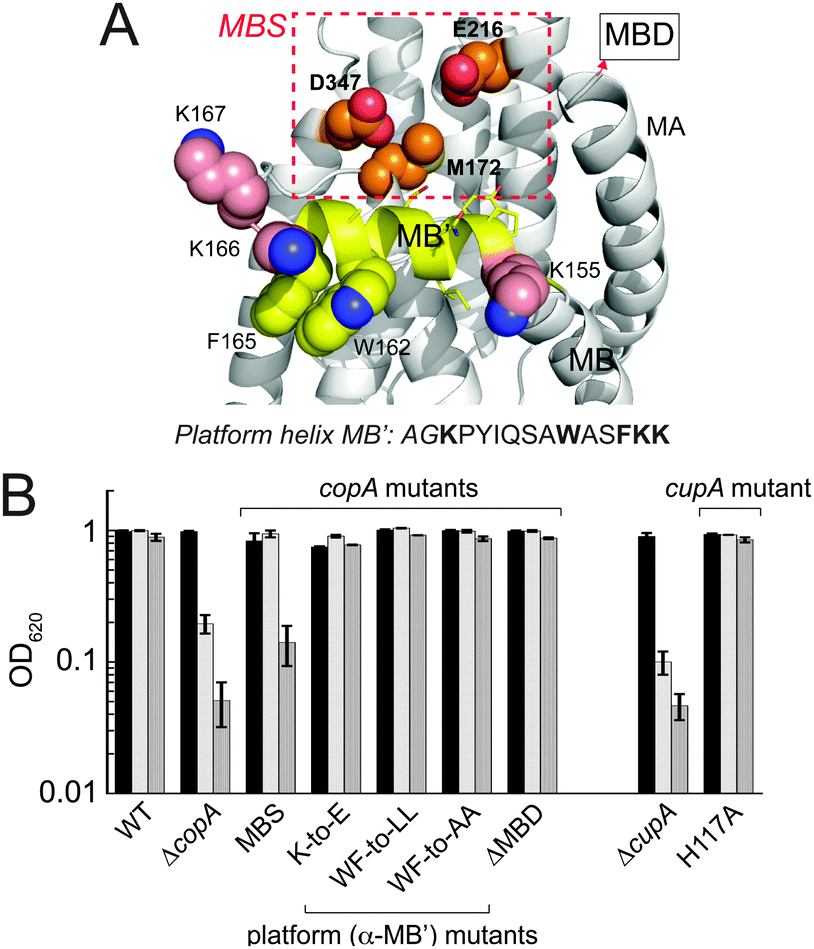 | ||
| Fig. 7 Functional characterization of the S. pneumoniae D39 copAΔMBS strain. (A) Homology model of the platform helix and entry metal binding site (MBS) region of S. pneumoniae CopA obtained by threading the Spn CopA sequence through the apo L. pneumophila CopA structure.19 The C-terminal half of the MB or platform helix, is shaded yellow with targeted residues in spacefill; the proposed entry metal binding site (MBS)19 based on an alignment with Lp CopA are boxed in red and shown in spacefill (M172, E216, D347). The N-terminal MA helix and the position at which the MBD would emerge from this structure are highlighted. The amino acid sequence of the platform helix portion of the MB helix is shown, with residues subjected to mutagenesis (see Fig. S5 and S6, ESI†) highlighted in bold. (B) Replicate growth yields of mutant S. pneumoniae D39 strains expressing various CopA and CupA alleles relative to ΔcopA and ΔcupA strains in BHI medium in the absence (black bars) or presence of 0.2 mM Cu (light grey bars) or 0.5 mM Cu (grey bars). | ||
We prepared a strain encoding a FLAG-tagged triple Ala substitution mutant of copA, M172A/E216A/D347A, denoted copAMBS in a C49S CopA background, and tested the viability of this strain with 0, 0.2 and 0.5 mM Cu added to the BHI growth medium (MBS, Fig. 7B). Although this strain grows with no clear growth phenotype relative to the wild-type strain at 0.2 mM Cu, it accumulates more total copper than the WT strain and the corresponding cupAC111S strain (Fig. 5B). In contrast to the cupAC111S strain, the copAMBS strain nearly fails to grow at 0.5 mM Cu and accumulates significantly more Cu than the WT strain (Fig. S5, ESI†).
In other CopA transporters, the platform helix is rich in positively charged residues and these residues are proposed to form a docking site stabilized by electrostatic interactions required for chaperone-mediated Cu(I) transport.30 In S. pneumoniae CopA, the platform helix is not particularly basic, and a triple glutamate substitution mutant of the three lysines that flank the platform helix (K155E/K166E/K167E) (see Fig. 7A) has no effect on cellular Cu resistance (K-to-E, Fig. 7B and Fig. S6A, ESI†). In addition, the conserved W162-x-x-F165 motif also appears to play no role in cellular Cu resistance since both the W162L/F165L (WF-to-LL; Fig. 7B) and W162A/F165A (WF-to-AA; Fig. 7B) grow with no growth phenotype up to 0.5 mM Cu (Fig. 7B) and the WF-to-AA strain accumulates Cu to the same degree as the WT strain (Fig. S5, ESI†). Finally as expected from previous studies of C49S CopA,17 deletion of the entire N-terminal metal-binding domain (ΔMBD) does not result in a growth phenotype (Fig. 7B) and this strain fails to accumulate significantly more Cu than the WT strain (Fig. S5, ESI†). These data taken collectively are consistent with the hypothesis that the MBS provides a docking site for CupA in stimulating extracellular Cu(I) efflux, but energetically important electrostatic or hydrophobic interactions for CupA S2 Cu(I) transfer, if present, lie beyond this region of the platform helix itself (see Fig. S6B, ESI†).30
Discussion
This work provides new insights into the mechanism of cellular Cu resistance in the Gram-positive respiratory pathogen Streptococcus pneumoniae, and builds significantly on previous structural studies of the novel, membrane-anchored cupredoxin fold protein, CupA.17The physical properties of CupA are optimized for Cu(I) transfer
Our solution structure and dynamics characterization of metal-free apo-sCupA collectively reveal that the S2 Cu(I) site region, notably M113 and M115, exhibits physical properties consistent with a role in intermolecular Cu(I) transfer. In contrast, the S1 Cu site is largely preorganized so that filling it quenches backbone disorder, but enhances side chain flexibility which collectively increase the S2 site Cu(I) binding affinity (Table 1). The S2 Cu site in CupA is necessary and sufficient for resistance against chronic Cu toxicity, while the S1 site is largely dispensable for this resistance. The S1 site provides some protection against Cu toxicity, and may well function to buffer very low levels of Cu in the absence of significant Cu stress. The N-terminal MBD of CopA, in contrast, plays no clear role in chronic cellular Cu resistance. A regulatory role remains possible, as described previously for many other transition metal-transporting P1B-ATPases11,28,30–32 and biochemical reconstitution30 studies will be required to assess a mechanistic role of the MBD in modulating the ATPase activity and/or rates of Cu(I) transport.Our findings reveal that the Met pair in the S2 Cu site (M113, M115) optimizes the dynamics, rather than the stability, of the site in a way that kinetically facilitates intermolecular Cu transfer, perhaps directly to the MBS of CopA, which is on pathway for Cu efflux.30,33 Intermolecular Cu transfer via ligand exchange at the S2 site28 is plausible given the fact this site is at the protein surface, and is capable of adding a fourth ligand from CopA to initiate Cu transfer as shown by coordination of Cl− or Br− in the isolated sCupA domain (see Fig. 1).17 In this model, Cu transfer to a partner protein (P, Scheme 1), e.g., CopA, can potentially harness a significant entropic driving force. We note that this is a highly simplified scheme that does not take into account the Cu(I) stoichiometry or affinity of P, which for S. pneumoniae CopA is not known.
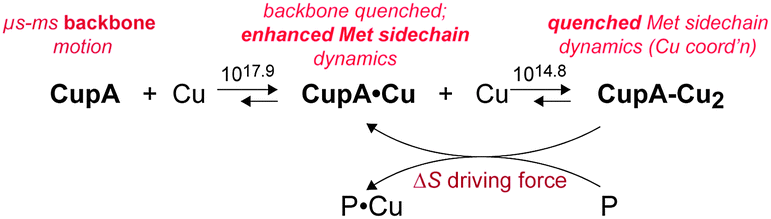 | ||
| Scheme 1 | ||
Is CupA a metallochaperone for CopA?
Most Cu resistance models posit that the copper chaperone physically, albeit transiently, docks onto some site on the effluxer CopA, thereby enhancing the rate of Cu(I)-efflux from the cell. In other words, P in Scheme 1 is CopA. The fact that cellular Cu resistance requires the structural integrity of the CupA S2 and CopA MBS Cu sites, is fully consistent30 with this model. The additional finding that a non-cognate chaperone, CopZ, tethered to the membrane and capable of binding Cu in the cell does not provide resistance against Cu toxicity is also strongly consistent with a direct and specific protein–protein interaction model.26 However, another possible explanation for the severe growth phenotype of the CupA–CopZ chimera is that the Cu(I) binding affinity (1018 M−1) and/or kinetic lability of Cu(I)-bound CopZ, the former on par with that of the CupA S1 site, is too high or too slow, respectively, to effect efficient Cu efflux from the cell.One argument against a strict co-dependence of CopA activity on CupA function is the recent finding that a ΔcopA S. pneumoniae TIGR4 (serotype 4) strain is significantly less virulent in a number of mouse models of infection relative to the ΔcupA strain, despite the fact each fails to grow under conditions of copper toxicity in liquid culture34 as previously reported.17 This suggests the possibility that under some conditions, perhaps under moderate Cu toxicity or acute stress, CopA can function independently of CupA. In addition, a clearly defined protein–protein interaction site(s) between these two proteins remains elusive. Mutagenesis of conserved positively charged and aromatic residues on the projected solvent-exposed side of the platform helix, the former of which were previously implicated in chaperone-stimulated ATPase activity of the transporter,30 has no impact on cellular Cu resistance in the pneumococcus. Furthermore, substitution of H117 on CupA, conserved, but only as a positively charged residue (H/K/R), also does not impact cellular Cu resistance (Fig. 7B).
These findings suggest that the interaction surfaces of CupA and CopA may be multipartite, with the loss of one determinant insufficient to measurably impact cellular Cu resistance. For example, a physical interaction between transmembrane helices of CupA and CopA can not be ruled out, particularly since plasma membrane localization of CupA is required for its function.17 In addition, a PY sequence immediately following K155 in the platform helix (see Fig. 7A) is essentially invariant (P-Y/F) and in our homology model, Y157 makes close approach to the MBS. Another possible CupA docking site is the conserved cluster of K/R residues on the N-terminal region of the immediately adjacent MA helix (Fig. S6, ESI†), including R102, K103, S106 and R110, just C-terminal to the MBD. These regions are currently being targeted in functional studies. Finally, it is also possible that some other CupA partner protein beyond CopA exists in the cell. The copper binding protein, CutC, previously characterized in Enterococcus faecalis, whose expression is not Cu-inducible in S. pneumoniae,17,35 may function in some way in CupA-mediated Cu resistance since a ΔcutC E. faecalis strain has been reported to hyperaccumulate Cu relative to a wild-type strain.36 The role of CutC in Cu-resistance in S. pneumoniae D39 (serotype 2) has not yet been investigated.
Is the Cu-binding affinity of the CupA S2 site consistent with a “set-point” model of Cu resistance?
We and others have speculated that the Cu(I) binding affinity of the copper-sensing repressor CopY when expressed as an equilibrium dissociation constant, 1/KCu, defines a threshold [Cu]free, below which pneumococcal cells efficiently buffer free Cu, and above which, the CopY-induced expression of the cop operon is required to mitigate the effects of cellular Cu(I) toxicity.1,2 In this model, this set point would be 10−16 M Cu, which corresponds to the affinity of CopY for Cu(I) (H. Glauninger and Y. Fu, unpublished data). As intracellular [Cu]free reaches ≥10−15 M, both Cu sites in CupA would be occupied, poised to facilitate Cu efflux from the cell. However, the viability of the cupAC111S strain suggests that even a CupA mutant with ≈200-fold lower Cu(I) binding affinity provides full protection against Cu toxicity. Thus, based on KCu values measured for other bacterial Cu chaperones, S. pneumoniae may be more tolerant of cytoplasmic Cu, and chelating this Cu from the bioavailable pool might be a primary role played by the S1 Cu site in CupA. This role is analogous to that played by low molecular weight thiols like glutathione which have been shown to protect S. pneumoniae from heavy metal toxicity.37 We emphasize, however, that a KCu of 1012 M−1 is not all that is required since a cupAC74S strain encoding C74S CupA, which disrupts Cu binding to both S1 and S2 sites yet retains a non-native Cu-binding site of 1012 M−1 affinity, is as sensitive to Cu stress as a ΔcupA strain.17In the event that CupA and CopA do indeed physically interact with one another, it is tempting to speculate that one of the MBS ligands, e.g., M172,29 enters into the first coordination sphere of the trigonal pyramidal S2 site on CupA, displacing the solvent-derived anion, thereby initiating Cu transfer to the MBS via ligand exchange. Efforts are underway to trap wild-type and potentially longer-lived CupA–CopA complexes by covalent crosslinking, as are biochemical reconstitution studies30 and NMR structural studies of intact CupA.
Materials & methods
Protein purification
Recombinant sCupA was expressed and purified as described previously.17 For the NMR experiments, 13C, 15N-labeled sCupA was expressed in Escherichia coli BL21(DE3) in M9 minimal media and purified as previously described.17NMR data acquisition and resonance assignments
For NMR structure calculations, sCupA was prepared in 50 mM sodium phosphate, pH 6.0, 50 mM NaCl, 5 mM EDTA and 5 mM tris(2-carboxyethyl)phosphine (TCEP) in 10% 2H2O/90% H2O or 100% 2H2O at a protein concentration of 400 μM to 600 μM. For the dynamics studies, sCupA was prepared in 50 mM potassium phosphate, pH 7.0, 50 mM NaCl, 5 mM EDTA and 5 mM TCEP in 10% 2H2O/90% H2O or 100% 2H2O at 400–600 μM protein. Varian DDR 600 and 800 MHz spectrometers equipped with cryogenic probes in the METACyt Biomolecular NMR Laboratory were used to acquire data for sCupA samples at 298 K. NMR spectra were referenced to H2O. NMR data were processed using NMRPipe38 and was analyzed using Sparky39 and CcpNmr Analysis.40 Backbone assignments were obtained in previous work.1715N relaxation experiments were acquired using two-dimensional, proton-detected heteronuclear NMR experiments.41R1 values were measured from the spectra recorded with ten different duration of delay T: T = 10, 50, 110, 190, 310, 500, 650, 1000, 1500 and 1900 ms. R2 values were determined from the spectra recorded with ten different durations of delay time, T: T = 10, 30, 50, 90, 110, 150, 190, 210, 230 and 250 ms. 15N–(1H} heteronuclear NOE values were determined from spectra recorded in the absence and presence of a 3 s 1H pre-saturation period. Uncertainties in measured peak heights were estimated from the baseline noise level and propagated to measured hNOE values. The rotational correlation time (τc) of apo sCupA was estimated from the data analysis according to model-free formalism using Tensor V2.0.42 to be 4.9 ns. Assignments of side chain resonances were made using three-dimensional C(CO)NH-TOCSY,43 H(CCO)NH-TOCSY,43,44 HCCH-TOCSY45 and HCCH-COSY46 experiments. Distance restrains were derived from 3D 15N-edited NOESY-HSQC (τm = 200 ms) and 13C-edited NOESY-HSQC (τm = 150 ms) experiments.Structure ensemble calculations
Backbone dihedral angle restraints for three-dimensional structure calculation were generated from apo sCupA backbone assignments using TALOS+.47 Initial NOE cross-peak assignments were obtained using automatic NOE assignment and structure calculation algorithm implemented in CYANA 2.1.48,49 Partial assigned peak lists were then further manually assigned and confirmed during subsequent CYANA calculations. In total, 100 structures were calculated using CYANA and subsequently refined using a simulated annealing protocol with Xplor-NIH.50 After iterative refinement and editing of the distance restraints based on the NOESY spectra to remove incorrect and ambiguous assignments, a final ensemble of 18 structures with the lowest energies were chosen for analysis using protein structure validation tools (PSVS 1.5;51 see Table 1) and were deposited in the Protein Data Bank (entry 2MRY). Substantially complete resonance assignments for apo-sCupA have been deposited in the BMRB under accession number 25098.Measurement of intra-methyl 1H–1H cross-correlated relaxation rates
We used the methyl groups of Met residues as a probe for side chain fast timescale (ps–ns) dynamics. To measure the intra-methyl 1H–1H dipole–dipole cross-correlated relaxation rate (η),23,52 pairs of experiments were recorded. The signal intensity (Ia) in the first experiment during a variable relaxation delay (T) depends on spin-dynamics in the I = 3/2 manifold of the Met ε-methyl 1H spin-system. The signal intensity (Ib) in the second experiment is determined by spin-dynamics in the I = 3/2 and I = 1/2 manifolds. The cross-correlated rate (η) is related to the intensity ratio (Ia/Ib) in the given equation at each time T: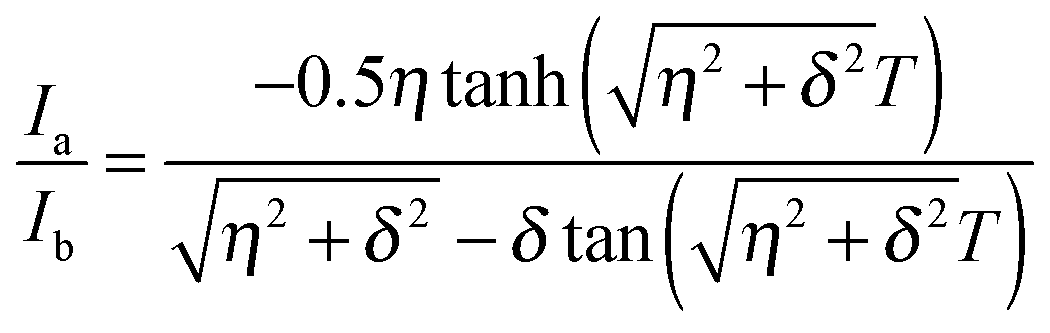 | (1) |
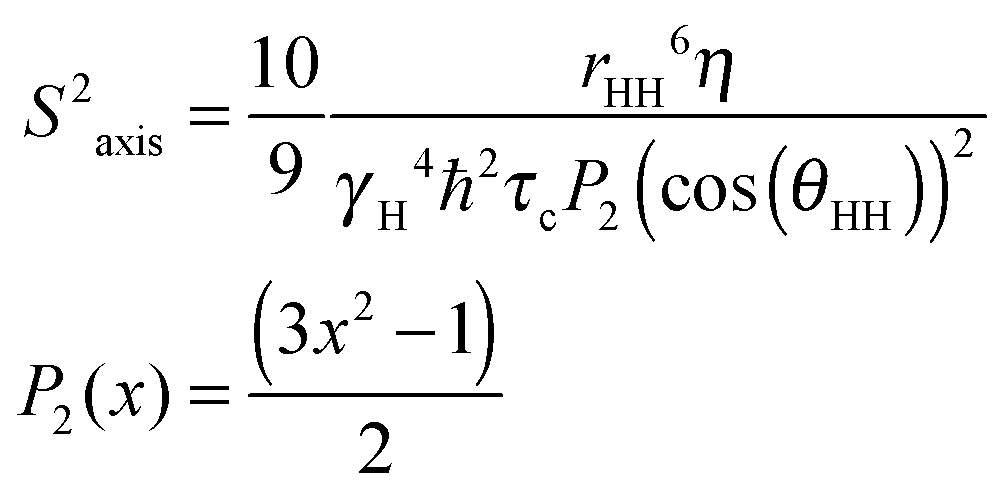 | (2) |
Bacterial strain construction
All strains used S. pneumoniae serotype 2 strain D39 as a parent strain. For all strains, linear DNA amplicons were constructed using overlapping fusion PCR and transformed into competent S. pneumoniae cells. Strains containing markerless cupA or copA mutant alleles in the native loci (Table S1, ESI†) were constructed using the Pc-[kanR-rpsL+] (Janus cassette) allele replacement method, as described previously.17 The ΔcupA strain (IU5971) was constructed by deleting the cupA gene sequence except for 42 base pairs at the 5′ end and 60 base pairs at the 3′ end. The cupATM–copZBsu-FLAG strain (IU7487) was constructed by combining the sequence encoding for amino acids 1–27 of the S. pneumoniae cupA gene with the sequence encoding for amino acids 2–69 of the Bacillus subtilis copZ gene (NC_000964), along with a FLAG sequence in the native locus of the cupA gene of S. pneumoniae. Mutant strains were confirmed by sequencing through the relevant portions of the genome. Whole genome sequencing was also performed on IU1781, IU5923, IU5921 and IU10148 strains (see Table S1, ESI†) to confirm the absence of any additional mutations. Strains were grown on plates containing Trypticase Soy Agar II (modified) (Becton-Dickinson) with 5% (v/v) defibrinated sheep blood at 37 °C with 5% CO2. Blood plates were supplemented with 250 μg mL−1 streptomycin or kanamycin for antibiotic resistance. For liquid cultures, strains were grown in Brain Heart Infusion (BHI) broth (Becton-Dickinson) at 37 °C with 5% CO2 without shaking (microaerophilic conditions).Growth rate analysis
Strains were serially diluted from frozen stocks into 3 mL of BHI broth and grown 14–16 h in microaerophilic conditions in 17 mm diameter polystyrene tubes. The optical density at 620 nm (OD) of the serial dilutions was determined using a Spectronic 20 Genesys spectrophotometer, and a serial dilution in log-phase growth (OD ∼0.1) was diluted to an OD of 0.005 in 5 mL BHI in a glass 17 mm diameter tube and grown microaerophilically. Additional Cu, if needed, was added to the cultures at the time of dilution.Measurement of total cell-associated copper
Total cell-associated copper was measured by inductively-coupled plasma mass spectrometry (ICP-MS) essentially as described in previous work17,53 using triplicate biological replicates. Briefly, overnight cultures, obtained as described above, in log-phase growth were diluted to OD 0.1 with BHI and then diluted to OD 0.005 in 2 tubes containing 5 mL BHI each. Cultures were incubated at 37 °C with 5% CO2 until OD reached ≈0.03, when Cu was added to one set of tubes to a final concentration of 0.2 or 0.5 mM. Cultures were incubated until OD reached ≈0.2, then centrifuged 20 min at 4000g at 4 °C. Samples were washed in 1 mL PBS with 2 mM nitrilotriacetic acid (NTA; Sigma Aldrich) and centrifuged 7 min at 16100g at 4 °C. Pellets were washed in 800 μL of PBS treated with Chelex-100 (Biorad) and centrifuged again. These pellets were then washed in 1 mL Chelex-treated PBS, split into 2 × 500 μL aliquots and centrifuged again. Supernatant was removed and one aliquot was resuspended in 100 μL lysis buffer (1% SDS and 0.1% Triton X-100) and used to assay total protein concentration (mg mL−1) using a Bradford DC assay (BioRad). The other aliquot was dried overnight and treated with 400 μL 30% nitric acid (Ultrapure, Sigma Aldrich) with 0.1% Triton X-100. Cells were lysed by heating at 95 °C with shaking at 500 rpm for 10 min, then vortexed for 20 s. 300 μL of lysate was added to 2.7 mL of 2.5% nitric acid and inductively coupled plasma mass spectrometry (ICP-MS) analysis was performed using a Perkin Elmer ELAN DRCII ICP-MS, giving a metal concentration in μg L−1. μg L−1 metal × 0.003 L sample × 1000 × 4/3 = ng total metal in the aliquot. The final Cu concentration is expressed as ng Cu per mg total protein.
Cellular fractionation and 4-acetamido-4-maleimidylstilbene-2,2-disulfonic acid (AMS) labeling
Cell fractionation was performed as described.54 IU6041 (cupA-(C)-FLAG) was induced with 0.3 mM Cu, and IU7487 (cupATM–copZBsu-(C)-FLAG) was induced with 0.15 mM Cu. For AMS labeling, overnight cultures in log-phase growth were diluted to OD 0.005 in 30 mL BHI with 0.15 mM Cu. Cultures were grown to OD ≈0.25 and centrifuged at 8000 g at 4 °C for 10 min. Pellets were resuspended in 5 mL PBS with 1 mM bathcuproinedisulfonic acid (BCS; Sigma Aldrich) and centrifuged again. Pellets were resuspended in 1 mL ice cold lysis buffer (100 mM Tris-HCl pH 8, 1 mM CaCl2, 3 mM EDTA, 18% sucrose, 1 mM BCS, 4 M urea, 8 μL mL−1 Protease Inhibitor Cocktail III (Calbiochem)) and transferred to prechilled 2 mL lysing Matrix B tube (MP Biomedicals). Cells were lysed using a FastPrep-24 (MP Biomedicals) at 4 °C by running at 6.0 m s−1 for 40 s for 3 consecutive runs. Samples were centrifuged at 8000g at 4 °C for 1 min, and 3 × 50 μL aliquots were made. Aliquots were treated immediately with one of the following: 20 mM 4-acetamido-4-maleimidylstilbene-2,2-disulfonic acid (AMS; Sigma Aldrich), 5 mM tris (2-carboxyethyl) phosphine (TCEP; Sigma Aldrich), or 0.2 mM 4,4′-dithiopyridine (DPS; Sigma Aldrich). 30 min after the addition of TCEP or DPS, 20 mM AMS was added to those aliquots and alkylation was allowed to proceed at room temperature for an additional 2 h. 60 μL 2× Laemmli SDS sample buffer (BioRad) was added and 20 μL of samples were loaded onto a 15% SDS-PAGE gel and run for 2 h at 150 V. Proteins were transferred to a nitrocellulose blot for 90 min at 0.35 amps at 4 °C. Blots were blocked overnight at 4 °C in 10 mL PBS with 3% BSA (Sigma Aldrich) and 5% skim milk powder (BBL). Primary antibody labeling in PBS with 0.05% Tween 20 (PBST) with 1% BSA used 1:1000 monoclonal mouse anti-FLAG M2 antibody (Sigma, F1804-50UG) for 3 h. Secondary antibody labeling in PBST with 1% BSA used 1:2000 donkey anti-mouse HRP (Sigma, SA1-100) for 2 h. FLAG-tagged proteins were detected with ECL detection reagent (GE Healthcare, RPN2109) and an IVIS imaging system.
Acknowledgements
The authors gratefully acknowledge support from US National Institutes of Health (R01 GM042569). We also thank Prof. Malcolm Winkler, Department of Biology, Indiana University for many helpful conversations and his support of this work, John Lisher for his assistance with the ICP-MS analysis, and Dr Katie Edmonds for comments on the manuscript. The NMR instrumentation in the METACyt Biomolecular NMR Laboratory at Indiana University was supported by the Indiana Metabolomics and Cytomics Initiative funded by the Lilly Endowment.Notes and references
- H. Reyes-Caballero, G. C. Campanello and D. P. Giedroc, Biophys. Chem., 2011, 156, 103–114 CrossRef CAS PubMed.
- A. W. Foster, D. Osman and N. J. Robinson, J. Biol. Chem., 2014, 289, 28095–28103 CrossRef CAS PubMed.
- Z. Ma, P. Chandrangsu, T. C. Helmann, A. Romsang, A. Gaballa and J. D. Helmann, Mol. Microbiol., 2014, 94, 756–770 CrossRef CAS PubMed.
- J. J. Braymer and D. P. Giedroc, Curr. Opin. Chem. Biol., 2014, 19, 59–66 CrossRef CAS PubMed.
- L. Macomber and J. A. Imlay, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8344–8349 CrossRef CAS PubMed.
- Y. Fu, F. M. Chang and D. P. Giedroc, Acc. Chem. Res., 2014, 47, 3605–3613 CrossRef CAS PubMed.
- K. Y. Djoko and A. G. McEwan, ACS Chem. Biol., 2013, 8, 2217–2223 CrossRef CAS PubMed.
- C. White, J. Lee, T. Kambe, K. Fritsche and M. J. Petris, J. Biol. Chem., 2009, 284, 33949–33956 CrossRef CAS PubMed.
- S. L. Stafford, N. J. Bokil, M. E. Achard, R. Kapetanovic, M. A. Schembri, A. G. McEwan and M. J. Sweet, Biosci. Rep., 2013, 33, pii: e00049 CrossRef PubMed.
- A. C. Rosenzweig and T. V. O'Halloran, Curr. Opin. Chem. Biol., 2000, 4, 140–147 CrossRef CAS PubMed.
- A. K. Boal and A. C. Rosenzweig, Chem. Rev., 2009, 109, 4760–4779 CrossRef CAS PubMed.
- P. Cobine, W. A. Wickramasinghe, M. D. Harrison, T. Weber, M. Solioz and C. T. Dameron, FEBS Lett., 1999, 445, 27–30 CrossRef CAS PubMed.
- L. Banci, I. Bertini, R. Del Conte, J. Markey and F. J. Ruiz-Duenas, Biochemistry, 2001, 40, 15660–15668 CrossRef CAS PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, R. Del Conte and L. Gonnelli, Biochemistry, 2003, 42, 1939–1949 CrossRef CAS PubMed.
- S. Tottey, C. J. Patterson, L. Banci, I. Bertini, I. C. Felli, A. Pavelkova, S. J. Dainty, R. Pernil, K. J. Waldron, A. W. Foster and N. J. Robinson, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 95–100 CrossRef CAS PubMed.
- D. Corbett, S. Schuler, S. Glenn, P. W. Andrew, J. S. Cavet and I. S. Roberts, Mol. Microbiol., 2011, 80, 14–21 CrossRef PubMed.
- Y. Fu, H.-C. T. Tsui, K. E. Bruce, L.-T. Sham, K. A. Higgins, J. P. Lisher, K. M. Kazmierczak, M. J. Maroney, C. E. Dann, M. E. Winkler and D. P. Giedroc, Nat. Chem. Biol., 2013, 9, 177–183 CrossRef CAS PubMed.
- P. A. Williams, N. J. Blackburn, D. Sanders, H. Bellamy, E. A. Stura, J. A. Fee and D. E. McRee, Nat. Struct. Biol., 1999, 6, 509–516 CrossRef CAS PubMed.
- P. Gourdon, X. Y. Liu, T. Skjorringe, J. P. Morth, L. B. Moller, B. P. Pedersen and P. Nissen, Nature, 2011, 475, 59–64 CrossRef CAS PubMed.
- A. J. Wand, Curr. Opin. Struct. Biol., 2013, 23, 75–81 CrossRef CAS PubMed.
- M. S. Marlow, J. Dogan, K. K. Frederick, K. G. Valentine and A. J. Wand, Nat. Chem. Biol., 2010, 6, 352–358 CrossRef CAS PubMed.
- V. Tugarinov, J. E. Ollerenshaw and L. E. Kay, J. Am. Chem. Soc., 2005, 127, 8214–8225 CrossRef CAS PubMed.
- V. Tugarinov, R. Sprangers and L. E. Kay, J. Am. Chem. Soc., 2007, 129, 1743–1750 CrossRef CAS PubMed.
- T. I. Igumenova, K. K. Frederick and A. J. Wand, Chem. Rev., 2006, 106, 1672–1699 CrossRef CAS PubMed.
- B. Mitra and R. Sharma, Biochemistry, 2001, 40, 7694–7699 CrossRef CAS PubMed.
- C. E. Blaby-Haas, T. Padilla-Benavides, R. Stube, J. M. Arguello and S. S. Merchant, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E5480–E5487 CrossRef CAS PubMed.
- Z. Xiao, J. Brose, S. Schimo, S. M. Ackland, S. La Fontaine and A. G. Wedd, J. Biol. Chem., 2011, 286, 11047–11055 CrossRef CAS PubMed.
- N. J. Robinson and D. R. Winge, Annu. Rev. Biochem., 2010, 79, 537–562 CrossRef CAS PubMed.
- D. Mattle, L. Zhang, O. Sitsel, L. T. Pedersen, M. R. Moncelli, F. Tadini-Buoninsegni, P. Gourdon, D. C. Rees, P. Nissen and G. Meloni, EMBO Rep., 2015, 16, 728–740 CrossRef CAS PubMed.
- T. Padilla-Benavides, C. J. McCann and J. M. Arguello, J. Biol. Chem., 2013, 288, 69–78 CrossRef CAS PubMed.
- A. T. Smith, D. Barupala, T. L. Stemmler and A. C. Rosenzweig, Nat. Chem. Biol., 2015, 11, 678–684 CrossRef CAS PubMed.
- Z. Hou and B. Mitra, J. Biol. Chem., 2003, 278, 28455–28461 CrossRef CAS PubMed.
- M. Gonzalez-Guerrero and J. M. Arguello, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5992–5997 CrossRef CAS PubMed.
- M. D. Johnson, T. E. Kehl-Fie, R. Klein, J. Kelly, C. Burnham, B. Mann and J. W. Rosch, Infect. Immun., 2015, 83, 1684–1694 CrossRef CAS PubMed.
- S. Shafeeq, H. Yesilkaya, T. G. Kloosterman, G. Narayanan, M. Wandel, P. W. Andrew, O. P. Kuipers and J. A. Morrissey, Mol. Microbiol., 2011, 81, 1255–1270 CrossRef CAS PubMed.
- M. Latorre, F. Olivares, A. Reyes-Jara, G. Lopez and M. Gonzalez, Biochem. Biophys. Res. Commun., 2011, 406, 633–637 CrossRef CAS PubMed.
- A. J. Potter, C. Trappetti and J. C. Paton, J. Bacteriol., 2012, 194, 6248–6254 CrossRef CAS PubMed.
- F. Delaglio, S. Grzesiek, G. Vuister, G. Zhu, J. Pfeifer and A. Bax, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS PubMed.
- T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco.
- W. F. Vranken, W. Boucher, T. J. Stevens, R. H. Fogh, A. Pajon, M. Llinas, E. L. Ulrich, J. L. Markley, J. Ionides and E. D. Laue, Proteins, 2005, 59, 687–696 CrossRef CAS PubMed.
- N. A. Farrow, R. Muhandiram, A. U. Singer, S. M. Pascal, C. M. Kay, G. Gish, S. E. Shoelson, T. Pawson, J. D. Forman-Kay and L. E. Kay, Biochemistry, 1994, 33, 5984–6003 CrossRef CAS PubMed.
- P. Dosset, J. C. Hus, M. Blackledge and D. Marion, J. Biomol. NMR, 2000, 16, 23–28 CrossRef CAS PubMed.
- S. Grzesiek, J. Anglister and A. Bax, J. Magn. Reson., Ser. B, 1993, 101, 114–119 CrossRef CAS.
- G. T. Montelione, B. A. Lyons, S. D. Emerson and M. Tashiro, J. Am. Chem. Soc., 1992, 114, 10974–10975 CrossRef CAS.
- A. Bax, G. M. Clore and A. M. Gronenborn, J. Magn. Reson., 1990, 88, 425–431 CAS.
- L. E. Kay, M. Ikura and A. Bax, J. Am. Chem. Soc., 1990, 112, 888–889 CrossRef CAS.
- Y. Shen, F. Delaglio, G. Cornilescu and A. Bax, J. Biomol. NMR, 2009, 44, 213–223 CrossRef CAS PubMed.
- P. Güntert, C. Mumenthaler and K. Wüthrich, J. Mol. Biol., 1997, 273, 283–298 CrossRef PubMed.
- T. Herrmann, P. Güntert and K. Wüthrich, J. Mol. Biol., 2002, 319, 209–227 CrossRef CAS PubMed.
- C. D. Schwieters, J. J. Kuszewski, N. Tjandra and G. M. Clore, J. Magn. Reson., 2003, 160, 65–73 CrossRef CAS PubMed.
- A. Bhattacharya, R. Tejero and G. T. Montelione, Proteins, 2007, 66, 778–795 CrossRef CAS PubMed.
- V. Tugarinov and L. E. Kay, J. Am. Chem. Soc., 2006, 128, 7299–7308 CrossRef CAS PubMed.
- F. E. Jacobsen, K. M. Kazmierczak, J. P. Lisher, M. E. Winkler and D. P. Giedroc, Metallomics, 2011, 3, 38–41 RSC.
- K. J. Wayne, L. T. Sham, H. C. Tsui, A. D. Gutu, S. M. Barendt, S. K. Keen and M. E. Winkler, J. Bacteriol., 2010, 192, 4388–4394 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplemental Tables S1, S2 and Fig. S1–S6. See DOI: 10.1039/c5mt00221d |
| ‡ Current address: Department of Early Discovery Biochemistry, Genentech, 1 DNA Way, South San Francisco, California 94080 USA. |
| § The molecular coordinates of apo-CupA have been deposited in the Protein Data Bank under accession code 2MRY. |
| This journal is © The Royal Society of Chemistry 2016 |