
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zinc is a transmembrane agonist that induces platelet activation in a tyrosine phosphorylation-dependent manner
Ben R.
Watson
a,
Nathan A.
White
b,
Kirk A.
Taylor
b,
Joanna-Marie
Howes
a,
Jean-Daniel M.
Malcor
a,
Dominique
Bihan
a,
Stewart O.
Sage
c,
Richard W.
Farndale
a and
Nicholas
Pugh
*ab
aDepartment of Biochemistry, University of Cambridge, Downing Site, Cambridge, UK
bDepartment of Biomedical and Forensic Sciences, Anglia Ruskin University, East Road, Cambridge, UK. E-mail: nicholas.pugh@anglia.ac.uk; Tel: +44 (0)845 196 2661
cDepartment of Physiology, Development and Neuroscience, University of Cambridge, Downing Street, Cambridge, UK
First published on 28th August 2015
Abstract
Following platelet adhesion and primary activation at sites of vascular injury, secondary platelet activation is induced by soluble platelet agonists, such as ADP, ATP, thrombin and thromboxane. Zinc ions are also released from platelets and damaged cells and have been shown to act as a platelet agonist. However, the mechanism of zinc-induced platelet activation is not well understood. Here we show that exogenous zinc gains access to the platelet cytosol and induces full platelet aggregation that is dependent on platelet protein tyrosine phosphorylation, PKC and integrin αIIbβ3 activity and is mediated by granule release and secondary signalling. ZnSO4 increased the binding affinity of GpVI, but not integrin α2β1. Low concentrations of ZnSO4 potentiated platelet aggregation by collagen-related peptide (CRP-XL), thrombin and adrenaline. Chelation of intracellular zinc reduced platelet aggregation induced by a number of different agonists, inhibited zinc-induced tyrosine phosphorylation and inhibited platelet activation in whole blood under physiologically relevant flow conditions. Our data are consistent with a transmembrane signalling role for zinc in platelet activation during thrombus formation.
Introduction
Platelets are the principal cellular determinants of haemostasis and pathological thrombus formation leading to myocardial infarction and stroke. Following adhesion to sites of vascular damage, platelets undergo primary activation leading to shape change, upregulation of integrin αIIbβ3 activity and secretion of alpha and dense granules.1 Granule release introduces bioactive molecules into the immediate environment of a growing thrombus. Such molecules include adhesion proteins, coagulation factors and soluble platelet agonists, such as ADP, ATP, thromboxane-A2 and zinc.2–4Plasma zinc concentration ranges from 10 to 20 μM, however, binding of zinc to plasma proteins lowers the free zinc concentration to 0.5 to 1 μM.5 Zinc is present in the cytosol and alpha granules of platelets at concentrations up to 60-fold higher than in the plasma.3,5,6 Upon platelet activation, zinc is released from platelets, contributing to a rise in the free zinc concentrations of the plasma and in the microenvironment of a growing thrombus.7,8
Endogenous zinc is also released or secreted from damaged cells and can act as a paracrine agonist of zinc-dependent signalling pathways in epithelial cells.9 Zinc has an acknowledged role in haemostasis. Low zinc diets are associated with platelet-related bleeding disorders in humans and rodents.10,11 Rats with acute and long term zinc deficiency have impaired platelet aggregation responses to ADP and thrombin,11,12 and rats fed with a high zinc diet show increased platelet responsiveness to collagen, ADP, thrombin and adrenaline.13 Additionally, zinc-deficient cancer patients exhibit cutaneous bleeding and platelet dysfunction that is reversed by zinc supplementation.14
Zinc has previously been shown to be a platelet agonist, with sub-millimolar concentrations of zinc being able to induce aggregation. Low concentrations of zinc potentiate ADP-induced platelet aggregation in an integrin αIIbβ3 dependent manner.15,16 However, the mechanism by which zinc causes platelet aggregation and its relative contribution to pathophysiological thrombus formation remains unstudied.
Here we confirm that zinc is a platelet agonist at millimolar concentrations, and is able to potentiate platelet activation at low micromolar concentrations. Furthermore, we investigated the mechanism of zinc-induced platelet activation using millimolar zinc. Zinc gains access to the platelet cytosol and induces aggregation in a manner that is dependent on secondary activators, intracellular calcium release and tyrosine phosphorylation. The pattern of phosphorylated proteins is suggestive of a novel signalling pathway.
Our data are consistent with a role for zinc in platelet activation during haemostasis and pathogenic thrombus formation.
Results
Zinc is a platelet agonist
Previous studies have shown that 0.5 mM ZnCl2 induces aggregation in both platelet rich plasma (PRP) and washed platelets, and that increasing the concentration further results in a reduced response.15,16 We confirmed this observation with the use of ZnSO4 (Fig. 1). Complete aggregation of washed platelets was induced by 300 μM of ZnSO4 (Fig. 1A and B). Aggregation was biphasic, indicating a reliance on secondary signalling. In agreement with previous reports, increasing zinc above 1 mM led to a reduced response. Of the other exogenously added cations tested (NiSO4, LiSO4, CuSO4, CoSO4, MgSO4, MnCl2), only CuSO4 induced a partial response (Fig. 1B).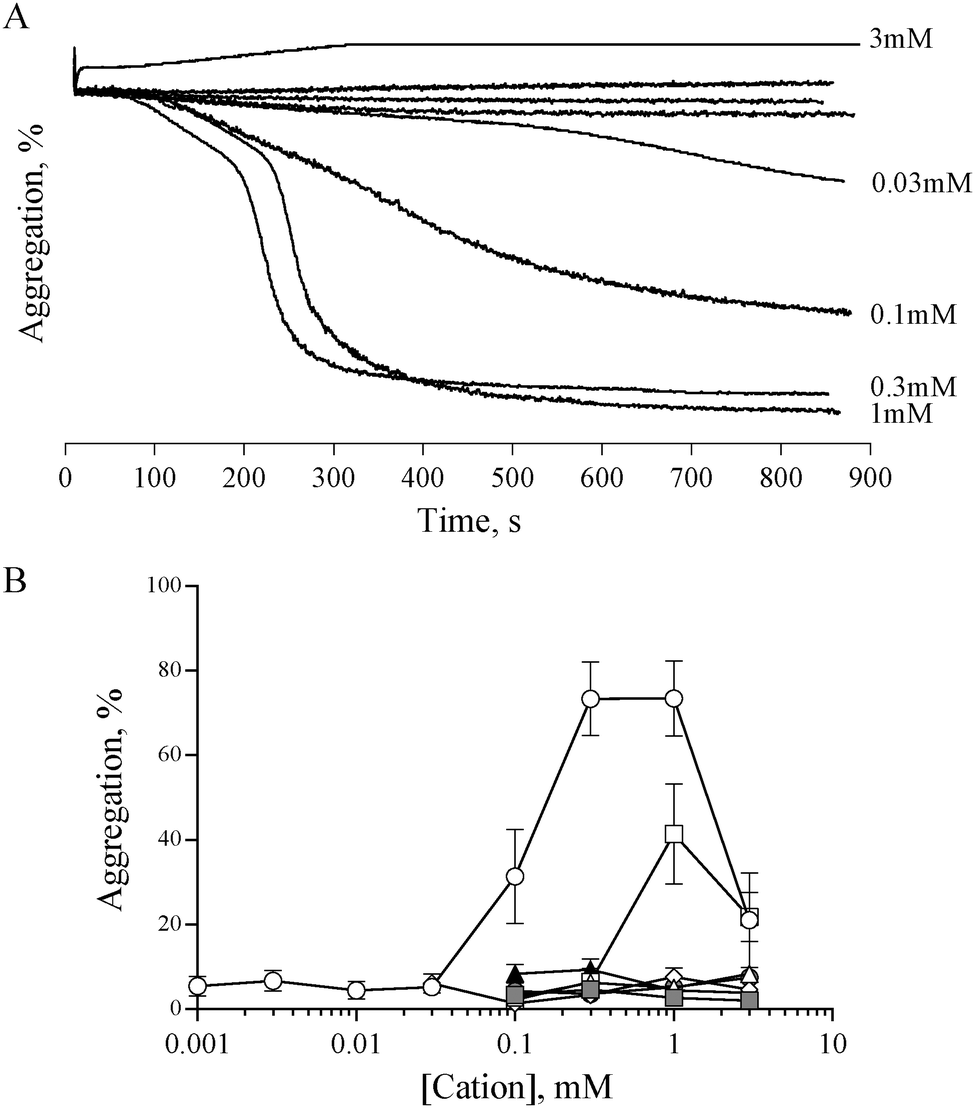 | ||
Fig. 1 Zinc, but not other cations, induces platelet aggregation. The effect of exogenous zinc on platelet activation was investigated using light transmission aggregometry. (A) Representative experiment showing platelet responses to differing concentrations of ZnSO4. At intermediate concentrations, ZnSO4 induced a biphasic aggregation response, suggestive of secondary signalling-mediated activation. High concentrations of ZnSO4 (3 mM) resulted in a reduced response. Similar aggregation traces were observed using ZnCl2 (not shown). (B) Quantitation of aggregation traces indicates that maximal aggregation was induced by 0.3 mM to 1 mM ZnSO4 (○). CuSO4 induced partial aggregation at 1 mM (□). Other cations (NiSO4 △, LiSO4 ⋄, CoSO4 , MgSO4 , MgSO4 , MnCl2 ▲) did not induce platelet aggregation. , MnCl2 ▲) did not induce platelet aggregation. | ||
Zinc-induced platelet aggregation involves secondary mediators of platelet activation
In order to assess the influence of secondary signalling on zinc-induced aggregation, platelets were pre-treated with antagonists of secondary signalling pathways before being challenged with different concentrations of ZnSO4.Pre-treatment with the αIIbβ3 antagonist, GR144053 (2 μM) abolished platelet aggregation in response to 1 mM ZnSO4 (Fig. 2A). Thus, zinc-induced activation results in αIIbβ3-dependent aggregation and is not dependent on cation-induced agglutination. Pre-treatment for 15 min with aspirin (1 mM), NF449 (P2X1 antagonist, 100 μM), 2-MeSAMP (P2Y12 antagonist, 100 μM) or PGE1 (PG receptor antagonist, 2 μM) reduced zinc-induced aggregation (Fig. 2A). This indicates roles for secondary activation pathways (thromboxane A2 and purinergic signalling) in zinc-induced aggregation, in addition to a role for cAMP in zinc-mediated signalling.
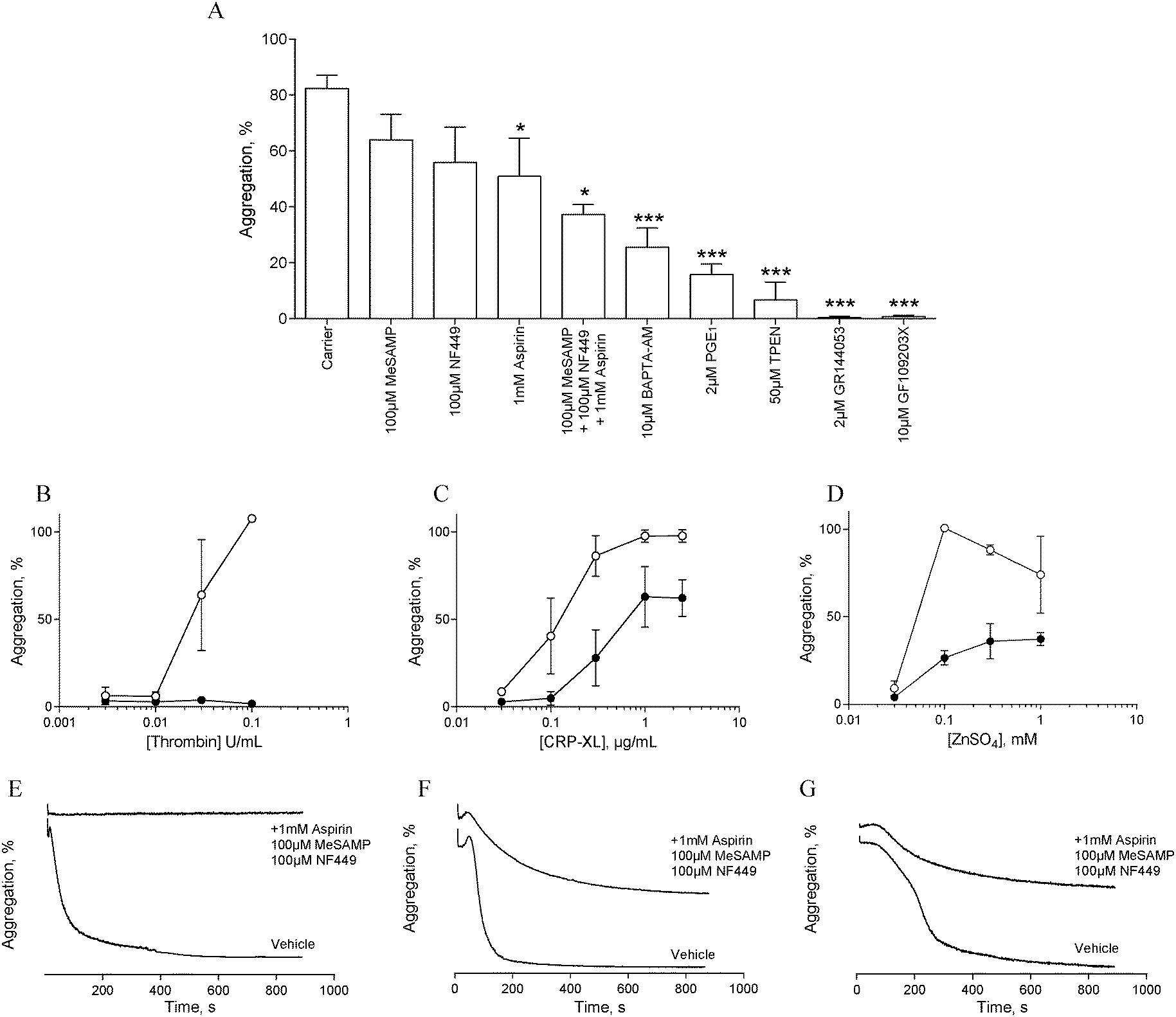 | ||
| Fig. 2 Addition of exogenous ZnSO4 to platelets pre-treated with specific antagonists reveals a role for secondary signalling in zinc-induced aggregation. (A) Platelets were treated with inhibitors before being challenged with 1 mM ZnSO4. Untreated platelets (○) or platelets pre-treated with a combination of inhibitors (1 mM aspirin, 100 μM MeSAMP and 100 μM NF449, ●) were challenged with different concentrations of thrombin (B), CRP-XL (C), or ZnSO4 (D). Representative aggregation traces following activation with 0.1 U mL−1 thrombin (E), 1 μg mL−1 CRP-XL (F), 1 mM ZnSO4 (G). | ||
The profile of aggregation traces following aspirin, MeSAMP or NF449 treatment is suggestive of partial aggregation without secondary activation (not shown). DM-BAPTA-AM pre-treatment (10 μM) inhibited zinc-induced aggregation (Fig. 2A). This is consistent with a requirement for intracellular signalling which may include exogenous cation entry into the platelet cytosol or cation release from intracellular stores. Pre-treatment with the PKC antagonist GF109203X fully inhibited zinc-induced aggregation (Fig. 2A), indicating that zinc-dependent signalling pathways converge on PKC. We further investigated the influence of zinc on platelet activation with the membrane permeable zinc chelator, TPEN. TPEN pre-treatment inhibited zinc-induced aggregation to a similar degree to that caused by PGE1, indicating that intracellular zinc is necessary for zinc-induced aggregation. The concentration of TPEN used here (50 μM) was effective when platelets were activated with 1 mM ZnSO4, indicating that the inhibitory effect of TPEN was intracellular, and not caused by chelation of extracellular zinc ions prior to platelet activation.
To further investigate the involvement of secondary signalling, platelets were pre-treated for 15 min with a combination of aspirin (1 mM), NF449 (100 μM) and MeSAMP (100 μM) before being challenged with increasing concentrations of CRP-XL, thrombin or ZnSO4. Whilst thrombin-induced aggregation was abolished (Fig. 2B and E), CRP-XL (Fig. 2C and F) and zinc-induced aggregation (Fig. 2D and G) were only partially inhibited. Thus, zinc-induced activation relies on secondary platelet signalling for a full aggregation response.
Zinc potentiates platelet reactivity to CRP-XL, thrombin and adrenaline
The data described in Fig. 1 and 2 suggest that zinc-induced platelet aggregation requires secondary agonists that are likely to be released in a PKC-dependent manner. However, the concentrations of zinc used here (0.1 to 1 mM) are higher than the local concentrations reported to occur during thrombus formation.7 Therefore, we investigated the potential of sub-activatory, pathophysiologically relevant zinc levels to potentiate platelet activation to different agonists. Sub-activatory concentrations of 10 μM and 30 μM ZnSO4 were used as these are only an order of magnitude greater than previously reported plasma free zinc levels.5Platelets potentiated with 30 μM ZnSO4 showed an increased response to low concentrations of all agonists tested (Fig. 3). Pre-treatment with 30 μM ZnSO4 followed by challenge with 0.01 μg mL−1 CRP-XL resulted in 48.1 ± 10.6% aggregation, whereas non-potentiated platelets resulted in 10.1 ± 5.0% aggregation (Fig. 3A and D). Similarly, 0.03 U mL−1 of thrombin failed to induce aggregation whilst potentiation with zinc restored aggregation to 60.6 ± 16.2% (Fig. 3B and E). Treatment of control platelets with adrenaline resulted in full aggregation following a lag period. Preincubation with zinc shortened this lag period at all adrenaline concentrations tested (Fig. 3C and F). For example, 1 μM adrenaline induced aggregation after a lag period of 586.6 ± 35.4 s. ZnSO4 potentiation reduced this time to 333.3 ± 29.1 s. Pre-treatment with 10 μM ZnSO4 did not potentiate platelet activation to CRP-XL (not shown).
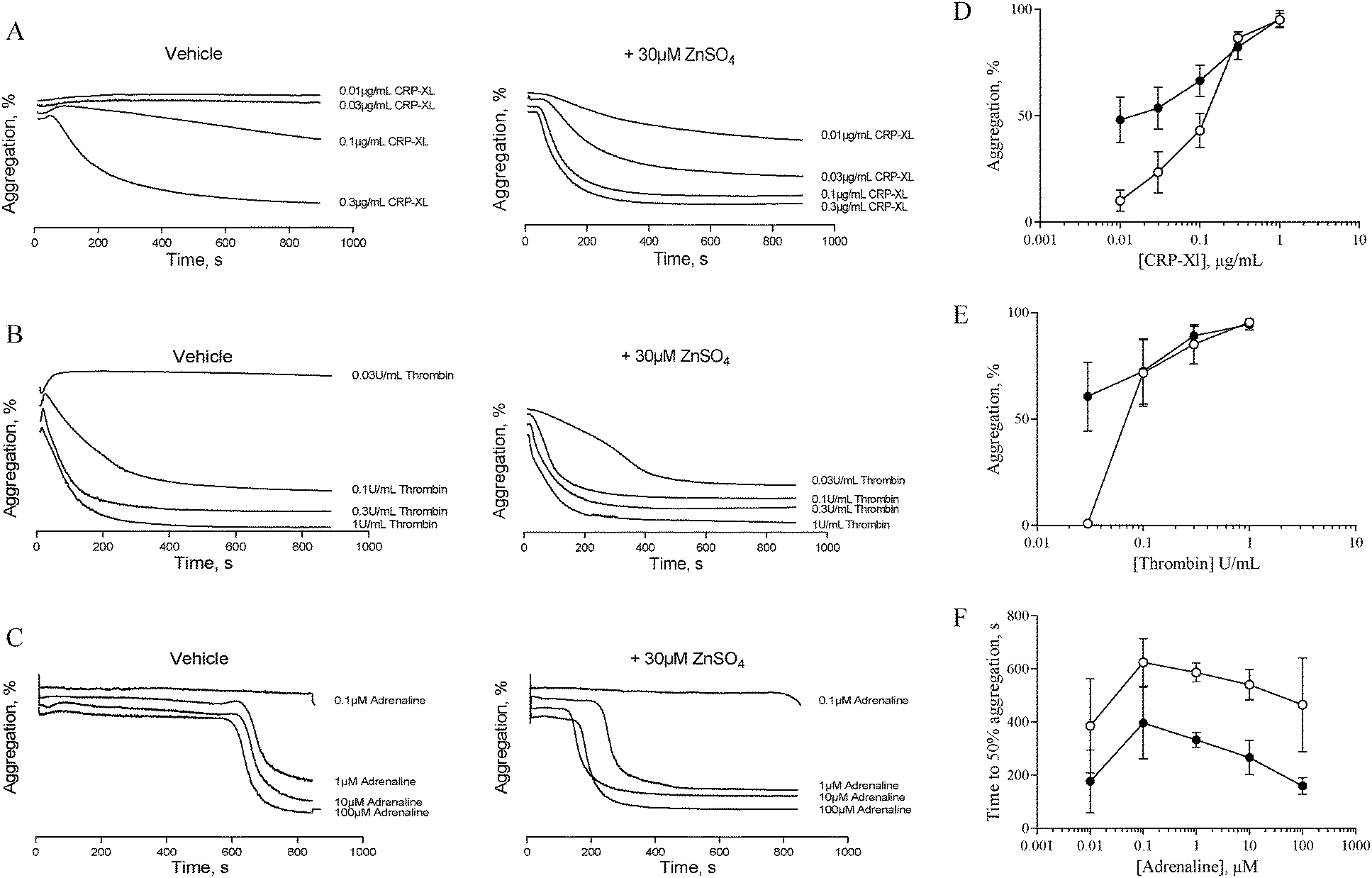 | ||
| Fig. 3 Sub-activatory concentrations of ZnSO4 potentiate platelet activation by other agonists. Platelet suspensions were pre-treated for 15 min with sub-activatory concentrations (30 μM) of ZnSO4 before being challenged with different concentrations of agonist. Representative aggregation traces following CRP-XL- (A), thrombin- (B) or adrenaline- (C) stimulation of 30 μM ZnSO4 pre-treated platelets. Maximal aggregation was calculated for CRP-XL- and thrombin-induced activation (D and E), and time to 50% aggregation was calculated for adrenaline-induced activation (F). ○ Untreated platelets and ● 30 μM ZnSO4 treated platelets. | ||
Zinc induces tyrosine phosphorylation of platelet proteins
Tyrosine phosphorylation is a platelet activatory signalling event strongly induced by collagen and CRP-XL,17–19 and to a lesser extent induced by other platelet agonists. To provide further insight into the mechanisms responsible for zinc-induced aggregation, the extent of tyrosine phosphorylation in the platelet suspensions following treatment with zinc was examined. Exogenous zinc (1 mM) induced tyrosine phosphorylation in a time-dependent manner (Fig. 4A). Increased phosphorylation of high molecular weight proteins was visible after 30 s. Thrombin (0.1 U mL−1) induced few tyrosine phosphorylation events, as previously reported20 (Fig. 4B). Interestingly, ZnSO4 treatment (0.1 mM) induced a pattern of tyrosine phosphorylation which differed from that observed in response to CRP-XL or thrombin. High MW proteins phosphorylated by ZnSO4 treatment were absent following CRP-XL treatment, whilst a low MW protein (consistent in size with the FcγR19) was phosphorylated by CRP-XL but not ZnSO4. Additionally, pre-treatment of platelets with 50 μM TPEN resulted in increased tyrosine phosphorylation of a high molecular weight protein (approximately 95 kDa). TPEN reduced phosphorylation of platelet proteins following stimulation with 0.1 mM ZnSO4 and 0.1 U mL−1 thrombin, but had little effect on phosphorylation initiated by 1 μg mL−1 CRP-XL.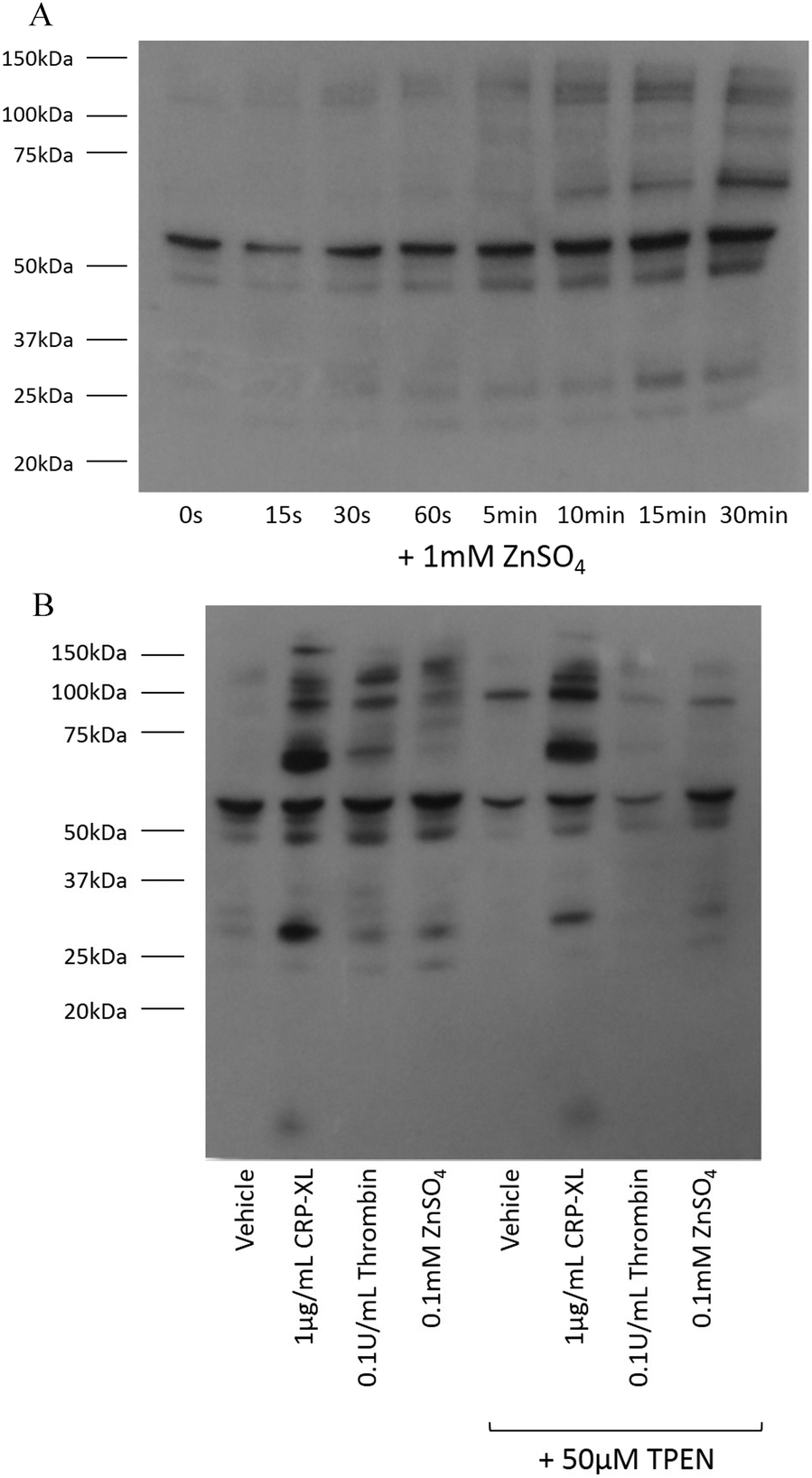 | ||
| Fig. 4 Zinc induces tyrosine phosphorylation of platelet proteins. (A) Platelet suspensions were incubated with 1 mM ZnSO4 for increasing time periods. Tyrosine phosphorylation of platelet proteins could be observed 30 s after incubation. Platelet suspensions were activated for 15 min with 0.1 μg mL−1 CRP-XL, 0.1 U mL−1 thrombin or 0.1 mM ZnSO4 with stirring (B). Substantially more tyrosine phosphorylation events were observed following GpVI-dependent activation with CRP-XL than were observed following thrombin activation. ZnSO4 also induced tyrosine phosphorylation of platelet proteins, although the pattern was different to that seen with CRP-XL. No phosphorylation of FcγR was observed following ZnSO4 treatment. TPEN treatment (50 μM) resulted in increased phosphorylation of a high molecular weight protein. TPEN inhibited ZnSO4- but not CRP-XL-induced phosphorylation. | ||
Zinc-induced aggregation is independent of GpVI-mediated activation, but upregulates GpVI activity
The mechanism by which zinc causes platelet aggregation is unclear. As PKC and Ca2+ signalling are downstream of GpVI engagement, we examined the involvement of GpVI in zinc-induced activation. Pre-treatment of platelet suspensions with the GpVI-blocking antibody, 1C3, resulted in a reduced response to CRP-XL, whilst zinc-induced aggregation was unaffected (Fig. 5A). 1C3 reduced aggregation induced by 0.1 μg mL−1 CRP-XL to 9.5 ± 5.6% whilst 300 μM ZnSO4 treatment resulted in 104.7 ± 8.0% aggregation (Fig. 5A and B). Thus, GpVI-functionality is not a prerequisite for zinc-induced aggregation.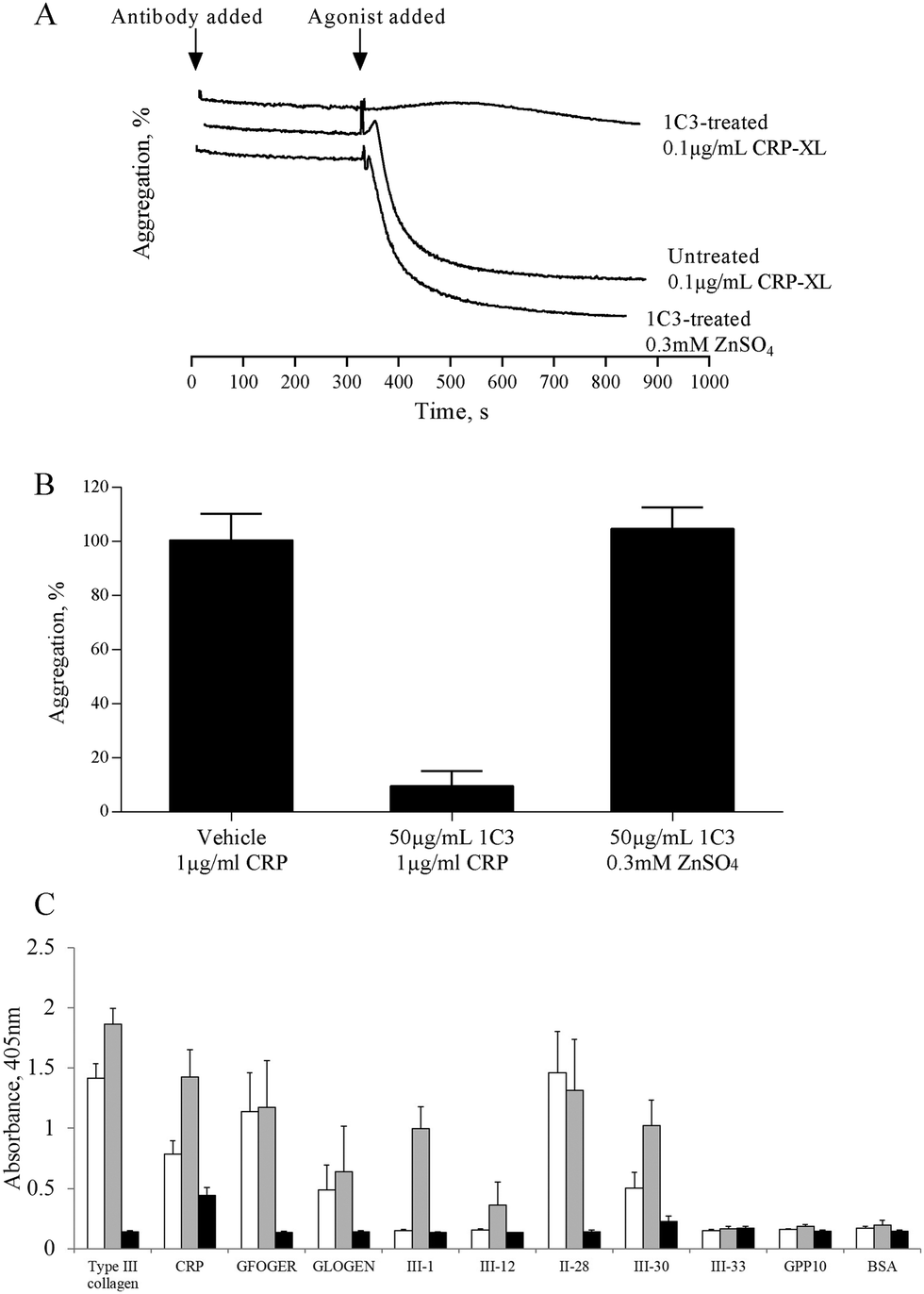 | ||
Fig. 5 Zinc-induced platelet activation is independent of GpVI activity, but upregulates GpVI-dependent platelet adhesion. Platelet suspensions were treated with the GpVI blocking antibody, 1C3 for 5 min before being challenged with 0.3 mM ZnSO4 (A and B). Antibody treatment inhibited aggregation to CRP-XL but not ZnSO4, indicating that GpVI activity is not required for zinc-induced aggregation. (C) GR144053-treated platelet suspensions activated by 100 μM ZnSO4 displayed a significant increase in binding activity to GpVI-adhesive peptides (CRP, III-1 and III-30), whilst binding to α2β1-adhesive peptides (GFOGER, GLOGEN and II-28) was not affected. □ GR144053-treated platelets,  GR144053-treated platelets activated by 100 μM ZnSO4- and ■ GR144053-treated platelets pre-treated with 2 mM EDTA. GR144053-treated platelets activated by 100 μM ZnSO4- and ■ GR144053-treated platelets pre-treated with 2 mM EDTA. | ||
The influence of zinc on platelet adhesive receptor activity was further investigated by examining receptor-specific platelet adhesion using collagen-mimetic peptides. GR144053-treated platelet suspensions treated with 100 μM ZnSO4 displayed a significant increase in binding activity to GpVI-binding peptides (CRP, III-1 and III-30), whilst binding to α2β1-binding peptides (GFOGER, GLOGEN and II-28) was not affected (Fig. 5C). For adhesion to GpVI-adhesive peptides, A405 values were 0.8 ± 0.1 and 1.4 ± 0.2 for untreated and 100 μM zinc treated platelets respectively on CRP, and 0.5 ± 0.1 and 1.0 ± 0.2 on III-30. On GFOGER, absorbance values were 1.1 ± 0.3 and 1.2 ± 0.4 respectively. These data indicate a selective role for zinc in increasing the affinity of GpVI for its cognate ligand.
Exogenous zinc gains access to the platelet cytosol
Zinc-induced platelet activation may occur as a result of zinc entry into the platelet cytosol. To investigate this possibility we loaded platelets with the zinc-sensitive indicator Fluozin-3 and assessed changes of fluorescence in response to exogenously applied ZnSO4. Using fluorometry, concentration dependent increases of fluorescence were observed following stimulation of platelet suspensions by extracellular zinc (Fig. 6A). To further assess the localisation of Fluozin-3 fluorescence confocal microscopy was employed. Fluozin-3-loaded platelets were adhered to collagen-coated coverslips and changes of intracellular fluorescence were imaged in response to exogenous zinc. Treatment with 10 μM ZnSO4 following platelet adhesion resulted in observable increases of intracellular fluorescence over time (Fig. 6B). This is consistent with increases in cytosolic zinc, demonstrating that exogenous zinc gains access to the platelet cytosol.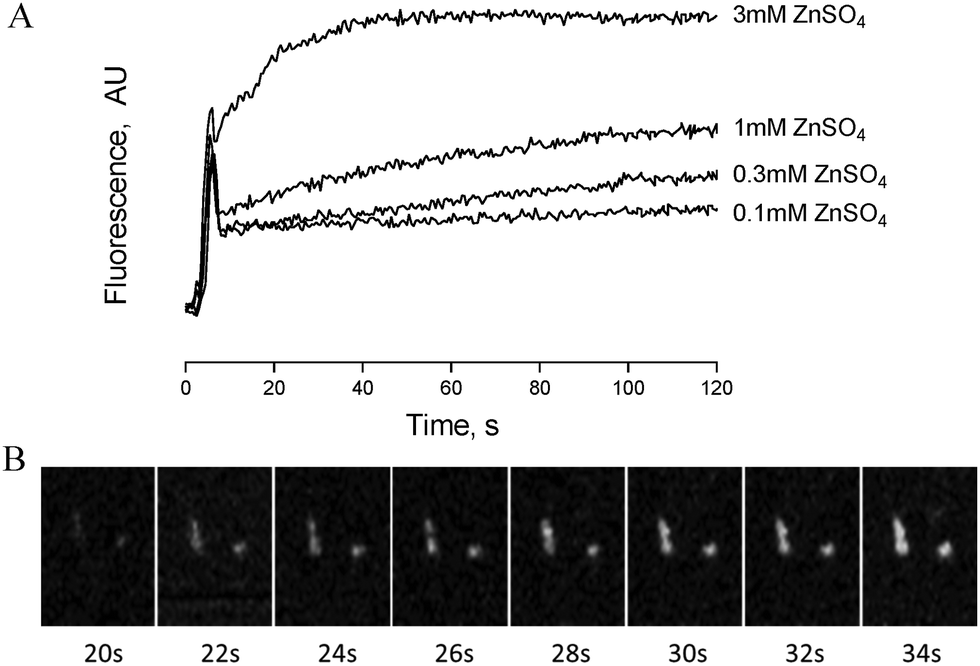 | ||
| Fig. 6 Exogenous zinc gains access to the platelet cytosol. (A) Fluorometric imaging of Fluozin-3-stained platelet suspensions following addition of increasing concentrations of ZnSO4. (B) Collagen-adhered Fluozin-3-loaded platelets were perfused with 10 μM ZnSO4 and imaged by confocal microscopy. This representative time-lapse image sequence shows increases in cytosolic zinc approximately 20 s after onset of perfusion. | ||
TPEN inhibits platelet activation induced by multiple agonists
To investigate the role of zinc in intracellular signalling processes, we examined aggregation of TPEN-treated platelets. Platelet aggregation induced by CRP-XL, U46619 or ZnSO4 was fully abrogated in TPEN-treated platelets, whilst aggregation induced by thrombin or A23187 was inhibited at low agonist concentrations (Fig. 7A–E).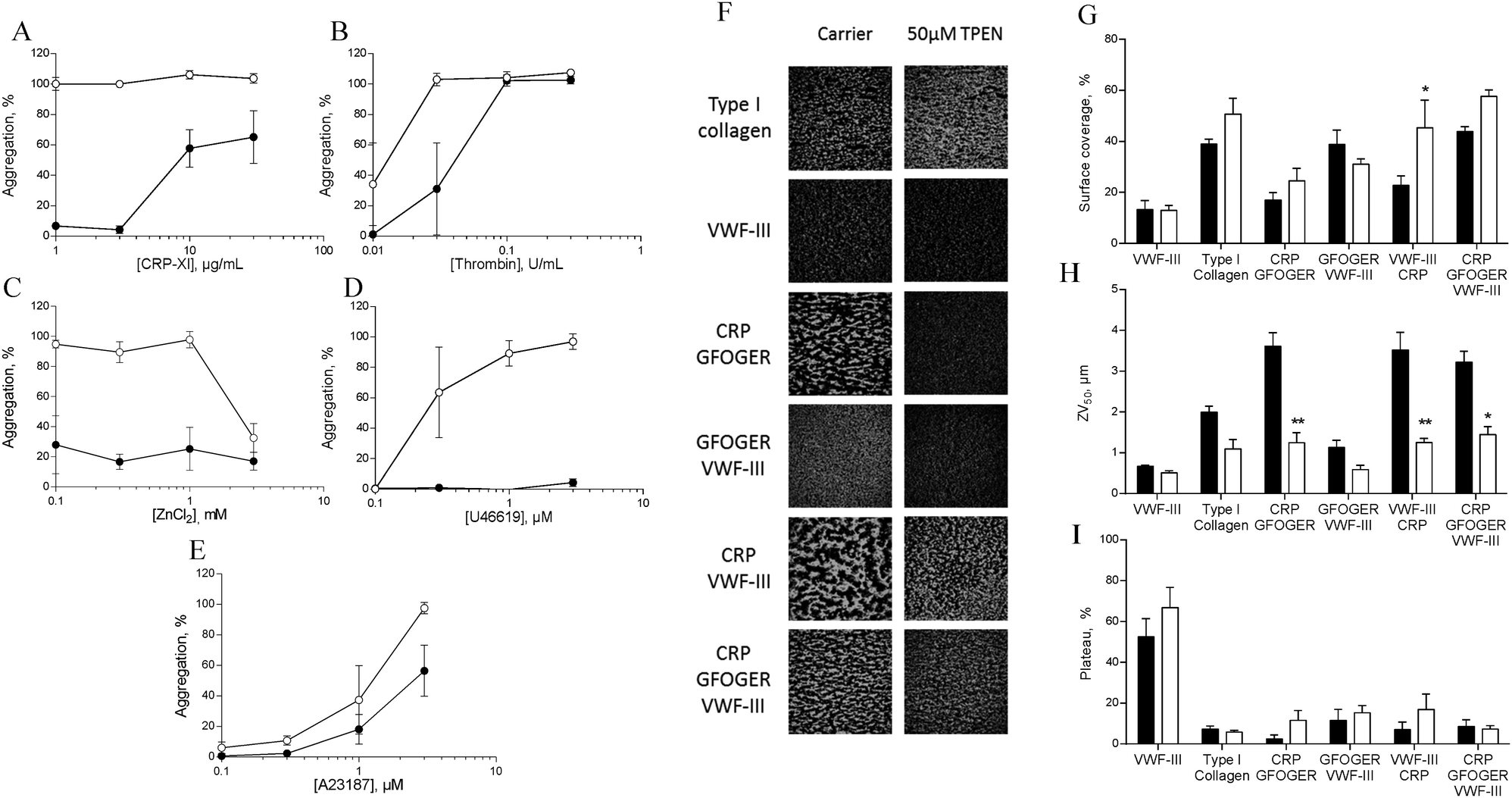 | ||
| Fig. 7 TPEN inhibits platelet activation. Platelets were pre-treated with 50 μM TPEN (●) or vehicle (○) before being challenged with different concentrations of platelet agonists during aggregometry. (A) CRP-XL, (B) thrombin, (C) ZnCl2, (D) U46619, (E) A23187. In all cases a reduction in aggregation as a result of TPEN treatment was observed. Platelets in whole blood were treated with 50 μM TPEN before being perfused across fibrillar collagen, or combinations of collagen mimetic peptide as described in the text. (F) Representative images of platelet thrombi formed following pre-treatment with vehicle (left panel) or 50 μM TPEN (right panel). Quantitative assessment of images of thrombi forming on thrombogenic surfaces under flow conditions. (G) Thrombus or platelet surface coverage, (H) ZV50 and (I) plateau, derived from platelet mobility (PM, see Methods). | ||
We used a whole-blood perfusion system with collagen or collagen-mimetic peptides as substrates to examine receptor-specific platelet adhesion and activation under physiologically relevant flow conditions (Fig. 7F–I).21
Surface coverage was relatively unaffected by TPEN treatment, with an increase only being seen on a VWF-III/CRP surface (from 22.7 ± 3.8% to 45.3 ± 10.8%, Fig. 7G).
The end-point parameter ZV50 reports platelet–platelet interactions, quantifying platelet activation under flow conditions (Fig. 7H).21,22 On CRP/GFOGER, 50 μM TPEN reduced ZV50 from 3.6 ± 0.3 μm to 1.2 ± 0.2 μm, whilst on CRP/GFOGER/VWF-III, ZV50 was reduced from 3.2 ± 0.3 μm to 1.4 ± 0.2 μm. On VWF-III/CRP, ZV50 was reduced from 3.5 ± 1.3 μm to 1.3 ± 0.1 μm. TPEN treatment did not affect ZV50 on any other surfaces. We used “platelet mobility” (PM) as a real time parameter to quantify the effect of TPEN on stable platelet adhesion.22 On all surfaces tested, PM was unaffected by TPEN treatment (Fig. 7I). Thus, TPEN reduces platelet thrombus formation, without affecting the ability of platelets to achieve stable adhesion.
Conclusions
The role of zinc in platelet activation and pathophysiological thrombus formation has received little attention, despite having been shown to be an important and physiologically relevant cofactor in haemostasis.5,10,11,13,15,16 Zinc as a signalling factor has been overlooked in favour of Ca2+, which is considered the more important cation for platelet function owing to its higher concentration and more readily observable fluctuations during platelet activation.23 Whilst the work presented here does not challenge this view, it should be noted that commonly used Ca2+ chelating agents and fluorophores have higher affinities for zinc (for example, the Kds of BAPTA for calcium and zinc are 160 nM and 0.5 nM respectively, whilst Fura-2 has Kds of 145 nM and 3 nM). Use of such reagents may have resulted in an underestimation of the role and significance of zinc in platelet activation.Here we show that exogenous zinc gains access to the platelet cytosol and is able to activate platelets. Chelation of intracellular zinc results in a reduction of platelet aggregation, tyrosine phosphorylation and thrombus formation under flow conditions. These data support a transmembrane signalling role for zinc in platelet activation.
We confirm previous observations that zinc is a potent platelet agonist at millimolar concentrations, and can potentiate platelet aggregation by other agonists at lower concentrations.
Free zinc is present in blood at a concentration of 0.5–1 μM rising to 6–10 μM in the close proximity of platelet membranes.5 However, zinc is released from activated platelets and damaged cells6–9 so is likely to be concentrated at sites of vascular injury. It is not unreasonable to suggest that higher local concentrations of zinc may be achieved in a growing thrombus, indicating that the potentiating effect observed here and by others2,15 may be pathophysiologically relevant.
Additionally, under conditions of extensive platelet activation, zinc concentrations may reach levels that could activate platelets directly, contributing to thrombus formation.
In our experiments, we have used concentrations of zinc up to and including millimolar levels. Whilst unlikely to represent the physiological situation, these levels have allowed us to probe the mechanisms by which platelets respond to zinc. This strategy has been employed by others in the investigation of platelet signalling pathways with different agonists. For example, the physiological concentration of collagen experienced by platelets during vascular injury is not known, but high concentrations of CRP-XL have been used to investigate GpVI function.19,24,25 To address the pathophysiological significance of zinc, we also used lower concentrations of zinc to investigate potentiation of platelet activation. Zinc-induced platelet aggregation is sensitive to inhibition of αIIbβ3, demonstrating an activatory effect, rather than non-specific cation-induced agglutination. We provide evidence that zinc is not an extracellular stimulus, but gains access to the platelet cytosol to activate platelets. Zinc entry into platelets was observed with confocal microscopy and fluorometry using Fluozin-3 stained platelets. Other cations that have previously been shown to activate integrins (such as Mn2+)26 had no effect on platelet aggregation.
The effect of intracellular chelating agents, BAPTA-AM and TPEN, is consistent with an intracellular effect of zinc, either by directly accessing the cytosol or by activating intracellular signalling processes. Furthermore, complete blockade of zinc-induced aggregation following inhibition of PKC indicates that intracellular biological processes are required for zinc-induced aggregation. These data all provide strong evidence for an active, dynamic transmembrane signalling role for zinc in platelet activation.
The mechanism by which zinc gains access to the platelet cytosol is not known. Zinc, and other divalent cation channels and transporters are present in the platelet proteome, but have yet to be investigated in the context of zinc induced activation. Assessment of the roles of specific channels or transporters in zinc entry must await future studies utilising specific blocking agents. Identification of such blocking reagents would assist in the evaluation of the physiological relevance of zinc in platelet behaviour. It is important to note that our experiments have been carefully designed to preclude contributions by exogenous Ca2+ (i.e. nominally Ca2+-free salines). Whilst these conditions are somewhat removed from the physiological situation, we observe normal platelet aggregation responses to the major agonists CRP-XL, thrombin and U46619 (Fig. 2 and 7). However, we cannot exclude the possibility that zinc-induced aggregation coincides with the release of intracellular Ca2+ and activation of store operated Ca2+ entry. Extracellular Ca2+ has previously been shown to be required for platelet responses to phorbol-myristate acetate (PMA) in zinc deficient rats.27 It is intriguing to postulate that zinc and calcium may have a synergistic relationship. This remains an important area of study and will be the focus of future research.
Zinc-induced activation resulted in tyrosine phosphorylation of platelet proteins, granule release and secondary activation leading to αIIbβ3 activation, whilst zinc chelation inhibited phosphorylation and aggregation in response to multiple agonists. This indicates a correlation between zinc-induced phosphorylation and aggregation.
Similarities were observed between GpVI- and zinc-induced platelet activation. Both involve substantial protein tyrosine phosphorylation, and both are less dependent on secondary signalling than thrombin-induced aggregation. However, the pattern of protein phosphorylation induced by the two agonists is sufficiently different to suggest an alternative mechanism. The absence of clear FcγR phosphorylation following zinc treatment suggests that zinc acts downstream of GpVI dimerization and Src-dependent FcγR phosphorylation. However, this is not consistent with the observation that zinc causes upregulation of GpVI-dependent platelet adhesion. The nature of the increased adhesive response of GpVI is not known, but may involve promotion of receptor dimerization.25 This may be regulated by increases in cytosolic zinc concentrations or by a direct extracellular effect on GpVI. Our results are suggestive of an inside-out and outside-in scheme of GpVI regulation that is reminiscent of integrin behaviour in platelets and other cells. This is a novel and interesting possibility that warrants further research.
Involvement of PKC in zinc-induced aggregation is inferred from inhibition of aggregation by PKC inhibitors. Whether an increase in cytosolic calcium is required for zinc-induced activation is not known and would be difficult to assess empirically due to the higher affinity for zinc over calcium of conventional calcium indicators and chelators. However, zinc has also been shown to affect PKC activity in platelets and lymphocytes, by increasing association with the cell membrane.28 Additionally, the biphasic aggregometry response to zinc indicates a role for granule release in full activation, further implicating PKC in zinc-induced aggregation.29 This is further supported by the observation that combined inhibition of P2Y12, P2X1 and thromboxane signalling results in partial aggregation following zinc treatment (an inhibition-resistant activation profile) demonstrating elements of an activatory response that are independent of granule release. Again, this resembles GpVI-rather than thrombin-induced aggregation and may reflect partial increase of αIIbβ3 activity in the absence of secondary signalling.
The phosphorylation of high molecular weight proteins indicates that kinase or phosphatase activity is affected by exogenous zinc. The pattern of phosphorylation differed from that of GpVI- and protease activated receptor (PAR) dependent phosphorylation, suggesting the involvement of different kinases and substrates. Interestingly, chelation of intracellular zinc with TPEN resulted in the phosphorylation of a 95 kDa protein, indicating the activity of kinases or phosphatases that are sensitive to intracellular zinc levels. TPEN treatment inhibited zinc- and PAR-dependent protein tyrosine phosphorylation but had little effect on GpVI-mediated phosphorylation. This may indicate conservation of PAR and zinc-dependent signalling mechanisms.
Zinc is released from platelets and is likely to affect platelets within a growing thrombus.7 However, the extent of the influence of zinc on pathophysiological thrombus formation is not known. Here, we demonstrate that chelation of intracellular zinc inhibits thrombus formation under physiologically relevant flow conditions. This work does not directly investigate signalling, but highlights a role for zinc in platelet activation. Given the number and variety of zinc-binding proteins in the platelet cytosol, an effect on activation is not surprising.
Potentiation of platelet aggregation with sub-activatory levels of zinc may provide evidence for a physiological role of zinc in platelet activation. Potentiation increased the efficacy of low concentrations of platelet agonists that act via GpVI and PARs. Interestingly, potentiation of adrenaline-induced activation led to a reduction in the time taken to achieve aggregation, but not in the final extent of aggregation. We propose that zinc released from activated platelets or damaged cells will potentiate the activation of other platelets in a growing thrombus. This hypothesis will be testable following the identification of specific antagonists that block zinc entry into platelets.
In conclusion, our data confirm previous observations of a role for zinc as a platelet agonist. We provide data demonstrating a potentiating role for sub-activatory levels of zinc during platelet activation by other agonists. Zinc enters the platelet cytosol and regulates protein phosphorylation-dependent platelet signalling events, leading to granule release and full PKC-mediated αIIbβ3-dependent aggregation. Although similarities exist between zinc-activation and GpVI or PAR-mediated activation, sufficient differences remain to suggest a novel mechanism. Furthermore, chelation of platelet zinc inhibits platelet activation and tyrosine phosphorylation, indicating a role for intracellular zinc in platelet function. Whilst this work does not address the physiological relevance of zinc in thrombus formation, we speculate that zinc is acting as a transmembrane signalling ion.
Experimental
Ethical approval
Whole blood was obtained from healthy volunteers, free from medication for two weeks, after consent in accordance with the Declaration of Helsinki and our Institutional guidelines, as approved by the local ethics committee.Materials
NF449 (4,4′,4′′,4′′′-[carbonyl bis(imino-5,1,3-benzenetriyl-bis(carbonylimino))]tetrakis-1,3-benzenedisulfonic acid, octasodium salt), 2-MeSAMP (methylthioadenosine 5′-monophosphate triethylammonium salt hydrate) and GF109203X (2-[1-(3-dimethylaminopropyl)indol-3-yl]-3-(indol-3-yl) maleimide) were from Tocris, UK. Fluozin-3-AM was from Invitrogen, UK. GR144053 (4-[4-[4-(aminoiminomethyl)phenyl]-1-piperazinyl]-1-piperidineacetic acid trihydrochloride hydrate) was from Novabiochem, UK. PPACK (Phe-Pro-Arg-chloromethylketone) was from Cambridge Bioscience, UK. Bovine tendon collagen I fibres were the gift of Ethicon Corp (New Jersey, USA). The anti-GpVI ScFv 1C3 has previously been described.30 TPEN (N,N,N′,N′-tetrakis(2-pyridylmethyl) ethylenediamine), aspirin, prostaglandin E1 (PGE1), DiOC6 (3,3′-dihexyloxacarbocyanine iodide), DM-BAPTA-AM (dimethyl-1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid) and other chemicals were from Sigma-Aldrich, UK. Cross-linked CRP (CRP-XL) was prepared as previously described.17 Collagen-mimetic peptides were prepared as previously described.21,31–33 Peptides used in these experiments included:CRP: GCO(GPO)10GCOG-amide (where O indicates hydroxyproline);
GFOGER: GPC(GPP)5GFOGER(GPP)5GPC-amide
VWF-III: GPC(GPP)5GPRGQOGVMGFO(GPP)5GPC-amide
GLOGEN: GPC(GPP)5GLOGEN(GPP)5GPC-amide;
GPP10: GPC(GPP)10GPC-amide;
III-1: GPC(GPP)5GLAGYOGPAGPOGPOGPOGTSGHOGSO(GPP)5GPC-amide
III-12: GPC(GPP)5GQRGEOGPQGHAGAQGPOGPOGINGSO(GPP)5GPC-amide;
III-30: GPC(GPP)5GAOGLRGGAGPOGPEGGKGAAGPOGPO(GPP)5GPC-amide;
III-33: GPC(GPP)5GEOGGOGADGVOGKDGPRGPTGPIGPO(GPP)5GPC-amide.
Platelet preparation
Whole blood was collected into 11 mM sodium citrate and centrifuged to obtain PRP (240 g, 15 min). 2 μM prostaglandin E1 (PGE1, Sigma-Aldrich) was added and the PRP centrifuged (640 g, 15 min). The pellet was resuspended to 2 × 108 platelets per mL in calcium-free Tyrodes buffer (CFT, 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 5 mM glucose, 0.42 mM NaH2PO4, 12 mM NaHCO3, pH 7.4). Platelet suspensions were rested for 30 min at 37 °C before use.Aggregometry
Aggregation experiments were performed at 37 °C in an AggRam aggregometer (Helena Biosciences, UK). Briefly, 250 μL of platelet suspension was aliquotted into a cuvette along with a magnetic stir bar. 100% aggregation was calibrated using CFT. Agonists or antagonists were added (1% v/v) and aggregation was recorded with stirring (1000 rpm) for 15 min. Maximal aggregation and the time to 50% aggregation were calculated. Platelet suspensions were treated with inhibitors, potentiating concentrations of ZnSO4 (indicated in the text) or vehicle control for 15 min prior to each experiment.Western blotting
Western blotting was performed as described previously.34 Briefly, washed platelet suspensions (2 × 109 mL−1) were stimulated under stirring conditions in the presence of reagents stated in the text. At given time points, platelets were lysed and boiled for 5 minutes. Samples were resolved by 10% SDS-PAGE and transferred onto a nitrocellulose membrane. Membranes were probed with an anti-phosphotyrosine primary antibody (4G10; Fisher, UK) and anti-mouse HRP-conjugated secondary antibodies. Equal amounts of proteins were loaded onto each lane, and was assessed by coomassie blue staining of the nitrocellulose membrane. Data are representative of a minimum of three independent experiments using platelets from different donors; representative blots are shown.Intracellular zinc measurements of Fluozin-3-stained platelets
For fluorometry, PRP was incubated with 2 μM Fluozin-3-AM (Invitrogen) for 45 min at 37 °C. Platelets were collected by centrifugation (350 g, 20 min) and resuspended in CFT to 8 × 108 mL−1. Fluorescence was recorded from 1.5 mL stirred aliquots of platelet suspension at 37 °C using a Cairn Research Spectrophotometer (Cairn Research, UK) with excitation at 488 nm and emission at 516 nm. For confocal microscopy, Fluozin-3-stained platelets were adhered to type I collagen fibres on a coverslip for 15 min. Following washing, adhered platelets were perfused with 10 μM ZnSO4, during which images were acquired using FV300 laser-scanning confocal microscope (Olympus, UK) with excitation at 488 nm and emission at 510–530 nm.Static adhesion assay
The adhesion of platelets to collagen or collagen mimetic peptides adsorbed to microtitre plates under static conditions at 21 °C after 60 min was measured using a colorimetric assay as previously described.35,36 The peptides used included ligands for GpVI (CRP, III-1, III-30, type I collagen),36 integrin α2β1 (II-28, GLOGEN, type I collagen),35 and inert peptides (GPP10, III-12 and III-33).36Confocal microscopy
For perfusion studies, whole blood was taken into 40 μM PPACK, supplemented with 10 μM PPACK hourly. Platelets in whole blood were stained with 5 μM DIOC6 for 15 min before use. Perfusion studies were performed as described21,22 with some modification. Briefly, glass coverslips were coated with collagen fibres or combinations of collagen-mimetic peptides. Each peptide, dissolved in 0.01 M acetic acid, was coated at 0.1 mg mL−1 in a mixture containing a total of 0.3 mg mL−1 of peptide. For surfaces where only 1 or 2 active peptides were applied, the total peptide concentration was made up to 0.3 mg mL−1 with the inert homolog GPP10. Collagen was coated at 0.1 mg mL−1. Coverslips were covered with peptide solution and held in a humid chamber overnight at 4 °C. After removal of excess fluid, they were blocked with 1% bovine serum albumin in HEPES buffer (36 mM NaCl, 2.7 mM KCl, 5 mM HEPES, 10 mM glucose, 2 mM MgCl2, 2 mM CaCl2). The coverslip, in a 125 μm deep flow chamber, was mounted on an FV300 laser-scanning confocal microscope (Olympus, UK) and washed for 1 min with HEPES buffer. Blood was drawn through the chamber for 5 min using a syringe pump at a calculated wall shear rate of 1000 s−1 after which residual blood was flushed using HEPES buffer before image acquisition. Where stated, whole blood was pre-incubated with TPEN for 15 min at 37 °C. Images were acquired at 0.2 Hz (for morphology experiments) and subjected to image analysis to generate parameters of thrombus formation including surface coverage and ZV50. ZV50 quantifies the height of a field of thrombi at which the thrombus volume is half maximal. This value correlates with platelet activation.21 Additional dynamic parameters of platelet mobility were calculated as previously described.22 The parameter reported here (plateau) reports the relationship between the extent of stable adhesion achieved by a platelet population and perfusion time.Data analysis
Values are expressed as the mean ± standard error of the mean. Experiments were repeated on at least four occasions using blood from different donors. Differences among mean values were identified using analysis of variance, and post hoc comparisons used Student–Newman–Keuls or Student t tests with residual degrees of freedom and residual mean square to calculate pooled SD.Abbreviations
| PKC | Protein kinase C |
| CRP-XL | Cross linked collagen-related peptide |
| ADP | Adenosine diphosphate |
| ATP | Adenosine triphosphate |
| PRP | Platelet rich plasma |
| PPACK | Phe-Pro-Arg-chloromethylketone |
| PGE1 | Prostaglandin E1 |
| CFT | Calcium free Tyrodes buffer |
| αIIbβ3 | Integrin αIIbβ3. |
Acknowledgements
This work was funded by British Heart Foundation grants to NP (Project PG/14/47/30912), to RWF (Programme RG/09/003/27122), New Horizons (NH/11/1/28922), and Wellcome Trust support to RWF (094470/Z/10/Z).References
- Z. M. Ruggeri and G. L. Mendolicchio, Adhesion Mechanisms in Platelet Function, Circ. Res., 2007, 100, 1673–1685 CrossRef CAS PubMed.
- G. Marx, G. Korner, X. Mou and R. Gorodetsky, Packaging zinc, fibrinogen, and factor XIII in platelet alpha-granules, J. Cell. Physiol., 1993, 156, 437–442 CrossRef CAS PubMed.
- D. B. Milne, N. V. Ralston and J. C. Wallwork, Zinc content of cellular components of blood: methods for cell separation and analysis evaluated, Clin. Chem., 1985, 31, 65–69 CAS.
- A. Zufferey, P. Fontana, J. L. Reny, S. Nolli and J. C. Sanchez, Platelet proteomics, Mass Spectrom. Rev., 2012, 31, 331–351 CrossRef CAS PubMed.
- B. L. Vallee and K. H. Falchuk, The biochemical basis of zinc physiology, Physiol. Rev., 1993, 73, 79–118 CAS.
- R. Gorodetsky, X. Mou, A. Blankenfeld and G. Marx, Platelet multielemental composition, lability, and subcellular localization, Am. J. Hematol., 1993, 42, 278–283 CrossRef CAS PubMed.
- F. Mahdi, Z. S. Madar, C. D. Figueroa and A. H. Schmaier, Factor XII interacts with the multiprotein assembly of urokinase plasminogen activator receptor, gC1qR, and cytokeratin 1 on endothelial cell membranes, Blood, 2002, 99, 3585–3596 CrossRef CAS PubMed.
- N. Stadler, N. Stanley, S. Heeneman, V. Vacata, M. J. Daemen, P. G. Bannon, J. Waltenberger and M. J. Davies, Accumulation of zinc in human atherosclerotic lesions correlates with calcium levels but does not protect against protein oxidation, Arterioscler., Thromb., Vasc. Biol., 2008, 28, 1024–1030 CrossRef CAS PubMed.
- H. Sharir, A. Zinger, A. Nevo, I. Sekler and M. Hershfinkel, Zinc released from injured cells is acting via the Zn2+-sensing receptor, ZnR, to trigger signaling leading to epithelial repair, J. Biol. Chem., 2010, 285, 26097–26106 CrossRef CAS PubMed.
- P. R. Gordon, C. W. Woodruff, H. L. Anderson and B. L. O'Dell, Effect of acute zinc deprivation on plasma zinc and platelet aggregation in adult males, Am. J. Clin. Nutr., 1982, 35, 113–119 CAS.
- M. P. Emery and B. L. O'Dell, Low zinc status in rats impairs calcium uptake and aggregation of platelets stimulated by fluoride, Proc. Soc. Exp. Biol. Med., 1993, 203, 480–484 CrossRef CAS PubMed.
- P. Faure, P. Durand, D. Blache, A. Favier and A. M. Roussel, Influence of a long-term zinc-deficient diet on rat platelet function and fatty acid composition, Biometals, 1995, 8, 80–85 CrossRef CAS PubMed.
- G. Marx, J. Krugliak and M. Shaklai, Nutritional zinc increases platelet reactivity, Am. J. Hematol., 1991, 38, 161–165 CrossRef CAS PubMed.
- M. Stefanini, Cutaneous bleeding related to zinc deficiency in two cases of advanced cancer, Cancer, 1999, 86, 866–870 CrossRef CAS PubMed.
- P. Heyns Adu, A. Eldor, R. Yarom and G. Marx, Zinc-induced platelet aggregation is mediated by the fibrinogen receptor and is not accompanied by release or by thromboxane synthesis, Blood, 1985, 66, 213–219 Search PubMed.
- M. Trybulec, et al., Exposure of platelet fibrinogen receptors by zinc ions: role of protein kinase C., Proc. Soc. Exp. Biol. Med., 1993, 203, 108–116 CrossRef CAS PubMed.
- J. Asselin, et al., A collagen-like peptide stimulates tyrosine phosphorylation of syk and phospholipase C gamma2 in platelets independent of the integrin alpha 2 beta 1, Blood, 1997, 89, 1235–1242 CAS.
- R. A. Blake, G. L. Schieven and S. P. Watson, Collagen stimulates tyrosine phosphorylation of phospholipase C-gamma 2 but not phospholipase C-gamma 1 in human platelets, FEBS Lett., 1994, 353, 212–216 CrossRef CAS PubMed.
- J. Gibbins, et al., Tyrosine phosphorylation of the Fc receptor gamma-chain in collagen-stimulated platelets, J. Biol. Chem., 1996, 271, 18095–18099 CrossRef CAS PubMed.
- A. Golden and J. S. Brugge, Thrombin treatment induces rapid changes in tyrosine phosphorylation in platelets, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 901–905 CrossRef CAS.
- N. Pugh, et al., Synergism between platelet collagen receptors defined using receptor-specific collagen-mimetic peptide substrata in flowing blood, Blood, 2010, 115, 5069–5079 CrossRef CAS PubMed.
- N. Pugh, D. Bihan, D. J. Perry and R. W. Farndale, Dynamic analysis of platelet deposition to resolve platelet adhesion receptor activity in whole blood at arterial shear rate, Platelets, 2015, 26, 216–219 CrossRef CAS PubMed.
- D. Varga-Szabo, A. Braun and B. Nieswandt, Calcium signaling in platelets, J. Thromb. Haemostasis, 2009, 7, 1057–1066 CrossRef CAS PubMed.
- B. Kehrel, S. Wierwille, K. J. Clemetson, O. Anders, M. Steiner, C. G. Knight, R. W. Farndale, M. Okuma and M. J. Barnes, Glycoprotein VI is a major collagen receptor for platelet activation: it recognizes the platelet-activating quaternary structure of collagen, whereas CD36, glycoprotein IIb/IIIa, and von Willebrand factor do not, Blood, 1998, 91(2), 491–499 CAS.
- S. M. Jung, et al., Constitutive dimerization of glycoprotein VI (GPVI) in resting platelets is essential for binding to collagen and activation in flowing blood, J. Biol. Chem., 2012, 287, 30000–30013 CrossRef CAS PubMed.
- K. Zhang and J. Chen, The regulation of integrin function by divalent cations, Cell Adh. Migr., 2012, 6, 20–29 CrossRef PubMed.
- J. Xia, M. Emery and B. O'Dell, Zinc signals and distribution of protein kinase C in rat platelets, J. Nutr. Biochem., 1994, 536–541, DOI:10.1016/0955-2863(94)90051-5.
- I. J. Forbes, P. D. Zalewski, C. Giannakis, H. S. Petkoff and P. A. Cowled, Interaction between protein kinase C and regulatory ligand is enhanced by a chelatable pool of cellular zinc, Biochim. Biophys. Acta, 1990, 1053, 113–117 CrossRef CAS.
- O. Konopatskaya, et al., PKCalpha regulates platelet granule secretion and thrombus formation in mice, J. Clin. Invest., 2009, 119, 399–407 CAS.
- P. A. Smethurst, et al., Identification of the primary collagen-binding surface on human glycoprotein VI by site-directed mutagenesis and by a blocking phage antibody, Blood, 2004, 103, 903–911 CrossRef CAS PubMed.
- C. G. Knight, et al., The collagen-binding A-domains of integrins alpha(1)beta(1) and alpha(2)beta(1) recognize the same specific amino acid sequence, GFOGER, in native (triple-helical) collagens, J. Biol. Chem., 2000, 275, 35–40 CrossRef CAS PubMed.
- L. F. Morton, P. G. Hargreaves, R. W. Farndale, R. D. Young and M. J. Barnes, Integrin alpha 2 beta 1-independent activation of platelets by simple collagen-like peptides: collagen tertiary (triple-helical) and quaternary (polymeric) structures are sufficient alone for alpha 2 beta 1-independent platelet reactivity, Biochem. J., 1995, 306(Pt 2), 337–344 CrossRef CAS PubMed.
- N. Raynal, et al., Use of synthetic peptides to locate novel integrin alpha2beta1-binding motifs in human collagen III, J. Biol. Chem., 2006, 281, 3821–3831 CrossRef CAS PubMed.
- J. M. Gibbins, Techniques for analysis of proteins by SDS-polyacrylamide gel electrophoresis and Western blotting, Methods Mol. Biol., 2004, 273, 139–152 CAS.
- D. J. Onley, C. G. Knight, D. S. Tuckwell, M. J. Barnes and R. W. Farndale, Micromolar Ca2+ concentrations are essential for Mg2+-dependent binding of collagen by the integrin alpha 2 beta 1 in human platelets, J. Biol. Chem., 2000, 275, 24560–24564 CrossRef CAS PubMed.
- G. E. Jarvis, et al., Identification of a major GpVI-binding locus in human type III collagen, Blood, 2008, 111, 4986–4996 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |