
Designing multi-layer graphene-based assemblies for enhanced toughness in nacre-inspired nanocomposites
Wenjie
Xia
a,
Jake
Song
b,
Zhaoxu
Meng
a,
Chen
Shao
c and
Sinan
Keten
*ac
aDepartment of Civil & Environmental Engineering, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA. E-mail: s-keten@northwestern.edu; Tel: 847 491 5282
bDepartment of Materials Science and Engineering, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
cDepartment of Mechanical Engineering, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
First published on 23rd May 2016
Abstract
Polymers reinforced with multi-layer graphene (MLG) phases are promising candidates for new materials with high modulus, strength and toughness. Drawing inspiration from nacre's layered “brick and mortar” structure, here we propose molecular scale design strategies to improve the mechanical performance of MLG–polymer layer-by-layer nanocomposites. We present a coarse-grained molecular dynamics (CG-MD) study of interfacial failure mechanisms of MLG domains embedded in a poly(methyl methacrylate) (PMMA) matrix through pull-out simulations. Our simulations reveal two distinct deformation and failure mechanisms that greatly influence the toughness and energy dissipation of the system: pull-out failure, which occurs along the MLG–PMMA interface, and yielding failure, which occurs within the graphitic phase through the sliding of staggered graphene sheets. For any length of the graphitic assembly, the energy dissipated per layer from MLG yielding is greater than that of MLG pull-out. Theoretical continuum analysis further reveals that there exists a critical number of layers of graphene, beyond which the failure mode changes from yielding to pull-out. Our modeling framework provides effective strategies to design graphene–polymer layered nanocomposites with optimal toughness, and advance the mechanical performance of nanomaterials.
Design, System, ApplicationGraphene, in addition to its impressive thermal and electrical transport properties, is a promising structural material with remarkable stiffness and strength. However, to harness graphene in load-bearing applications, its low fracture toughness must be overcome. A promising strategy is to use layered nanocomposite architectures found in natural materials such as nacre, which are known to have high toughness due to their ability to dissipate tremendous amounts of energy under large deformation. However, to fully utilize this design strategy, nanoscale mechanisms that govern mechanical properties of graphene-based nanocomposites must be understood. Using multi-scale molecular models of multi-layer graphene and polymer nanocomposites, we probe these molecular failure mechanisms as a function of design parameters such as interfacial interaction and graphene nanostructure configurations. Our work lends key insight into structural failure mechanisms broadly pertaining to multi-layered assemblies of 2D nanomaterials with polymers, and provides guidelines on optimal design of such nanocomposites. |
Introduction
The optimization of diametric properties such as strength and toughness is an important step towards designing novel materials with high mechanical performance. This is a naturally challenging task for many applications, such as nanoelectronics and structural protection,1,2 as strong materials often exhibit low fracture toughness and vice versa, which categorically limit their engineering applications.3 To overcome this issue, we can draw inspiration from biomaterials, which can optimize strength and toughness via unique hierarchical architectures that can dissipate significant amounts of energy under loading.4 For example, nacre, which is abundantly available in the inner layer of mollusk shells, features a layer-by-layer arrangement of soft organic polymers and hard aragonite platelets that form a nanoscopic ‘brick-and-mortar’ structure.5,6 This layered nanostructure – through various proposed mechanisms such as transfer of shear stresses,6 confinement of cracks upon reaching the polymeric matrix,5 resistance to shear from frictional asperities,7 and pull-out of the crystalline platelets – preserves the strength of the stiff crystalline plates, while having toughness that is orders of magnitude higher than its constituents.Considerable efforts have been engaged in designing high performance nanocomposites that employ a layer-by-layer assembly or similar lamellar structures, mostly involving polymer-clay or ceramic (e.g. alumina) microphases.8–10 The focus has recently been turned to graphene and its derivatives such as graphene oxide (GO) to take advantage of their exceptional strength and modulus.10–15 The layered structuring of graphene nanocomposites is advantageous in many ways. For one, the intercalation of polymeric domains with hard graphene-based sheets can significantly increase its mechanical properties while retaining the ductility of the polymer matrix.16–18 Moreover, the brick-and-mortar arrangement of graphene within a staggered multi-layer graphene (MLG) architecture can dramatically improve the toughness of graphene-based nanocomposites. This is due to their self-healing van der Waals interfaces, which allow MLGs to dissipate significant amounts of energy upon large deformation,19 in stark contrast to single continuous graphene sheets that undergo brittle failure.14,20,21 A recent study has illustrated a proof-of-concept demonstration of MLG for armor applications, where micro-scale projectile experiments established the superior energy dissipation capacity of MLG compared to conventional protective materials such as steel and Kevlar.2
In order to harness the superior properties of nacre-inspired graphene structures in polymer nanocomposites, it is crucial to understand the interfacial mechanics between staggered graphene layers and polymer layers. Although prior studies have investigated the interfacial shearing mechanisms of other fibrous materials such as carbon and glass fibers,22,23 cellulose nanocrystals and fibers,24–26 and single and multi-walled carbon nanotubes,27–30 the interfacial shear responses that occur between polymer matrices and staggered 2-D materials such as MLGs remain to be fully explored. As such, the present study aims to characterize the interfacial shearing mechanisms of layer-by-layer MLG and polymer assemblies to optimize the design process of mechanically robust materials. Taking the advantages of our recently developed coarse-grained molecular dynamics (CG-MD) models of MLG31 and poly(methyl methacrylate) (PMMA),32 we design a nacre-inspired layered architecture of MLG–PMMA nanocomposites (Fig. 1a) as recently studied in experiments and simulations.12,33 Utilizing the developed CG techniques and by performing pull-out test simulations, we are able to characterize the interfacial behaviors of the designed system as well as identify critical sizes of MLG that govern deformational behaviors. We discuss these results in the context of theoretical framework based on continuum mechanics models and CG-MD simulations, and present effective optimization strategies to design mechanically robust nacre-inspired polymeric nanocomposite systems.
 | ||
| Fig. 1 (a) Computational design of nacre-inspired layer-by-layer assembly of multi-layer graphene (MLG) and poly(methyl methacrylate) (PMMA) nanocomposites. (b) All-atomistic (AA) to coarse-grained (CG) mapping schemes for graphene (top) and PMMA (bottom). (c) Illustration of the microscopic picture of the nanocomposite system under loading. (d) Schematic setup of PMMA–MLG system investigated in our study. The MLG consists of discrete sheets stacked in a staggered configuration. H (∼20 nm) is the thickness of the polymer layer, L is the total or embedded length, and N is the number of layers of graphene sheets in the MLG phase. | ||
Simulation methods
Overview of coarse-grained models
The nacre-inspired nanocomposite consists of a layered nanostructure of CG PMMA and graphene models (Fig. 1b). The CG graphene model follows a 4-to-1 mapping scheme, where 4 carbon atoms are represented by one CG bead. The hexagonal symmetry of the atomic lattice is conserved to capture the interlayer shear response between graphene, including superlubricity effects. The CG force-field is developed based on a strain energy conservation approach, and the developed MLG model has been shown to quantitatively capture complex mechanical properties such as tensile and shear modulus, and failure properties.10,31 For the CG PMMA model, we employ a two-bead mapping scheme for each monomer with one bead representing the backbone group and the second bead representing the sidechain methyl group. The bonded interactions, including bond, angle and dihedral potentials, are parametrized using the inverse Boltzmann method (IBM)34 to match the atomistic probability distributions. The non-bonded interactions take the form of a 12-6 Lennard-Jones (LJ) potential, and are parametrized to capture the bulk density, glass transition temperature Tg and elastic modulus that are in good agreement with experiments. The resulting CG models of MLG and PMMA are roughly 2–3 orders of magnitude more computationally efficient than the corresponding atomistic models. Detailed procedures for developing the CG models can be found in our original publications.31,32Coarse-grained molecular dynamics simulations
The investigated system consists of one PMMA layer and one MLG layer, which constitute a representative volume element (RVE) in the microscopic layered nanocomposites (illustrated in Fig. 1c). Fig. 1d shows the setup of the simulated system. The PMMA layer with a thickness of H ∼ 20 nm consists of a block of polymer chains with a chain length of 100 monomers per chain. The MLG layer consists of varying number of layers N of graphene with two discrete finite graphene sheets per layer; the sheets are stacked in a staggered architecture with an overlap ratio of 50% and an overlap length Lo of ∼12.5 nm. The total length L of the system is thus ∼50 nm in the y axis. The width of the system is ∼10 nm in the x direction. Periodic boundary conditions (PBC) are applied in the x and z directions and non-PBC are applied in the y direction. The non-bonded interaction between graphene and polymer is captured by the LJ potential: | (1) |
To investigate the interfacial mechanics and deformational behaviors of the MLG–PMMA nanocomposite, we perform steered molecular dynamics (SMD) simulations. All simulations are carried out using LAMMPS.35 The energy of the system is first minimized using the conjugate gradient algorithm. The system is then equilibrated through a dynamics run for ∼2 ns at 300 K. During the SMD simulation, one edge of the graphene sheet at each layer in the MLG is pulled by applying a force F generated by a stiff harmonic spring: F = kSMD [vt − y(t)], where kSMD = 1000 kcal mol−1 Å−2 is the spring constant and v = 0.0001 Å fs−1 is the pulling velocity. Our previous work has shown the adopted spring constant and velocity to be reasonable in determining accurate force readings from SMD simulations.19,36 All SMD simulations are carried out at a temperature of 300 K, and therefore the PMMA layer is well below its glass transition temperature (Tg) of about 380 K as measured from our simulations. Throughout the SMD simulations, we confine the PMMA layer on both sides of the y-axis by a repulsive harmonic potential wall in the x–z plane. The reason for applying the wall is to keep the layered geometry and prevent large deformations in the PMMA phase during MLG pull-out test, ensuring clean interfacial shearing between MLG and PMMA phases. This is similar to the constraints applied on the polymer matrix by fixing polymer atoms as reported in previous pull-out simulation studies.37 This is reasonable as the force contribution from the deformation of a glassy polymer matrix is negligible compared to that arising from the graphene–polymer interface. It should be noted, however, that the polymer deformation becomes significant when the graphene–polymer interfacial interaction is very strong or when the temperature is close to or above the Tg, and thus this assumption needs to be revisited accordingly.
Results and discussion
We first discuss the tensile properties of multi-layer graphene (MLG) with a staggered architecture. The simulation details have been reported in our previous study,19 and we present a brief summary of our key results here, as they serve as a rationale for our design and analysis of MLG–PMMA nanocomposites. For MLG, tensile stress is transferred through shear between graphene sheets in adjacent layers, which makes the overlap length Lo an important parameter. Both the tensile stress σm and the effective modulus Em can be predicted by the continuum shear-lag model:38–40 | (2) |
 | (3) |
 is a parameter that represents the length scale over which most of the stress is transferred in the graphene–graphene interface, Eg (∼ 950 GPa) is the Young's modulus of an individual graphene sheet, G (∼ 1 GPa) is the shear modulus of the interface, hg (= 3.35 Å) is the thickness of each graphene layer, and γcrs (0.35) is the critical interlayer shear strain. The values of these fundamental parameters are defined based on the properties of the CG model, and thus they are not variable fitting parameters. To validate the shear-lag model predictions, we performed uniaxial tensile simulations on MLG to obtain σm and Em as a function of Lo. Our simulation results are shown in Fig. 2a and b, respectively (the inset in Fig. 2a illustrates the schematic of the tensile simulation). The shear-lag model predictions (solid lines) are in good agreement with the CG-MD simulation results (diamond markers). Both σm and Em increase with increasing the overlap length Lo and then reach a plateau. The strength σm saturates as Lo is beyond ∼3l, corresponding to a value of ∼17 nm. The elastic modulus Em also saturates at a slightly larger overlap length of ∼40 nm and converges to the Eg of individual graphene sheets.
is a parameter that represents the length scale over which most of the stress is transferred in the graphene–graphene interface, Eg (∼ 950 GPa) is the Young's modulus of an individual graphene sheet, G (∼ 1 GPa) is the shear modulus of the interface, hg (= 3.35 Å) is the thickness of each graphene layer, and γcrs (0.35) is the critical interlayer shear strain. The values of these fundamental parameters are defined based on the properties of the CG model, and thus they are not variable fitting parameters. To validate the shear-lag model predictions, we performed uniaxial tensile simulations on MLG to obtain σm and Em as a function of Lo. Our simulation results are shown in Fig. 2a and b, respectively (the inset in Fig. 2a illustrates the schematic of the tensile simulation). The shear-lag model predictions (solid lines) are in good agreement with the CG-MD simulation results (diamond markers). Both σm and Em increase with increasing the overlap length Lo and then reach a plateau. The strength σm saturates as Lo is beyond ∼3l, corresponding to a value of ∼17 nm. The elastic modulus Em also saturates at a slightly larger overlap length of ∼40 nm and converges to the Eg of individual graphene sheets.
 | ||
| Fig. 2 (a) Tensile strength σm and (b) effective elastic modulus Em of MLG as a function of overlap length Lo from tensile deformation simulations (inset). The solid lines are prediction from the shear-lag model. (c) Shear stress–displacement curve from pulling tests on a small single graphene sheet interacting with a polymer layer (inset). (d) Interfacial shear strength τs as a function of interfacial interaction strength εgp between graphene and polymer. The dashed line shows the linear fit of the data. | ||
Next, we calculate the interfacial shear strength τs between the graphene and polymer through pull-out test simulations of a single small graphene flake on top of a polymer layer. During the pulling process, a spring force is applied to one end of the graphene sheet and the atoms of the bottom polymer layer are fixed (inset in Fig. 2c). The interfacial shear strength τs can be obtained from the stress–displacement response as shown in Fig. 2c. At the critical displacement of ∼10 Å, the shear stress reaches the maximum point corresponding to τs ∼ 0.11 GPa. As displacement increases, the shear stress starts to decay. This arises from a static-to-dynamic transition in the stick-slip friction, where strength is always higher in the first slippage event. This constitutive interfacial behavior is similar to those observed in the fiber pull-out experiments. The interfacial shear stiffness K can be determined from the initial slope of the stress–displacement curve as shown in Fig. 2c. We have also calculated τs with varying the interfacial interaction strength εgp between graphene and polymer (Fig. 2d). Our results indicate that the value of τs increases with increasing εgp with nearly a linear scaling relationship for the range of values tested. The interfacial interaction parameter can be tuned by surface modifications on graphene, and this results simply shows that increasing the interfacial energy will result in greater shear stiffness and shear strength.
We address two questions pertaining to the interfacial mechanical response of the MLG–PMMA layered nanocomposites: (1) what are the failure mechanisms of such layered nanostructure systems? (2) What are the strategies of optimizing the toughness of such systems? To answer these questions, we carry out pull-out simulations on our MLG–PMMA nanocomposite system to get insight into the interfacial properties. Our simulations reveal two different modes of failure: a pull-out mode which corresponds to MLG–PMMA interfacial failure in the case of thicker MLG assemblies, and a yielding mode which occurs within the MLG through the sliding of staggered graphene sheets (Fig. 3a). This observation is also schematically illustrated in Fig. 3b. We observe that for a given interfacial interaction strength εgp and embedded length L, the failure mode of MLG changes from yielding to pull-out as the number of layers N increases. This is quantitatively shown in the force–displacement (F–d) curves from our simulations (Fig. 4a). As N increases, peak force initially increases until it saturates after a critical number of layers, Ncr. This indicates the transition of failure modes from yielding to pull-out occurring at Ncr. The peak force remains constant for large N since the pull-out force is independent of the number of layers and instead depends on interfacial shear properties between graphene and PMMA. For all F–d curves, we observe an initial increase in force, followed by a gradual decay upon transitioning from static to dynamic friction.
 | ||
| Fig. 3 (a) Snapshots of the SMD pulling simulations for MLG yielding failure mode (top) and MLG pull-out failure mode (bottom). (b) Schematic of the different failure modes identified in the simulations. | ||
 | ||
| Fig. 4 (a) Force–displacement curves from the SMD pulling simulations for varying number of graphene layers N. (b) The normalized work of fracture per layer (W/N) as a function of N. The dashed lines show the trend. The different color regions correspond to different failure modes as identified in the simulations. | ||
The failure mode transition as a function of N can also be predicted from continuum theory. Neglecting the force contribution from the axial deformation of polymer (polymer stiffness is two orders of magnitude lower than that of MLG), the force per unit width F required to propagate interfacial crack along the MLG–PMMA interface can be determined from the continuum fiber pull-out model:41
| F = F∞ tanh(αL) | (4) |
 is a length scale parameter governing the shear stress transfer along the MLG–PMMA interface, Em and h (= N × hg) are the modulus and thickness of MLG, respectively, and
is a length scale parameter governing the shear stress transfer along the MLG–PMMA interface, Em and h (= N × hg) are the modulus and thickness of MLG, respectively, and 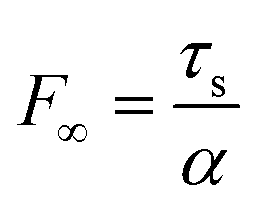 is the maximum pull-out force per unit width at an infinitely large L. By knowing the tensile strength σm of MLG and F, the critical condition for each failure mode can be determined by solving the force balance equation:
is the maximum pull-out force per unit width at an infinitely large L. By knowing the tensile strength σm of MLG and F, the critical condition for each failure mode can be determined by solving the force balance equation:| 2F∞ tanh(αL) = σmNh | (5) |
From simulations and theoretical predictions, it is clear that failure modes have a great impact on energy dissipation in the nanocomposite system, which directly controls the toughness. Fig. 4b shows the results of the work of fracture (W) normalized by N for different N, which corresponds to energy dissipation per layer. We observe that W/N is maximized at N values that are in the yielding regime. This is because the yielding mode of failure involves the sliding of graphene surfaces – precisely due to the staggered architecture of the MLGs – which can dissipate tremendous amount of energy upon tensile loading. In recent supersonic ballistic experiments by Lee et al.,2 it has been demonstrated that the energy dissipation of MLG is roughly an order of magnitude higher than protective materials such as steel and Kevlar. Our recent study also shows that by arranging the MLG in a staggered architecture, the energy dissipated through interlayer sliding can be manifolds higher than that of continuous sheets that fail due to covalent bond breaking.19 By contrast, in pull-out mode, the work required to fracture the interface between MLG and PMMA is mainly governed by the surface energy, which is weakly dependent on N and strongly dependent on interfacial interaction strength εgp. Therefore, as N increases beyond Ncr, W/N decreases with increasing N (roughly following a scaling of 1/N as shown by the dashed curve), indicating a loss in energy dissipation efficiency upon entering the pull-out failure regime.
Given the reasonable agreement between continuum theory and simulations, we can use theory to extend this analysis to arbitrary values of L and εgp to understand how Ncr will depend on these parameters. Using data we have on the shear strength and shear stiffness as a function of εgp for different values of L, we can predict the critical number of layers Ncr as a function of L for εgp. As shown in Fig. 5, the value of Ncr increases initially with the length L, and then saturates at very large L ∼ 400 nm. This saturation effect at large L is due to the non-uniform shear stress distribution along the MLG–PMMA interface as predicted by the continuum model. Increasing εgp will increase Ncr for a given value of L. However, the length scale where Ncr starts to saturate is almost invariant with εgp. Considering these effects, tuning key parameters N, L and εgp is central for designing systems with superior toughness.
 | ||
| Fig. 5 Theoretical prediction of the critical number of layers Ncr of graphene in the MLG as a function of total length L for different interfacial interaction strength εgp (unit in kcal mol−1). The solid diamond data set highlights the specific interfacial interaction strength that was studied in our simulations. | ||
Our results provide several potential strategies to optimize performance of graphene-based nacre-inspired nanocomposites. As graphene nanocomposites are often strong but brittle, we may employ MLG components and tailor N to be below Ncr to optimize energy dissipation, which triggers yielding failure and therefore increases the toughness of the nanocomposite. This may be achieved by dispersing larger aggregates of MLG, for instance by sonication or other processing methods. The greater energy dissipation capacity of MLG-based nanocomposites may make them preferable over more brittle nanocomposite systems such as carbon nanotube (CNT)-based nanocomposites when toughness is a key criterion. Additionally, we may tailor N to be above Ncr such that the graphene–polymer interface would be the yielding area, allowing for the graphene layer to remain intact even at large deformations. This may have some advantages in applications where the graphitic phase needs to remain intact even at large deformation to maintain good thermal/electrical properties. Ncr can also be tailored by changing the overall length of the system L, and of course, energy dissipation can generally be enhanced by increasing εgpvia methods such as surface functionalization. For systems with very large L, MLG yielding may eventually lead to fragmentation of the continuous layers into short fibers, which then promote further energy dissipation via pull-out in a cascade fashion. This is a more complicated scenario that would involve evolutionary models, but the current study already lays out the foundation for tackling these issues.
In addition to shear-related mechanisms, other factors may contribute to the mechanical performance of graphene nanocomposites as well. For instance, nanoscale interphase formation can significantly influence the chain dynamics of the confined polymer, as observed in many studies.42–46 In the MLG–PMMA system, this implies that thermomechanical properties such as Tg and modulus can be tuned by modifying the polymer thickness H and interfacial interaction strength εgp through surface functionalization. Additionally, strain localization within MLG and fracture behavior of polymers can affect bulk mechanical properties, along with crack-propagation mechanisms and the overall distribution of the phases. Particularly, large deformation behavior of the polymers may become very important when the interfacial shear strength is greater than the strength of polymer. This could be achieved by the covalent crosslinks at the interface leading to strong interfacial interactions. While our investigation into the interfacial properties is by no means exhaustive, it provides a good starting point for establishing strategies to harness the dissipation mechanisms intrinsic to multi-layer graphene for diverse engineering applications. We anticipate that the models developed here could also be extended to other multilayer fillers, such as cellulose nanocrystals and 2D nanomaterials.47,48
Conclusions
In summary, we have investigated interfacial mechanical behaviors of nacre-inspired MLG–PMMA layered nanocomposite systems by performing pull-out simulations using coarse-grained molecular dynamics (CG-MD) approach. Our simulations uncover two different deformation and failure mechanisms, which greatly influence the toughness and energy dissipation of the system: pull-out failure, which occurs along the MLG–PMMA interface, and yielding failure, which occur along graphene–graphene interfaces. A theoretical model validated by the simulation data is proposed to determine the critical number of layers Ncr of graphene that governs the mode of failures as a function of MLG length and graphene-polymer interfacial interactions. We find that when the number of graphene layers N ≤ Ncr, significant energy dissipation is observed via yielding failure mode, a direct result of the staggered arrangement of MLG. This staggered architecture allows sliding between graphene sheets, resulting in higher toughness compared to that of pull-out failure mode when N > Ncr. We also find that increasing the system length L and the interfacial interaction strength εgp between the layers will enhance the energy dissipation of the nanocomposite, which is a direct result of the nacre-like layer-by-layer arrangement of hard and soft phases.Our prediction of failure modes has important implications for designing MLG nanocomposites. In the case where the MLG domains or “platelets” are too thick (large N) or too short (small L), our analysis reveals two routes towards improving composite toughness. The first route is to break down the MLG into smaller assemblies such that N ≤ Ncr, and the second route is to carry out surface functionalization to create stronger, compatible interfaces with the polymer matrix. Both methods activate MLG yielding as an energy dissipation mechanism, which effectively maximizes the amount of new surface area created during the failure process. Our findings advocate the use of nacre-inspired architectures in order to improve the strength and toughness of graphene-based nanocomposites, while providing ideal design strategies within this framework.
Acknowledgements
The authors acknowledge support by the National Institute of Standards and Technology through the Center for Hierarchical Materials Design (CHiMaD) and a grant by the Army Research Office award (#W911NF-13-1-0241). The authors acknowledge funding from the National Science Foundation (#CMMI-1235480). C. S. acknowledges the support by the National Science Foundation (#MSS150013) through the Extreme Science and Engineering Discovery Environment (XSEDE). The authors also acknowledge the support from the Department of Civil and Environmental Engineering and the Department of Mechanical Engineering at Northwestern University, as well as the Northwestern University High Performance Computing Center (QUEST) for a supercomputing grant.References
- M. Zhang, K. R. Atkinson and R. H. Baughman, Science, 2004, 306, 1358–1361 CrossRef CAS PubMed.
- J.-H. Lee, P. E. Loya, J. Lou and E. L. Thomas, Science, 2014, 346, 1092–1096 CrossRef CAS PubMed.
- R. O. Ritchie, Nat. Mater., 2011, 10, 817–822 CrossRef CAS PubMed.
- P. Egan, R. Sinko, P. R. LeDuc and S. Keten, Nat. Commun., 2015, 6, 1–12 Search PubMed.
- H. D. Espinosa, J. E. Rim, F. Barthelat and M. J. Buehler, Prog. Mater. Sci., 2009, 54, 1059–1100 CrossRef CAS.
- T. Giesa and M. J. Buehler, Annu. Rev. Biophys., 2013, 42, 651–673 CrossRef CAS PubMed.
- R. Wang, Z. Suo, A. Evans, N. Yao and I. Aksay, J. Mater. Res., 2001, 16, 2485–2493 CrossRef CAS.
- T. Verho, M. Karesoja, P. Das, L. Martikainen, R. Lund, A. Alegría, A. Walther and O. Ikkala, Adv. Mater., 2013, 25, 5055–5059 CrossRef CAS PubMed.
- E. Munch, M. E. Launey, D. H. Alsem, E. Saiz, A. P. Tomsia and R. O. Ritchie, Science, 2008, 322, 1516–1520 CrossRef CAS PubMed.
- X. Wei, Z. Meng, L. Ruiz, W. Xia, C. Lee, J. W. Kysar, J. C. Hone, S. Keten and H. D. Espinosa, ACS Nano, 2016, 10, 1820–1828 CrossRef CAS PubMed.
- H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515–6530 CrossRef CAS.
- L. Gong, R. J. Young, I. A. Kinloch, I. Riaz, R. Jalil and K. S. Novoselov, ACS Nano, 2012, 6, 2086–2095 CrossRef CAS PubMed.
- Z. Li, R. J. Young and I. A. Kinloch, ACS Appl. Mater. Interfaces, 2013, 5, 456–463 CAS.
- C. Lee, X. D. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- O. C. Compton, S. W. Cranford, K. W. Putz, Z. An, L. C. Brinson, M. J. Buehler and S. T. Nguyen, ACS Nano, 2012, 6, 2008–2019 CrossRef CAS PubMed.
- W. Lu and C. M. Lieber, Nat. Mater., 2007, 6, 841–850 CrossRef CAS PubMed.
- T. Ramanathan, A. Abdala, S. Stankovich, D. Dikin, M. Herrera-Alonso, R. Piner, D. Adamson, H. Schniepp, X. Chen and R. Ruoff, Nat. Nanotechnol., 2008, 3, 327–331 CrossRef CAS PubMed.
- L. Gong, I. A. Kinloch, R. J. Young, I. Riaz, R. Jalil and K. S. Novoselov, Adv. Mater., 2010, 22, 2694–2697 CrossRef CAS PubMed.
- W. Xia, L. Ruiz, N. M. Pugno and S. Keten, Nanoscale, 2016, 8, 6456–6462 RSC.
- T. Belytschko, S. P. Xiao, G. C. Schatz and R. S. Ruoff, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 235430 CrossRef.
- H. Zhao, K. Min and N. R. Aluru, Nano Lett., 2009, 9, 3012–3015 CrossRef CAS PubMed.
- Y. Wang, V. C. Li and S. Backer, Int. J. Cem. Compos. Lightweight Concr., 1988, 10, 143–149 CrossRef CAS.
- V. C. Li, Y. Wang and S. Backer, J. Mech. Phys. Solids, 1991, 39, 607–625 CrossRef.
- M. Brahmakumar, C. Pavithran and R. Pillai, Compos. Sci. Technol., 2005, 65, 563–569 CrossRef CAS.
- R. Sinko and S. Keten, J. Mech. Phys. Solids, 2015, 78, 526–539 CrossRef CAS.
- H. L. Zhu, S. Z. Zhu, Z. Jia, S. Parvinian, Y. Y. Li, O. Vaaland, L. B. Hu and T. Li, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8971–8976 CrossRef CAS PubMed.
- Y. Li, Y. Liu, X. Peng, C. Yan, S. Liu and N. Hu, Comput. Mater. Sci., 2011, 50, 1854–1860 CrossRef CAS.
- X. M. Chen, M. Zheng, C. Park and C. H. Ke, Small, 2013, 9, 3345–3351 CAS.
- Y. L. Chen, B. Liu, X. Q. He, Y. Huang and K. C. Hwang, Compos. Sci. Technol., 2010, 70, 1360–1367 CrossRef CAS.
- M. Naraghi, G. H. Bratzel, T. Filleter, Z. An, X. Wei, S. T. Nguyen, M. J. Buehler and H. D. Espinosa, Adv. Funct. Mater., 2013, 23, 1883–1892 CrossRef CAS.
- L. Ruiz, W. Xia, Z. Meng and S. Keten, Carbon, 2015, 82, 103–115 CrossRef CAS.
- D. D. Hsu, W. Xia, S. G. Arturo and S. Keten, J. Chem. Theory Comput., 2014, 10, 2514–2527 CrossRef CAS PubMed.
- C. Shao and S. Keten, Sci. Rep., 2015, 5, 16452 CrossRef CAS PubMed.
- F. Müller-Plathe, ChemPhysChem, 2002, 3, 754–769 CrossRef.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- W. Xia, D. D. Hsu and S. Keten, Macromolecules, 2014, 47, 5286–5294 CrossRef CAS.
- Y. A. Li, Y. L. Liu, X. H. Peng, C. Yan, S. Liu and N. Hu, Comput. Mater. Sci., 2011, 50, 1854–1860 CrossRef CAS.
- H. L. Cox, Br. J. Appl. Phys., 1952, 3, 72–79 CrossRef.
- Y. Liu, B. Xie, Z. Zhang, Q. Zheng and Z. Xu, J. Mech. Phys. Solids, 2012, 60, 591–605 CrossRef CAS.
- X. Wei, M. Naraghi and H. D. Espinosa, ACS Nano, 2012, 6, 2333–2344 CrossRef CAS PubMed.
- P. Lawrence, J. Mater. Sci., 1972, 7, 1–6 CrossRef CAS.
- W. Xia, S. Mishra and S. Keten, Polymer, 2013, 54, 5942–5951 CrossRef CAS.
- W. Xia and S. Keten, Extreme Mech. Lett., 2015, 4, 89–95 CrossRef.
- D. S. Fryer, R. D. Peters, E. J. Kim, J. E. Tomaszewski, J. J. de Pablo, P. F. Nealey, C. C. White and W. L. Wu, Macromolecules, 2001, 34, 5627–5634 CrossRef CAS.
- X. Cheng, K. W. Putz, C. D. Wood and L. C. Brinson, Macromol. Rapid Commun., 2015, 36, 391–397 CrossRef CAS PubMed.
- Y. Li, M. Kroger and W. K. Liu, Phys. Rev. Lett., 2012, 109, 118001 CrossRef PubMed.
- R. Sinko, S. Mishra, L. Ruiz, N. Brandis and S. Keten, ACS Macro Lett., 2014, 3, 64–69 CrossRef CAS.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |