
Exploring inhibitor structural features required to engage the 216-loop of human parainfluenza virus type-3 hemagglutinin-neuraminidase†‡
Ibrahim M.
El-Deeb§
*,
Patrice
Guillon
*,
Larissa
Dirr
and
Mark
von Itzstein
*
Institute for Glycomics, Griffith University, Gold Coast Campus, Queensland 4222, Australia. E-mail: m.vonitzstein@griffith.edu.au
First published on 5th October 2016
Abstract
Human parainfluenza virus type-3 is a leading cause of acute respiratory infection in infants and children. There is currently neither vaccine nor clinically effective treatment for parainfluenza virus infection. Hemagglutinin-neuraminidase glycoprotein is a key protein in viral infection, and its inhibition has been a target for inhibitor development. In this study, we explore the structural features required for Neu2en derivatives to efficiently lock-open the 216-loop of the human parainfluenza virus type-3 hemagglutinin-neuraminidase protein.
Introduction
Human parainfluenza viruses (hPIVs) are a group of respiratory viruses that belong to the Paramyxoviridae family and cause a range of human respiratory diseases in infants, children, immunocompromised patients and elderly people.1 Human parainfluenza virus type-3 (hPIV-3) is an enveloped, single-stranded, negative-sense RNA virus that affects children worldwide causing croup, bronchiolitis, and pneumonia.2 It comes second only to respiratory syncytial virus (RSV) as the leading cause of acute respiratory infections that require hospitalization of infants and children.3 It is also an important cause of morbidity and occasional mortality in immunocompromised patients.4,5 Despite continuing efforts, there are currently no effective vaccines or specific therapies to control or treat parainfluenza virus infection.The envelope of hPIV-3 contains two viral glycoproteins, the hemagglutinin-neuraminidase (HN) and the fusion protein (F).2 The HN protein recognizes sialic acid-containing receptors on respiratory epithelial cell surfaces and uses these sialic acid residues to anchor the virus to the host cell. A second function for the HN protein is promoting F-mediated fusion to allow penetration of the virus genome into the host cell. In addition to its receptor attachment and fusion promotion functions, HN possesses neuraminidase activity that promotes the release of newly formed virions from the cell surface and prevents them from self-agglutination.2,6 These three functions of the HN protein have made it a major target for anti-hPIV inhibitor design.
Over the past decades, numerous inhibitors that target sialidase proteins of other related viruses, such as influenza A virus (IAV), have been developed. Representative examples are the naturally occurring inhibitor 2-deoxy-2,3-didehydro-N-acetylneuraminic acid (Neu5Ac2en, 1, Fig. 1)7 and the potent anti-IAV 4-deoxy-4-guanidino-Neu5Ac2en inhibitor (zanamivir, 2, Fig. 1).8,9 However, as a result of the structural differences between hPIV-HN and other sialidases, inhibitory potencies for these unsaturated sialic acid-based inhibitors were very weak against hPIV-HN.10,11 The first potent Neu2en-based inhibitors to be rationally designed to specifically target hPIV-HN were BCX 2798 (3) and BCX 2855 (4).12–14 In spite of their in vitro potency against hPIV, these two inhibitions failed to continue in development as a result of their poor in vivo efficiency.12–14 We have recently reported11 the development of potent Neu2en-based inhibitors (5 and 6) that carry bulky C-4 substituents. These bulky C-4 substituents target a unique feature in the hPIV-3 HN protein, the flexible 216-loop, locking open the loop and efficiently occupying the created 216-cavity.11 This efficient binding to the HN protein results in potent inhibition of both HN functions, as well as significant blockade of virus propagation.
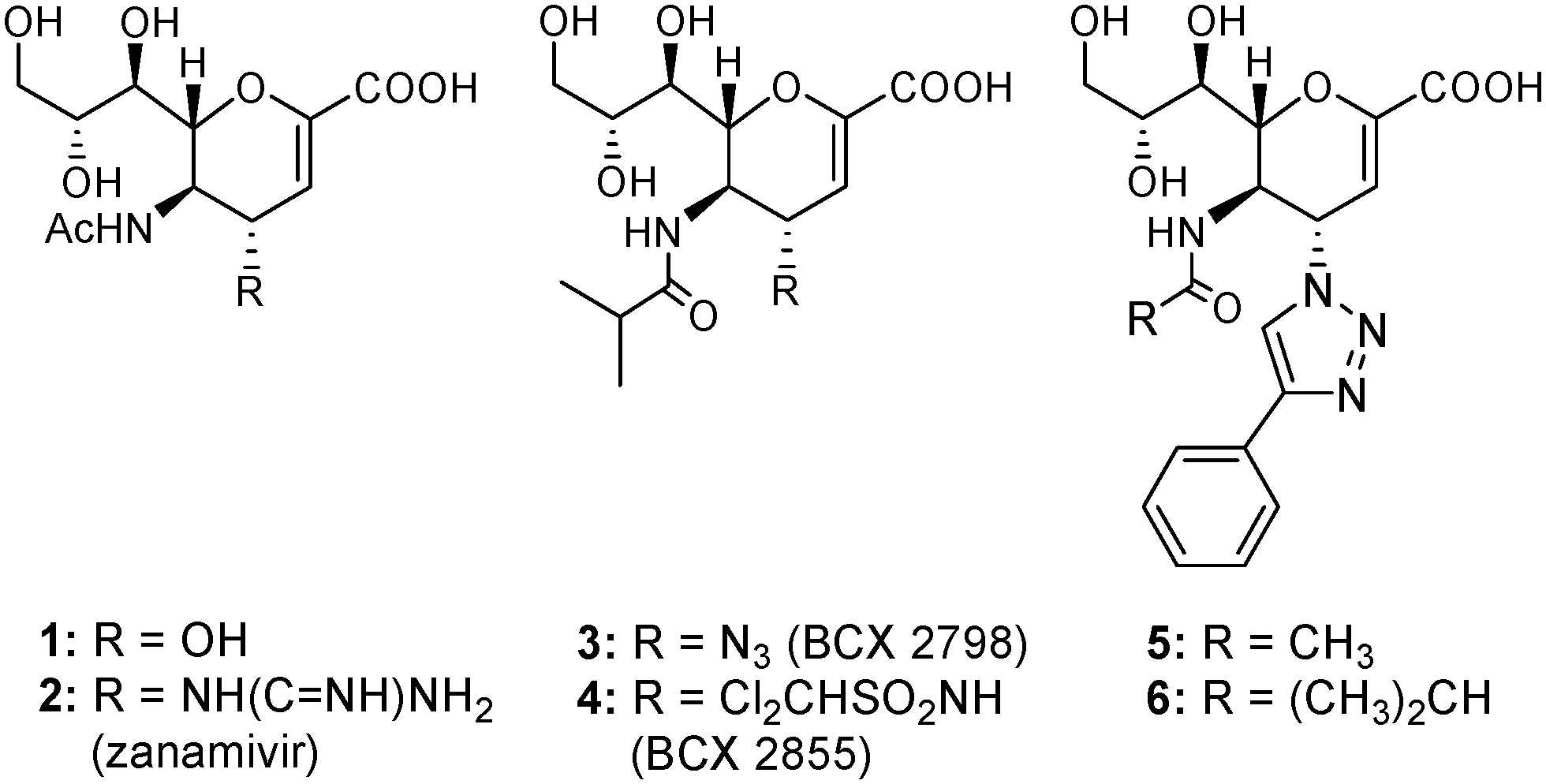 | ||
| Fig. 1 Structures of the anti-influenza sialidase inhibitors (1 and 2), the hPIV inhibitors (3 and 4), and the potent hPIV-3 designer inhibitors (5 and 6). | ||
The ability of these new designer inhibitors (5 and 6) to force the 216-loop open and reveal the 216-cavity is attributed to their bulky C-4 functionality (the 4-phenyl-1,2,3-triazole group). This outcome has proved the notion that the binding pocket of the hPIV-3 HN protein can efficiently accommodate Neu2en derivatives carrying bulky, hydrophobic C-4 substituents.15 Such bulky inhibitors are not only able to fit into the binding pocket but also benefit from the extra hydrophobic binding interactions exerted by their C-4 bulky substituents, resulting finally in more potent inhibition of HN functions.
The STD NMR experiments carried out for these triazole inhibitors in complex with either the intact virus or the recombinant HN protein have shown that the most significant interactions with the protein were exerted by the phenyl moiety carried by the 1,2,3-triazole ring.11 Accordingly, in this study, we verify the importance of the triazole ring as a spacer carrying the hydrophobic phenyl group at the C-4 position of 4-deoxy-Neu2en, by replacing it with other acyclic spacers (urea,16 amide and sulfonamide functionalities), in order to explore the structural features of the C-4 substituent and define the essential inhibitor characteristics required to force the 216-loop open to reveal and efficiently occupy the 216-cavity.
Results and discussion
Computational chemistry17
In the lead inhibitors (5 and 6), the distance between the triazole N-1 and C-1 of the phenyl ring in the energy minimized structures was found to be 3.58 Å. Accordingly, the triazole ring was replaced by a number of acyclic spacers that hold the phenyl ring at comparable distances from the 4-deoxy-NeuAc2en skeleton. The selected spacers, which can be readily synthesized using common 4-deoxy-Neu5Ac2en intermediates, are urea-, amide- and sulfonamide-based. These three linkers were found to generate distances of 3.65 Å, 2.44 Å and 2.87 Å, respectively, between the 4-amino-4-deoxy-Neu5Ac2en C-4 nitrogen and C-1 of the phenyl ring (Fig. 2). | ||
| Fig. 2 Distance between the nitrogen atom at C-4 of the 4-deoxy-Neu5Ac2en skeleton and C-1 of the phenyl ring in triazoles (a), ureas (b), amides (c) and sulfonamides (d). | ||
As predicted from these preliminary calculations and by aligning the structures of these 4-deoxy-Neu5Ac2en derivatives that contain these three acyclic linkers, the urea-based linker creates the most comparable distance between the 4-deoxy-Neu5Ac2en skeleton and the phenyl ring in the triazole inhibitor 5 (Fig. 3).
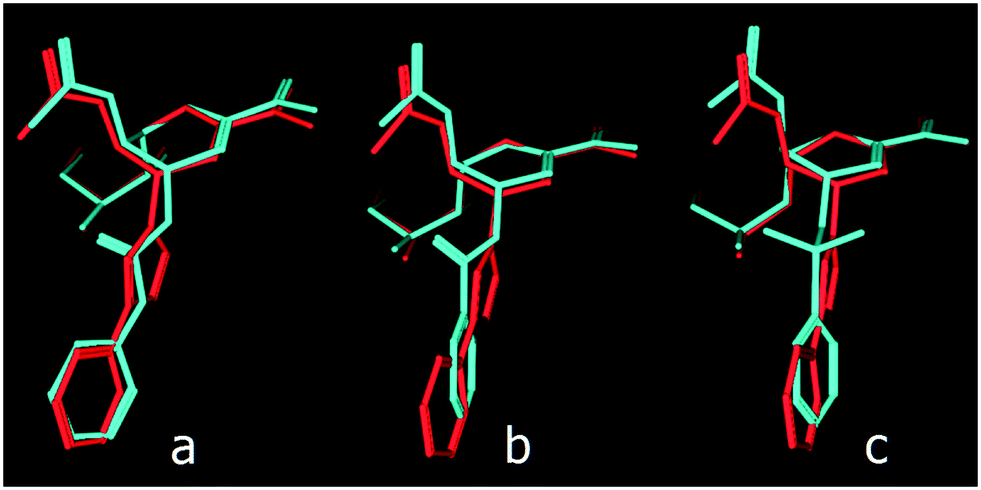 | ||
| Fig. 3 Alignment of 4-deoxy-4-phenylureido-Neu5Ac2en (a), 4-deoxy-4-phenylamido-Neu5Ac2en (b) and 4-deoxy-4-phenylsulfonamido-Neu5Ac2en (c) to the 4-deoxy-4-(4-phenyl-1,2,3-triazol-1-yl)-Neu5Ac2en inhibitor (5). | ||
Chemistry
Based on the initial computational chemistry calculations, a series of 4-deoxy-4-phenylureido-Neu5Ac2en-based inhibitors that contain variable substituents at the phenyl group (9a–g, Scheme 1) was synthesized. The synthesis of these urea derivatives was achieved by using the known intermediate 7.10 As shown in Scheme 1, the fully protected 4-amino-4-deoxy-Neu5Ac2en intermediate (7) was reacted with a number of substituted phenyl and benzyl isocyanate reagents in dichloromethane, in the presence of a catalytic amount of DMAP, to yield the protected urea derivatives 8a–g in excellent yields. The deprotection of compounds 8a–g by treatment with NaOH in MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1), provided the deprotected target 4-deoxy-4-arylureido-Neu5Ac2en derivatives 9a–g.
:H2O (1:1), provided the deprotected target 4-deoxy-4-arylureido-Neu5Ac2en derivatives 9a–g.
 | ||
| Scheme 1 Synthesis of compounds 9a–g. Reagents and conditions: (a) R-NCO, DMAP, DCM, rt, o/n, (8a, 87%; 8b, 96%; 8c, 90%; 8d, 84%; 8e, 89%; 8f, 91%; 8g, 82%); (b) NaOH, MeOH/H2O (1:1), rt, o/n (9a, 80%; 9b, 91%; 9c, 78%; 9d, 87%; 9e, 77%; 9f, 88%; 9g, 85%). | ||
The same intermediate 7 was utilized (Scheme 2) for the synthesis of amides 11a–c by reaction with three different benzoyl chlorides in the presence of triethylamine to yield the fully protected amides 10a–c, which upon stirring in a 1:1 mixture of methanol:water at pH 13–14 (adjusted by NaOH) afforded the final deprotected amides 11a–c.
 | ||
| Scheme 2 Synthesis of compounds 11a–c. Reagents and conditions: (a) R-COCl, Et3N, DCM, argon, rt, o/n (10a, 80%; 10b, 77%; 10c, 89%); (b) NaOH, MeOH:H2O (1:1), rt, o/n (11a, 89%; 11b, 85%; 11c, 91%). | ||
In Scheme 3, amine 7 was converted to the protected sulfonamides 12a–d by reaction with the appropriate sulfonyl chloride in the presence of triethylamine. The resultant sulfonamides 12a–d were then deprotected by treatment with NaOH in 50% MeOH to afford the target sulfonamides 13a–d.
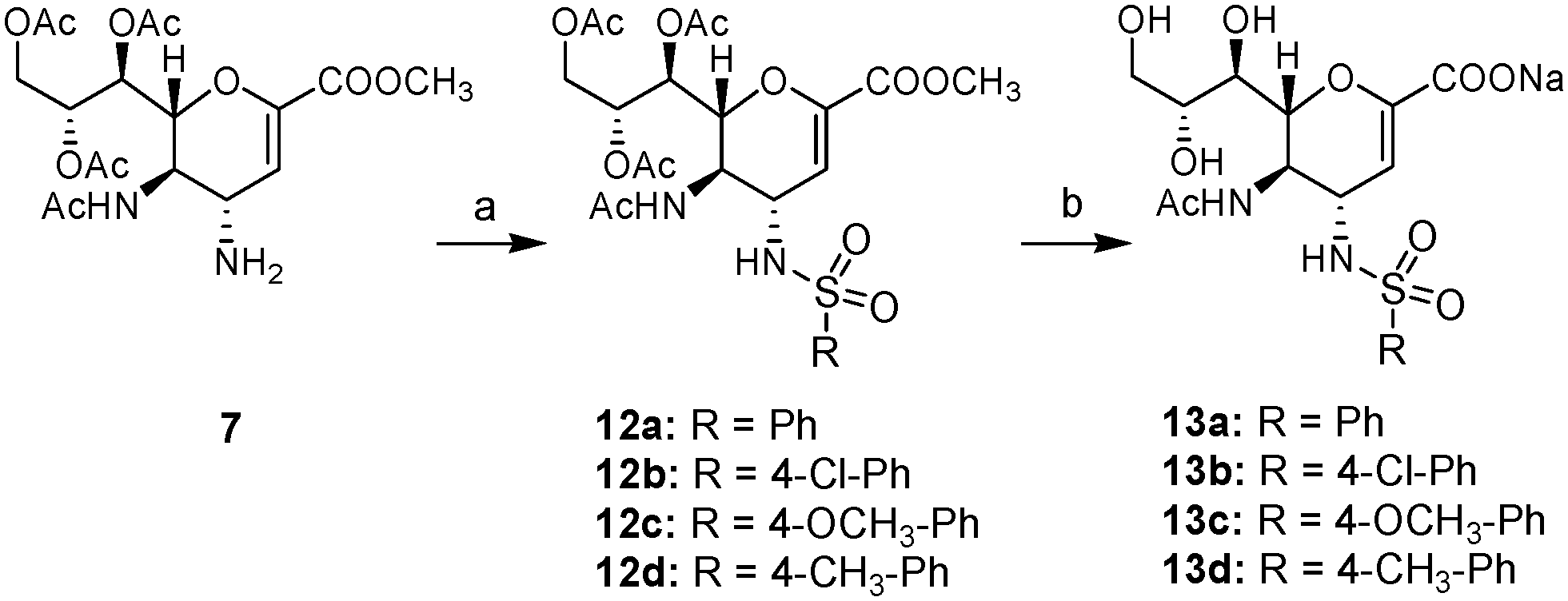 | ||
| Scheme 3 Synthesis of compounds 13a–d. Reagents and conditions: (a) R-SO2Cl, Et3N, DCM, argon, rt, o/n (12a, 83%; 12b, 89%; 12c, 82%; 12d, 74%); (b) NaOH, MeOH:H2O (1:1), rt, o/n (13a, 85%; 13b, 92%; 13c, 89%; 13d, 88%). | ||
In order to evaluate the effect of a C-5 modification on the inhibitory potency of these derivatives, the reported 4-amino-4-deoxy-5-isobutyramido-Neu2en intermediate (14) (ref. 10) was utilized to synthesize amide 15 as shown in Scheme 4. Thus, stirring amine 14 in the presence of triethylamine with 4-chlorobenzoyl chloride afforded amide 15 in 85% yield. Deprotection of the resultant amide by exposure to NaOH in aq. MeOH yielded the final deprotected inhibitor 16 in 94% yield.
 | ||
| Scheme 4 Synthesis of compound 16. Reagents and conditions: (a) 4-Cl-Ph-COCl, Et3N, DCM, argon, rt, o/n, 85%; (b) NaOH, MeOH:H2O (1:1), rt, o/n, 94%. | ||
Biological screening
Evaluation of the synthesized urea-, amide- and sulfonamide-based 4-deoxy-Neu2en derivatives for their capacity to block the neuraminidase function of hPIV-3 HN, as well as a comparison of their potencies to those of the lead triazole inhibitors 5 and 6, was carried out by using a standard neuraminidase inhibition (NI) assay (Fig. 4).11 | ||
| Fig. 4 NI IC50 values of inhibitors 11a–c, 13a–d and 16. Plain bars represent 4-deoxy-Neu2en derivatives with a C-5 acetamido group, while bars with stripes represent 4-deoxy-Neu2en derivatives with a C-5 isobutyramido group. The IC50 values are the result of 3 independent experiments performed in duplicate. Error bars represent the SEM of the NI IC50 values of the compounds. | ||
Surprisingly, and in spite of the close spacer length measurements between the triazole ring and the urea functionalities, all of the synthesized C-4 ureido-Neu5Ac2en derivatives failed to show NI IC50 values below the maximal-threshold concentration used in this assay (150 μM, data not shown). On the other hand, the neuraminidase inhibition activities exerted by amide and sulfonamide derivatives were relatively more potent and comparable to each other. Within each of these two series (amides and sulfonamides), the most potent neuraminidase inhibition activity was exerted by the 4-chlorophenyl derivatives (11b and 13b), with the amide 11b showing an IC50 value of 45.1 μM and the sulfonamide 13b showing an IC50 value of 48.7 μM. By comparing the three amides, the next potent neuraminidase inhibitor within this group was found to be the phenylamido derivative 11a with an IC50 value of 103.5 μM, while derivative 11c with a 4-methoxyphenylamido group failed to show an IC50 value below the detection threshold. Within the sulfonamide series, the 4-toluenesulfonamido derivative 13d was a poorer inhibitor compared to the 4-chlorophenylsulfonamide 13b with an IC50 value of 64.8 μM. The 4-methoxyphenylsulfonamide 13c was found to be slightly less potent with an IC50 of 71.0 μM, while the weakest neuraminidase inhibitor within the sulfonamides was the phenylsulfonamido derivative 13a with an IC50 of 99.6 μM.
The unexpected weak inhibition observed for all of the urea derivatives can be rationalized on the basis of the significant flexibility of the phenyl moiety-bearing urea linker in these derivatives, in contrast to the rigid triazole ring in the potent inhibitors 5 and 6. It is reasonable to conclude from these results that the linker that carries the bulky phenyl substituent at the C-4 position of Neu2en needs to be rigid enough to keep the phenyl ring maximally oriented towards the 216-loop and occupy the open loop 216-cavity. In both the amide and the sulfonamide derivatives, the linker holds the phenyl group at a shorter distance relative to that in the C-4 urea derivatives. Moreover, these linkers are less flexible and direct the C-4 phenyl moiety towards the open loop 216-cavity to provide the observed improved potency within these two series compared with the C-4 urea derivatives. Conversely, in the inhibitor series 11 and 13 the combination of C-4 phenyl moieties positioned at a shorter distance from the 216 loop with the use of more flexible linkers, compared to the rigid triazole linker in inhibitors 5 and 6, resulted in a decreased potency. This combination presumably limits the inhibitors' capability to lock open the 216-loop and efficiently occupy the 216-cavity.
In order to evaluate the effect of C-5 substitution in these new inhibitors on their NI potencies, the C-5 acetamido group of the derivative that showed the most potent inhibition (the 4-chlorophenylamide 11b) was replaced by the isobutyramido group that has been reported in previous studies to further improve the inhibition of Neu2en derivatives against hPIV HN.10,11,18 Accordingly, inhibitor 16 that contains a C-4 4-chlorophenylamido substituent and a C-5 isobutyramido group was synthesized and tested for its capacity to inhibit hPIV-3 HN neuraminidase activity. Indeed, the new C-5 isobutyramido-based inhibitor proved to be slightly more potent with a NI IC50 value of 38.9 μM (vs. 45.1 μM for the parent C-5 acetamido derivative 11b), suggesting a similar binding mode to that of the triazole derivatives.
Conclusions
In conclusion, a group of C-4 phenylureido, phenylamido and phenylsulfonamido 4-deoxy-Neu2en derivatives, in which the triazole linker carrying the phenyl moiety in inhibitors 5 and 6 was replaced by more flexible linkers, have been synthesized and evaluated for their capacity to inhibit hPIV-3 HN. The results have shown that the triazole ring in inhibitors 5 and 6 plays an important role in potency. Indeed, the rigidity of the triazole ring is critical for inhibitor potency, as it suitably orients the phenyl ring towards the 216-loop. This optimal linker can induce 216-loop movement to reveal the 216-cavity, allowing the inhibitor to efficiently occupy the cavity and tightly bind HN. Importantly, the more flexible the linker carrying the phenyl ring is, the less capable the inhibitor is to induce the 216-loop movement, resulting in weaker inhibition of hPIV-3 HN functions. This was illustrated by the NI IC50 values of the urea, amide and sulfonamide derivatives evaluated in this study, where the weakest inhibition was observed for the most flexible derivatives (the urea series), while intermediate inhibition was found for the derivatives that have linkers with moderate rigidity (the amides and sulfonamides). These results set the basis for the design of novel Neu2en derivatives carrying bulky C-4 substituents targeting hPIV-3 HN with the aim of reorienting the 216-loop and occupying the 216-cavity.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. I. M. E.-D. and P. G. contributed equally and are joint senior authors.Acknowledgements
The Australian Research Council (DP1094549) is gratefully acknowledged for its financial support (MvI) and for the award of an Australian Postdoctoral Award (PG). The National Health and Medical Research Council (1047824 and 1071659) is thanked for its financial support (MvI). Griffith University is gratefully acknowledged for the award of a Griffith University Postdoctoral Award (IMED).Notes and references
- G. M. Loughlin and A. Moscona, Pediatr. Clin. North Am., 2006, 53, 929–959 CrossRef PubMed , ix–x.
- A. Moscona, J. Clin. Invest., 2005, 115, 1688–1698 CrossRef CAS PubMed.
- C. A. Heilman, J. Infect. Dis., 1990, 161, 402–406 CrossRef CAS PubMed.
- K. J. Cortez, D. D. Erdman, T. C. Peret, V. J. Gill, R. Childs, A. J. Barrett and J. E. Bennett, J. Infect. Dis., 2001, 184, 1093–1097 CrossRef CAS PubMed.
- S. A. Madhi, N. Ramasamy, K. Petersen, A. Madhi and K. P. Klugman, Eur. J. Clin. Microbiol. Infect. Dis., 2002, 21, 499–505 CrossRef CAS PubMed.
- K. Huberman, R. W. Peluso and A. Moscona, Virology, 1995, 214, 294–300 CrossRef CAS PubMed.
- C. T. Holzer, M. von Itzstein, B. Jin, M. S. Pegg, W. P. Stewart and W. Y. Wu, Glycoconjugate J., 1993, 10, 40–44 CrossRef CAS PubMed.
- M. von Itzstein, W. Y. Wu, G. B. Kok, M. S. Pegg, J. C. Dyason, B. Jin, T. Van Phan, M. L. Smythe, H. F. White and S. W. Oliver, et al. , Nature, 1993, 363, 418–423 CrossRef CAS PubMed.
- B. Bailly, L. Dirr, I. M. El-Deeb, R. Altmeyer, P. Guillon and M. von Itzstein, Sci. Rep., 2016, 6, 24138 CrossRef CAS PubMed.
- I. M. El-Deeb, P. Guillon, M. Winger, T. Eveno, T. Haselhorst, J. C. Dyason and M. von Itzstein, J. Med. Chem., 2014, 57, 7613–7623 CrossRef CAS PubMed.
- P. Guillon, L. Dirr, I. M. El-Deeb, M. Winger, B. Bailly, T. Haselhorst, J. C. Dyason and M. von Itzstein, Nat. Commun., 2014, 5, 5268 CrossRef CAS PubMed.
- M. Watanabe, V. P. Mishin, S. A. Brown, C. J. Russell, K. Boyd, Y. S. Babu, G. Taylor, X. Xiong, X. Yan, A. Portner and I. V. Alymova, Antimicrob. Agents Chemother., 2009, 53, 3942–3951 CrossRef CAS PubMed.
- I. V. Alymova, G. Taylor, T. Takimoto, T. H. Lin, P. Chand, Y. S. Babu, C. Li, X. Xiong and A. Portner, Antimicrob. Agents Chemother., 2004, 48, 1495–1502 CrossRef CAS PubMed.
- I. V. Alymova, M. Watanabe, K. L. Boyd, P. Chand, Y. S. Babu and A. Portner, Antiviral Ther., 2009, 14, 891–898 CrossRef CAS PubMed.
- M. Winger and M. von Itzstein, J. Am. Chem. Soc., 2012, 134, 18447–18452 CrossRef CAS PubMed.
- D. Ye, W. J. Shin, N. Li, W. Tang, E. Feng, J. Li, P. L. He, J. P. Zuo, H. Kim, K. Y. Nam, W. Zhu, B. L. Seong, K. T. No, H. Jiang and H. Liu, Eur. J. Med. Chem., 2012, 54, 764–770 CrossRef CAS PubMed.
- Computational Chemistry calculations were carried out using ‘Molecular Operating Environment (MOE) version 2008.10’, Chemical Computing Group Inc., 1010 Sherbrooke Street West, Suite 910, Montreal, H3A 2R7, Canada Search PubMed.
- L. Dirr, I. M. El-Deeb, P. Guillon, C. J. Carroux, L. M. Chavas and M. von Itzstein, Angew. Chem., Int. Ed., 2015, 54, 2936–2940 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing financial interest. |
| ‡ Electronic supplementary information (ESI) available: Complete experimental procedures, characterization data, and 1H and 13C NMR spectra of new intermediates and final compounds. See DOI: 10.1039/c6md00519e |
| § Current address: RCSI Bahrain, Adilya, Bahrain. |
| This journal is © The Royal Society of Chemistry 2017 |