
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical epigenetics to assess the role of HDAC1–3 inhibition in macrophage pro-inflammatory gene expression†‡
Maria E.
Ourailidou
a,
Niek G. J.
Leus
a,
Kim
Krist
a,
Alessia
Lenoci
b,
Antonello
Mai
*bc and
Frank J.
Dekker
*a
aDepartment of Chemical and Pharmaceutical Biology, Groningen Research Institute of Pharmacy, University of Groningen, Antonius Deusinglaan 1, Groningen 9713 AV, The Netherlands. E-mail: f.j.dekker@rug.nl; Fax: +31 50 3637953; Tel: +31 50 3638030
bDepartment of Drug Chemistry and Technologies, ‘Sapienza’ University, P.le A. Moro 5, 00185 Rome, Italy
cPasteur Institute, Cenci Bolognetti Foundation, ‘Sapienza’ University, P.le A. Moro 5, 00185 Rome, Italy
First published on 7th September 2016
Abstract
Histone deacetylases (HDACs) have been used as pharmacological targets for the treatment of various diseases. Some non-selective HDAC inhibitors (HDACi) have been clinically-used as therapeutic agents for treatment of hematological cancers but their cytotoxic side effects are an important downside. The discovery of more selective inhibitors has certified the involvement of individual HDACs in pathological processes but the elucidation of the role of specific family members in inflammatory responses still remains a challenge. Here, we report the development of closely related, structural analogues of the clinically-used HDACi Entinostat via a chemical epigenetic approach. Three compounds were designed and synthesized in which the cap moiety of Entinostat was replaced by an azobenzene group that is either para, meta or ortho substituted. The compounds were then evaluated for selectivity towards HDACs 1–3 and their effect on pro-inflammatory gene expression in macrophages. One analogue, compound 4, lacked selectivity and demonstrated inhibition of NF-κB reporter gene activity and pro-inflammatory gene expression in RAW264.7 macrophages, thus indicating that there is a delicate balance between the selectivity of HDACi over specific family members and their pro- or anti-inflammatory effects.
Introduction
Reversible lysine acetylation has been extensively proven to play a critical role in epigenetic regulation of gene expression.1–5 Lysine acetylation is catalyzed by histone acetyltransferases (HATs) (writers), leading to transcriptionally active chromatin, while the reverse process is catalyzed by histone deacetylases (HDACs) (erasers) resulting in condensed chromatin and transcriptional silencing. Over the past decades, eighteen mammalian HDACs have been identified and divided into four classes (I–IV) according to their sequence homology to yeast orthologs.6 Among them, class I, containing the zinc-dependent HDACs 1–3 and 8, is crucial for normal cell survival, homeostasis, proliferation and gene expression.7 Consequently, mounting evidence points to a link between abnormal HDAC activity and inflammatory8 and neurological diseases,9 tumorigenesis,9,10 and metabolic disorders.11There is a tremendously increasing interest in the development of HDAC inhibitors (HDACi) as potential therapeutic agents for several pathological conditions. To date, a wide variety of small molecules has been reported to target HDACs, with differences in their structure, potency, selectivity and, thus, pharmacological effects.7,12 However, a limited number has reached clinical trials and has been approved for medical use, mainly for treatment of cancer patients.13,14 The toxicity of the currently available chemotherapeutic agents hamper their further applications. Nevertheless, more selective HDAC inhibitors such as Entinostat targeting only HDAC1–3 is less toxic in clinical trials compared to the non-selective HDAC inhibitors thus providing a perspective towards clinical applications of these inhibitors.15 Recently, significant efforts in the field of medicinal chemistry have led to the discovery of more selective inhibitors9,16–21 that have shed some light on the role of individual HDACs in specific diseases. Yet, the important question that remains to be answered is whether inhibition of specific HDAC family members enables achievement of the desired biological effects. In order to address this issue it is imperative to develop novel methods to investigate the inhibition pattern needed for each disease model.
Our interest lies in the better understanding of the role of class I HDACs in inflammatory signaling. It is known that HDACs influence the activation of NF-κB pathway, a key mediator in inflammatory diseases like COPD and asthma.22,23 Studies indicate that acetylations of specific lysine residues of the NF-κB p65 transcription factor affect its transcriptional capacity, duration of action and DNA binding.22,24 The alterations observed in NF-κB-mediated gene expression after treatment with HDACi confirm a deacetylation-dependent mechanism of activation.25–27 Several groups have reported the pivotal role of HDACs 1–3 in inflammatory responses, such as Schwann cell myelination, intestinal epithelial cell homeostasis and allergic reaction.28–32 Recently, we explored the function of HDACs 1–3 in inflammatory gene expression in LPS/IFNγ-stimulated murine macrophages by si-RNA-mediated downregulation.26 Our data suggested a pro-inflammatory role for HDAC1 and 3. Furthermore, selective pharmacological HDAC3 inhibition by RGFP966 demonstrated an attenuation of pro-inflammatory signals in models for inflammatory lung diseases. In another study, HDACi highly potent against HDAC3 and/or HDAC6 have been shown to downregulate mRNA expression of pro-inflammatory cytokines (IL-1β and IL-6) and to promote the transcription of IL-10 gene in KB31, C2C12, and 3T3-J2 cell lines, without affecting the cell viability.33 Yet, further optimization is required in the development of novel HDACi, mainly against HDAC1 and 3, as anti-inflammatory therapeutics in order to obtain a clearer picture on which HDAC family member to target.
A very promising approach towards this purpose is the use of chemical epigenetic tools, where a parental clinically available agent is slightly modified to generate closely related inhibitors with a different selectivity profile among biological family members. This approach aims at the fine-tuning of the selectivity of inhibitors over specific family members with respect to their pharmacological effects.34 Therefore, we set out to design closely related analogues of Entinostat (MS-275), an ortho-aminoanilide containing HDAC1–3 inhibitor with proven therapeutic properties, currently undergoing clinical trials for treatment of various cancers.35 These analogues embody isomeric ortho-aminoanilide-type HDAC inhibitors with an azobenzene cap. Inhibition and kinetic studies against class I HDACs were performed in order to evaluate their potency and binding kinetic parameters. Next, we tested the effects of the inhibitors on transcriptional activity and intracellular localization of NF-κB p65 as well as inflammatory gene expression in LPS/IFNγ-stimulated murine macrophages. Interestingly, one analogue (compound 4, para isomer) demonstrated anti-inflammatory characteristics providing insight in the selectivity profile required to attenuate macrophage-mediated pro-inflammatory responses.
Results and discussion
Chemical epigenetic design
For our purposes, we chose Entinostat as a starting point to design novel HDACi with refined characteristics. The ortho-aminoanilide group, that serves for zinc binding, is advantageous compared to the hydroxamic acid moiety (like suberanilohydroxamic acid, SAHA, pan-HDACi) as it assures for selective inhibition among class I HDACs 1–3, reducing cytotoxicity.36–38 Our modifications were limited to the cap region of Entinostat, where an azobenzene group was introduced to a para, meta and ortho position with respect to the amine group of the connection unit of the parental inhibitor (compounds 4, 7, and 9 respectively, Fig. 1). Azobenzenes are often used in photopharmacology as photoswitches that allow for two isomeric forms, preferably the cis being more potent than the trans analogue.39–41 Previously, we developed successfully SAHA photoswitchable derivatives as potential antitumor agents.42 Our data demonstrated that substitution of the cap of the original drug with an azobenzene group in the para, meta and ortho position led to notable differences in terms of potency of the trans form (meta and ortho isomers more potent than para). Additionally, para and ortho trans isomers provided an improved selectivity towards HDACs 1 and 3. These findings encouraged us to employ the same structural alterations in Entinostat in an attempt to improve potency and specificity among HDACs 1–3 and gain useful information on the selectivity profile required to inhibit pro-inflammatory gene expression in macrophages.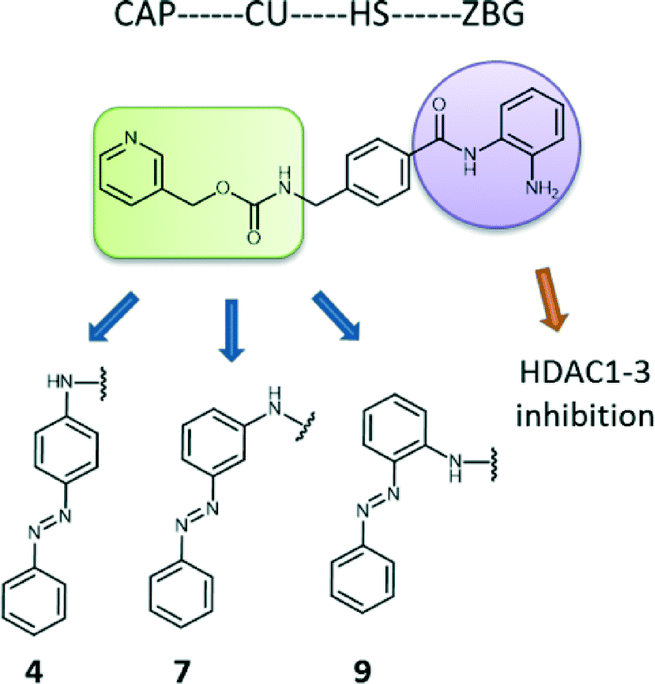 | ||
| Fig. 1 Design of structural analogues of Entinostat. An azobenzene moiety was introduced into the CAP region in para (4), meta (7) and ortho (9) position in respect to the amine group of the CU of Entinostat. CU; connection unit, HS; hydrophobic spacer, ZBG; zinc-binding group. | ||
Synthesis
The starting materials were synthesized as described previously.42 Two synthetic routes were followed for the production of the Entinostat analogues (see ESI‡). Compound 4 was synthesized by a reductive amination reaction between methyl-4-formylbenzoate and (E)-4-(phenyldiazenyl)aniline. Subsequent hydrolysis to the corresponding ester allowed for coupling to the mono tert-butyloxycarbonyl-protected 2-aminoaniline. Boc deprotection using a solution of 4 N HCl afforded compound 4. As for compounds 7 and 9, the peptide coupling between the mono Boc-protected 2-aminoaniline and 4-formylbenzoic acid was performed first followed by reductive amination using the corresponding (phenyldiazenyl)aniline. For compound 7, the deprotection of Boc group was performed as for compound 4, while the same conditions led to benzyl cleavage due the basic nature of the amino group at the ortho position of the azobenzene in compound 9. In this case, milder acidic conditions were used by employing trifluoroacetic acid.IC50 values reveal limited selectivity among HDACs 1–3
The resulting collection of structurally related HDACi was tested for HDAC inhibition via an assay based on the measurement of the deacetylation of a pro-fluorogenic substrate.42 Prior to testing, the stock solutions of the inhibitors (10 mM in DMSO) were heated at 60 °C for 10 min to ensure occurrence of the azobenzene in its trans isomeric form. Various concentrations of each inhibitor were incubated with HDAC1, 2, 3 or 8 in presence of the substrate Boc-Lys(Ac)-AMC at room temperature. After one hour, the reaction was stopped by the addition of SAHA and trypsin to cleave the deacetylated lysine, resulting in the release of fluorescent 7-amino-4-methylcoumarin (AMC). As expected for ortho-aminoanilide-type inhibitors, no inhibition was observed against HDAC8 at concentrations up to 50 μM. It is known that Entinostat inhibits HDACs 1, 2 and 3 with a potency of 0.18, 1.2 and 2.3 μM correspondigly.43 The IC50 values of our isomeric set of compounds were found to be in the low μM range with no meaningful differences among individual family members (see ESI‡ Fig. S1). We then set out to perform kinetic studies in order to determine the affinity rate constant (Ki) values, known to be more representative for the inhibitory potencies.Binding kinetic analysis reaffirms a non-family member-selective profile
Previous studies have shown that enzyme binding kinetics are more informative on the selectivity and behavior of the inhibitors inside the biological targets.44 Therefore, we expanded our investigations with determination of the enzyme kinetics on HDAC1, 2 and 3. There is strong evidence that ortho-aminoanilides are slow, tight-binding inhibitors, following the mechanism of enzyme inhibition.34,45 In contrast to hydroxamate-based inhibitors, they act via a slow on/slow off binding; they are characterized by low association rate constants (kon) and form a tight complex with the enzyme (EI), exhibiting long residence times. In order to determine the kinetic parameters of each inhibitor, Lys-C peptidase was used as a developer instead of trypsin as described by Chou et al. to prevent enzyme degradation.46 Moreover, the deacetylation reaction was not stopped by the addition of SAHA but HDAC activity was monitored over time to generate progress curves allowing for the investigation of the rate constants.46According to a previously reported protocol, the measurement of the association rate constants (kon) involved the incubation of HDACs 1–3 with different concentrations of the inhibitors in presence of Lys-C and high amounts of the substrate (5 × KM).46 The fluorescence was monitored for 60–90 min at room temperature. The progress curves of all the inhibitors confirmed a time-dependent mechanism of HDAC inhibition and a linear relationship between kobs and inhibitor concentration as observed before (Fig. S2,‡kon values were calculated as described in the ESI‡ section).46 The off-rate constants (koff) were determined after 100-fold dilution of samples containing high concentrations of HDACs 1–3 pre-incubated for 1 h with each inhibitor, and subsequent measurement of HDAC activity as described above.46
In terms of selectivity among family members, all compounds appeared to bind faster in HDAC1, less fast in HDAC2 and even slower in HDAC3 (compound 7 presented similar kon values in HDAC2 and 3) (Fig. 2, ESI‡ Table S1). More specifically, regarding HDAC1, compounds 4 and 7 were found to bind slightly faster than the reference Entinostat whereas the observed kon values in HDAC2 were lower. Compound 9 displayed the smallest kon values in all HDACs 1–3 (even 3.5× slower than Entinostat in the case of HDAC3). A certain pattern was also noticed during dissociation experiments, where we discovered that Entinostat and its azobenzene analogues are generally released rapidly from HDAC3, less fast from HDAC1 and remain longer in HDAC2 (compound 7 presented similar koff values in HDAC1 and 3) (Fig. 2). Compound 4 showed longer residence in HDAC1 and 3 than compounds 7 and 9, whereas the dissociation rate from HDAC2 was found to be similar for all the developed inhibitors. We then used the kon and koff values of the analogues to calculate the affinity rate contants Ki, using the following equation: Ki = koff/kon (see ESI,‡ Table S2). All compounds exhibited statistically significant differences in their potencies against HDACs 1 and 2 in comparison to HDAC3, while stronger inhibition was observed for compound 4. However, the differences in potencies among family members are still subtle in order to be further useful to explain any pharmacological variations. The subtle differences in these cases are clearly different from the 10-fold preference for HDAC3 inhibition observed for ortho-aminoanilide type HDAC inhibitors lacking the phenyl group in the linker region.46
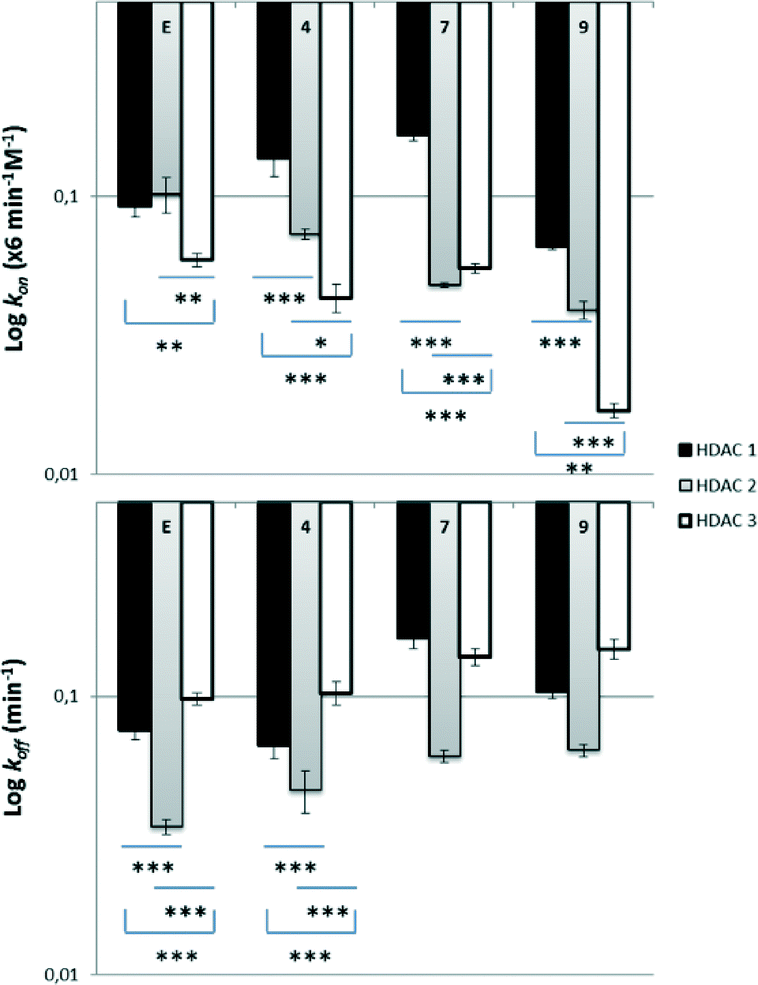 | ||
| Fig. 2 Comparison bar graphs of the kinetic values of Entinostat (E) and its para (4), meta (7) and ortho (9) azobenzene analogues on HDACs 1–3 expressed in logarithmic scale. The data are presented as mean values of three independent measurements with their respective standard deviations. *p < 0.05, **p < 0.01 ***p < 0.001. | ||
Compound 4 exhibits anti-inflammatory effects in LPS/IFNγ-stimulated murine macrophages
Extensive literature has established the connection between HDACs 1–3 and inflammatory signaling in macrophages.47–49 In order to investigate the effect of 1–3 selective HDAC inhibitors in inflammatory pathways, we performed a series of pharmacological studies in RAW264.7 murine macrophages. Each time, cells were incubated with the respective inhibitor for 20 h followed by inflammatory stimulation with LPS and IFNγ during the last 4 h to ensure activation towards M1 (pro-inflammatory) macrophages.50RAW264.7 cells were treated with compounds 4, 7 and 9 in a concentration range 1–10 μM. Interestingly, treatment with para (4) or ortho (9) isomers resulted in inhibition of NF-κB p65 transcriptional activity with the highest effect observed at 10 μM (up to 50% reduction, Fig. 3). On the contrary, a robust activation of NF-κB p65 was found in case of 10 μM of meta derivative (7), while lower concentrations gave no change.
 | ||
| Fig. 3 Effects of para (4), meta (7) and ortho (9) azobenzene analogues of Entinostat on LPS/IFNγ-induced NF-κB p65 transcriptional activity. RAW blue cells were treated with the respective HDAC inhibitors at the indicated concentrations for 20 h and stimulated with LPS/IFNγ for the last 4 h of the experiment. For vehicle treatment, cells were pre-treated with a proportional dilution of the inhibitor solvent DMSO. Data are shown as representative data set of 3 independent experiments with the respective standard deviations. *p < 0.05, ***p < 0.001 compared to vehicle-treated group. | ||
These findings provided the first indications that structural modifications and relatively small changes in inhibitory selectivity among HDACs 1–3 can result in considerable differences in pharmacological performance. Yet, considering that at 10 μM the HDAC activity is mostly inhibited, the differences in the biological behavior of the analogues cannot be entirely attributed to HDAC inhibition as other cellular targets may also be involved.
 | ||
| Fig. 4 Effects of para (4), meta (7) and ortho (9) azobenzene analogues of Entinostat on nuclear translocation profile of NF-kB p65 in RAW264.7 macrophages. After 20 h incubation with 1 μM of the respective inhibitors followed by 1 h LPS/IFNγ stimulation, RAW264.7 macrophages were prepared for immunofluorescence microscopy. Green signal represents NF-kB p65 protein, while the blue signal visualizes the Hoechst stained nuclei. The presented data set shows representative images of 2 independent experiments, original magnification 400×. All images were taken under identical instrumental conditions. | ||
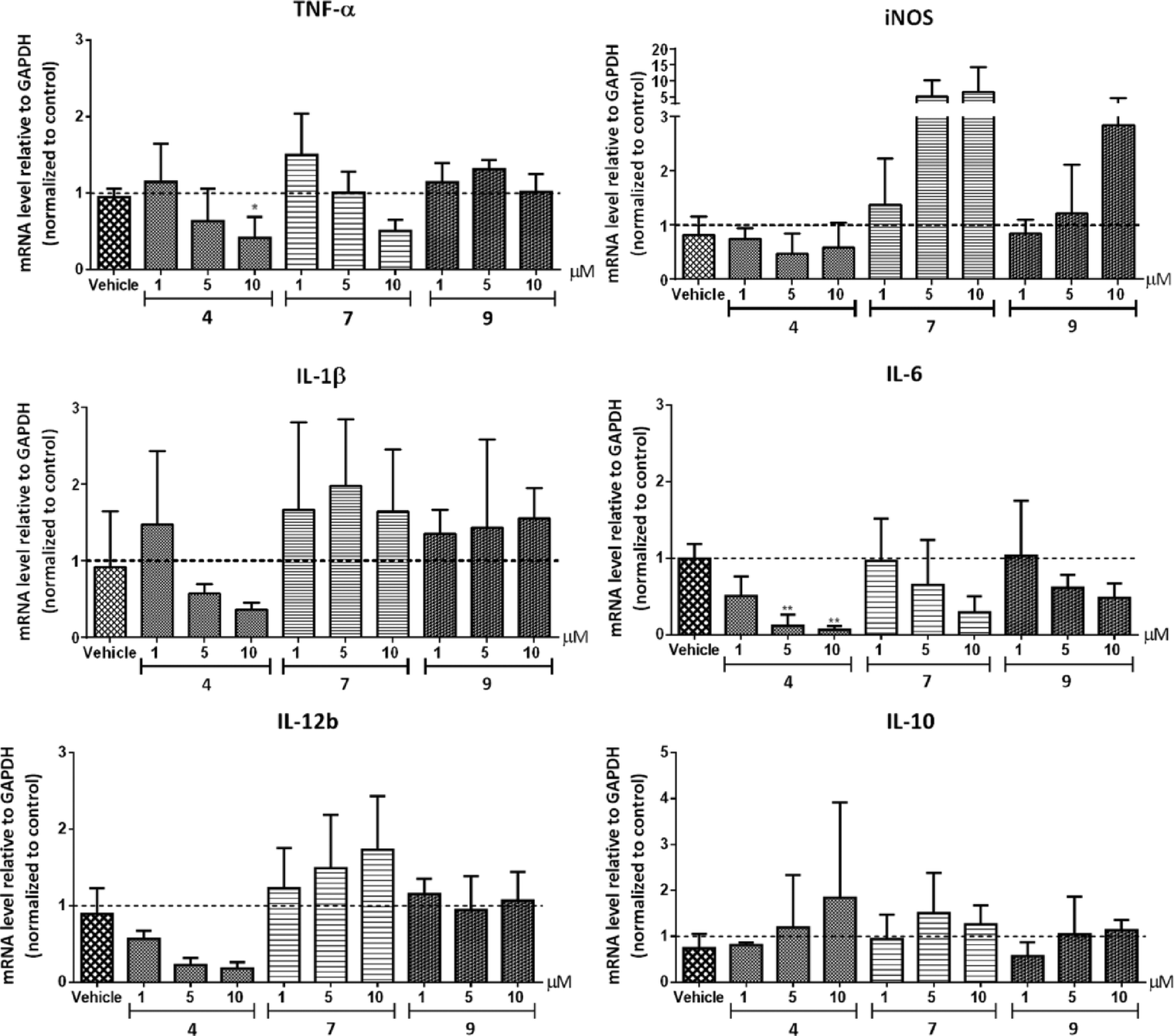 | ||
| Fig. 5 Effects of para (4), meta (7) and ortho (9) azobenzene analogues of Entinostat on pro- and anti-inflammatory gene expression in RAW264.7 macrophages. Cells were treated with the respective HDAC inhibitors at the indicated concentrations for 20 h and stimulated with LPS/IFNγ for the last 4 h of the experiment. Gene expression was analyzed by RT-q-PCR. For vehicle treatment, cells were pre-treated with a proportional dilution of the inhibitor solvent DMSO. Data represent the target gene expression normalized to the reference gene and are presented as mean values of 3–4 independent experiments with their respective standard deviations. *p < 0.05, **p < 0.01 compared to vehicle-treated group. | ||
Conclusions
In summary, we developed three structural analogues of the clinically-used ortho-aminoanilide type HDAC1–3 inhibitor Entinostat in an attempt to explore the role of class I HDACs in macrophage pro-inflammatory signaling. The compounds exhibited small differences in their HDAC inhibitory profile with a slight preference to HDAC1 and 2. Studies on the effect of the inhibitors on LPS/IFNγ-induced inflammatory responses in RAW264.7 macrophages revealed that compounds 4 and 9 impaired NF-κB p65 transcriptional activity and suppressed NF-κB p65 nuclear translocation, while an increase was observed in case of compound 7. These findings suggest that, additionally to overall HDAC inhibition, the designed compounds target other mechanisms that play a role in transcriptional activity of NF-κB p65. Further investigations of the impact of the analogues on the expression of pro-inflammatory genes corroborated the anti-inflammatory profile of compound 4.Our work supports that a non-selective inhibition of HDACs 1–3 offers a promising potential towards the desirable anti-inflammatory characteristics, taking into account that possible off-target effects may be significant as well. This sets the stage for further development and exploration of the contribution of HDAC1, 2 and 3 in drug discovery effort aimed at the development of anti-inflammatory therapeutics based on the ortho-aminoanilide scaffold. Moreover, we demonstrated that chemical epigenetics can be employed as a useful strategy in order to fine-tune the family member(s) selectivity with the ultimate goal to develop inhibitors that possess the desired pharmacological behavior.
Acknowledgements
This work was financially supported by a VIDI grant (723.012.005) from the Netherlands Organization for Scientific Research and an ERC starting grant (309782) from the European Union to F. J. D. Funds from RF-2010-2318330 and IIT-Sapienza Project grants are also acknowledged by A. M. We also thank W. Szymanski for providing the starting materials for the synthesis of the reported compounds.References
- C. A. Mizzen and C. D. Allis, Cell. Mol. Life Sci., 1998, 54, 6–20 CrossRef CAS PubMed.
- B. M. Turner, BioEssays, 2000, 22, 836–845 CrossRef CAS PubMed.
- M. Grunstein, Nature, 1997, 389, 349–352 CrossRef CAS PubMed.
- G. P. Delcuve, D. H. Khan and J. R. Davie, Clin. Epigenet., 2012, 4, 5 CrossRef CAS PubMed.
- E. E. Cameron, K. E. Bachman, S. Myöhänen, J. G. Herman and S. B. Baylin, Nat. Genet., 1999, 21, 103–107 CrossRef CAS PubMed.
- I. V. Gregoretti, Y. M. Lee and H. V. Goodson, J. Mol. Biol., 2004, 338, 17–31 CrossRef CAS PubMed.
- M. Dokmanovic, C. Clarke and P. A. Marks, Mol. Cancer Res., 2007, 5, 981–989 CrossRef CAS PubMed.
- M. R. Shakespear, M. A. Halili, K. M. Irvine, D. P. Fairlie and M. J. Sweet, Trends Immunol., 2011, 32, 335–343 CrossRef CAS PubMed.
- K. J. Falkenberg and R. W. Johnstone, Nat. Rev. Drug Discovery, 2014, 13, 673–691 CrossRef CAS PubMed.
- S. Ropero and M. Esteller, Mol. Oncol., 2007, 1, 19–25 CrossRef CAS PubMed.
- J. N. Sangshetti, N. S. Sakle, M. H. Dehghan and D. B. Shinde, Mini-Rev. Med. Chem., 2013, 13, 1005–1026 CrossRef CAS PubMed.
- S. Minucci and P. G. Pelicci, Nat. Rev. Cancer, 2006, 6, 38–51 CrossRef CAS PubMed.
- M. Mottamal, S. Zheng, T. L. Huang and G. Wang, Molecules, 2015, 20, 3898–3941 CrossRef CAS PubMed.
- A. C. West and R. W. Johnstone, J. Clin. Invest., 2014, 124, 30–39 CrossRef CAS PubMed.
- R. Pili, B. Salumbides, M. Zhao, S. Altiok, D. Qian, J. Zwiebel, M. A. Carducci and M. A. Rudek, Br. J. Cancer, 2012, 106, 77–84 CrossRef CAS PubMed.
- A. V. Bieliauskas and M. K. H. Pflum, Chem. Soc. Rev., 2008, 37, 1402–1413 RSC.
- T. A. McKinsey, J. Mol. Cell. Cardiol., 2011, 51, 491–496 CrossRef CAS PubMed.
- M. Fournel, C. Bonfils, Y. Hou, P. T. Yan, M.-C. Trachy-Bourget, A. Kalita, J. Liu, A.-H. Lu, N. Z. Zhou, M.-F. Robert, J. Gillespie, J. J. Wang, H. Ste-Croix, J. Rahil, S. Lefebvre, O. Moradei, D. Delorme, A. R. Macleod, J. M. Besterman and Z. Li, Mol. Cancer Ther., 2008, 7, 759–768 CrossRef CAS PubMed.
- J. H. Park, Y. Jung, T. Y. Kim, S. G. Kim, H. S. Jong, J. W. Lee, D. K. Kim, J. S. Lee, N. K. Kim, T. Y. Kim and Y. J. Bang, Clin. Cancer Res., 2004, 10, 5271–5281 CrossRef CAS PubMed.
- J. H. Lee, A. Mahendran, Y. Yao, L. Ngo, G. Venta-Perez, M. L. Choy, N. Kim, W.-S. Ham, R. Breslow and P. A. Marks, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15704–15709 CrossRef CAS PubMed.
- P. Gupta, R. C. Reid, A. Iyer, M. J. Sweet, D. P. Fairlie, R. C. Reid, A. Iyer, M. J. Sweet and D. P. Fairlie, Curr. Top. Med. Chem., 2012, 12, 1479–1499 CrossRef CAS PubMed.
- M. Ghizzoni, H. J. Haisma, H. Maarsingh and F. J. Dekker, Drug Discovery Today, 2011, 16, 504–511 CrossRef CAS PubMed.
- F. J. Dekker, T. van den Bosch and N. I. Martin, Drug Discovery Today, 2014, 19, 654–660 CrossRef CAS PubMed.
- L. F. Chen, W. Fischle, E. Verdin and W. C. Greene, Science, 2001, 293, 1653–1657 CrossRef CAS PubMed.
- T. van den Bosch, A. Boichenko, N. G. J. Leus, M. E. Ourailidou, H. Wapenaar, D. Rotili, A. Mai, A. Imhof, R. Bischoff, H. J. Haisma and F. J. Dekker, Biochem. Pharmacol., 2015, 102, 130–140 CrossRef PubMed.
- N. G. J. Leus, P. E. van der Wouden, T. van den Bosch, W. T. R. Hooghiemstra, M. E. Ourailidou, L. E. M. Kistemaker, R. Bischoff, R. Gosens, H. J. Haisma and F. J. Dekker, Biochem. Pharmacol., 2016, 108, 58–74 CrossRef CAS PubMed.
- V. Quivy and C. Van Lint, Biochem. Pharmacol., 2004, 68, 1221–1229 CrossRef CAS PubMed.
- B. P. Ashburner, S. D. Westerheide and A. S. Baldwin Jr., Mol. Cell. Biol., 2001, 21, 7065–7077 CrossRef CAS PubMed.
- Y. Chen, H. Wang, S. O. Yoon, X. Xu, M. O. Hottiger, J. Svaren, K. A. Nave, H. A. Kim, E. N. Olson and Q. R. Lu, Nat. Neurosci., 2011, 14, 437–441 CrossRef CAS PubMed.
- A. Gonneaud, J. M. Gagné, N. Turgeon and C. Asselin, J. Inflammation, 2014, 11, 43 CrossRef PubMed.
- X. Chen, I. Barozzi, A. Termanini, E. Prosperini, A. Recchiuti, J. Dalli, F. Mietton, G. Matteoli, S. Hiebert and G. Natoli, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E2865–2874 CrossRef CAS PubMed.
- Y. Kim, K. Kim, D. Park, E. Lee, H. Lee, Y. S. Lee, J. Choe and D. Jeoung, J. Biol. Chem., 2012, 287, 25844–25859 CrossRef CAS PubMed.
- R. Di Liddo, S. Valente, S. Taurone, C. Zwergel, B. Marrocco, R. Turchetta, M. T. Conconi, C. Scarpa, T. Bertalot, S. Schrenk, A. Mai and M. Artico, Autoimmunity, 2016, 1–11 CrossRef PubMed.
- F. F. Wagner, M. Lundh, T. Kaya, P. McCarren, Y.-L. Zhang, S. Chattopadhyay, J. P. Gale, T. Galbo, S. L. Fisher, B. C. Meier, A. Vetere, S. Richardson, N. G. Morgan, D. P. Christensen, T. J. Gilbert, J. M. Hooker, M. Leroy, D. Walpita, T. Mandrup-Poulsen, B. K. Wagner and E. B. Holson, ACS Chem. Biol., 2016, 11, 363–374 CrossRef CAS PubMed.
- D. A. Yardley, R. R. Ismail-Khan, B. Melichar, M. Lichinitser, P. N. Munster, P. M. Klein, S. Cruickshank, K. D. Miller, M. J. Lee and J. B. Trepel, J. Clin. Oncol., 2013, 31, 2128–2135 CrossRef CAS PubMed.
- O. M. Moradei, T. C. Mallais, S. Frechette, I. Paquin, P. E. Tessier, S. M. Leit, M. Fournel, C. Bonfils, M. C. Trachy-Bourget, J. Liu, T. P. Yan, A. H. Lu, J. Rahil, J. Wang, S. Lefebvre, Z. Li, A. F. Vaisburg and J. M. Besterman, J. Med. Chem., 2007, 50, 5543–5546 CrossRef CAS PubMed.
- A. Lu, H. Luo, M. Shi, G. Wu, Y. Yuan, J. Liu and F. Tang, Bioorg. Med. Chem. Lett., 2011, 21, 4924–4927 CrossRef CAS PubMed.
- M. Boissinot, M. Inman, A. Hempshall, S. R. James, J. H. Gill, P. Selby, D. T. Bowen, R. Grigg and P. N. Cockerill, Leuk. Res., 2012, 36, 1304–1310 CrossRef CAS PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- F. Lynen and S. Ochoa, Biochim. Biophys. Acta, 1953, 12, 299–314 CrossRef CAS.
- S. A. Reis, B. Ghosh, J. A. Hendricks, D. M. Szantai-Kis, L. Törk, K. N. Ross, J. Lamb, W. Read-Button, B. Zheng, H. Wang, C. Salthouse, S. J. Haggarty and R. Mazitschek, Nat. Chem. Biol., 2016, 12, 317–323 CrossRef CAS PubMed.
- W. Szymanski, M. E. Ourailidou, W. A. Velema, F. J. Dekker and B. L. Feringa, Chem. – Eur. J., 2015, 21, 16517–16524 CrossRef CAS PubMed.
- N. Khan, M. Jeffers, S. Kumar, C. Hackett, F. Boldog, N. Khramtsov, X. Qian, E. Mills, S. C. Berghs, N. Carey, P. W. Finn, L. S. Collins, A. Tumber, J. W. Ritchie, P. B. Jensen, H. S. Lichenstein and M. Sehested, Biochem. J., 2008, 409, 581–589 CrossRef CAS PubMed.
- F. F. Wagner, Y.-L. Zhang, D. M. Fass, N. Joseph, J. P. Gale, M. Weïwer, P. McCarren, S. L. Fisher, T. Kaya, W.-N. Zhao, S. A. Reis, K. M. Hennig, M. Thomas, B. C. Lemercier, M. C. Lewis, J. S. Guan, M. P. Moyer, E. Scolnick, S. J. Haggarty, L.-H. Tsai and E. B. Holson, Chem. Sci., 2015, 6, 804–815 RSC.
- B. E. L. Lauffer, R. Mintzer, R. Fong, S. Mukund, C. Tam, I. Zilberleyb, B. Flicke, A. Ritscher, G. Fedorowicz, R. Vallero, D. F. Ortwine, J. Gunzner, Z. Modrusan, L. Neumann, C. M. Koth, J. S. Kaminker, C. E. Heise and P. Steiner, J. Biol. Chem., 2013, 288, 26926–26943 CrossRef CAS PubMed.
- C. J. Chou, D. Herman and J. M. Gottesfeld, J. Biol. Chem., 2008, 283, 35402–35409 CrossRef CAS PubMed.
- K. Das Gupta, M. R. Shakespear, A. Iyer, D. P. Fairlie and M. J. Sweet, Clin. Transl. Immunol., 2016, 5, e62 CrossRef PubMed.
- A. M. Grabiec, S. Krausz, W. de Jager, T. Burakowski, D. Groot, M. E. Sanders, B. J. Prakken, W. Maslinski, E. Eldering, P. P. Tak and K. A. Reedquist, J. Immunol., 2010, 184, 2718–2728 CrossRef CAS PubMed.
- M. A. Halili, M. R. Andrews, L. I. Labzin, K. Schroder, G. Matthias, C. Cao, E. Lovelace, R. C. Reid, G. T. Le, D. A. Hume, K. M. Irvine, P. Matthias, D. P. Fairlie and M. J. Sweet, J. Leukocyte Biol., 2010, 87, 1103–1114 CrossRef CAS PubMed.
- A. Mantovani, A. Sica, S. Sozzani, P. Allavena, A. Vecchi and M. Locati, Trends Immunol., 2004, 25, 677–686 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00375c |
| This journal is © The Royal Society of Chemistry 2016 |