
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical probes and inhibitors of bromodomains outside the BET family†
Moses
Moustakim‡
 abc,
Peter G. K.
Clark‡
d,
Duncan A.
Hay‡
e,
Darren J.
Dixon
a and
Paul E.
Brennan
*bc
abc,
Peter G. K.
Clark‡
d,
Duncan A.
Hay‡
e,
Darren J.
Dixon
a and
Paul E.
Brennan
*bc
aDepartment of Chemistry, University of Oxford, Oxford OX1 3TA, UK
bStructural Genomics Consortium, University of Oxford, OX3 7DQ, UK. E-mail: paul.brennan@sgc.ox.ac.uk
cTarget Discovery Institute, Nuffield Department of Medicine, University of Oxford, OX3 7FZ, UK
dDepartment of Chemistry, Simon Fraser University, Burnaby V5A 1S6, Canada
eEvotec (UK) Ltd, 114 Innovation Drive, Milton Park, Abingdon, Oxfordshire OX14 4RZ, UK
First published on 7th September 2016
Abstract
In the last five years, the development of inhibitors of bromodomains has emerged as an area of intensive worldwide research. Emerging evidence has implicated a number of non-BET bromodomains in the onset and progression of diseases such as cancer, HIV infection and inflammation. The development and use of small molecule chemical probes has been fundamental to pre-clinical evaluation of bromodomains as targets. Recent efforts are described highlighting the development of potent, selective and cell active non-BET bromodomain inhibitors and their therapeutic potential. Over half of typical bromodomains now have reported ligands, but those with atypical binding site residues remain resistant to chemical probe discovery efforts.
 Moses Moustakim | Moses Moustakim is a second year DPhil student in the Synthesis for Biology and Medicine CDT at the University of Oxford. His current research focus is the development of epigenetic chemical probes and inhibitors through the use of contemporary synthetic methodology supervised by Prof. Paul E. Brennan (SGC) and Prof. Darren J. Dixon (University of Oxford). Moses obtained his MChem degree at the University of Leicester under the supervision of Dr Greg A. Solan and Prof Eric G. Hope (2014). |
 Peter G. K. Clark | Peter G. K. Clark received both a Bachelor's of Biomedical Science in Molecular Pharmacology and Medicinal Chemistry (2008) and a MSc in Chemistry (2011), under the supervision of Dr. Robert Keyzers, from Victoria University of Wellington in New Zealand. During these studies, Peter also completed internships at Industrial Research Ltd and the Malaghan Institute of Medical Research. In 2015, Peter completed his DPhil at the University of Oxford under the supervision of Prof. Darren J. Dixon (University of Oxford) and Prof. Paul E. Brennan (SGC), supported by a Woolf Fisher Scholarship. His research focused on the development of bromodomain inhibitors, culminating in the chemical probes LP99 and PNZ5. Peter is currently a CREATE ChemNET postdoctoral fellow in the laboratory of Prof. Robert Young at Simon Fraser University in Canada, performing medicinal chemistry, biochemistry and pre-clinical studies towards the development of inhibitors for the key autophagy enzyme ATG4B. |
 Duncan Hay | Duncan Hay is a medicinal chemist, currently working for Evotec at Abingdon, UK. Duncan received his M. Chem. from the University of Edinburgh. In 2003, he joined Discovery Chemistry at Pfizer's Sandwich laboratories. During this time, he worked on various preclinical medicinal chemistry projects, including antivirals and allergy and respiratory targets. In 2011, Duncan joined the Structural Genomics Consortium at the University of Oxford to develop chemical probes for epigenetic targets. Under the supervision of Prof. Paul E. Brennan (SGC) and Prof. Christopher J. Schofield (University of Oxford) his work was submitted as a DPhil thesis. Duncan completed his DPhil in 2014, then went on to join Evotec where he has worked on various preclinical projects in oncology and respiratory diseases. |
 Darren J. Dixon | Darren J. Dixon obtained his BA, MA and DPhil (supervised by Professor Stephen Davies) from the University of Oxford. After a postdoctoral appointment with Professor Steve Ley FRS he was appointed to the Staff of the Department of Chemistry, University of Cambridge in 2000. In 2004 he took a Senior Lectureship at The University of Manchester and was promoted to Reader in 2007. In 2008, he moved to his current position of Professor of Chemistry, University of Oxford. His honors include an EPSRC Leadership Fellowship, the RSC Catalysis in Organic Chemistry Award, the AstraZeneca Research Award and Novartis Lectureship. He is a Director of the newly announced EPSRC CDT in Synthesis for Biology and Medicine. |
 Paul E. Brennan | Paul E. Brennan received his PhD in organic chemistry from the University of California, Berkeley under the mentorship of Paul Bartlett working on synthetic methodology for combinatorial chemistry and synthesizing inhibitors for new anti-bacterial targets. Following three years of post-doctoral research with Steve Ley in Cambridge University on the total synthesis of rapamycin, Paul returned to California to take a position at Amgen. His research was focussed on designing and synthesizing kinase inhibitors for oncology. After two years at Amgen, Paul accepted a position as medicinal chemistry design lead at Pfizer in Sandwich, UK. Over the next six years Paul designed and synthesized compounds for most major drug classes: GPCR's, CNS-targets, ion-channels and metabolic enzymes. In 2011 Paul joined the Structural Genomics Consortium as a principal investigator to discover chemical probes for epigenetic proteins. He is currently an Associate Professor of Medicinal Chemistry at the SGC and head of chemistry of the Alzheimer's Research UK Oxford Drug Discovery Institute at the University of Oxford. |
Introduction
Epigenetics describes the network of mechanisms that modulates gene expression without directly affecting gene sequence.1 A number of proteins are involved in epigenetic control and referred to as readers (bromodomains, chromodomains, tudor domains, etc.), writers (lysine acetyltransferases, lysine methyltransferases and DNA methyltransferases) and erasers (lysine deacetylases and lysine demethylases). These proteins interact with and act upon DNA or histones (large nuclear proteins which DNA is packaged around), and non-histone proteins such as transcription factors. They function by adding, removing and interacting with epigenetic marks (post-translational modifications to histone proteins). Aberrant regulation of a number of these epigenetic proteins is linked with the onset and progression of multiple disease states including cancer2–4 and inflammation.5 Lysine acetylation, which is effected by the lysine acetyltransferases (KATs) and removed by lysine deacetylases (KDACs), is an epigenetic mark that has been the subject of a plethora of research.6–9 Bromodomains (Brds) bind to acetylated lysines (KAc) in histones and other proteins through the bromodomain KAc binding site which is also the binding site of most Brd ligands (Fig. 1A). | ||
| Fig. 1 Bromodomains with reported inhibitors. A. Brd inhibitors such as LP99 (pale ball and stick) bind in the acetyl lysine binding pocket (green ribbon) to a common Asn residue (green ball and stick) and a network of water molecules (blue CPK) (LP99 and BRD9 from PDB ID 5IGN). B. Distribution of acetyl lysine binding residues in Brd pockets. Brds are colored by their acetyl lysine binding residue (red: Asn, blue: Tyr, green: Thr, purple: Asp). All reported Brd inhibitors (grey sections of bars) target Brds with a typical Asn residue with the exception of PHIP(2) which has a Thr. C. Brds in grey boxes have reported inhibitors. Brds in colored typeface have no reported inhibitors (colors as in B). | ||
Once bound to acetylated histones, bromodomains recruit other nuclear proteins to form large chromatin modelling and transcriptional regulation complexes. To this end, bromodomains are increasingly being considered as attractive therapeutic targets for a variety of disease states due to the critical role they play in control of target genes that are difficult to modulate directly with small molecules.10
The 61 Brds in the human genome can be divided into eight subfamilies based on their sequence and structures (Fig. 1C).8 Brds can be further classified by a single key residue in the binding site. The majority of Brds have an Asn as the KAc recognition residue (48 examples), but a minority have either a Tyr (8), Thr (4) or Asp (1) residue (Fig. 1B). To date, all potent inhibitors target one of the typical Asn-containing Brds (Fig. 1B and C).
To date a large majority of the work in validating the pharmacological relevance of Brds as therapeutic targets has relied upon genetic manipulation of an entire Brd containing gene via knock-out or RNAi knock down. It should be noted that this does not imply modulation of individual Brds will deliver a pharmacological effect or phenotype. However, the development of chemical probes and inhibitors for Brds will afford the scientific community with an additional ‘go/no-go’ checkpoint on implicated Brds in target validation. Use of chemical probes from multiple chemotypes will also be of benefit as this will allow for a more robust analysis of Brd inhibition and pharmacological effect due to the likely orthogonal off-target activity of different chemical series.
A chemical probe has been defined to be an entity capable of binding to a given target with in vitro potency <100 nM (KD or IC50), selectivity >30-fold against other families and evidence of cellular target engagement <1 μM compound concentration.11 As research in this area has progressed, additional desirable features are becoming necessary for qualification of a small molecule entity as a chemical probe such as availability of a negative control compound, favourable toxicity profiles, higher selectivity (e.g. intra-family and >100 fold over BET Brds – for non-BET bromodomain chemical probes).12 It is expected that as the field develops, the delivery of chemical tools that satisfy these criteria to a greater extent may drive more demanding criteria for what is deemed a chemical probe. Brds that have few to no chemical probes may be associated with slightly relaxed chemical probe criteria, to allow for rapid dissemination of early chemical probe material and associated data (thereby promoting developments in the understudied target). Where compounds have fallen short of satisfying one or more key chemical probe criterion – owing to deficiencies in the chemical entity or missing data, they are termed ‘inhibitors’ (the application of this term may be applied more regularly for target areas where higher quality chemical probes already exist). Collectively in this review both chemical probes and inhibitors are referred to as ‘chemical tools’.
Early studies into the development of chemical tools useful in interrogating bromodomain function yielded many probes and inhibitors of the BET bromodomains (sub-family II: BRD2, BRD3, BRD4, BRDT) (Fig. 1C). Previously written reviews have discussed the discovery and impact of such chemical tools in some depth, in part owing to the significant pharmacological relevance of targeting the BET bromodomains.7,13–18 This review will focus on chemical probes11,12 and inhibitors of the remaining non-BET bromodomains14,17,18 of the Brd sub-families I and III–VIII. Where possible selectivity over BET Brds will be discussed which is deemed a critical factor in interpreting the effects of Brd inhibitors, this can be rapidly ascertained through a number of tractable assays including Differential Scanning Fluorimetry (DSF) selectivity panels.19 An additional desirable feature of chemical probes/inhibitors would be achieving different degrees of selectivity: family-wide inhibition and intra-family selectivity as comparisons of inhibition profiles would allow for an accurate analysis of Brd pharmacological relevance (family-wide relevance vs. specific Brd relevance). Other properties of chemical probes and inhibitors are discussed herein if known, such as cellular activity, pharmacokinetics and solubility.
Sub-family I
PCAF
A number of reports have provided support for the therapeutic potential in the development of inhibitors of the PCAF Brd (p300/CBP Associated Factor) owing to the link with a variety of diseases including cancer,20–22 HIV,20,23–26 and neuro-inflammation20,27 PCAF has been predicted to be a highly druggable target by Vidler et al.28Early reports of PCAF bromodomain inhibitors were disclosed by Wang et al.25 It was shown that interactions between the HIV-1 Tat peptide (a viral factor essential for replication) and PCAF Brd were disrupted through competitive binding to PCAF bromodomain by compound 1 (reference compound 16) (PCAF IC50 1.60 μM, Fig. 2). Compound 1 was also shown to be effective at perturbing HIV-1 replication (EC50 2.76 μM). Hu et al.29 later reported on more PCAF bromodomain inhibitors including compound 2 (reference compound 20) which displayed moderate to good inhibition against PCAF Brd/Tat association (IC50 0.93 μM, Fig. 2) and viral replication (EC50 11.52 μM). Recently Chaikuad et al. at the Structural Genomics Consortium (SGC) discovered fragment leads for the PCAF Brd. Compound 3 (reference compound 14) showed moderate binding activity using Isothermal Titration Calorimetry (ITC) (PCAF KD 6.80 μM, Fig. 2).30 More recently Genentech and Constellation pharmaceuticals disclosed the structures of highly potent compounds 4, 5 and 6 (reference examples 20, 65B and 18 respectively) (IC50 19 nM and 70 nM respectively, Fig. 2) for the treatment of PCAF mediated diseases including cancer.21,22,31
 | ||
| Fig. 2 PCAF bromodomain inhibitors. | ||
CECR2
CECR2 has been predicted to be a highly druggable target.28 A highly potent and selective chemical probe for the bromodomain containing CECR2 (Cat Eye Syndrome Chromosome Region, candidate 2) has recently been developed by Novartis and the SGC (NVS-CECR2-1, Fig. 3).32 Details of the development of this probe is yet to be published, however NVS-CECR2-1 is reported to have high affinity for CECR2 (CECR2 IC50 47 nM, CECR2 KD 80 nM). NVS-CECR2-1 also displays robust in-cell target engagement in a Fluorescence Recovery After Photobleaching (FRAP) assay at 0.1 μM against full-length CECR2, despite being poorly soluble.32 Co-workers from Genentech and Constellation pharmaceuticals have recently reported the development of inhibitors of TAF1(2), CECR2, BRD4(1) and BRD9 from a common N-methyl pyrrolopyridone structural motif.33 Compound 7 (reference compound 3, Fig. 3) was shown to inhibit the CECR2 bromodomain with good potency (CECR2 IC50 0.17 μM) and exhibited novel interactions stemming from the rearrangement of the conserved solvent network. Compound 7 was profiled for selectivity across a broad range of bromodomain targets (DiscoveRx BROMOscan)34 showing significant off-target potency for BRD9 (BRD9 KD < 0.1 μM) as confirmed by a Time-resolved Förster resonance energy transfer assay (TR-FRET) (BRD9 IC50 0.29 μM).35 | ||
| Fig. 3 CECR2/BPTF inhibitors. | ||
BPTF
BPTF (Bromodomain and PHD Finger Transcription Factor/FALZ) has been linked to various cancers including bladder,36 colorectal,37 melanoma,38 and leukemia.39 BPTF has been predicted to be a highly druggable target.28 Until recently no known inhibitors of BPTF had been available to interrogate its role in the onset and progression of cancer. Urick et al. have recently reported the first BPTF inhibitor (AU1, Fig. 3) discovered through a 19F NMR assay.40 Moderate potency (BPTF KD 2.8 μM) was displayed by AU1in vitro and in a cell-based reporter assay. Further optimisation of the chemical scaffold of AU1 may serve as a good strategy towards more potent BPTF inhibitors.Sub-family III
CBP/p300
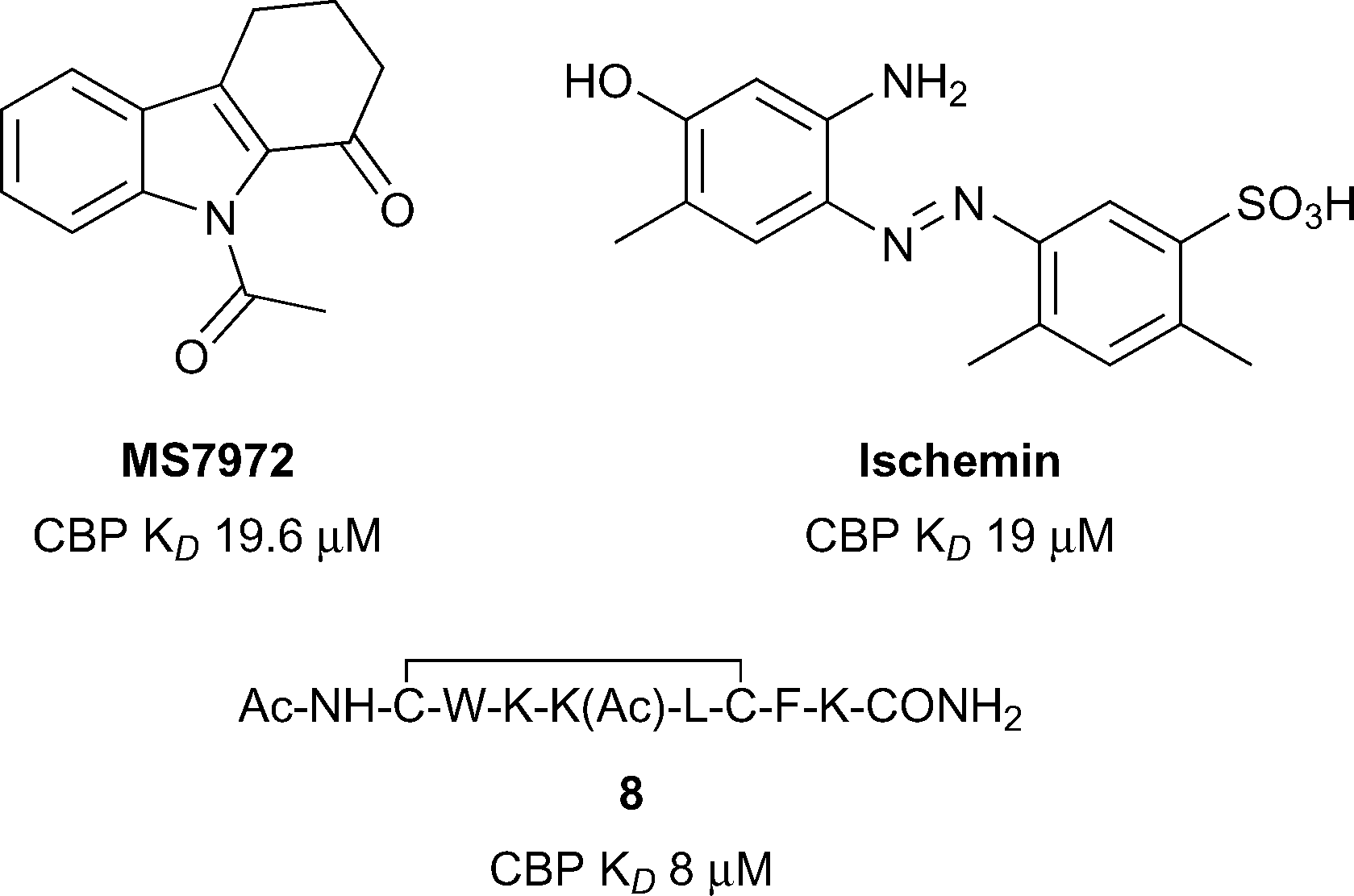
The lysine acetyltransferases CBP (also known as CREBBP and KAT3A) and p300 (also known as EP300 and KAT3B) are among the most studied bromodomain-containing proteins outside of the BET sub-family.41–45 CBP and p300 share a high degree of sequence similarity, particularly in their bromodomains (96% similar).46 Interest in developing chemical probes for CBP/p300 has been fuelled by the large and diverse nature of cellular processes which utilise these transcriptional coactivators, and by the strong links of CBP and p300 dysfunction with human disorders and diseases. In particular, CBP and p300 are implicated in the developmental disorder Rubinstein–Taybi syndrome,47 and are strongly linked to cancer, especially haematological malignancies,48 inflammation,49 and neuropsychiatric disorders.50The Zhou group pioneered the development of inhibitors of the CBP bromodomain (Fig. 4).51 The N-acetyl indole, MS7972, had modest CBP bromodomain affinity (CBP KD 19.6 μM) and inhibited the association of acetylated p53 with the CBP bromodomain at 50 μM.51–53 A biarylazo inhibitor, Ischemin, also displayed modest CBP bromodomain affinity (CBP KD 19 μM) and inhibited p53-induced p21 activation in a reporter-gene assay (p21 IC50 5 μM).52 The cyclic peptide 8 (reference compound 4) has also been shown to bind the CBP Brd (KD 8 μM) and to inhibit p53 activation in a reporter assay.52 The inhibitors reported by the Zhou group demonstrated that the CBP bromodomain could be targeted by multiple diverse chemotypes. However, the utility of these early ligands was limited by their relatively low affinity and lack of reported selectivity data for other bromodomain subfamilies.
 | ||
| Fig. 4 Early CBP inhibitors from the Zhou group. | ||
The first reported sub-micromolar CBP ligands were described by Rooney and co-workers.54 A series of dihydroquinoxalinones was developed from a fragment hit. The fragment itself was discovered after compound 9 was found to be a weak (CBP IC50 1.9 mM, Fig. 5) but efficient ligand for CBP (ligand efficiency 0.54). Screening of N-methylpyrrolidinone (NMP) analogues led to two series being pursued: benzoxazinones and dihydroquinoxalinones. An optimised dihydroquinoxalinone inhibitor, compound 11 (reference compound (R)-2) was shown to have sub-micromolar affinity for CBP (KD 0.39 μM), albeit with modest selectivity over BRD4(1) (KD 1.4 μM). An X-ray structure of compound 10 (reference compound (R)-1) bound to CBP revealed that the dihydroquinoxalinone moiety mimics the acetyl lysine binding interactions, whilst an internal hydrogen bond helps to direct the tetrahydroquinoline moiety into an induced-fit pocket created by the movement of R1173 to allow a cation–π interaction arginine side chain. Molecular dynamics were used to calculate the contribution of the cation–π interaction (3.2–4.7 kcal mol−1). On target cellular activity was shown in a FRAP assay, where dose-dependent inhibition of the FRAP signal of a GFP-tagged CBP construct with SAHA-stimulated hyper-acetylated chromatin was observed for compound 11 at low micromolar concentrations.
 | ||
| Fig. 5 Dihydroquinoxalinone CBP inhibitors. | ||
Hewings and co-workers gave further encouragement that selective CBP bromodomain inhibition was possible with small molecules (Fig. 6).55 A series of 4-aryl-3,5-dimethylisoxazoles was described with differing selectivity for the Brd sub-families. Compound 12 inhibited a histone peptide–Brd interaction at micromolar concentrations in an AlphaScreen assay (CBP IC50 32.2 μM) and had modest selectivity (1.6-fold) for CBP over the first Brd of BRD4 (BRD4(1)). X-ray crystallography revealed that the dimethylisoxazole acts as the KAc mimic via a direct hydrogen bond between the isoxazole oxygen and the amide side-chain of N1168, and a water-mediated hydrogen-bond to Y1125 from the isoxazole nitrogen. The ethoxy oxygen of compound 12 forms a hydrogen bond to another structured water in the ZA channel. Possible weak electrostatic interactions between the carboxylate of 12 and R1173 may be partly responsible for the CBP selectivity. The discovery of more potent and selective CBP/p300 inhibitors soon followed.
 | ||
| Fig. 6 Isoxazole and related CBP/p300 inhibitors. | ||
The discovery of the CBP/p300 Brd chemical probe SGC-CBP30 began with a 5-isoxazolyl-benzimidazole fragment 13 (Fig. 6) which was unselective for CBP over BRD4(1).46N-1 and C-2 substituents were introduced to target regions of structural difference between CBP and BRD4(1). The combination of a phenethyl group at the C-2 position and an ethylene linked morpholine at N-1 gave compound 14 (reference compound 17) displaying sub-micromolar affinity for CBP and p300 as measured by ITC (CBP KD 0.32 μM and p300 KD 0.35 μM). However 14 also displayed off-target activity against BRD4(1) (3-fold selectivity against CBP vs. BRD4(1)). Analysis of the X-ray structure of compound 14 bound to CBP and BRD4(1) guided the design of more potent and selective inhibitors. Potency was initially enhanced through variation of the substitution on the phenyl ring. Attempts at rigidifying the scaffold to increase selectivity led to the observation that introduction of a methyl branch on the N-1 ethylene linker led to potent and selective analogues. When synthesised as single enantiomers, it was found that the (S)-methyl analogues were favourable for CBP binding. The optimal compound (SGC-CBP30) was found to have low nanomolar affinity for CBP and p300 (CBP KD 21 nM and p300 KD 32 nM) and also displayed 40-fold selectivity for CBP over BRD4(1). SGC-CBP30 was also shown to be selective against a wide panel of bromodomain subfamilies in a Differential Scanning Fluorimetry (DSF) assay.19 X-ray crystallography of SGC-CBP30 bound to CBP revealed the expected dimethylisoxazole interactions with the acetyl lysine binding residues, whilst the aryl group formed a cation–π interaction with R1173 in an induced pocket analogous to that observed by Rooney et al.54 On-target cellular activity was demonstrated in a FRAP assay and a p53 reporter assay (p53 IC50 1.54 μM). More recently, SGC-CBP30 has been shown to reduce immune cell production of pro-inflammatory cytokines, including IL-17A, and to inhibit IL-17A secretion from Th17 cells.56 Transcriptional profiling of SGC-CBP30 in T cells indicated that the effects of CBP/p300 bromodomain inhibition were more limited than those of pan-BET inhibitor JQ1. The report suggests that inhibition of CBP and p300 bromodomains warrants further investigation as a potential therapeutic strategy to combat ankylosing spondylitis, psoriatic arthritis and other human type-17-mediated autoimmune diseases.
A team at Pfizer modified the SGC-CBP30 scaffold with the aim of improving the selectivity for CBP over BRD4(1).57 A propoxy analogue of SGC-CBP30, PF-CBP1 (Fig. 6), was found to have good affinity for CBP (CBP KD 0.19 μM) and was selective over BRD4(1) (BRD4(1) KD 20 μM). BROMOscan (DiscoveRx) profiling confirmed the broader selectivity of PF-CBP1 for CBP and p300 over other bromodomain subfamilies. The report also describes a dual CBP/BRD4(1) inhibitor, ISOX-DUAL (CBP IC50 0.65 μM BRD4(1) IC50 1.5 μM). A negative control was developed by introduction of two methyl groups, flanking the dimethylisoxazole head group, which cause an unfavourable change in the isoxazole-benzimidazole torsion angle. The resulting compound, ISOX-INACT, was a very weak inhibitor of CBP and was inactive against BRD4(1) (CBP IC50 10.6 μM, BRD4(1) IC50 > 120 μM). Computational techniques have been used to search for bioisosteric replacements for the dimethylisoxazole head group on the SGC-CBP30 scaffold.58 It was found that tropolone benzimidazole, compound 15 (reference compound 3) was a sub-micromolar inhibitor of CBP and BRD4(1) (CBP IC50 0.208 μM, BRD4(1) IC50 0.343 μM). An X-ray crystal structure of 15 bound to CBP and BRD4(1) confirmed that the tropolone head group was acting as the acetyl lysine mimic. The tropolone head group was seen as an attractive photoreactive handle for capture of bromodomains. Treatment of CBP and BRD4(1) with the para-methoxy derivative 16 (reference compound 4) with irradiation at 365 nm led to the observation of a MS adduct of protein plus inhibitor, minus 14 Da, presumably formed by photoreaction of the ligand with the protein and demethylation. The alkyne derivative 17 was prepared (reference compounds 7) in order to give a ‘clickable’ handle and was used to photolabel BRD4(1) spiked into K562 cells. Subsequent copper-mediated azide-alkyne cycloaddition to an azide-biotin tag and streptavidin-enrichment led to visualisation of the labelled BRD4(1) by Western blot. Although no native BRD4 could be isolated from live cells using the same method, the approach shows the potential for modification of chemical probes to provide photoreactive tools for the capture of bromodomain-containing proteins. A complementary CBP/p300 bromodomain chemical probe from an alternative chemotype has been disclosed by the SGC and GSK.
Oxazepine I-CBP112 (Fig. 7), was discovered through the analysis of inhibitors which were structurally related to the BET-selective inhibitors JQ1 and I-BET762.59I-CBP112 was found to be highly selective in a DSF selectivity panel of 41 other bromodomains, and in a biolayer interferometry (BLI) panel of 42 bromodomains. Weak off-target activity was noted only for the BET sub-family of bromodomains. The affinity of I-CBP112 for CBP and p300 was measured by ITC (CBP KD 0.151 μM, p300 KD 0.167 μM). I-CBP112 displayed 37-fold selectivity for CBP over BRD4(1) (BRD4(1) KD 5.59 μM). An X-ray crystal structure of the (R)-enantiomer of I-CBP112 bound to CBP confirmed that the carbonyl group mimicked the KAc binding interactions. The aromatic group formed a π–π interaction with R1173 in an induced pocket also seen for SGC-CBP30 and 14. On target cellular activity was demonstrated in a FRAP assay, where I-CBP112 significantly reduced the recovery time using a GFP-tagged triple-CBP bromodomain substrate. In a nanoBRET assay I-CBP112 inhibited the interaction of a nanoLuc luciferase CBP bromodomain construct with a Halo-tagged histone H3.3 construct (CBP IC50 0.6 μM).60I-CBP112 was also screened for novel phenotypes in the DiscoveRx BioMAP Diversity PLUS panel where altered expression of the anti-inflammatory cytokine IL10 and VCAM1 was observed.61I-CBP112 was found to reduce the clonogenic growth of MLL-CBP immortalised murine bone marrow cells, but did not significantly affect the cell survival. Treatment of MLL-AF9+ leukemic myeloblasts with I-CBP112 reduced the number of leukemic stem cells. Additionally, transplantation of MLL-AF9+ cells pre-treated with I-CBP112 delayed the disease initiation. Furthermore I-CBP112 was shown to impair the clonogenic growth of 12 cells from 12 human leukemic cell lines and to reduce the number of colonies in primary human AML cells. In combination with BET inhibitor JQ1, I-CBP112 was also found to enhance the cytotoxic effects of doxorubicin in human leukemic cells.
 | ||
| Fig. 7 CBP/p300 chemical probe I-CBP112. | ||
The availability of multiple chemotypes with the ability to inhibit a particular target affords greater confidence in the interpretation of results from cellular and in vivo studies because the chances of spurious off-target effects are reduced. Reports have begun to emerge utilising SGC-CBP30 and I-CBP112 in concert to interrogate CBP/p300 biology. Researchers at Genentech demonstrated that SGC-CBP30 and I-CBP112 caused a slowdown in the proliferation of multiple myeloma cells through arrest in the G1 cell cycle phase.10 RNA sequencing of SGC-CBP30 treated LP-1 cells indicated that IRF4 (interferon regulatory factor 4) target genes were down-regulated. Notably, the gene encoding for Myc was down-regulated which has been shown to be important for cell division and growth. IRF4 is essential for the survival of multiple myeloma cells, indicating that the IRF4/Myc transcription pathway is being affected by CBP/p300 bromodomain inhibition. The results suggest that CBP/p300 bromodomain inhibition has therapeutic potential for the treatment of multiple myeloma. A Genentech patent also investigated the effects of SGC-CBP30 and I-CBP112 BET inhibitor resistant cells.62 The BET inhibitor resistant cells were generated by treating acute myeloid leukemia cells (NOMO-1) with increasing concentrations of a BET inhibitor. Treatment of the BET inhibitor resistant cells with SGC-CBP30 or I-CBP112 inhibited MYC expression and cell growth. The claims in the patent include independent and synergistic dosing of CBP/p300 and BET inhibitors to treat a number of human malignancies.
Xu et al. utilised a computational technique coined ALTA (anchor-based library tailoring) to virtually screen a library of fragments against the CBP Brd.63 Two X-ray structures of CBP bearing different orientations of the sidechains of V1174 (the gatekeeper) and R1173 were used for the virtual screen. Molecules which contained the top ranking fragments were then docked. After clustering based on the ‘head groups’ which interacted with the key acetyl lysine-binding residues, 20 compounds were selected for molecular dynamics (MD) simulations. The simulations eliminated 3 out of the 20 compounds which moved out of the binding site in <100 ns. The remaining 17 compounds were assessed by BROMOscan. Two compounds, 18 and 19 (Fig. 8, reference compounds 1 and 9), containing an acylaryl head group had low micromolar affinities (CBP KD 13 μM, 17 CBP KD 29 μM respectively). Further MD simulations with compound 18 suggested that replacement of the oxadiazole ring with a negatively charged group may enhance the CBP affinity through the gain of electrostatic interactions with the guanidinium sidechain of R1173. By analogy a benzoic acid derivative, compound 20, (reference compound 6) displayed improved CBP potency (CBP KD 4.2 μM). Due to its synthetic versatility, compound 18 was selected for further optimization.64 Screening of commercially available analogues of the initial docking hit identified an inhibitor containing a fumaric acid derived amide. Replacing the fumaric acid moiety with an isophthalic group led to sub-micromolar potency in a competition assay. X-ray crystallography confirmed the acidic group forms favourable polar interactions with R1173 on CBP. Interaction with the Brd was further enhanced through substitution on the benzene ring ortho to the acid moiety. The elaborated inhibitors showed improved potency epitomised by the furan-containing inhibitor 21 (Fig. 9), (reference compound 19), which had sub-micromolar affinity as measured by ITC (CBP KD 0.3 μM). The optimised inhibitors were selective for CBP and p300 in a panel of 7 bromodomains and, along with their methyl ester derivatives, demonstrated growth inhibition in MOLM-13, ML2 and HL-60 leukemia lines.
 | ||
| Fig. 8 CBP inhibitors from Nevado and Caflisch. | ||
 | ||
| Fig. 9 Optimized CBP inhibitor from Nevado and Caflisch. | ||
In another application of a fragment-based approach, Taylor et al. developed a benzodiazepinone fragment into a potent and selective inhibitor.65 The initial hit 22 (reference compound 1, Fig. 10) was active against CBP (IC50 32 μM) in a TR-FRET assay. Compound 22 was optimised while attempting to maintain a lipophilic ligand efficiency (LLE) consistent with moderate in vivo clearance. Substitution at the 6-position was used to orientate substituents along the LPF shelf (residues L1109-P1110-F1111), an approach which was hoped may lead to beneficial interactions with CBP and detrimental steric clashes with the corresponding WPF shelf in BRD4(1). In particular, it was found that 6-aryl substituents were beneficial for CBP potency and selectivity over BRD4(1). The optimised inhibitor CPI-637 comprised a substituted indazole in the 6-position. The IC50 of CPI-637versus CBP and p300 was 30 nM and 51 nM respectively, while the selectivity over BRD4(1) was 367-fold and 215-fold. However off-target potency was observed against BRD9 (IC50 0.73 μM). Target-related cellular activity was demonstrated in a CBP nanoBRET assay (EC50 0.3 μM) and in the inhibition of MYC expression in AMO-1 cells (EC50 0.60 μM).
 | ||
| Fig. 10 CBP/p300 inhibitors from Constellation and Genentech. | ||
Progress has clearly been made in the development of selective CBP/300 bromodomain inhibitors and there are are numerous chemotypes emerging as tool compounds. A common challenge that persists in the development of CBP/p300 inhibitors is selectivity over the BET subfamily. Non-selective compounds or poorly characterised compounds could confound interpretation of cellular studies as the effects of inhibiting the BET phenotype could be misattributed to CBP/p300 inhibition. The development of compounds with higher selectivity will help the field progress further. Nevertheless, careful cellular studies with SGC-CBP30 have revealed a distinct CBP/p300 phenotype.10,46,56
PHIP
The only atypical Brd with a reported inhibitor is the second Brd of PHIP (PHIP(2)) which has a Thr instead of the typical Asn as the KAc recognition residue (Fig. 1B and C). PHIP is the most upregulated protein in metastatic melanoma and has potential as a therapeutic target and diagnostic marker.66 PHIP(2) has been predicted to be highly druggable.28 A high concentration crystallographic fragment was used to identify compounds 23–25 (reference compounds 4, 12 and 11, Fig. 11).67 Although binding of fragments 23–25 is very weak, they show that it is possible to find hits for the atypical Brds. | ||
| Fig. 11 PHIP(2) fragment hits. | ||
Sub-family IV
BRD7/9
Elucidation of the biological roles of BRD7 and BRD9 is an emerging area of research. Both BRD7 and BRD9 have been identified as members of the SWI/SNF nucleosome remodeling complex,68,69 which has emerged as an attractive target for developing anti-cancer agents.70 BRD7 is frequently found as a tumour suppressor,71–73 whereas BRD9 has been found to be mutated,74 upregulated,75 or over-expressed76 in various cancers. Additionally, BRD9 has been recently reported as a biomarker for Sézary syndrome,77 and for prediction of patient response to various forms of chemotherapy.78 Although the overall sequences of BRD7 and BRD9 share low similarity (36%), the homology in their Brds is significant (72%), a potential obstacle to the development of specific ligands. BRD9 has an intermediate druggability score according to analysis by Vidler et al., however BRD7 was not assessed.28,34 The development of early BRD9 inhibitors has been recently reviewed,79,80 however further application of these probes in interrogating the activity of BRD7 and BRD9, in addition to the development of novel probes, has been reported subsequently.An initial dual BRD7/9 probe was developed by Clark and co-workers at the University of Oxford and the SGC. Fragment screening identified a quinolone lead that was subjected to a structure-based drug discovery program, culminating in the discovery of LP99 (Fig. 12).81 This probe showed a high affinity for BRD9 (KD 99 nM) with moderate activity against BRD7 (KD 0.91 μM). Interactions with BRD9 were shown to be enthalpically driven with a net loss in entropy upon binding (ΔH −11 kcal mol−1, TΔS −2.0 kcal mol−1), a finding supported by the determination of multiple H-bonding interactions in a co-crystal structure (Fig. 1A, PDB ID 5IGN): the carbonyl of the quinolone replicates the key H-bonds to N216 and Y173 of KAc recognition, with additional H-bonding via both the lactam carbonyl and the sulfonamide NH. One benefit of the chirality of LP99 was the ready amenability of the opposite enantiomer as a negative control, as confirmed by ITC with no detectable inhibition of BRD9 observed. The selectivity of LP99 was determined by DSF against all expressible Brds, in which, besides BRD9 and BRD7, no thermal shift >1.0 °C was observed. Cellular permeability and chromatin binding activity was confirmed using a FRAP assay, with U2OS cells expressing a full length BRD9-GFP fusion protein showing a dose-dependent decrease in fluorescence recovery times after LP99 treatment. Cellular activity was further profiled through a nanoBRET assay with HEK293 cells expressing combinations of BRD7- or BRD9-NanoLuc fusion proteins and H3.3- or H4-HaloTag proteins: LP99 decreased the BRET ratio for all combinations of BRD7/9 and H3.3/H4 in a dose-dependent manner (BRD7/9 IC50 3.3–6.2 μM). Finally, a cytotoxicity assay performed with U2OS cells showed no effect of LP99 on proliferation at concentrations below 33 μM.
 | ||
| Fig. 12 BRD7 and BRD9 inhibitors. | ||
Preliminary screening with LP99 identified a role of BRD7/9 in inflammatory pathways. Initially, LP99 was assessed for in vitro anti-cancer activity in the US National Cancer Institute human tumour cell line anticancer drug screen,82 although no growth inhibition of greater than 40% was observed for any cell line at a 10 μM concentration. Screening in a BioMAP panel, however, showed an effect of LP99 on the secretion of pro-inflammatory cytokines. Interleukin 6 (IL-6) secretion from lipopolysaccharide-stimulated THP-1 monocytes was measured through an ELISA assay, with LP99 resulting in a dose-dependent decrease in IL-6 secretion (IL-6 IC50 < 10 μM). This role of BRD7/9 in inflammatory pathways has been supported by a recent patent describing inhibition of BRD7 or BRD9 as a method for treating TH2 cytokine-mediated diseases.83
A BRD9-specific probe was concurrently developed by Theodoulou and co-workers at GlaxoSmithKline and the University of Strathclyde. An initial library screening revealed a thienopyridone lead against BRD9, which, through a structure-based drug discovery program, resulted in the development of I-BRD9 (Fig. 12).84 A high affinity for BRD9 was observed through both a TR-FRET assay (IC50 50 nM) and a BROMOscan assay (IC50 1.9 nM), with the latter also demonstrating the selectivity of I-BRD9; >70-fold selectivity was seen against 34 Brds, including the homolog BRD7 (KD 0.38 μM). Wider selectivity against 49 unrelated proteins, including ion channels, GPCRs, transporters, kinases, nuclear receptors and other enzymes, revealed IC50 values predominantly >10 μM, with limited activity seen on a serotonin receptor (IC50 6 μM) and a norepinephrine transporter (IC50 8 μM). An X-ray co-crystal structure of I-BRD9 with BRD9 demonstrated the carbonyl of the thienopyridone replicating the key KAc interactions to N216 and Y173, with additional H-bonding to the protein by the amidine and a sulfone oxygen (PDB ID 4UIW). The previous characterisation utilised a truncated BRD9 protein, however, a chemoproteomic assay using cell lysate revealed comparable activity with the native protein (IC50 80 nM). Cellular activity was confirmed using a nanoBRET assay with the BRD9 Brd (IC50 0.158 μM), and cellular permeability further demonstrated in an artificial membrane assay.
I-BRD9 was found to lead to the selective modulation of a range of genes. Kasumi-1 cells were treated with I-BRD9 (10 μM) or the BET inhibitor I-BET151 (ref. 85) (1 μM) to investigate the difference between BRD9 and BET inhibition. 700 genes showed up- or down-regulation (>1.5-fold expression change relative to DMSO vehicle) by I-BRD9 treatment but not by I-BET151. Of these, the modulation of four genes implicated in cancer and immunological pathways (CLEC1, DUSP6, FES and SAMSN1) were subsequently confirmed by qPCR. This broad modulation of gene expression by inhibition of Brd binding confirms the epigenetic nature of the BRD9 protein and warrants further exploration.
A further dual BRD7/9 probe was developed by Hay et al. at the University of Oxford and the SGC. An indolizine active against the Brd of BAZ2B was also found to have activity against BRD9; following a biophysical assay-guided SAR program, compound 26 was identified (reference compound 28, Fig. 12).86 This compound displayed a high affinity for BRD9 (KD 68 nM) and BRD7 (KD 0.368 μM), with no affinity for the original target BAZ2B. Selectivity amongst Brds was further assessed by a DSF assay, where only BRD9 (ΔTm 4.5 °C) and BRD7 (ΔTm 5.6 °C) demonstrated a thermal shift >2.0 °C after treatment with compound 26. For BRD9, binding was found to be driven by enthalpic contributions (ΔH −10.7 kcal mol−1, TΔS −1.32 kcal mol−1) whereas entropy dominated BRD7 binding (ΔH −2.64 kcal mol−1, TΔS 5.99 kcal mol−1). An X-ray co-crystal structure of compound 26 with BRD9 revealed the methyl ketone formed the conserved H-bonds of KAc recognition to N216 and Y163, with additional π–π stacking interactions between the indolizine system and Y222 (PDB ID 5E9V). Final characterisation through a FRAP assay showed compound 26 decreased the recovery time of BRD9-GFP in a dose-dependent manner, towards the t1/2 observed with a non-binding N216F mutant, confirming cellular permeability and activity.
A pair of related BRD9 and BRD7/9 probes were developed by Martin and co-workers at Boehringer Ingelheim and the SGC. Parallel screening by biophysical assays and computational docking identified a dimethylpyridinone lead, which, after a structure-based drug discovery program, resulted in the discovery of the naphthyridinone-based probes BI-7273 and BI-9564 (Fig. 12).87BI-9564 showed selectivity for BRD9 (KD 5.9 nM) over BRD7 (KD 0.239 μM), whereas BI-7273 showed a decreased selectivity profile (BRD9 IC50 19 nM; BRD7 IC50 0.117 μM). Wider selectivity screening by DSF with BI-7273 showed significant binding to BRD7 (ΔTm 9.7 °C), BRD9 (ΔTm 11.4 °C) and CECR2 (ΔTm 8.2 °C), an observation mirrored by BI-9564 (BRD7 ΔTm 6.5 °C; BRD9 ΔTm 9.2 °C; CECR2 ΔTm 5.6 °C). The off-target affinity for CECR2 was confirmed by ITC for both BI-7273 (CECR2 KD 0.187 μM) and BI-9564 (CECR2 KD 0.2 μM), although the latter affinity was over 30-times higher than that for BRD9. Aside from these three domains, both compounds showed low affinity for all other Brds assessed, including for the BET family of proteins. In wider target screening, some limited activity was observed with a number of kinases (IC50 3.8–5.1 μM) and GPCRs (GPCRs IC50 ≥ 10 μM). Co-crystal structures of BI-9564 (PDB ID 5F1H) and BI-7273 (PDB ID 5EU1) with BRD9 were determined by X-ray crystallography, demonstrating the carbonyl of the naphthyridinone replicating the key H-bonds of KAc recognition to N216 and Y173, in addition to numerous π-interactions with the protein. Cellular target engagement was demonstrated through a FRAP assay, with complete inhibition of BRD7/9-chromatin binding by both probes at 1 μM. Importantly, no cellular activity against the off-target Brd CECR2 was observed at this concentration. Further evidence of cellular activity of BI-7273 was demonstrated through disruption of BRD9-histone H3 interactions in a nanoBRET assay at submicromolar concentrations.88 A final cytotoxicity assay of the compounds showed no effect on proliferation after 24 hours of exposure.
BI-7273 and BI-9564 were found to have activity against acute myeloid leukemia (AML) cell lines. Whilst screening the probes against a range of cancer cells lines, BI-9564 was found to induce growth inhibition in a number of AML cell lines, with exposure to BI-7273 found to result in partial but significant inhibition of Myc expression in these cells.87 The role of BRD9 in supporting leukemia-maintenance through the SWI/SNF complex and Myc expression has been subsequently confirmed through genetic knockdown studies.88 To confirm the role of Brd binding in this antiproliferative activity, domain-swapped alleles expressing BRD9 with the Brd of another protein were developed. It was fortuitiously discovered that swapping the Brd with BRD4(1) led to a retention of the chromatin binding specificity and activity of the native protein. AML cells expressing these domain-swapped proteins showed full BRD9 function, despite the different Brd architecture. Treatment of these cells with BI-7273 showed a complete resistance to the antiproliferative effects of the probe, confirming both the on-target selectivity of BI-7273 and the identification of BRD9 as the sole mediator of this anti-proliferative activity. In the same experiment, this allele only partially reduced the anti-proliferative activity of I-BRD9, and had a minimal effect on sensitivity to LP99, suggesting some off-target effects of both these probes. These data show the power of domain-swap experiments as a general strategy to demonstrate on- and off-target activity of probes in cells, and should be incorporated into the routine characterisation of Brd probes where possible. Human acute myeloid eosinophilic leukemia cell line EOL-1 proved to be the most susceptible to inhibition with BI-7273 (EC50 0.8 μM) and BI-9564 (EC50 1.4 μM), with BRD9 confirmed as the biological target through a domain-swap experiment that mitigated all antiproliferative effects of BI-9564 up to concentrations of 5 μM.87
Towards application in in vivo studies, the pharmacokinetic parameters of BI-7273 and BI-9564 were assessed. Good solubility, moderate to low hepatic clearance across different model systems, low plasma protein binding and no cytochrome P450 inhibition at concentrations below 50 μM were observed for both compounds, although significant efflux ratios in a Caco-2 transporter assay were observed. Initial testing in mouse models with twice daily p.o. dosing, at both 20 mg kg−1 and 180 mg kg−1, resulted in dose-dependent blood levels in excess of the EC50 determined for antiproliferative activity against EOL-1 cells. Of the two probes, BI-9564 achieved both a higher exposure and bioavailability than BI-7273. A seven day tolerability study was performed on CIEA-NOG mice with daily p.o. dosing of BI-9564 at a 180 mg kg−1 loading, which showed good tolerance and minimum weight change observed.
The efficacy of BI-9564 as a treatment for AML was explored further in a disseminated mouse model. EOL-1 cells, transduced with a luciferase-expressing vector to allow for a bioluminescent assessment of tumour load, were injected into CIEA-NOG mice, and BI-9564 was subsequently administered p.o. daily. Plasma samples revealed a high systemic exposure of BI-9564, with mean total plasma concentrations in excess of the EC50 required for EOL-1 cellular proliferation inhibition, for 20 h after dosing. A statistically significant reduction in tumour growth, measured in average radiance, was observed, resulting in median tumour growth inhibition of 52%. This was confirmed by imaging data in which the disease burden was visually reduced. In addition to decreased tumour growth, there was a small increase in median survival of the treatment group compared to the vehicle animals. Although only moderate effects on AML proliferation were observed, these experiments demonstrated the suitability of this probe for further in vivo assessment of BRD7 and BRD9 inhibition.
Recently a novel BRD7/9 probe has been added to the SGC chemical probes list with limited data. TP-472, which features a pyrrolo[1,2-a]pyrimidine scaffold and a methyl ketone as a KAc bioisostere, was developed collaboratively between Takeda and the SGC (Fig. 12).89TP-472 has reportedly demonstrated a high potency for BRD9 (KD 33 nM) and BRD7 (KD 0.34 μM), with >30-fold selectivity over other Brds. Cellular activity has been demonstrated through a nanoBRET assay with BRD9 (EC50 0.32 μM). A structurally-related negative control compound (BRD9 KD > 20 μM) has also been developed for use in parallel in biological assays to corroborate on-target activity. A full report of the development and characterisation of TP-472 is awaited in the scientific literature.
Other potent ligands of BRD7 and BRD9 have been reported, however these lack the potency, selectivity and/or cellular activity requisite for use as chemical probes. From fragment screening, the purine 27 (reference compound 11, Fig. 12) was developed from a structure-based drug discovery program.90 This ligand displayed a reasonable affinity for BRD9 (KD 0.278 μM), and good selectivity over the representative BET protein BRD4(1) (KD 1.4 μM). Cellular activity of the ligand was confirmed through a nanoBRET assay (IC50 0.477 μM), and no cytotoxicity in HEK293 cells were seen up to concentrations of 33 μM. Conversely, a platform-based approach screening derivatives of the known KAc bioisostere [1,2,4]triazolo[4,3-a]phthalazine resulted in the identification of compound 28 (reference compound 51, Fig. 12).91 Moderate affinities were observed equally for BRD9 (IC50 0.199 μM), CBP (IC50 0.199 μM) and BRD4(1) (IC50 0.158 μM). Inhibition of the interaction of a CBP Brd-GFP fusion protein with chromatin was demonstrated with a FRAP assay, confirming cellular permeability and activity of compound 28.
BRPF1/2/3
The bromodomain-PHD finger protein (BRPF) aids the complex assembly of MYST-family histone acetyltransferases (HATs).92 BRPF1 forms a subunit of the monocytic leukemic zinc finger (MOZ) complex in which translocations have been linked to aggressive forms of myeloid leukemia.93 Relatively little is known about the biological function or therapeutic potential of the Brd of BRPF1, however BRPF1B has been predicted to be a highly druggable target.28,94 GSK recently reported the discovery of BRPF1B inhibitor N,N-dimethylbenzimidazolone compound 29 (reference compound 34, Fig. 13).95 | ||
| Fig. 13 BRPF1B inhibitors. | ||
Compound 29 was further optimized to give the a potent and selective BRPF1 Brd inhibitor GSK6853 with improved solubility.96GSK6853 (BRPF1B IC50 8 nM) showed 1600-fold selectivity against other bromodomains in the DiscoveRx BROMOscan panel. Furthermore, GSK6853 showed excellent cellular activity in a BRPF1B NanoBRET assay against NanoLuc tagged full-length BRPF1 bromodomain (IC50 20 nM) and a favourable solubility profile (140 μg mL−1) supporting its potential utility for in vivo work. Earlier reports of BRPF inhibitors also include the structurally related PFI-4 and OF-1.97,98 Another BRPF chemical probe NI-57 features an orthogonal 1,3-dimethylquinolin-2(1H)-one chemotype.99
ATAD2
Overexpression of the Brd-containing protein ATAD2 (ATPase family, AAA domain containing 2) has been linked to a variety of cancers including breast,100,101 prostate,102 liver,103 lung,100 osteosarcoma104 among others.105 Little is known of the role that the ATAD2 bromodomain plays in these indications. ATAD2(A/B) have been predicted to be difficult in terms of druggability analysis.28 Early work towards the development of inhibitors of ATAD2 focused on the use of crystal transfer/soaking approaches in parallel to NMR screening of nucleoside derived fragments (Fig. 14).106 Chaikuad et al. obtained nine crystal structures of ATAD2 in complex with fragments such as thymidine (ATAD2 KD 10 mM). These nucleoside analogues represent chemical starting points for the development of more potent ATAD2 inhibitors. More recent work from Demont et al. describes the development of the first known micromolar inhibitors of the ATAD2 bromodomain through a focused fragment based screen.105 Optimisation of this fragment series led to compounds 30 and 31 (reference compounds 38 and 46, Fig. 14) displaying very good potency for the ATAD2 bromodomain (30 ATAD2 KD 0.125 μM, 31 ATAD2 KD 0.316 μM) (Fig. 14).107 Compounds 30 and 31 showed good selectivity against the therapeutically implicated BET bromodomains however cell permability needs to be improved. Compounds 30 and 31 were used as lead compounds in the development of a cell-permeable, potent and chemical probe of the ATAD2 bromodomain resulting in compound 32 (reference compound 16) reported by the same group from GSK.108 Compound 32 displays good potency (ATAD2 KD 8 nM), selectivity (2.8 log selectivity against BET Brds) and moderate cellular activity (IC50 2.7 μM) in a NanoBRET assay against NanoLuc tagged truncated ATAD2 bromodomain. Compound 32 was shown to target both ATAD2A and ATAD2B using DiscoveRx's BROMOScan (ATAD2A KD 1.3 nM, ATAD2B KD 1 nM). The enantiomer of compound 32 displays much weaker activity against ATAD2 (ATAD2 KD 3.1 μM) and so is a potential negative control, also displayed in a much weaker inhibition profile in the same NanoBRET assay. Novel use of a –CF2 group as a polar hydrophobic isostere of the sulfone groups seen in compounds 30 and 31 allowed for improvements in both selectivity, solubility and cell permeability. It is noteworthy that although discovered independently, the ATAD2 Brd inhibitors in Fig. 14 share a common unsaturated, 3-methyl substituted lactam KAc mimetic motif. | ||
| Fig. 14 ATAD2 inhibitors. | ||
Sub-family V
BAZ2A/B
The BAZ proteins (Bromodomain Adjacent to Zinc finger) represents a diverse set of proteins including BAZ1A, BAZ1B, BAZ2A and BAZ2B.109 Both BAZ2A and BAZ2B have been shown to be involved in chromatin remodelling110 and regulation of non-coding RNAs.111 Mutations in the BAZ2B gene has been linked to sudden cardiac death112 and over expression of BAZ2B negatively affects the outcome of pediatric B cell acute lymphoblastic leukemia (B-ALL).113Aberrant overexpression of BAZ2A correlates well with recurrence in prostate cancer and is also linked with maintaining prostate cancer cell growth.114 BAZ2B has been predicted to be difficult in terms of druggability analysis.28 Owing to the significant therapeutic potential in developing BAZ2A/BAZ2B inhibitors/chemical Drouin et al.113 sought to develop a potent, selective and cell active chemical probe through structure based discovery starting from hit compound 33 (reference compound 1, Fig. 15). Through an iterative process of structure based design BAZ2A/BAZ2B inhibitor, BAZ2-ICR was discovered displaying potency (BAZ2A IC50 0.13 μM, BAZ2B IC50 0.18 μM) and good selectivity against a wide panel of Brds.
 | ||
| Fig. 15 BAZ2A/BAZ2B inhibitors. | ||
Furthermore, BAZ2-ICR showed accelerated recovery in a FRAP assay at 1 μM for GFP-tagged full length BAZ2A comparable to a non-histone binding mutant negative control. A chemically distinct BAZ2A/BAZ2B inhibitor was disclosed in the form of compound GSK2801 (Fig. 15). GSK2801 was discovered through structure based design starting from initial hit compound 34 (reference compound 1, Fig. 15) and displays high potency for BAZ2A/BAZ2B (BAZ2A IC50 0.257 μM, BAZ2B IC50 0.136 μM), good selectivity against other Brds and target engagement in a cellular FRAP assay using full-length GFP tagged BAZ2A.115 In addition, GSK2801 displayed favourable pharmacokinetic properties after intraperitoneal and oral dosing to male CD1 mice providing rationale for GSK2801 to be used as an in vivo as well as in vitro BAZ2A/BAZ2B inhibitor.
To date, no inhibitors have been reported for the remaining BAZ proteins BAZ1A and BAZ1B. Although BAZ1A and BAZ1B have a similar domain architecture, their Brds are actually very different from BAZ2A/2B and each other.
TRIM24
A family of bromodomain containing proteins, the tripartite motif containing proteins (TRIMs), more specifically TRIM24, TRIM28 and TRIM33, represent interesting targets due to their role in a variety of cancers including breast,116,117 head and neck,118 non-small-cell lung,119 hepatocellular,120 and glioblastoma.121 TRIM24 has been predicted to be difficult in terms of druggability analysis.28 Recent work from the SGC and Bayer led to the development of a dual BRPF1B/TRIM24 Brd inhibitor.122 Screening of commercial 1,3-benzimidazolones led to the discovery of compound 35 (reference compound 34, Fig. 16) showing good binding affinity for BRPF1B and TRIM24 Brds (BRPF1B KD 0.14 μM, TRIM24 KD 0.22 μM). Compound 35 displayed cellular target engagement for TRIM24 via a FRAP assay at 1 μM concentration for full length TRIM24 fused to GFP. Another report by Palmer et al.123 showed optimisation of the same 1,3-benzimidazolone motif of compound 35 leading to the discovery of IACS-9571. IACS-9571 displayed excellent potency against both BRPF1B and TRIM24 (BRPF1B KD 14 nM, TRIM24 KD 31 nM), highly potent cellular activity for TRIM24 (TRIM24 EC50 50 nM – Cellular AlphaLisa using Flag-tagged TRIM24-PHD-Bromo construct) and good pharmacokinetic properties (F 29% after oral dosing). Despite progress in the area, a selective TRIM24 Brd inhibitor has not yet been reported. | ||
| Fig. 16 Dual TRIM24/BRPF1 inhibitors. | ||
Sub-family VI
The 6th sub-family of Brds is composed of only two members, MLL and TRIM28. MLL is a large protein with many functional domains including a Brd and is one of the most frequently mutated genes in cancer.124 TRIM28 has been associated with regulation of mitophagy125 and HCMV latency.126 Neither of these Brds has any reported inhibitors despite their interesting links to disease, presumably due to the challenge in finding hit compounds for their atypical KAc binding residues (Asp in MLL and Thr in TRIM28).Sub-family VII
TAF1 has been predicted to be difficult in terms of druggability analysis.28 From the previously mentioned report by Genentech and Constellation pharmaceuticals33 a highly potent TAF1(2) inhibitor, compound 36 (reference compound 5, Fig. 17), was accessed through the development of N-methyl pyrrolopyridones. Compound 36 was shown to inhibit TAF1(2) Brd with excellent potency (IC50 46 nM), 30-fold selectivity over BRD9 (IC50 1.4 μM) and displayed novel interactions stemming from the rearrangement of the conserved solvent network. The 1-butenyl substituent extends into the water channel between Tyr1540 and lipophilic shelf residues Pro1527 and Phe1528 displacing conserved water 4 in the KAc binding site. Compound 36 represents an attractive lead for development of additional TAF1 and TAF1L inhibitors. | ||
| Fig. 17 TAF1(2) inhibitor compound 40 sub-family VIII. | ||
SMARCA/PB1
The final bromodomain sub-family is composed of PB1 (Polybromodomain protein 1), SMARCA2 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A 2; BRM) and SMARCA4 (BRG1). These three Brd-containing proteins are components of human SWI/SNF (switch/sucrose nonfermentable) chromatin remodelling complexes and have been linked genetically to a number of cancers.127,128PB1 is a multidomain protein containing six different Brds whereas SMARCA2 and SMARCA4 each have a single Brd and an ATPase domain. In order to dissect the role of the bromodomain contribution to SWI/SNF mediated processes, chemical inhibitors targeting the bromodomains of SMARCA2/SMARCA4/PB1 represent useful entities. PB1(A/B/C/5) has been predicted to be intermediate in terms of druggability analysis, SMARCA4/28 have been predicted to be difficult.28 Gerstenberger et al. reported the discovery of PFI-3 (Fig. 18), a broadly selective, potent and cellular active inhibitor of family VIII bromodomains including SMARCA2/SMARCA4/PB1.129PFI-3 showed excellent binding affinities for PB1 (PB1(5) KD 54 nM), and SMARCA2/SMARCA4 (KD < 0.1 μM) and excellent selectivity for these sub-family VIII bromodomains in a panel against >40 bromodomains from other families.130–132PFI-3 also displayed an increase in the half recovery time using a FRAP assay (1 μM PFI-3) comparing recovery times of wild-type full-length SMARCA2 vs. a mutant incapable of binding chromatin (N1464F) providing evidence for cellular target engagement. No inhibitory effects were seen against a range of cellular endpoints in 12 primary human cell based systems when incubated with PFI-3.130 More recently Sutherell et al. at the University of Cambridge and the SGC reported the structure guided discovery of compound 37 (reference compound 26, Fig. 18) which showed good potency for PB1(5) (KD 0.126 μM), SMARCA2B (KD 0.262 μM), and SMARCA4 (KD 0.417 μM).133 Compound 42 showed reasonable selectivity over other bromodomain families, cellular target engagement at 1 μM with full-length SMARCA2 (FRAP assay) and revealed a new interaction in a crystal structure with PB1(5) through potential halogen bonding with Met731. Used in concert with PFI-3 these inhibitors may be used to decipher the role of sub-family VIII Brds.
 | ||
| Fig. 18 PB1/SMARCA2/SMARCA4 inhibitors. | ||
Conclusions
Since the initial disclosure of the first BET inhibitors, Brds have been a target class of great interest in drug discovery. The success in discovering small molecule inhibitors targeting half of the Brd family in less than a decade has greatly expanded our understanding of the biological role of the individual Brds and jump-started translational efforts, primarily in oncology.Although half of the Brds have succumbed to inhibitor discovery efforts, another half remains. For many of these, potent inhibitors may already exist and broader cross-screening may uncover additional Brd activity e.g. ATAD2A inhibitor 32 also inhibits ATAD2B; and the PCAF inhibitor 4 may also inhibit the closely related GCN5. For other typical Brds (BRD8, WRD(1), PHIP(1), BRWD3(1)) cross-screening of focused Brd inhibitor sets may deliver hits. But there remain a further twelve atypical Brds (Fig. 1B and C) for which no hits exist. New unbiased screening efforts using fragments,67 HTS,134 DNA-encoded libraries,135 or in silico screening,63 will be needed to find new chemotypes targeting Tyr, Thr and Asp-containing Brds.
Acknowledgements
The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA [ULTRA-DD grant no. 115766], Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome Trust [092809/Z/10/Z]. P. G. K. C. gratefully acknowledges the Woolf Fisher Trust, the CREATE ChemNET programme and the Centre for Drug Research and Development through a grant awarded to Prof Robert Young, SFU. M. M. is supported by the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1).References
- C. H. Arrowsmith, C. Bountra, P. V. Fish, K. Lee and M. Schapira, Nat. Rev. Drug Discovery, 2012, 11, 384–400 CrossRef CAS PubMed.
- M. Esteller, N. Engl. J. Med., 2008, 358, 1148–1159 CrossRef CAS PubMed.
- P. A. Jones and P. W. Laird, Nat. Genet., 1999, 21, 163–167 CrossRef CAS PubMed.
- M. A. Dawson, T. Kouzarides and B. J. P. Huntly, N. Engl. J. Med., 2012, 367, 647–657 CrossRef CAS PubMed.
- M. K. Shanmugam and G. Sethi, Subcell. Biochem., 2013, 61, 627–657 CAS.
- C.-W. Chung, H. Coste, J. H. White, O. Mirguet, J. Wilde, R. L. Gosmini, C. Delves, S. M. Magny, R. Woodward, S. A. Hughes, E. V. Boursier, H. Flynn, A. M. Bouillot, P. Bamborough, J.-M. G. Brusq, F. J. Gellibert, E. J. Jones, A. M. Riou, P. Homes, S. L. Martin, I. J. Uings, J. Toum, C. A. Clement, A.-B. Boullay, R. L. Grimley, F. M. Blandel, R. K. Prinjha, K. Lee, J. Kirilovsky and E. Nicodeme, J. Med. Chem., 2011, 54, 3827–3838 CrossRef CAS PubMed.
- P. Filippakopoulos and S. Knapp, Nat. Rev. Drug Discovery, 2014, 13, 337–356 CrossRef CAS PubMed.
- P. Filippakopoulos, S. Picaud, M. Mangos, T. Keates, J.-P. Lambert, D. Barsyte-Lovejoy, I. Felletar, R. Volkmer, S. Muller, T. Pawson, A.-C. Gingras, C. H. Arrowsmith and S. Knapp, Cell, 2012, 149, 214–231 CrossRef CAS PubMed.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067–1073 CrossRef CAS PubMed.
- A. R. Conery, R. C. Centore, A. Neiss, P. J. Keller, S. Joshi, K. L. Spillane, P. Sandy, C. Hatton, E. Pardo, L. Zawadzke, A. Bommi-Reddy, K. E. Gascoigne, B. M. Bryant, J. A. Mertz and R. J. Sims III, eLife, 2016, 5, e10483 Search PubMed.
- http://www.thesgc.org/chemical-probes .
- M. E. Bunnage, E. L. P. Chekler and L. H. Jones, Nat. Chem. Biol., 2013, 9, 195–199 CrossRef CAS PubMed.
- J. M. Garnier, P. P. Sharp and C. J. Burns, Expert Opin. Ther. Pat., 2014, 24, 185–199 CrossRef CAS PubMed.
- F. A. Romero, A. M. Taylor, T. D. Crawford, V. Tsui, A. Côté and S. Magnuson, J. Med. Chem., 2016, 59, 1271–1298 CrossRef CAS PubMed.
- S. G. Smith and M.-M. Zhou, ACS Chem. Biol., 2016, 11, 598–608 CrossRef CAS PubMed.
- N. H. Theodoulou, N. C. O. Tomkinson, R. K. Prinjha and P. G. Humphreys, Curr. Opin. Chem. Biol., 2016, 33, 58–66 CrossRef CAS PubMed.
- N. H. Theodoulou, N. C. O. Tomkinson, R. K. Prinjha and P. G. Humphreys, ChemMedChem, 2016, 11, 477–487 CrossRef CAS PubMed.
- G. Zhang, S. G. Smith and M.-M. Zhou, Chem. Rev., 2015, 115, 11625–11668 CrossRef CAS PubMed.
- M. W. Pantoliano, E. C. Petrella, J. D. Kwasnoski, V. S. Lobanov, J. Myslik, E. Graf, T. Carver, E. Asel, B. A. Springer, P. Lane and F. R. Salemme, J. Biomol. Screening, 2001, 6, 429–440 CrossRef CAS PubMed.
- L. Kruidenier, K. Lee, D. F. Tough and D. M. Wilson, UK Pat., WO2014037362A1, 2014 Search PubMed.
- B. K. Albrecht, A. Cote, T. Crawford, M. Duplessis, A. C. Good, Y. Leblanc, S. Magnuson, C. G. Nasveschuk, R. Pastor, F. A. Romero and A. M. Taylor, US Pat., WO2016036954A1, 2016 Search PubMed.
- B. K. Albrecht, A. Cote, T. Crawford, M. Duplessis, A. C. Good, Y. Leblanc, S. Magnuson, C. G. Nasveschuk, R. Pastor, F. A. Romero and A. M. Taylor, US Pat., WO2016036873A1, 2016 Search PubMed.
- S. Mujtaba, Y. He, L. Zeng, A. Farooq, J. E. Carlson, M. Ott, E. Verdin and M.-M. Zhou, Mol. Cell, 2002, 9, 575–586 CrossRef CAS PubMed.
- V. C. Quy, S. Pantano, G. Rossetti, M. Giacca and P. Carloni, Biology, 2012, 1, 277–296 CrossRef CAS PubMed.
- Q. Wang, R. Wang, B. Zhang, S. Zhang, Y. Zheng and Z. Wang, Med. Chem. Commun., 2013, 4, 737–740 RSC.
- A. Dorr, V. Kiermer, A. Pedal, H.-R. Rackwitz, P. Henklein, U. Schubert, M.-M. Zhou, E. Verdin and M. Ott, EMBO J., 2002, 21, 2715–2723 CrossRef CAS PubMed.
- M.-M. Zhou, G. Gerona-Navarro, Y. Rodriguez-Fernandez and P. Casaccia, US Pat., WO2015184257A2, 2015 Search PubMed.
- L. R. Vidler, N. Brown, S. Knapp and S. Hoelder, J. Med. Chem., 2012, 55, 7346–7359 CrossRef CAS PubMed.
- P. Hu, X. Wang, B. Zhang, S. Zhang, Q. Wang and Z. Wang, ChemMedChem, 2014, 9, 928–931 CrossRef CAS PubMed.
- A. Chaikuad, S. Lang, P. E. Brennan, C. Temperini, O. Fedorov, J. Hollander, R. Nachane, C. Abell, S. Muller, G. Siegal and S. Knapp, J. Med. Chem., 2016, 59, 1648–1653 CrossRef CAS PubMed.
- B. K. Albrecht, D. J. Burdick, A. Cote, M. Duplessis, C. G. Nasveschuk and A. M. Taylor, US Pat., WO2016112298A1, 2016 Search PubMed.
- http://www.thesgc.org/chemical-probes/NVS-1 .
- T. D. Crawford, V. Tsui, E. M. Flynn, S. Wang, A. M. Taylor, A. Côté, J. E. Audia, M. H. Beresini, D. J. Burdick, R. Cummings, L. A. Dakin, M. Duplessis, A. C. Good, M. C. Hewitt, H.-R. Huang, H. Jayaram, J. R. Kiefer, Y. Jiang, J. Murray, C. G. Nasveschuk, E. Pardo, F. Poy, F. A. Romero, Y. Tang, J. Wang, Z. Xu, L. E. Zawadzke, X. Zhu, B. K. Albrecht, S. R. Magnuson, S. Bellon and A. G. Cochran, J. Med. Chem., 2016, 59, 5391–5402 CrossRef CAS PubMed.
- E. Quinn, L. Wodicka, P. Ciceri, G. Pallares, E. Pickle, A. Torrey, M. Floyd, J. Hunt and D. Treiber, Cancer Res., 2013, 73, 4238–4238 Search PubMed.
- D. J. Moshinsky, L. Ruslim, R. A. Blake and F. Tang, J. Biomol. Screening, 2003, 8, 447–452 CrossRef CAS PubMed.
- K. Kim, V. Punj, J. Choi, K. Heo, J.-M. Kim, P. W. Laird and W. An, Epigenet. Chromatin, 2013, 6, 34 CrossRef CAS PubMed.
- S. Xiao, L. Liu, X. Lu, J. Long, X. Zhou and M. Fang, J. Cancer Res. Clin. Oncol., 2015, 141, 1465–1474 CrossRef CAS PubMed.
- A. A. Dar, M. Nosrati, V. Bezrookove, D. de Semir, S. Majid, S. Thummala, V. Sun, S. Tong, S. P. L. Leong, D. Minor, P. R. Billings, L. Soroceanu, R. Debs, J. R. Miller, R. W. Sagebiel and M. Kashani-Sabet, JNCI, J. Natl. Cancer Inst., 2015, 107, djv034 CrossRef PubMed.
- Y. Buganim, I. Goldstein, D. Lipson, M. Milyavsky, S. Polak-Charcon, C. Mardoukh, H. Solomon, E. Kalo, S. Madar, R. Brosh, M. Perelman, R. Navon, N. Goldfinger, I. Barshack, Z. Yakhini and V. Rotter, PLoS One, 2010, 5, e9657 Search PubMed.
- A. K. Urick, L. M. L. Hawk, M. K. Cassel, N. K. Mishra, S. Liu, N. Adhikari, W. Zhang, C. O. dos Santos, J. L. Hall and W. C. K. Pomerantz, ACS Chem. Biol., 2015, 10, 2246–2256 CrossRef CAS PubMed.
- R. H. Goodman and S. Smolik, Genes Dev., 2000, 14, 1553–1577 CAS.
- N. Vo and R. H. Goodman, J. Biol. Chem., 2001, 276, 13505–13508 CrossRef CAS PubMed.
- E. Kalkhoven, Biochem. Pharmacol., 2004, 68, 1145–1155 CrossRef CAS PubMed.
- H. M. Chan and T. N. B. La, J. Cell Sci., 2001, 114, 2363–2373 CAS.
- D. C. Bedford, L. H. Kasper, T. Fukuyama and P. K. Brindle, Epigenetics, 2010, 5, 9–15 CrossRef CAS PubMed.
- D. A. Hay, O. Fedorov, S. Martin, D. C. Singleton, C. Tallant, C. Wells, S. Picaud, M. Philpott, O. P. Monteiro, C. M. Rogers, S. J. Conway, T. P. C. Rooney, A. Tumber, C. Yapp, P. Filippakopoulos, M. E. Bunnage, S. Müller, S. Knapp, C. J. Schofield and P. E. Brennan, J. Am. Chem. Soc., 2014, 136, 9308–9319 CrossRef CAS PubMed.
- R. C. M. Hennekam, Eur. J. Hum. Genet., 2006, 14, 981–985 CAS.
- F. Wang, C. B. Marshall and M. Ikura, Cell. Mol. Life Sci., 2013, 70, 3989–4008 CrossRef CAS PubMed.
- Y. Revilla and A. G. Granja, Crit. Rev. Immunol., 2009, 29, 131–154 CrossRef CAS PubMed.
- L. M. Valor, J. Viosca, J. P. Lopez-Atalaya and A. Barco, Curr. Pharm. Des., 2013, 19, 5051–5064 CrossRef CAS PubMed.
- Sachchidanand, L. Resnick-Silverman, S. Yan, S. Mutjaba, W.-J. Liu, L. Zeng, J. J. Manfredi and M.-M. Zhou, Chem. Biol., 2006, 13, 81–90 CrossRef CAS PubMed.
- J. C. Borah, S. Mujtaba, I. Karakikes, L. Zeng, M. Muller, J. Patel, N. Moshkina, K. Morohashi, W. Zhang, G. Gerona-Navarro, R. J. Hajjar and M.-M. Zhou, Chem. Biol., 2011, 18, 531–541 CrossRef CAS PubMed.
- G. Gerona-Navarro, S. Mujtaba, A. Frasca, J. Patel, L. Zeng, A. N. Plotnikov, R. Osman and M. M. Zhou, J. Am. Chem. Soc., 2011, 133, 2040–2043 CrossRef CAS PubMed.
- T. P. C. Rooney, P. Filippakopoulos, O. Fedorov, S. Picaud, W. A. Cortopassi, D. A. Hay, S. Martin, A. Tumber, C. M. Rogers, M. Philpott, M. Wang, A. L. Thompson, T. D. Heightman, D. C. Pryde, A. Cook, R. S. Paton, S. Müller, S. Knapp, P. E. Brennan and S. J. Conway, Angew. Chem., Int. Ed., 2014, 53, 6126–6130 CrossRef CAS PubMed.
- D. S. Hewings, M. Wang, M. Philpott, O. Fedorov, S. Uttarkar, P. Filippakopoulos, S. Picaud, C. Vuppusetty, B. Marsden, S. Knapp, S. J. Conway and T. D. Heightman, J. Med. Chem., 2011, 54, 6761–6770 CrossRef CAS PubMed.
- A. Hammitzsch, C. Tallant, O. Fedorov, A. O'Mahony, P. E. Brennan, D. A. Hay, F. O. Martinez, M. H. Al-Mossawi, J. de Wit, M. Vecellio, C. Wells, P. Wordsworth, S. Müller, S. Knapp and P. Bowness, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10768–10773 CrossRef CAS PubMed.
- E. L. P. Chekler, J. A. Pellegrino, T. A. Lanz, R. A. Denny, A. C. Flick, J. Coe, J. Langille, A. Basak, S. Liu, I. A. Stock, P. Sahasrabudhe, P. D. Bonin, K. Lee, M. T. Pletcher and L. H. Jones, Chem. Biol., 2015, 22, 1588–1596 CrossRef CAS PubMed.
- E. C. Hett, E. L. Piatnitski Chekler, A. Basak, P. D. Bonin, R. A. Denny, A. C. Flick, K. F. Geoghegan, S. Liu, M. T. Pletcher, P. Sahasrabudhe, S. C. Salter, I. A. Stock, A. P. Taylor and L. H. Jones, Med. Chem. Commun., 2015, 6, 1018–1023 RSC.
- S. Picaud, O. Fedorov, A. Thanasopoulou, K. Leonards, K. Jones, J. Meier, H. Olzscha, O. Monteiro, S. Martin, M. Philpott, A. Tumber, P. Filippakopoulos, C. Yapp, C. Wells, K. H. Che, A. Bannister, S. Robson, U. Kumar, N. Parr, K. Lee, D. Lugo, P. Jeffrey, S. Taylor, M. L. Vecellio, C. Bountra, P. E. Brennan, A. O'Mahony, S. Velichko, S. Müller, D. Hay, D. L. Daniels, M. Urh, N. B. La Thangue, T. Kouzarides, R. Prinjha, J. Schwaller and S. Knapp, Cancer Res., 2015, 75, 5106–5119 CrossRef CAS PubMed.
- T. Machleidt, C. C. Woodroofe, M. K. Schwinn, J. Méndez, M. B. Robers, K. Zimmerman, P. Otto, D. L. Daniels, T. A. Kirkland and K. V. Wood, ACS Chem. Biol., 2015, 10, 1797–1804 CrossRef CAS PubMed.
- E. L. Berg, J. Yang and M. A. Polokoff, J. Biomol. Screening, 2013, 18, 1260–1269 CrossRef PubMed , 1210 pp.
- R. C. Centore, A. R. Conery, K. Gascoigne and I. I. I. R. J. Sims, US Pat., WO2016044694A1, 2016 Search PubMed.
- M. Xu, A. Unzue, J. Dong, D. Spiliotopoulos, C. Nevado and A. Caflisch, J. Med. Chem., 2016, 59, 1340–1349 CrossRef CAS PubMed.
- A. Unzue, M. Xu, J. Dong, L. Wiedmer, D. Spiliotopoulos, A. Caflisch and C. Nevado, J. Med. Chem., 2016, 59, 1350–1356 CrossRef CAS PubMed.
- A. M. Taylor, A. Côté, M. C. Hewitt, R. Pastor, Y. Leblanc, C. G. Nasveschuk, F. A. Romero, T. D. Crawford, N. Cantone, H. Jayaram, J. Setser, J. Murray, M. H. Beresini, G. de Leon Boenig, Z. Chen, A. R. Conery, R. T. Cummings, L. A. Dakin, E. M. Flynn, O. W. Huang, S. Kaufman, P. J. Keller, J. R. Kiefer, T. Lai, Y. Li, J. Liao, W. Liu, H. Lu, E. Pardo, V. Tsui, J. Wang, Y. Wang, Z. Xu, F. Yan, D. Yu, L. Zawadzke, X. Zhu, X. Zhu, R. J. Sims, A. G. Cochran, S. Bellon, J. E. Audia, S. Magnuson and B. K. Albrecht, ACS Med. Chem. Lett., 2016, 7, 531–536 CrossRef CAS PubMed.
- D. De Semir, M. Nosrati, V. Bezrookove, A. A. Dar, S. Federman, G. Bienvenu, S. Venna, J. Rangel, J. Climent, T. M. Meyer Tamgüney, S. Thummala, S. Tong, S. P. L. Leong, C. Haqq, P. Billings, J. R. Miller, R. W. Sagebiel, R. Debs and M. Kashani-Sabet, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7067–7072 CrossRef CAS PubMed.
- O. B. Cox, T. Krojer, P. Collins, O. Monteiro, R. Talon, A. Bradley, O. Fedorov, J. Amin, B. D. Marsden, J. Spencer, F. von Delft and P. E. Brennan, Chem. Sci., 2016, 7, 2322–2330 RSC.
- E. Middeljans, X. Wan, P. W. Jansen, V. Sharma, H. G. Stunnenberg and C. Logie, PLoS One, 2012, 7, e33834 CAS.
- C. Kadoch, D. C. Hargreaves, C. Hodges, L. Elias, L. Ho, J. Ranish and G. R. Crabtree, Nat. Genet., 2013, 45, 592–601 CrossRef CAS PubMed.
- G. Zinzalla, ChemBioChem, 2016, 17, 677–682 CrossRef CAS PubMed.
- Y. Liu, R. Zhao, H. Wang, Y. Luo, X. Wang, W. Niu, Y. Zhou, Q. Wen, S. Fan, X. Li, W. Xiong, J. Ma, X. Li, M. Tan, G. Li and M. Zhou, Cell Death Dis., 2016, 7, e2156 CrossRef CAS PubMed.
- X. Yu, Z. Li and J. Shen, Am. J. Transl. Res., 2016, 8, 742–748 Search PubMed.
- C.-L. Chen, Y. Wang, Q.-Z. Pan, Y. Tang, Q.-J. Wang, K. Pan, L.-X. Huang, J. He, J.-J. Zhao, S.-S. Jiang, X.-F. Zhang, H.-X. Zhang, Z.-Q. Zhou, D.-S. Weng and J.-C. Xia, Oncotarget, 2016, 7, 16248–16261 Search PubMed.
- H.-T. Huang, S.-M. Chen, L.-B. Pan, J. Yao and H.-T. Ma, Oncol. Rep., 2015, 33, 283–291 CAS.
- S. P. Cleary, W. R. Jeck, X. Zhao, K. Chen, S. R. Selitsky, G. L. Savich, T. X. Tan, M. C. Wu, G. Getz, M. S. Lawrence, J. S. Parker, J. Li, S. Powers, H. Kim, S. Fischer, M. Guindi, A. Ghanekar and D. Y. Chiang, Hepatology, 2013, 58, 1693–1702 CrossRef CAS PubMed.
- L. Scotto, G. Narayan, S. V. Nandula, S. Subramaniyam, A. M. Kaufmann, J. D. Wright, B. Pothuri, M. Mansukhani, A. Schneider, H. Arias-Pulido and V. V. Murty, Mol. Cancer, 2008, 7, 58–67 CrossRef PubMed.
- A. C. da Silva Almeida, F. Abate, H. Khiabanian, E. Martinez-Escala, J. Guitart, C. P. Tensen, M. H. Vermeer, R. Rabadan, A. Ferrando and T. Palomero, Nat. Genet., 2015, 47, 1465–1470 CrossRef CAS PubMed.
- J. G. Bosquet and J. M. Lancaster, US Pat., WO2016004387A1, 2016 Search PubMed.
- R. M. Karim and E. Schönbrunn, J. Med. Chem., 2016, 59, 4459–4461 CrossRef CAS PubMed.
- P. G. K. Clark, D. J. Dixon and P. E. Brennan, Drug Discovery Today: Technol., 2016 DOI:10.1016/j.ddtec.2016.05.002.
- P. G. K. Clark, L. C. C. Vieira, C. Tallant, O. Fedorov, D. C. Singleton, C. M. Rogers, O. P. Monteiro, J. M. Bennett, R. Baronio, S. Müller, D. L. Daniels, J. Méndez, S. Knapp, P. E. Brennan and D. J. Dixon, Angew. Chem., Int. Ed., 2015, 54, 6217–6221 CrossRef CAS PubMed.
- R. H. Shoemaker and R. H. Shoemaker, Nat. Rev. Cancer, 2006, 6, 813–823 CrossRef CAS PubMed.
- B. K. Albrecht, A. Cote, T. Crawford, B. Fauber, H.-R. Huang, J. M. Lora, S. Magnuson, C. G. Nasveschuk, A. Salmeron, R. J. Sims III and A. M. Taylor, US Pat., WO2014144721A2, 2014 Search PubMed.
- N. H. Theodoulou, P. Bamborough, A. J. Bannister, I. Becher, R. A. Bit, K. H. Che, C.-W. Chung, A. Dittmann, G. Drewes, D. H. Drewry, L. Gordon, P. Grandi, M. Leveridge, M. Lindon, A.-M. Michon, J. Molnar, S. C. Robson, N. C. O. Tomkinson, T. Kouzarides, R. K. Prinjha and P. G. Humphreys, J. Med. Chem., 2015, 59, 1425–1439 CrossRef PubMed.
- O. Mirguet, Y. Lamotte, F. Donche, J. Toum, F. Gellibert, A. Bouillot, R. Gosmini, V.-L. Nguyen, D. Delannée, J. Seal, F. Blandel, A.-B. Boullay, E. Boursier, S. Martin, J.-M. Brusq, G. Krysa, A. Riou, R. Tellier, A. Costaz, P. Huet, Y. Dudit, L. Trottet, J. Kirilovsky and E. Nicodeme, Bioorg. Med. Chem. Lett., 2012, 22, 2963–2967 CrossRef CAS PubMed.
- D. A. Hay, C. M. Rogers, O. Fedorov, C. Tallant, S. Martin, O. P. Monteiro, S. Muller, S. Knapp, C. J. Schofield and P. E. Brennan, Med. Chem. Commun., 2015, 6, 1381–1386 RSC.
- L. J. Martin, M. Koegl, G. Bader, X.-L. Cockcroft, O. Fedorov, D. Fiegen, T. Gerstberger, M. H. Hofmann, A. F. Hohmann, D. Kessler, S. Knapp, P. Knesl, S. Kornigg, S. Müller, H. Nar, C. Rogers, K. Rumpel, O. Schaaf, S. Steurer, C. Tallant, C. R. Vakoc, M. Zeeb, A. Zoephel, M. Pearson, G. Boehmelt and D. McConnell, J. Med. Chem., 2016, 59, 4462–4475 CrossRef CAS PubMed.
- A. F. Hohmann, L. J. Martin, J. L. Minder, J.-S. Roe, J. Shi, S. Steurer, G. Bader, D. McConnell, M. Pearson, T. Gerstberger, T. Gottschamel, D. Thompson, Y. Suzuki, M. Koegl and C. R. Vakoc, Nat. Chem. Biol., 2016, 12, 672–679 CrossRef CAS PubMed.
- http://www.thesgc.org/chemical-probes/TP-472 .
- S. Picaud, M. Strocchia, S. Terracciano, G. Lauro, J. Mendez, D. L. Daniels, R. Riccio, G. Bifulco, I. Bruno and P. Filippakopoulos, J. Med. Chem., 2015, 58, 2718–2736 CrossRef CAS PubMed.
- O. Fedorov, H. Lingard, C. Wells, O. P. Monteiro, S. Picaud, T. Keates, C. Yapp, M. Philpott, S. J. Martin, I. Felletar, B. D. Marsden, P. Filippakopoulos, S. Mu, S. Knapp and P. E. Brennan, J. Med. Chem., 2014, 57, 462–476 CrossRef CAS PubMed.
- S. Carlson and K. C. Glass, J. Cell. Physiol., 2014, 229, 1571–1574 CrossRef CAS PubMed.
- T. Brown, J. Swansbury and M. M. Taj, Leuk. Lymphoma, 2012, 53, 338–341 CrossRef PubMed.
- M. Y. Lubula, B. E. Eckenroth, S. Carlson, A. Poplawski, M. Chruszcz and K. C. Glass, FEBS Lett., 2014, 588, 3844–3854 CrossRef CAS PubMed.
- E. H. Demont, P. Bamborough, C.-W. Chung, P. D. Craggs, D. Fallon, L. J. Gordon, P. Grandi, C. I. Hobbs, J. Hussain, E. J. Jones, A. Le Gall, A.-M. Michon, D. J. Mitchell, R. K. Prinjha, A. D. Roberts, R. J. Sheppard and R. J. Watson, ACS Med. Chem. Lett., 2014, 5, 1190–1195 CrossRef CAS PubMed.
- P. Bamborough, H. A. Barnett, I. Becher, M. J. Bird, C.-W. Chung, P. D. Craggs, E. H. Demont, H. Diallo, D. J. Fallon, L. J. Gordon, P. Grandi, C. I. Hobbs, E. Hooper-Greenhill, E. J. Jones, R. P. Law, A. Le Gall, D. Lugo, A.-M. Michon, D. J. Mitchell, R. K. Prinjha, R. J. Sheppard, A. J. B. Watson and R. J. Watson, ACS Med. Chem. Lett., 2016, 7, 552–557 CrossRef CAS PubMed.
- http://www.thesgc.org/chemical-probes/PFI-4 .
- http://www.thesgc.org/chemical-probes/OF-1 .
- http://www.thesgc.org/chemical-probes/NI-57 .
- C. Caron, C. Lestrat, S. Marsal, E. Escoffier, S. Curtet, V. Virolle, P. Barbry, A. Debernardi, C. Brambilla, E. Brambilla, S. Rousseaux and S. Khochbin, Oncogene, 2010, 29, 5171–5181 CrossRef CAS PubMed.
- E. Y. C. Hsia, E. V. Kalashnikova, A. S. Revenko, J. X. Zou, A. D. Borowsky and H.-W. Chen, Mol. Cancer Res., 2010, 8, 183–193 CrossRef CAS PubMed.
- J. X. Zou, L. Guo, A. S. Revenko, C. G. Tepper, A. T. Gemo, H.-J. Kung and H.-W. Chen, Cancer Res., 2009, 69, 3339–3346 CrossRef CAS PubMed.
- X. Chen, S. T. Cheung, S. So, S. T. Fan, C. Barry, J. Higgins, K.-M. Lai, J. Ji, S. Dudoit, I. O. L. Ng, M. Van de Rijn, D. Botstein and P. O. Brown, Mol. Biol. Cell, 2002, 13, 1929–1939 CrossRef CAS PubMed.
- J. Fellenberg, L. Bernd, G. Delling, D. Witte and A. Zahlten-Hinguranage, Mod. Pathol., 2007, 20, 1085–1094 CrossRef CAS PubMed.
- E. H. Demont, C.-W. Chung, R. C. Furze, P. Grandi, A.-M. Michon, C. Wellaway, N. Barrett, A. M. Bridges, P. D. Craggs, H. Diallo, D. P. Dixon, C. Douault, A. J. Emmons, E. J. Jones, B. V. Karamshi, K. Locke, D. J. Mitchell, B. H. Mouzon, R. K. Prinjha, A. D. Roberts, R. J. Sheppard, R. J. Watson and P. Bamborough, J. Med. Chem., 2015, 58, 5649–5673 CrossRef CAS PubMed.
- A. Chaikuad, A. M. Petros, O. Fedorov, J. Xu and S. Knapp, Med. Chem. Commun., 2014, 5, 1843–1848 RSC.
- P. Bamborough, C.-w. Chung, R. C. Furze, P. Grandi, A.-M. Michon, R. J. Sheppard, H. Barnett, H. Diallo, D. P. Dixon, C. Douault, E. J. Jones, B. Karamshi, D. J. Mitchell, R. K. Prinjha, C. Rau, R. J. Watson, T. Werner and E. H. Demont, J. Med. Chem., 2015, 58, 6151–6178 CrossRef CAS PubMed.
- P. Bamborough, C.-W. Chung, E. H. Demont, R. C. Furze, A. J. Bannister, K. H. Che, H. Diallo, C. Douault, P. Grandi, T. Kouzarides, A.-M. Michon, D. J. Mitchell, R. K. Prinjha, C. Rau, S. Robson, R. J. Sheppard, R. Upton and R. J. Watson, Angew. Chem., Int. Ed., 2016, 55, 11382–11386 CrossRef CAS PubMed.
- M. H. Jones, N. Hamana, J.-I. Nezu and M. Shimane, Genomics, 2000, 63, 40–45 CrossRef CAS PubMed.
- R. Strohner, A. Nemeth, P. Jansa, U. Hofmann-Rohrer, R. Santoro, G. Langst and I. Grummt, EMBO J., 2001, 20, 4892–4900 CrossRef CAS PubMed.
- C. Mayer, M. Neubert and I. Grummt, EMBO Rep., 2008, 9, 774–780 CrossRef CAS PubMed.
- D. E. Arking, M. J. Junttila, P. Goyette, A. Huertas-Vazquez, M. Eijgelsheim, M. T. Blom, C. Newton-Cheh, K. Reinier, C. Teodorescu, A. Uy-Evanado, N. Carter-Monroe, K. S. Kaikkonen, M.-L. Kortelainen, G. Boucher, C. Lagace, A. Moes, X. Zhao, F. Kolodgie, F. Rivadeneira, A. Hofman, J. C. M. Witteman, A. G. Uitterlinden, R. F. Marsman, R. Pazoki, A. Bardai, R. W. Koster, A. Dehghan, S.-J. Hwang, P. Bhatnagar, W. Post, G. Hilton, R. J. Prineas, M. Li, A. Kottgen, G. Ehret, E. Boerwinkle, J. Coresh, W. H. L. Kao, B. M. Psaty, G. F. Tomaselli, N. Sotoodehnia, D. S. Siscovick, G. L. Burke, E. Marban, P. M. Spooner, L. A. Cupples, J. Jui, K. Gunson, Y. A. Kesaniemi, A. A. M. Wilde, J.-C. Tardif, C. J. O'Donnell, C. R. Bezzina, R. Virmani, B. H. C. Stricker, H. L. Tan, C. M. Albert, A. Chakravarti, J. D. Rioux, H. V. Huikuri and S. S. Chugh, PLoS Genet., 2011, 7, e1002158 CAS.
- L. Drouin, S. McGrath, L. R. Vidler, A. Chaikuad, O. Monteiro, C. Tallant, M. Philpott, C. Rogers, O. Fedorov, M. Liu, W. Akhtar, A. Hayes, F. Raynaud, S. Muller, S. Knapp and S. Hoelder, J. Med. Chem., 2015, 58, 2553–2559 CrossRef CAS PubMed.
- L. Gu, S. C. Frommel, C. C. Oakes, R. Simon, K. Grupp, C. Y. Gerig, D. Baer, M. D. Robinson, C. Baer, M. Weiss, Z. Gu, M. Schapira, R. Kuner, H. Sueltmann, M. Provenzano, M.-L. Yaspo, B. Brors, J. Korbel, T. Schlomm, G. Sauter, R. Eils, C. Plass and R. Santoro, Nat. Genet., 2015, 47, 22–30 CrossRef CAS PubMed.
- P. Chen, A. Chaikuad, P. Bamborough, M. Bantscheff, C. Bountra, C.-W. Chung, O. Fedorov, P. Grandi, D. Jung, R. Lesniak, M. Lindon, S. Müller, M. Philpott, R. Prinjha, C. Rogers, C. Selenski, C. Tallant, T. Werner, T. M. Willson, S. Knapp and D. H. Drewry, J. Med. Chem., 2016, 59, 1410–1424 CrossRef CAS PubMed.
- W.-W. Tsai, Z. Wang, T. T. Yiu, K. C. Akdemir, W. Xia, S. Winter, C.-Y. Tsai, X. Shi, D. Schwarzer, W. Plunkett, B. Aronow, O. Gozani, W. Fischle, M.-C. Hung, D. J. Patel and M. C. Barton, Nature, 2010, 468, 927–932 CrossRef CAS PubMed.
- M. Chambon, B. Orsetti, M.-L. Berthe, C. Bascoul-Mollevi, C. Rodriguez, V. Duong, M. Gleizes, S. Thenot, F. Bibeau, C. Theillet and V. Cavailles, Am. J. Pathol., 2011, 178, 1461–1469 CrossRef CAS PubMed.
- Z. Cui, W. Cao, J. Li, X. Song, L. Mao and W. Chen, PLoS One, 2013, 8, e63887 Search PubMed.
- H. Li, L. Sun, Z. Tang, L. Fu, Y. Xu, Z. Li, W. Luo, X. Qiu and E. Wang, PLoS One, 2012, 7, e37657 CAS.
- X. Liu, Y. Huang, D. Yang, X. Li, J. Liang, L. Lin, M. Zhang, K. Zhong, B. Liang and J. Li, PLoS One, 2014, 9, e85462 Search PubMed.
- J. Chen and W. A. Weiss, Oncogene, 2015, 34, 1–14 CrossRef CAS PubMed.
- J. Bennett, O. Fedorov, C. Tallant, O. Monteiro, J. Meier, V. Gamble, P. Savitsky, G. A. Nunez-Alonso, B. Haendler, C. Rogers, P. E. Brennan, S. Müller and S. Knapp, J. Med. Chem., 2016, 59, 1642–1647 CrossRef CAS PubMed.
- W. S. Palmer, G. Poncet-Montange, G. Liu, A. Petrocchi, N. Reyna, G. Subramanian, J. Theroff, A. Yau, M. Kost-Alimova, J. P. Bardenhagen, E. Leo, H. E. Shepard, T. N. Tieu, X. Shi, Y. Zhan, S. Zhao, M. C. Barton, G. Draetta, C. Toniatti, P. Jones, M. Geck Do and J. N. Andersen, J. Med. Chem., 2016, 59, 1440–1454 CrossRef CAS PubMed.
- R. C. Rao and Y. Dou, Nat. Rev. Cancer, 2015, 15, 334–346 CrossRef CAS PubMed.
- I. Barde, B. Rauwel, R. M. Marin-Florez, A. Corsinotti, E. Laurenti, S. Verp, S. Offner, J. Marquis, A. Kapopoulou, J. Vanicek and D. Trono, Science, 2013, 340, 350–353 CrossRef CAS PubMed.
- B. Rauwel, S. M. Jang, M. Cassano, A. Kapopoulou, I. Barde and D. Trono, eLife, 2015, 4, e06068 Search PubMed.
- M. Singh, G. M. Popowicz, M. Krajewski and T. A. Holak, ChemBioChem, 2007, 8, 1308–1316 CrossRef CAS PubMed.
- J. Masliah-Planchon, I. Bieche, J.-M. Guinebretiere, F. Bourdeaut and O. Delattre, Annu. Rev. Pathol.: Mech. Dis., 2015, 10, 145–171 CrossRef CAS PubMed.
- B. S. Gerstenberger, J. D. Trzupek, C. Tallant, O. Fedorov, P. Filippakopoulos, P. E. Brennan, V. Fedele, S. Martin, S. Picaud, C. M. Rogers, M. D. Parikh, A. Taylor, B. M. Samas, A. O'Mahony, E. L. Berg, G. Pallares, A. V. Torrey, D. K. Treiber, I. J. Samardjiev, B. T. Nasipak, T. Padilla-Benavides, Q. Wu, A. N. Imbalzano, J. A. Nickerson, M. E. Bunnage, S. Muller, S. Knapp and D. R. Owen, J. Med. Chem., 2016, 59, 4800–4811 CrossRef CAS PubMed.
- O. Fedorov, C. Tallant, S. Martin, O. Monteiro, C. Bountra, C. Wells, M. Philpott, C. Rogers, P. E. Brennan, S. Muller, J. Castex, D. Willmann, T. Gunther, D. R. Owen, J. D. Trzupek, B. S. Gerstenberger, M. E. Bunnage, M. Aldeghi, P. Filippakopoulos, S. Picaud, P. C. Biggin, R. Schule and S. Knapp, Sci. Adv., 2015, 1, e1500723 Search PubMed.
- M. Philpott, J. Yang, T. Tumber, O. Fedorov, S. Uttarkar, P. Filippakopoulos, S. Picaud, T. Keates, I. Felletar, A. Ciulli, S. Knapp and T. D. Heightman, Mol. BioSyst., 2011, 7, 2899–2908 RSC.
- P. Ciceri, S. Mueller, A. O'Mahony, O. Fedorov, P. Filippakopoulos, J. P. Hunt, E. A. Lasater, G. Pallares, S. Picaud, C. Wells, S. Martin, L. M. Wodicka, N. P. Shah, D. K. Treiber and S. Knapp, Nat. Chem. Biol., 2014, 10, 305–312 CrossRef CAS PubMed.
- C. L. Sutherell, C. Tallant, O. P. Monteiro, C. Yapp, J. E. Fuchs, O. Fedorov, P. Siejka, S. Muller, S. Knapp, J. D. Brenton, P. E. Brennan and S. V. Ley, J. Med. Chem., 2016, 59, 5095–5101 CrossRef CAS PubMed.
- L. Kruidenier, C.-W. Chung, Z. Cheng, J. Liddle, K. Che, G. Joberty, M. Bantscheff, C. Bountra, A. Bridges, H. Diallo, D. Eberhard, S. Hutchinson, E. Jones, R. Katso, M. Leveridge, P. K. Mander, J. Mosley, C. Ramirez-Molina, P. Rowland, C. J. Schofield, R. J. Sheppard, J. E. Smith, C. Swales, R. Tanner, P. Thomas, A. Tumber, G. Drewes, U. Oppermann, D. J. Patel, K. Lee and D. M. Wilson, Nature, 2012, 488, 404–408 CrossRef CAS PubMed.
- H. D. Lewis, J. Liddle, J. E. Coote, S. J. Atkinson, M. D. Barker, B. D. Bax, K. L. Bicker, R. P. Bingham, M. Campbell, Y. H. Chen, C.-W. Chung, P. D. Craggs, R. P. Davis, D. Eberhard, G. Joberty, K. E. Lind, K. Locke, C. Maller, K. Martinod, C. Patten, O. Polyakova, C. E. Rise, M. Rüdiger, R. J. Sheppard, D. J. Slade, P. Thomas, J. Thorpe, G. Yao, G. Drewes, D. D. Wagner, P. R. Thompson, R. K. Prinjha and D. M. Wilson, Nat. Chem. Biol., 2015, 11, 189–191 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |