
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSAR and identification of 2-(quinolin-4-yloxy)acetamides as Mycobacterium tuberculosis cytochrome bc1 inhibitors†‡
Narisa
Phummarin
a,
Helena I.
Boshoff
b,
Patricia S.
Tsang
b,
James
Dalton
c,
Siouxsie
Wiles
cd,
Clifton E.
Barry 3rd
b and
Brent R.
Copp
 *a
*a
aSchool of Chemical Sciences, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand. E-mail: b.copp@auckland.ac.nz; Fax: +64 9 3737422; Tel: +64 9 3737599
bTuberculosis Research Section, Laboratory of Clinical Infectious Diseases, National Institute of Allergy and Infectious Disease, National Institutes of Health, Bethesda, Maryland, USA
cBioluminescent Superbugs Lab, Department of Molecular Medicine and Pathology, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
dTe Pūnaha Matatini, c/o University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
First published on 22nd August 2016
Abstract
A previous phenotypic screen by GSK identified 2-(quinolin-4-yloxy)acetamides as potent growth inhibitors of Mycobacterium tuberculosis (Mtb). We report the results of a preliminary structure–activity relationship (SAR) study of the compound class which has yielded more potent inhibitors. An Mtb cytochrome bd oxidase deletion mutant (cydKO) was found to be hypersensitive to most members of the compound library, while strains carrying single-nucleotide polymorphisms of the qcrB gene, which encodes a subunit of the menaquinol cytochrome c oxidoreductase (bc1) complex, were resistant to the library. These results identify that the 2-(quinolin-4-yloxy)acetamide class of Mtb growth inhibitors can be added to the growing number of scaffolds that target the M. tuberculosis bc1 complex.
1. Introduction
The current paradigm for the discovery of antibiotics that can be developed into much-needed new treatments for tuberculosis typically starts with a whole-cell phenotypic screening campaign.1,2 The majority of these screens use aerobic culture conditions and so by necessity, lead to the discovery of growth inhibitors of replicating Mycobacterium tuberculosis (Mtb). Recent efforts have also been directed towards undertaking screening using culture conditions considered more relevant to the in vivo disease state,3,4 or towards Mtb inside host macrophages.5 Defining the mechanism of action of screening hits obtained in such an agnostic manner can be challenging, however with significant cost reductions, sequencing of resistant mutants has become a productive pathway. Two recent examples of screening campaign hits are the imidazo[1,2-a]pyridine amides (IPAs),2,3,6–8 exemplified by the clinical candidate Q203 (1) identified during an Mtb/macrophage assay,9 and lansoprazole (2, Prevacid), identified in a pharmaceutical re-purposing screen designed to identify compounds that protect lung fibroblasts from Mtb-induced cytotoxicity (Fig. 1).10 Genome sequencing of spontaneous resistant mutants generated for both 1 and 2 revealed a common target of QcrB (Rv2196), which encodes the b subunit of the cytochrome bc1 complex, an essential component of the electron transport chain. In the case of Q203, a mutation of T313 A or T313I in QcrB was observed to cause resistance, while for 2, a L176P mutation was responsible.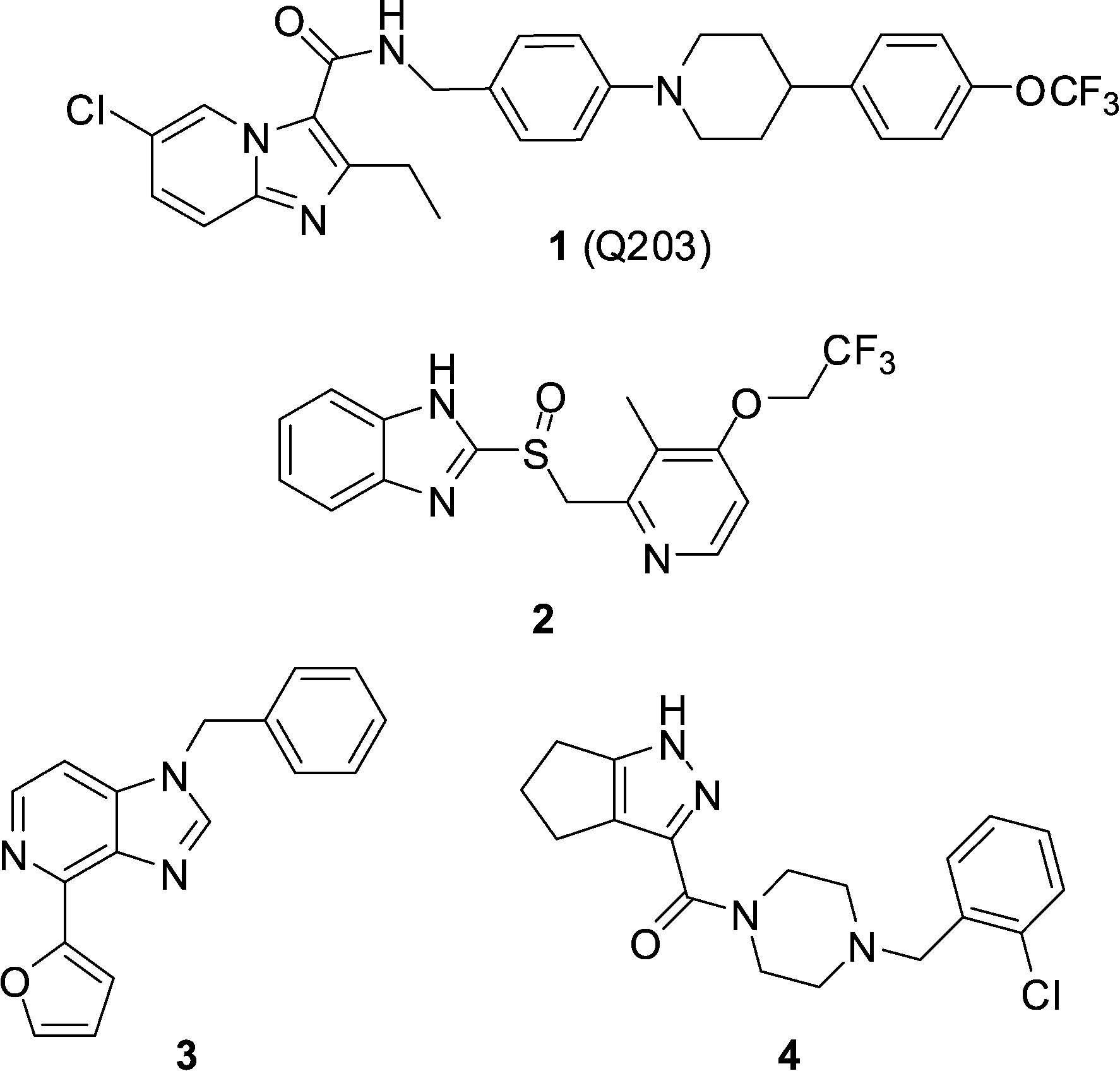 | ||
| Fig. 1 Structures of cytochrome bc1 oxidase inhibitors 1–4. | ||
In addition to the chemtypes encompassed by 1 and 2, further chemically diverse scaffolds have been identified to bind to QcrB,11 including imidazo[4,5-c]pyridine 3 and pyrazole 4,12 suggesting the target is somewhat promiscuous (Fig. 1).
GSK recently made publically available the results of a phenotypic screening campaign, whereby a large compound library was assessed for the ability to inhibit the growth of Mycobacterium bovis BCG and M. tuberculosis H37Rv. The curated results led to the identification of 177 hits covering a number of different structural classes.13 Amongst this set were five 2-(quinolin-4-yloxy)acetamides (QOAs) (5–9, Fig. 2) exhibiting favourable Mtb growth inhibition properties with MIC90 Mtb H37Rv 0.3–3.3 μM.
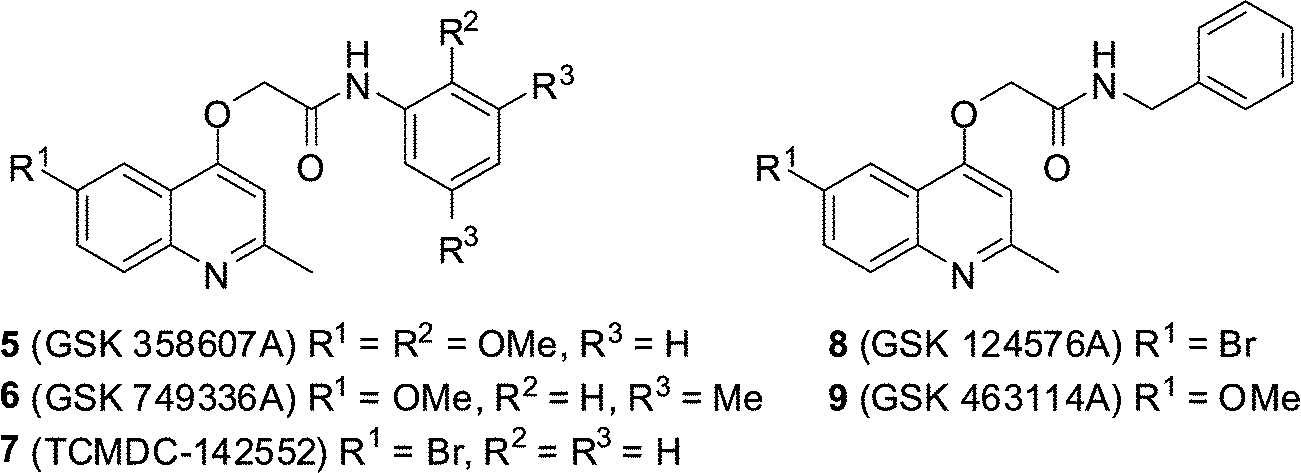 | ||
| Fig. 2 Structures of GSK hit compounds 5–9. | ||
The potency of activity and structural simplicity of this series makes them an attractive target, prompting efforts to explore the structure–activity relationship of the class. During the preparation of this manuscript, two groups have reported the results of their studies of the QOA class, confirming compound potency, selectivity for M. tuberculosis and lack of toxicity in a Danio rerio (zebrafish) model.14 Herein we report our efforts in optimizing the in vitro anti-tuberculosis activity of the more potent GSK QOA analogue 5 and also identify, via use of selective mutant strains, their cellular target as involving the cytochrome bc1 complex.
2. Results and discussion
Our preliminary structure–activity relationship study of GSK 358607A (5) focused on changes to the (i) 2-methoxyphenylacetamide moiety and (ii) the quinoline 6-position. The target 2-(quinolin-4-yloxy)acetamide library (12a–12aa), in addition to GSK compounds 5 and 9, were prepared in three steps from commercially available materials (Scheme 1). The synthesis began by preparing the required 4-hydroxyquinolines by heating the appropriate aniline with ethyl acetoacetate in polyphosphoric acid to afford, after purification, 10a–f in yields of 25–77%. Bromoacetamides 11a–x were prepared in 27–100% yields from the acylation, at −77 °C, of various substituted anilines or alkylamines with bromoacetyl bromide. In the final step, 2-(quinolin-4-yloxy)acetamides 5, 9 and 12a–12aa were prepared, in yields of 16–89%, by reaction of 4-hydroxyquinolines with bromoacetamides in either acetone or DMF solvent in the presence of K2CO3 base.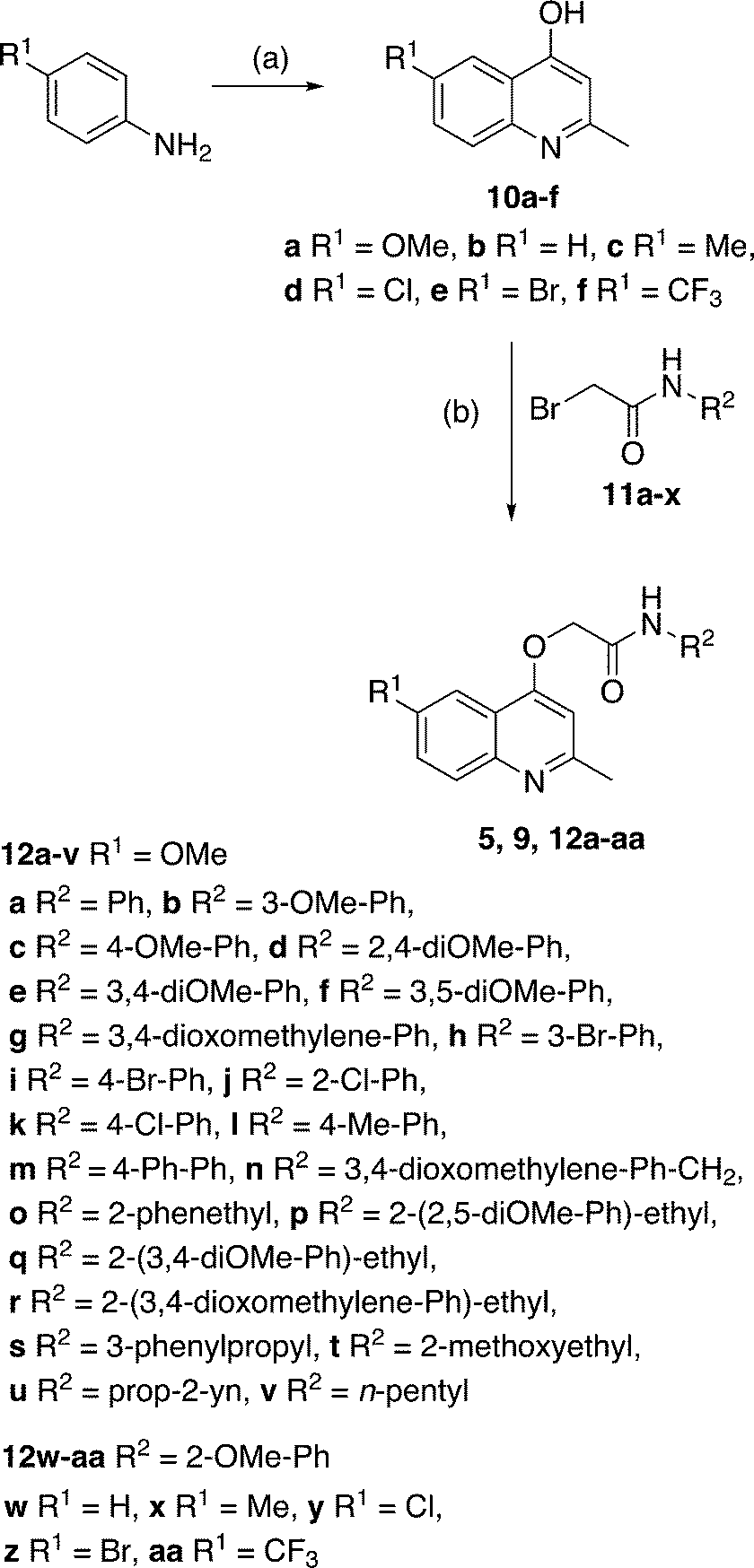 | ||
| Scheme 1 Synthesis of target compounds 5, 9, 12a–12aa. Reagents and conditions: (a) ethylacetoacetate, PPA, 130 °C, 2 h, yield: 25–77%; (b) bromide 11a–x, K2CO3, DMF or acetone, 5–21 h, yield: 16–89%. | ||
The library of analogues were tested for whole-cell growth inhibition of Mycobacterium tuberculosis (Mtb) H37Rv under a variety of growth conditions and different readouts of growth and minimum inhibitory concentration (MIC) values were determined at either 7 or 14 days (Table 1). Using laboratory-adapted strain Mtb H37Rv grown in Middlebrook 7H9 media with microplate Alamar Blue assay (MABA) readout after 14 days (Table 1, data column 1) revealed that the GSK hits 5 and 9 exhibited MICs of 1.1 and 2.3 μM respectively (entries 1 and 2). Of the phenylacetamide sidechain analogues prepared, examples containing either no substituent (12a, entry 3), or 2-chloro (12j, entry 12) or 4-methyl (12l, entry 14) substitution were slightly more potent than GSK compounds 5 and 9. The remaining phenylacetamide sidechain analogues (12b–12i, 12k, 12m, entries 4–11, 13, 15) were either equipotent or less active than the original hits. In most cases introduction of conformational flexibility in the form of 2-phenethyl (12o, 12p, 12q, 12r) or 3-phenylpropyl (12s) acetamide sidechains yielded compounds that were poor growth inhibitors. The exception to this trend was 12n, a 3,4-dioxomethylenebenzyl derivative, which exhibited an MIC of 0.53 μM, being slightly more potent than the unsubstituted GSK benzyl analogue 9. Acetamide sidechains that incorporated no aryl moiety (i.e.12t 2-methoxyethyl, 12u prop-2-yn and 12vn-pentyl, entries 22–24) were also significantly less active than the original hit compounds. Overall, four compounds 12a, 12j, 12l, 12n were identified as being more potent growth inhibitors than the original GSK hit compound. In general, bulky, lipophilic substituents of limited conformational flexibility at R2 improved the antimycobacterial activity of the compounds, while R1 being methoxyl was critical for activity. Similar trends in overall structure–activity relationship were observed by Pissinate et al., where they arrived at conclusions regarding the requirement for a bulky lipophilic group at R2, finding a 2-naphthyl derivative to be potently antimycobacterial.14a
| MIC (μM) | |||||
|---|---|---|---|---|---|
| Entry | Compound | H37Rv MABA 2 weeka | H37Rv 1 weekb | H37Rv 2 weekc | cydKO 2 weekd |
| MIC values are the average of two independent assays. Assay protocols are described in ref. 8. a MIC against H37Rv grown in 7H9/ADC/Tween media. MIC determination using microplate Alamar Blue assay (MABA) after 2 weeks post compound addition. b MIC against H37Rv grown in 7H9/ADC/Tween media. MIC determination after 1 week post compound addition. c MIC against H37Rv grown in 7H9/ADC/Tween media. MIC determination after 2 weeks post compound addition. d MIC against cyd knock-out strain of H37Rv grown in 7H9/ADC/Tween media. MIC determination after 2 weeks post-compound addition. e Linezolid, PAS (p-aminosalicylic acid) and 3 were used as positive controls. | |||||
| 1 | 5 | 1.11 | 2.21 | 53.92 | 0.14 |
| 2 | 9 | 2.32 | 3.57 | >148 | 0.59 |
| 3 | 12a | 0.62 | 1.21 | >155 | 0.12 |
| 4 | 12b | 6.53 | 141.88 | >142 | 0.57 |
| 5 | 12c | 1.11 | 2.21 | >142 | 0.14 |
| 6 | 12d | 2.04 | 24.58 | 32.69 | 0.18 |
| 7 | 12e | 24.58 | >131 | >131 | 3.14 |
| 8 | 12f | 24.58 | >131 | >131 | 2.04 |
| 9 | 12g | 2.13 | 4.26 | >136 | 0.19 |
| 10 | 12h | >125 | 92.20 | >125 | 0.37 |
| 11 | 12i | 1.94 | >125 | >125 | 0.17 |
| 12 | 12j | 0.56 | 26.35 | 53.25 | <0.067 |
| 13 | 12k | 3.36 | 103.70 | >140 | 0.20 |
| 14 | 12l | 0.45 | 0.45 | >149 | <0.071 |
| 15 | 12m | 1.51 | >125 | >125 | 0.12 |
| 16 | 12n | 0.53 | 0.53 | 24.71 | <0.063 |
| 17 | 12o | 54.22 | 142.69 | >143 | 4.45 |
| 18 | 12p | 60.90 | 121.80 | >122 | 7.62 |
| 19 | 12q | 122.00 | >122 | >122 | 90.13 |
| 20 | 12r | 31.69 | ≧127 | >127 | 3.04 |
| 21 | 12s | 25.79 | 68.59 | 68.59 | 2.14 |
| 22 | 12t | 121.55 | >164 | >164 | 41.06 |
| 23 | 12u | >176 | >176 | >176 | 21.98 |
| 24 | 12v | 19.75 | 158.03 | 158.03 | 0.95 |
| 25 | 12w | >78 | 38.77 | >155 | 7.13 |
| 26 | 12x | 9.30 | 56.48 | >74 | 0.45 |
| 27 | 12y | 103.70 | 26.35 | >140 | 1.09 |
| 28 | 12z | >125 | 23.42 | >125 | 0.50 |
| 29 | 12aa | >128 | >128 | >128 | >128 |
| PASe | 0.3 | 0.3 | 0.6 | 0.6 | |
| Linezolide | 2.3 | 2.3 | 2.3 | 1.56 | |
| 3 | 1.56 | 12.5 | >25 | 0.31 | |
When Mtb H37Rv was grown in the same media (7H9/ADC/Tween) but using optical density as the growth readout, there was apparent bacterial outgrowth as evidenced by time-dependent outgrowth of the cells in the presence of the compound resulting in a dramatic shift in apparent MIC (Table 1, data columns 2 and 3). Similar outgrowth was also observed for M. tuberculosis BSG001 (M. tuberculosis H37Rv transformed with the bacterial luciferase-encoding vector pMV306hsp + LuxAB + G13 + CDE)15 grown in Middlebrook 7H9 media (data not shown). This discordance between growth MIC and the Alamar Blue MIC has been previously reported to be a characteristic of compounds that inhibit the respiratory bc1 complex of Mycobacterium tuberculosis.12 The ability of cells to overcome the growth inhibitory effect of these compounds could at least in part be driven by compensatory upregulation of the alternate oxygen-dependent cytochrome bd oxidase pathway.16 Evidence for this upregulation-based protection model is based upon the findings of a number of groups, whereby cytochrome bd oxidase knock-out strains (ΔcydKO) of mycobacteria are hyper-susceptible to electron transport chain inhibitors17 including those compounds that target bc1.8,11,12 As summarized in Table 1 (data column 4), most of the test set of compounds did indeed exhibit enhanced MIC potency towards the Mtb ΔcydKO strain versus laboratory-adapted Mtb H37Rv. Based upon the magnitude of MIC enhancement evident in Table 1, it was concluded that the majority of the analogues, and GSK compounds 5 and 9, are inhibitors of cytochrome bc1 oxidase. Further direct evidence for the bc1 oxidase inhibiting properties of 5 and 9 and analogues 12a, 12c, 12g, 12l and 12n was obtained when the compounds were found to exhibit reduced potency against a series of QcrB subunit mutants of the cydKO strain of Mtb (Table 2). The seven defined Mtb cydKO qcrB amino acid mutants, A317V, M342T, W312G, A396T, M342I, A317T and S182P, were originally generated in response to imidazo[4,5-c]pyridine 3 with homology modeling of QcrB identifying all mutations to be located proximal to the binding site of stigmatellin.12 In the present study, the magnitudes of the fold-resistance observed for the test compounds against these seven cydKO mutants confirms cytochrome bc1 oxidase as a cellular target of 2-(quinolin-4-yloxy)acetamides. Compounds that inhibit cytochrome bc1 oxidase have previously been shown to deplete intracellular ATP levels in anaerobic cells.9 We found that our 2-(quinolin-4-yloxy)acetamides were able to reduce ATP levels within 24 hours of exposure under anaerobic conditions in a similar manner to TMC2073,9 (Bedaquiline, a F0F1 ATP synthase inhibitor) and 3, a known cytochrome bc1 oxidase inhibitor12 (Fig. 3) (and ESI‡ Fig. S1–S3). ATP levels were not reduced by the cell-wall synthesis inhibitor linezolid.
| Fold resistance to qcrB mutant | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Compound | A317V | M342T | W312G | A396T | M342I | A317T | S182P |
| a Compound 3 (positive control), PAS (p-aminosalicylic acid, negative control). | ||||||||
| 1 | 5 | 46.9 | 1.4 | 46.9 | 4.1 | 6.1 | 24.5 | 15.9 |
| 2 | 9 | >250 | 3.0 | 47.0 | 3.9 | 3.9 | 23.5 | 15.7 |
| 3 | 12a | >1250 | 3.8 | 235.0 | 5.0 | 1.8 | 30.0 | 19.5 |
| 4 | 12c | 63.9 | 4.1 | 24.5 | 3.1 | 3.1 | 24.5 | 15.9 |
| 5 | 12g | 134.3 | 2.1 | 17.1 | 5.6 | 1.4 | 32.9 | 32.9 |
| 6 | 12l | >32.5 | >2 | >16.3 | >2 | 1.0 | >12.5 | >16.3 |
| 7 | 12n | >130 | >12.5 | >25 | >6.3 | >12.5 | >32.5 | >95.8 |
| 8 | 3 | >81 | 20.2 | >81 | 20.2 | 20.2 | 10.1 | 61.3 |
| PASa | 0.5 | 0.3 | 0.5 | 0.3 | 0.5 | 0.2 | 0.5 | |
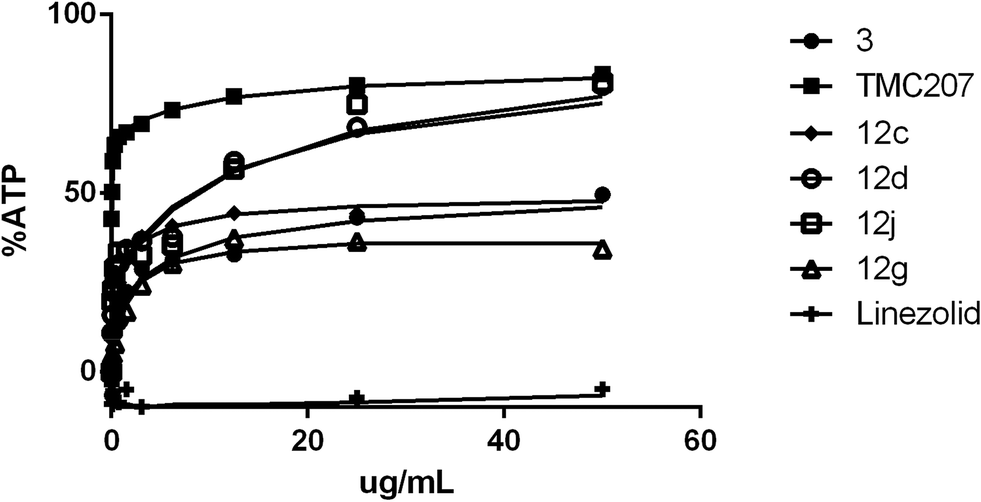 | ||
| Fig. 3 ATP depletion (%) under anaerobic conditions for test compounds 12c, 12d, 12g, and 12j. | ||
Previous groups have determined the selectivity of antitubercular cytochrome bc1 oxidase inhibitors towards Mycobacterium tuberculosis, with little or no detectable antibacterial activity being observed towards panels of Gram-positive or Gram-negative bacteria.8,10 We can confirm the recent report by Pissinate et al.,14a where they noted the absence of antibacterial activity of the 2-(quinolin-4-yloxy)acetamide compound class towards Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus and Acinetobacter baumannii. In the present study, no activity was observed towards Escherichia coli 25922, Staphylococcus aureus XEN36, Mycobacterium smegmatis and M. abscessus (data not shown).
The cytotoxicity of the 2-(quinolin-4-yloxy)acetamide library was determined using HepG2 cells, with the assay media containing either glucose or galactose. The use of galactose forces the cells to rely on mitochondrial oxidative phosphorylation rather than glycolysis for growth.18 While five of the test compounds (12b, 12g, 12k, 12o, 12w; Table 3, entries 4, 9, 13, 17, 25) were considered to be mildly cytotoxic independent of glucose/galactose-based media, two analogues (5, 12l) exhibited mild to moderate levels of cytotoxicity (CC50 < 50 μM) only towards cells grown in galactose-containing media. This latter result identifies these two compounds, which includes GSK hit 5, as being potential inhibitors of mitochondrial respiration.
| Entry | Compound | GalactoseCC50 (μM) | GlucoseCC50 (μM) |
|---|---|---|---|
| IC50 values are the average of two independent assays. Assay protocols are described in ref. 18. a Compound 3 and antimycin A were used as positive controls. | |||
| 1 | 5 | 19.6 | >142 |
| 2 | 9 | >149 | >149 |
| 3 | 12a | 118.1 | >155 |
| 4 | 12b | 24.7 | 43.0 |
| 5 | 12c | >142 | >142 |
| 6 | 12d | >131 | >131 |
| 7 | 12e | >131 | >131 |
| 8 | 12f | >131 | >131 |
| 9 | 12g | 39.6 | 80.0 |
| 10 | 12h | >125 | >125 |
| 11 | 12i | >125 | >125 |
| 12 | 12j | >140 | >140 |
| 13 | 12k | 71.1 | 89.9 |
| 14 | 12l | 41.5 | >149 |
| 15 | 12m | >126 | >126 |
| 16 | 12n | 86.9 | >131 |
| 17 | 12o | 83.9 | 61.4 |
| 18 | 12p | >122 | >122 |
| 19 | 12q | >122 | >122 |
| 20 | 12r | >127 | >127 |
| 21 | 12s | 50.7 | >137 |
| 22 | 12t | >164 | >164 |
| 23 | 12u | >176 | >176 |
| 24 | 12v | 106.3 | >158 |
| 25 | 12w | 8.64 | 43.9 |
| 26 | 12x | >74 | >74 |
| 27 | 12y | 99.2 | 140.1 |
| 28 | 12z | >125 | >125 |
| 29 | 12aa | >128 | >128 |
| 3 | >25 | >25 | |
| Antimycin Aa | 0.017 | >50 | |
3. Conclusions
In conclusion, our preliminary structure–activity relationship investigation of the anti-tuberculosis activity of 2-(quinolin-4-yloxy)acetamides has identified four analogues (12a, 12j, 12l, 12n) as being more potent growth inhibitors than the original GSK 358607A (5) hit compound, exhibiting sub-micromolar MIC values. We have established that this compound class targets QcrB, an essential component of the electron transport chain. Knowledge of the cellular target of 2-(quinolin-4-yloxy)acetamides will now facilitate structure-based drug design as we seek to optimize the antituberculosis potency of this series.Author contributions
N. P. and B. R. C. synthesized the compound library. H. I. B., C. E. B., J. D. and S. W. performed antibacterial testing. P. S. T. performed cytotoxicity testing. B. R. C. wrote the manuscript with contributions from all co-authors.Acknowledgements
We acknowledge funding from the University of Auckland (including in part from the Vice Chancellors Strategic Development Fund [23563]) and, in part, by the Intramural Research Program of NIAID, NIH. We thank Dr M. Schmitz for assistance with NMR data acquisition, Mr Tony Chen for MS data and V. de Guzman for preliminary studies.Notes and references
- K. Mdluli, T. Kaneko and A. Upton, Ann. N. Y. Acad. Sci., 2014, 1323, 56 CrossRef CAS PubMed.
- U. H. Manjunatha and P. W. Smith, Bioorg. Med. Chem., 2015, 23, 5087 CrossRef CAS PubMed.
- P. A. Mak, S. P. S. Rao, M. P. Tan, X. Lin, J. Chyba, J. Tay, S. H. Ng, B. H. Tan, J. Cherian, J. Duraiswamy, P. Bifani, V. Lim, B. H. Lee, N. L. Ma, D. Beer, P. Thayalan, K. Kuhen, A. Chatterjee, F. Supek, R. Glynne, J. Zheng, H. I. Boshoff, C. E. Barry 3rd, T. Dick, K. Pethe and L. R. Camacho, ACS Chem. Biol., 2012, 7, 1190 CrossRef CAS PubMed.
- C. M. Darby, H. I. Ingólfsson, X. Jiang, C. Shen, M. Sun, N. Zhao, K. Burns, G. Liu, S. Ehrt, J. D. Warren, O. S. Anderson, S. J. Brickner and C. Nathan, PLoS One, 2013, 8, e68942 CAS.
- J. Rybniker, J. M. Chen, C. Sala, R. C. Hartkoorn, A. Vocat, A. Benjak, S. Boy-Rottger, M. Zhang, R. Szekely, Z. Greff, L. Orfi, I. Szabadkai, J. Pato, G. Keri and S. T. Cole, Cell Host Microbe, 2014, 16, 538 CAS.
- G. C. Moraski, L. D. Markley, P. A. Hipskind, H. Boshoff, S. Cho, S. G. Franzblau and M. J. Miller, ACS Med. Chem. Lett., 2011, 2, 466 CrossRef CAS PubMed.
- K. A. Abrahams, J. A. G. Cox, V. L. Spivey, N. J. Loman, M. J. Pallen, C. Constantinidou, R. Fernández, C. Alemparte, M. J. Remuiñán, D. Barros, L. Ballell and G. S. Besra, PLoS One, 2012, 7, e52951 CAS.
- G. C. Moraski, P. A. Miller, M. A. Bailey, J. Ollinger, T. Parish, H. I. Boshoff, S. Cho, J. R. Anderson, S. Mulugeta, S. G. Franzblau and M. J. Miller, ACS Infect. Dis., 2015, 1, 85 CrossRef CAS PubMed.
- K. Pethe, P. Bifan, J. Jang, S. Kang, S. Park, S. Ahn, J. Jiricek, J. Jung, H. K. Jeon, J. Cechetto, T. Christophe, H. Lee, M. Kempf, M. Jackson, A. J. Lenaerts, H. Pham, V. Jones, M. J. Seo, Y. M. Kim, M. Seo, J. J. Seo, D. Park, Y. Ko, I. Cho, R. Kim, S. Y. Kim, S. B. Lim, S. Yim, J. Nam, H. Kang, H. Kwon, C.-T. Oh, Y. Cho, Y. Jang, J. Kim, A. Chua, B. H. Tan, M. B. Nanjundappa, S. P. S. Rao, W. S. Barnes, R. Wintjens, J. R. Walker, S. Alonso, S. Lee, J. Kim, S. Oh, T. Oh, U. Nehrbass, S.-J. Han, Z. No, J. Lee, P. Brodin, S.-N. Cho, K. Nam and J. Kim, Nat. Med., 2013, 19, 1157 CrossRef CAS PubMed.
- J. Rybniker, A. Vocat, C. Sala, P. Busso, F. Pojer, A. Benjak and S. T. Cole, Nat. Commun., 2015, 6, 7659 CrossRef PubMed.
- R. van der Westhuyzen, S. Winks, C. R. Wilson, G. A. Boyle, R. K. Gessner, C. S. de Melo, D. Taylor, C. de Kock, M. Njoroge, C. Brunschwig, N. Lawrence, S. P. S. Rao, F. Sirgel, P. van Helden, R. Seldon, A. Moosa, D. F. Warner, L. Arista, U. H. Manjunatha, P. W. Smith, L. J. Street and K. Chibale, J. Med. Chem., 2015, 58, 9371 CrossRef CAS PubMed.
- K. Arora, B. Ochoa-Montano, P. S. Tsang, T. L. Blundell, S. S. Dawes, V. Mizrahi, T. Bayliss, C. J. MacKenzie, L. A. T. Cleghorn, P. C. Ray, P. G. Wyatt, E. Uh, J. Lee, C. E. Barry 3rd and H. I. Boshoff, Antimicrob. Agents Chemother., 2014, 58, 6962 CrossRef CAS PubMed.
- L. Ballell, R. H. Bates, R. J. Young, D. Alvarez-Gomez, E. Alvarez-Ruiz, V. Barroso, D. Blanco, B. Crespo, J. Escribano, R. Gonzalez, S. Lozano, S. Huss, A. Santos-Villarejo, J. J. Martin-Plaza, A. Mendoza, M. J. Rebollo-Lopez, M. Remuinan-Blanco, J. L. Lavandera, E. Perez-Herran, F. J. Gamo-Benito, J. F. Garcia-Bustos, D. Barros, J. P. Castro and N. Cammack, ChemMedChem, 2013, 8, 313 CrossRef CAS PubMed.
- (a) K. Pissinate, A. D. Villela, V. Rodrigues-Junior, B. C. Giacobbo, E. S. Grams, B. L. Abbadi, R. V. Trindade, L. R. Nery, C. D. Bonan, D. F. Back, M. M. Campos, L. A. Basso, D. S. Santos and P. Machado, ACS Med. Chem. Lett., 2016, 7, 235 CrossRef CAS PubMed; (b) E. Pitta, M. K. Rogacki, O. Balabon, S. Huss, F. Cunningham, E. M. Lopez-Roman, J. Joossens, K. Augustyns, L. Ballell, R. H. Bates and P. Van der Veken, J. Med. Chem., 2016, 59, 6709 CrossRef CAS PubMed.
- N. Andreu, A. Zelmer, T. Fletcher, P. T. Elkington, T. H. Ward, J. Ripoll, T. Parish, G. J. Bancroft, U. Schaible, B. D. Robertson and S. Wiles, PLoS One, 2010, 5, e10777 Search PubMed.
- L. G. Matsoso, B. D. Kana, P. K. Crellin, D. J. Lea-Smith, A. Pelosi, D. Powell, S. S. Dawes, H. Rubin, R. L. Coppel and V. Mizrahi, J. Bacteriol., 2005, 187, 6300 CrossRef CAS PubMed.
- M. Berney, T. E. Hartman and W. R. Jacobs, mBio, 2014, 5, e01275–14 CrossRef PubMed.
- L. D. Marroquin, J. Hynes, J. A. Dykens, J. D. Jamieson and Y. Will, Toxicol. Sci., 2007, 97, 539 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available: Materials and methods, and characterisation of compounds 10a–f, 11a–x and 5, 9, 12a–12aa. See DOI: 10.1039/c6md00236f |
| This journal is © The Royal Society of Chemistry 2016 |