
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA potent trifluoromethyl ketone histone deacetylase inhibitor exhibits class-dependent mechanism of action
Andreas S.
Madsen
and
Christian A.
Olsen
*
Center for Biopharmaceuticals and Department of Drug Design & Pharmacology, University of Copenhagen, Universitetsparken 2, DK-2100, Copenhagen, Denmark. E-mail: cao@sund.ku.dk
First published on 15th December 2015
Abstract
Histone deacetylase (HDAC) enzymes are validated targets for treatment of certain cancers and have potential as targets for pharmacological intervention in a number of other diseases. Thus, inhibitors of these enzymes have received considerable attention, but these are often evaluated by IC50 value determination, which may vary significantly depending on assay conditions. In this work, we therefore performed detailed kinetic evaluation of inhibitors containing two fundamentally different zinc-binding chemotypes, hydroxamic acid or trifluoromethyl ketone. For the hydroxamic acids, a fast-on–fast-off mechanism was observed, but the trifluoromethyl ketone compound exhibited differential mechanisms depending on the enzyme isoform. The trifluoromethyl ketone compound displayed a fast-on–fast-off mechanism against class-IIa HDACs 4 and 7, but slow-binding mechanisms against class-I and class-IIb enzymes (HDACs 1–3, 6 and 8). Furthermore, different competitive, slow-binding mechanisms were observed for HDACs 1, 2, and 6 vs. HDACs 3 and 8, demonstrating the power of kinetic experiments for characterisation of enzyme inhibitors.
Introduction
The Zn2+-dependent histone deacetylase (HDAC) enzymes catalyse hydrolytic removal of acetyl functionalities from ε-amino groups of lysine residues in a variety of proteins including histones.1 Posttranslational modification (PTM) plays major roles in regulation of protein function, and histone acetylation affects chromatin packing and recruitment of transcription factors resulting in effects on gene expression.2,3 Recent discoveries have underpinned lysine acetylation as a general PTM in proteins,4 and a growing list of non-histone proteins are identified as substrates for the HDACs,5–8 thereby extending their potential impact on cellular function.2,3,9,10 Eleven HDAC isoforms are encoded by the human genome, classified according to sequence similarity:11 class-I (HDACs 1–3 and 8), class-IIa (HDACs 4, 5, 7 and 9), class-IIb (HDACs 6 and 10), and class-IV (HDAC11).Due to the importance of HDACs in a wide variety of disease states,12–15 selective HDAC inhibitors have potential as tool compounds and possibly as novel drugs. Several chemotypes have been described as HDAC inhibitors with the most extensively studied class being inhibitors featuring the strongly zinc-chelating hydroxamic acid moiety,16,17 including vorinostat (SAHA, 1) and trichostatin A (2) (Fig. 1). Due to the strong metal ion-chelating propensity of hydroxamic acids, inhibitors containing this functionality have been suspected to give rise to off-target effects, but recent experiments have indicated that this may be of minor concern.18,19 Nevertheless, hydroxamic acids have also been associated with rapid degradation and clearance, and investigation of other zinc-binding functionalities, such as thiols (as in romidepsin (3)),20 trifluoromethyl ketones (4),21–24 and o-aminoanilides (5)25,26 (Fig. 1), may therefore be of interest.
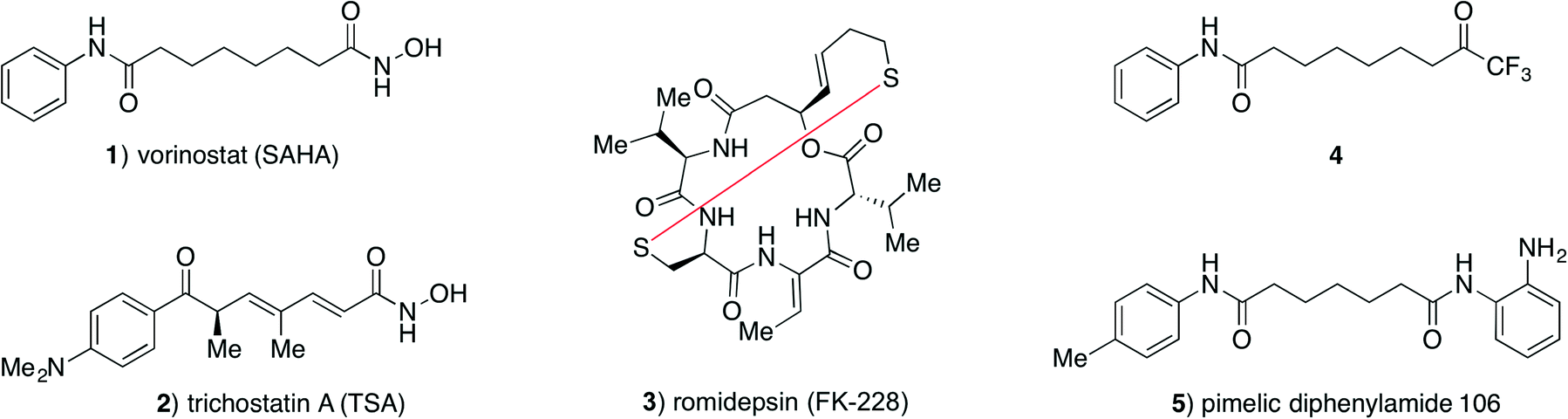 | ||
| Fig. 1 Structures of selected HDAC inhibitors. | ||
The binding mode of hydroxamic acids, which are generally considered potent class-I, -IIb and -IV inhibitors, but much poorer class-IIa inhibitors,27 is through bidentate coordination to the active site Zn2+ ion via the carbonyl oxygen and deprotonated nitrogen-bound oxygen atom.28
Trifluoromethyl ketones constitute another strongly zinc-binding functionality. This motif is readily hydrated in aqueous solution29 and presumably binds to the active site Zn2+ ion via bidentate coordination from the two resulting geminal hydroxy groups.30 Although trifluoromethyl ketones have also been reported to be metabolically labile by reduction to the corresponding alcohol in vivo,21 they may give rise to interesting tool compounds for investigation of HDAC inhibition in vitro. Interestingly, compounds containing this zinc-binding group have been reported to selectively inhibit class-IIa HDAC isoforms;22,23,31 however, class-IIa inhibition does not appear to be an inherent capacity of the functional group itself.32 We recently reinvestigated the inhibitory properties of a known trifluoromethyl ketone compound (4)21 and extended the previous studies to characterization of its inhibitory effect against all eleven zinc-dependent HDACs,32 thereby identifying compound 4 as a promising inhibitor of isozymes from all four HDAC classes.32 In our previous study, IC50 and Ki values were determined from standard endpoint dose–response assays assuming a fast-on–fast-off binding mechanism. In these experiments, the HDAC enzyme and fluorogenic substrate are incubated with varying concentrations of inhibitor for 1 h, followed by assay development by addition of trypsin to achieve fluorophore release. Thus, a constant rate over the entire incubation period has to be assumed in order to apply the Cheng–Prusoff equation for approximation of the corresponding Ki values.
While endpoint assays are convenient for screening large numbers of inhibitors, more information can be obtained from kinetic inhibition experiments, where progression of enzyme activity is monitored over time. By applying such assays, Gottesfeld and co-workers demonstrated that pimelic diphenylamide 106 (5), an o-aminoanilide based class-I HDAC inhibitor, inhibits HDACs 1–3 via different mechanisms not revealed by simple IC50 determination. They were instead able to show that compound 5 is a slow, tight-binding inhibitor of HDACs 1–3, but acting via a different mechanism for HDACs 1 and 2 compared to HDAC3.26 More recently, two groups have also reported on the development of kinetically selective HDAC2 inhibitors based on o-aminoanilide moieties, and considering differences in the active sites of HDACs 1–3.33,34
Interestingly, trifluoromethyl ketones have previously been described as slow-binding inhibitors for serine proteases,29,35,36 and we therefore found it interesting to perform a more elaborate investigation of the inhibitory mechanism of compound 4 on the different HDAC isoforms. We have previously used continuous rate experiments for investigating inhibitors of the NAD+-dependent deacylase sirtuin 537 as well as cyclic peptide HDAC inhibitors38 and thought we could modify those protocols for evaluation of compound 4 in continuous assays against a selection of the zinc-dependent HDACs.
Not surprisingly, our investigation showed that the hydroxamic acid-containing inhibitors acted via a fast-on–fast-off binding mechanism against all the tested enzymes. For the trifluoromethyl ketone inhibitor, on the other hand, the mechanism of inhibition proved to vary between enzyme isoforms. Thus, this study provides valuable mechanistic insight for future medicinal chemistry efforts involving HDAC inhibitors containing this zinc-binding group.
Results and discussion
Initially, we investigated the influence of preincubation of HDAC3 and trifluoromethyl ketone 4 on the determined IC50 values and found a 17-fold increase in apparent inhibitor potency at 2 h of preincubation time (Fig. 2). Further incubation lead to substantial decrease in enzyme activity, even without addition of inhibitor, thus precluding prolonged preincubation due to enzyme degradation in the assay buffer. This initial observation prompted us to investigate the kinetics of HDAC inhibition in more detail.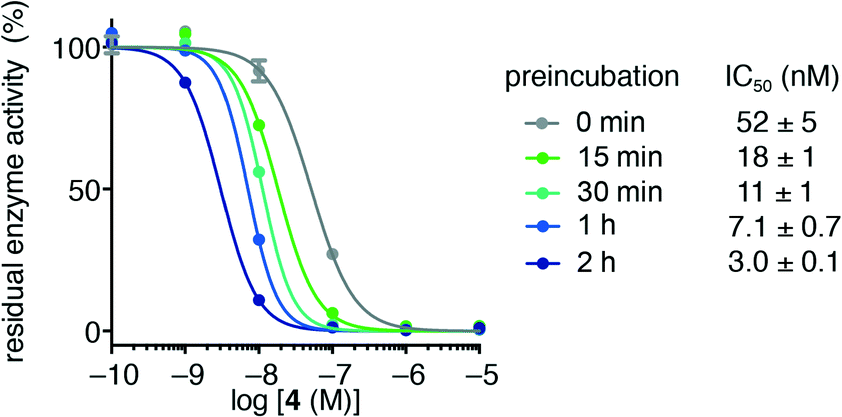 | ||
| Fig. 2 Residual activity of HDAC3 after preincubation with trifluoromethyl ketone 4. Preincubation times and apparent IC50 values are shown. | ||
Usually, the equilibrium of inhibitor binding to enzyme is assumed to be rapidly obtained, i.e., fast on and off rates for the enzyme–inhibitor equilibrium, which is therefore obtained prior to onset of the experiment. For this fast-on–fast-off mechanism, linear progression curves are observed when monitoring product formation continuously, and the enzyme–inhibitor equilibrium constant (Ki) can be determined by fitting a secondary plot of the relative rates (vi/vo) to eqn (1), where vi and vo are the observed rates with and without inhibitor, respectively.
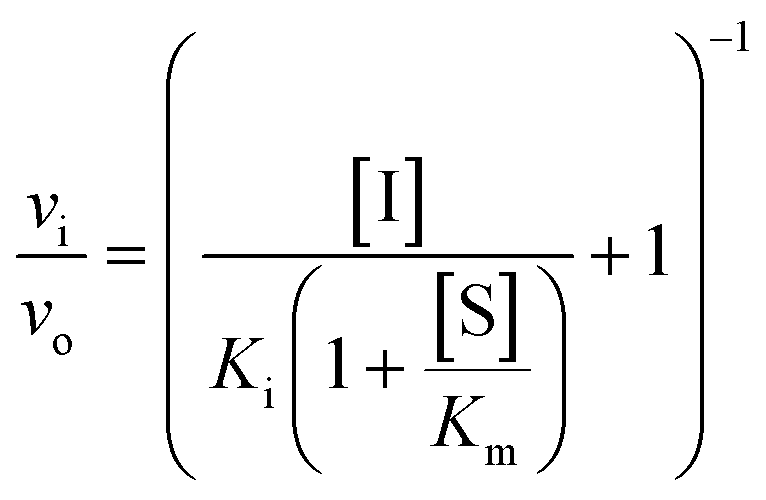 | (1) |
However, if the enzyme–inhibitor equilibrium is slow and thus not obtained prior to onset of the experiment, non-linear progression curves will be obtained. In such an event, the slow, competitive binding of inhibitor may be divided into two mechanisms as shown in Fig. 3. In mechanism A, the enzyme–inhibitor complex forms without kinetically observable intermediates, whereas in mechanism B, an equilibrium is rapidly established with an initial complex, which is then converted to a second stable complex displaying rate-determining kinetics.
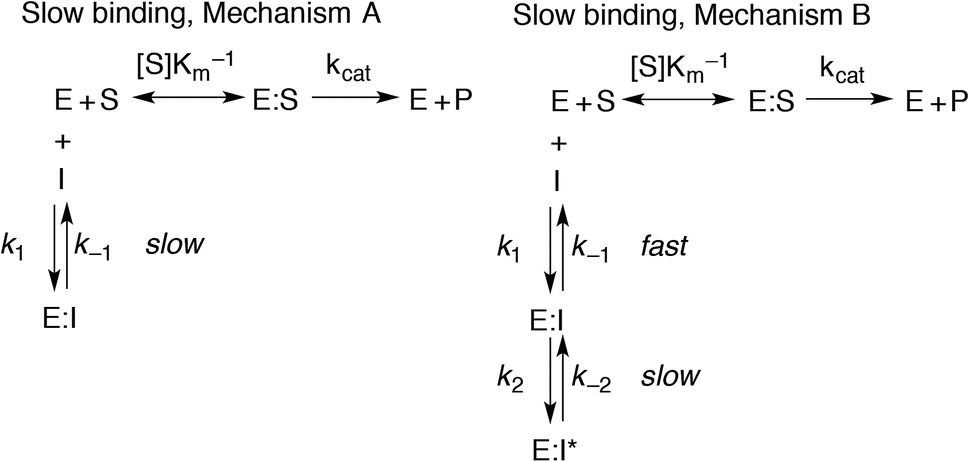 | ||
| Fig. 3 Mechanism of slow inhibitor binding with the corresponding rate constants. | ||
For both types of slow binding kinetics, product formation follows eqn (2), where [P] is the concentration of product at time t. The vo and vs are the initial and final steady-state velocities, respectively, and kobs is the apparent first-order rate constant for establishment of the enzyme–inhibitor equilibrium. It should be noted, that for the fast-on–fast-off mechanism, [P] = vst is the limiting case of mechanism A when kobs is very large.
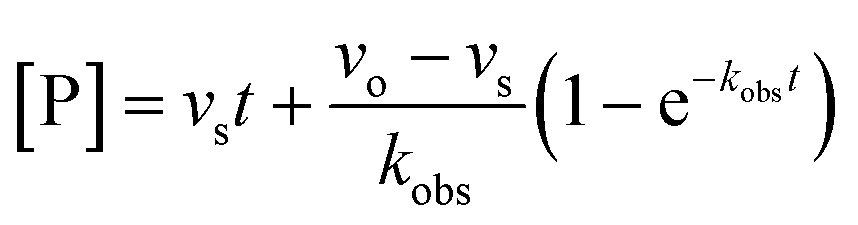 | (2) |
For mechanisms A and B, the relationship between kobs and [I] is linear (eqn (3)) and hyperbolic (eqn (4)), respectively. As a result, fitting of these equations to secondary plots of the kobs–[I] relationship allows determination of the binding mechanism, as well as calculation of the rate constants involved and the resulting enzyme–inhibitor equilibrium constants [Ki for mechanism A (eqn (5)) and Ki,1 and Ki for mechanism B (eqn (6) and (7), respectively)].
Slow binding, mechanism A
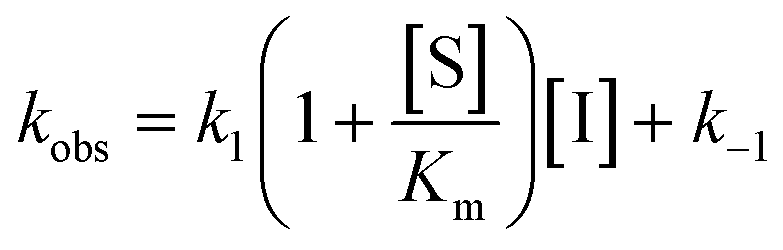 | (3) |
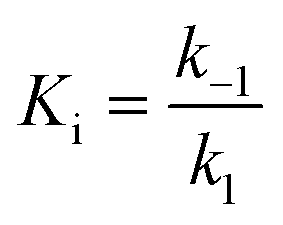 | (5) |
Slow binding, mechanism B
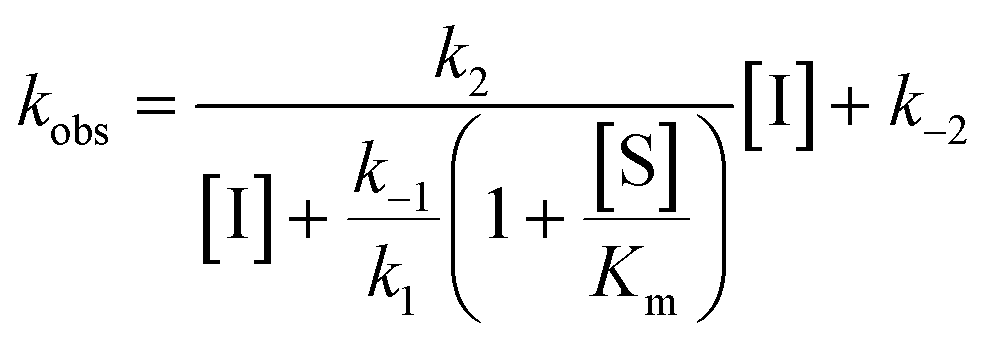 | (4) |
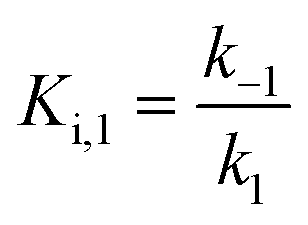 | (6) |
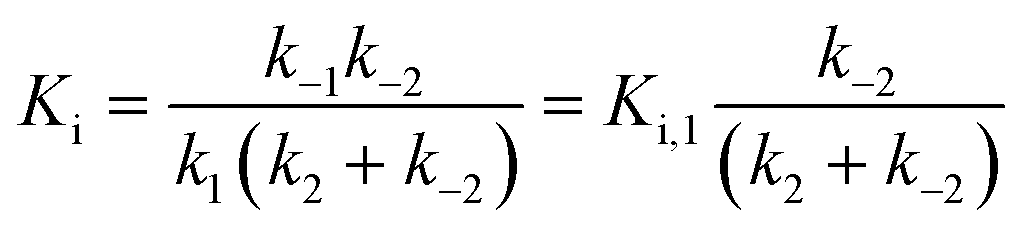 | (7) |
To determine the inhibitory mechanism and associated kinetic values, we initially established assay conditions that allowed monitoring of enzyme activity for 60 min. Progression curves were generated by incubating the enzyme–inhibitor combination with an appropriate fluorogenic substrate at concentrations greater than the Km [Ac-LGKac-AMC (20 μM) for HDACs 1–3 and 6 (Km values 6 μM, 3 μM, 6 μM, and 16 μM, respectively), Ac-LGKtfa-AMC (50 μM) for HDACs 4 and 7 (Km values 12 μM and 26 μM, respectively), and Ac-RHKacKac-AMC (400 μM) for HDAC8 (Km value 430 μM)]. The trypsin concentration was optimised to be high enough to ensure rapid establishment of steady state by cleaving the AMC fluorophore from the deacylated peptide intermediate at a rate that avoids lag time. At the same time, the trypsin concentration should be low enough to ensure a linear progress curve for 60 min without observable reduction of HDAC activity in the experiment without inhibitor added. Previously, Gottesfeld and co-workers reported low stability of HDAC8 under the employed conditions.26 In agreement with this, we also observed rapid loss of activity of HDAC8 when incubated in the standard Tris buffer. However, changing the buffer to either the buffer marketed for HDAC8 (a Tris buffer containing PEG8kDa) or the buffer described by Bradner, Mazitschek and co-workers (a HEPES buffer, data not shown),27 allowed monitoring of HDAC8 activity for 60 min as well.
Having established suitable conditions for all the investigated enzymes, a series of progress curves were generated for HDACs 1–4 and 6–8 against the three investigated inhibitors (1, 2 and 4).
SAHA (1) has previously been demonstrated to be a poor class-IIa inhibitor32 and was not included for HDACs 4 and 7. Instead, TSA was chosen as the hydroxamic acid inhibitor against class-IIa HDACs and HDAC3 was chosen as the representative for TSA inhibition of class-I HDACs. All seven HDACs were included for investigation of compound 4.
Hydroxamate inhibitors such as SAHA have been reported to display fast-on–fast-off kinetics against class-I HDACs,26 and in agreement with these reports, linear progress curves were obtained for inhibition of HDACs 1–3 and 8 as well as the class-IIb enzyme HDAC6 when monitored for 1 h (Fig. 4). Low nanomolar inhibition constants were observed for all investigated enzymes except for HDAC8 (230 ± 20 nM; Table 1), and all values are in agreement with the literature.27 Similarly, linear progress curves were obtained for TSA inhibition of all investigated enzymes, including class-IIa enzymes HDAC4 and HDAC7 (Fig. 4). Despite a marked span of potencies of almost 2000-fold (0.54 ± 0.02 nM vs. 980 ± 80 nM for HDACs 3 and 4, respectively), these results indicate that fast-on–fast-off kinetics is general for hydroxamate mediated HDAC inhibition regardless of enzyme class and potency.
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC6 | HDAC4 | HDAC7 | |
|---|---|---|---|---|---|---|---|
| 1 | 6.4 ± 0.2 | 4.2 ± 0.1 | 9.9 ± 0.3 | 230 ± 20 | 17.7 ± 0.3 | n.d. | n.d. |
| 2 | n.d. | n.d. | 0.54 ± 0.02 | 200 ± 20 | n.d. | 980 ± 80 | 300 ± 10 |
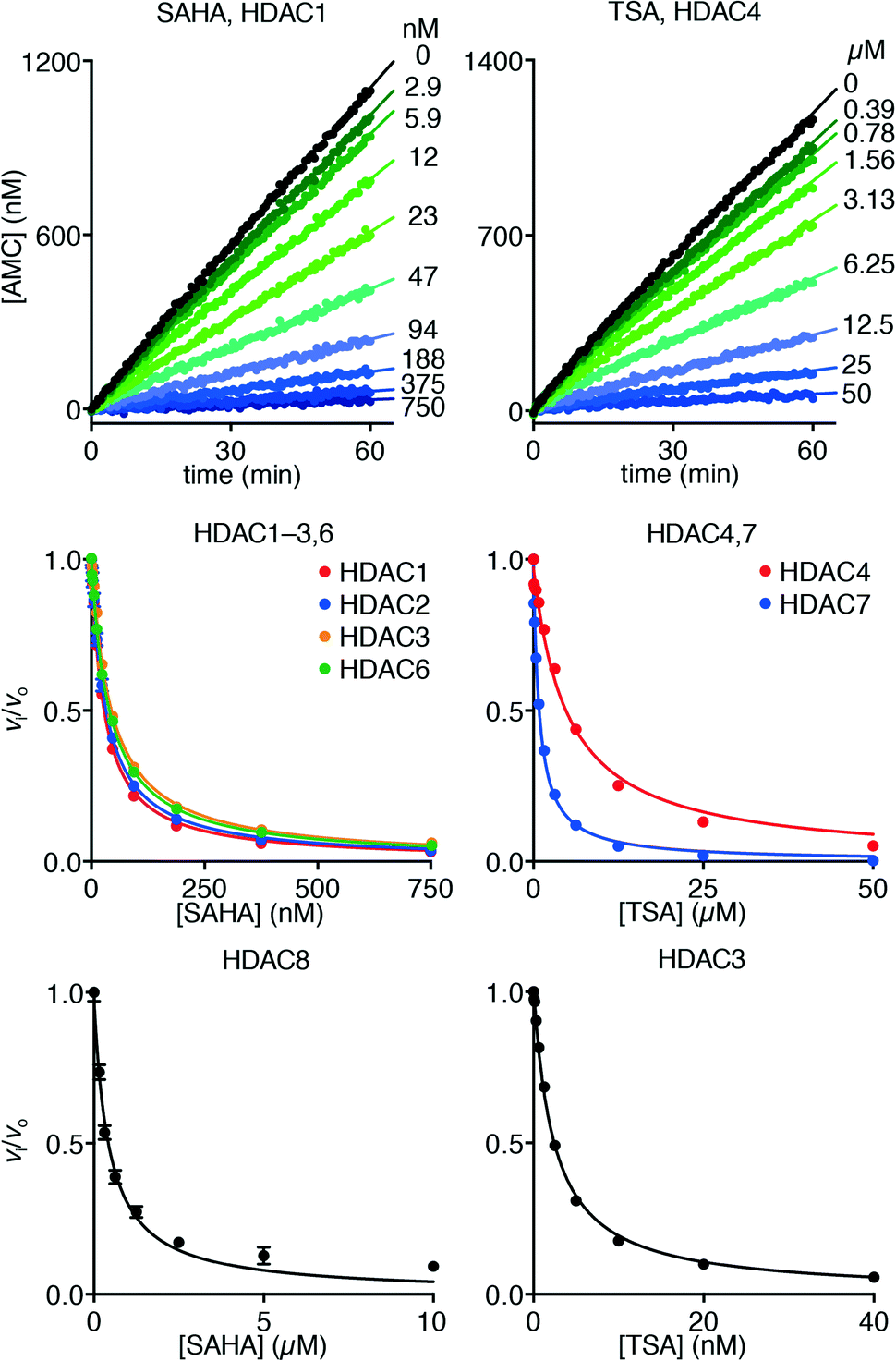 | ||
| Fig. 4 Progress curves of HDAC1 (LGKac) and HDAC4 (LGKtfa) deacylase activities under SAHA and TSA inhibition, respectively. Relative rates (vi/vo), fitted to eqn (1), for HDACs 1–3 and 6 (LGKac), and HDAC8 (RHKacKac) against concentrations of SAHA and HDACs 3, 4 and 7 (LGKtfa) against concentrations of TSA. | ||
Interestingly, trifluoromethyl ketone 4 displayed a differential inhibition mechanism depending on enzyme class. Thus compound 4 was found to be a potent inhibitor of class-IIa enzymes displaying a fast-on–fast-off mechanism with Ki values of 5.1 ± 0.2 nM and 4.7 ± 0.1 nM against HDACs 4 and 7, respectively (Fig. 5).
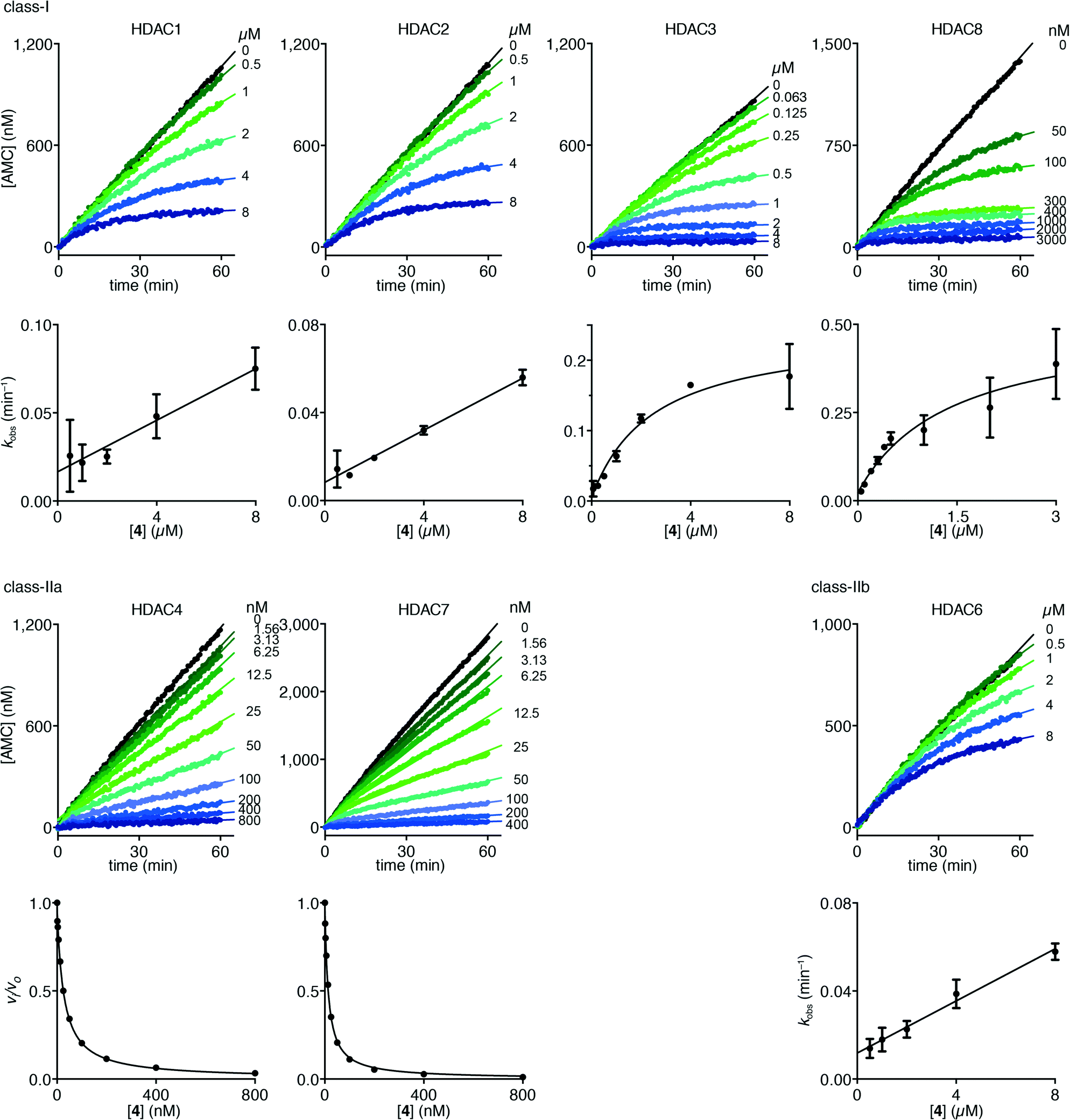 | ||
| Fig. 5 Progress curves of HDACs 1–3 and 6 (LGKac), HDAC8 (RHKacKac), and HDACs 4 and 7 (LGKtfa) deacylase activities under inhibition by trifluoromethyl ketone 4. Apparent first-order rate constants (kobs) for trifluoromethyl ketone 4 inhibition were fitted to eqn (3) for HDACs 1, 2 and 6 (LGKac) or eqn (4) for HDAC3 and relative rates (vi/vo) were fitted to eqn (1) for HDACs 4 and 7. | ||
In contrast, compound 4 displayed slow binding kinetics with non-linear progression curves when incubated with all the investigated class-I and class-IIb enzymes. The obtained progression curves were fitted to eqn (2) and secondary plots of the calculated kobs values against inhibitor concentration were created (Fig. 5). For HDACs 1, 2 and 6, a linear relationship was observed, consistent with competitive, slow-binding of the inhibitor following mechanism A, and the data were therefore fitted to eqn (3) to determine the kinetic parameters. The overall inhibition constants (Ki) were determined to be 9.8 μM, 11 μM and 4.5 μM, respectively. For HDACs 1 and 6, these are in good agreement with our previous findings using endpoint experiments, while the Ki value against HDAC2 is ∼30-fold higher than the previously determined.32 The overall trend that HDACs 4 and 7 are significantly more potently inhibited than HDACs 1, 2 and 6, however, corresponds well with previous findings.
For HDACs 3 and 8, on the other hand, a hyperbolic relationship between kobs and inhibitor concentration was indicated, which is consistent with competitive, slow binding of the inhibitor following mechanism B. Thus, fitting of the data to eqn (4) allowed determination of the accompanying kinetic parameters (Table 2). For HDAC3 the overall inhibition constant (Ki) was estimated to be 11 nM and the overall inhibition constant (Ki) for HDAC8 was estimated to be 33 nM, which is in the range of inhibition constants for class-IIa HDACs 4 and 7. For HDAC3, the Ki value is an order of magnitude lower than previously estimated from endpoint experiments using the Cheng–Prusoff equation,32 which may be explained by a low k−2 value indicating a highly stable enzyme–inhibitor complex (E:I*, Fig. 3 mechanism B). This also helps explain the data obtained by preincubation experiments discussed above (Fig. 2), and underlines the power of kinetic evaluation of inhibitors that do not follow standard fast-on–fast-off mechanism of action.
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC6 | HDAC4 | HDAC7 | |
|---|---|---|---|---|---|---|---|
| K i (nM) | (9.8 ± 6.7) × 103 | (1.1 ± 0.4) × 104 | ∼11 | ∼33 | (4.5 ± 1.6) × 103 | 5.1 ± 0.2 | 4.7 ± 0.1 |
| k 1 (M−1 min−1) | (1.7 ± 0.4) × 103 | (7.7 ± 0.8) × 102 | — | — | (2.6 ± 0.3) × 103 | — | — |
| k −1 (min−1) | (1.7 ± 0.7) × 10−2 | (8 ± 2) × 10−3 | — | — | (1.2 ± 0.3) × 10−2 | — | — |
| K i,1 (nM) | — | — | (5.7 ± 2.6) × 102 | (7.3 ± 5.7) × 103 | — | — | — |
| k 2 (min−1) | — | — | 0.24 ± 0.03 | 0.49 ± 0.13 | — | — | — |
| k −2 (min−1) | — | — | (5 ± 11) × 10−3 | (2.3 ± 3.7) × 10−2 | — | — | — |
Interestingly, potent inhibition of HDACs 3, 4, 7 and 8 by trifluoromethyl ketone 4 correlates well with the observation that these enzymes all efficiently cleave ε-N-trifluoroacetylated lysine substrates.39 Furthermore, the lower affinities for compound 4 against HDACs 1, 2 and 6 also agree with the low detrifluoroacetylase activities of these enzymes. Taken together, these observations suggest that certain HDACs accommodate the trifluoromethyl group better than others, and that this feature is not in accordance with isoform sub-classification.
Conclusions
In summary, we have evaluated hydroxamate and trifluoromethyl ketone HDAC inhibitors by continuous assays monitoring the progress of substrate conversion over time to enable kinetic insight. We selected enzymes from class-I (HDACs 1–3 and 8), class-IIa (HDACs 4 and 7), and class-IIb (HDAC6), and as suspected based on literature precedent, the hydroxamic acids all exhibited a fast-on–fast-off mechanism of inhibition. However, the SAHA-inspired trifluoromethyl ketone (4) only displayed a fast-on–fast-off mechanism against HDACs 4 and 7, whereas slow-binding mechanisms were observed against class-I and class-IIb enzymes (HDACs 1–3, 6 and 8). Furthermore, a more detailed investigation revealed similar mechanisms for HDACs 1, 2, and 6, which were different from the mechanism exhibited against HDACs 3 and 8. This insight is important for understanding the effects of trifluoromethyl ketone containing inhibitors, and should be taken into account when designing novel chemotypes containing this moiety. Furthermore, it will be interesting to investigate the possible effects of these mechanistic differences as well as kinetics of ketone reduction in cellular environments.Experimental
Materials
HDAC1, HDAC2, HDAC3–NCoR1, HDAC6 and HDAC8 were purchased from Enzo Life Sciences (Postfach, Switzerland). HDAC4 and HDAC7 were purchased from Millipore (Upstate, Temecula, CA). The HDAC assay buffers used were: Buffer A, Biomol International BML-KI-143 [Tris/Cl (50 mM), NaCl (137 μM), KCl (2.7 μM), MgCl2 (1 μM), pH 8.0] with BSA (0.5 mg mL−1) added; or Buffer B, Biomol International BML-KI-311 [Tris/Cl (50 mM), NaCl (137 μM), KCl (2.7 μM), MgCl2 (1 μM), PEG-8 kDa (10% w/v), pH 8.0] with BSA (0.5 mg mL−1) added. Trichostatin A and trypsin (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per mg, TPCK treated from bovine pancreas) were from Sigma-Aldrich (Steinheim, Germany), and syntheses of SAHA and compound 4 were previously reported.32 All chemicals and solvents were of analytical grade and were used without further purification as obtained from commercial suppliers.
000 units per mg, TPCK treated from bovine pancreas) were from Sigma-Aldrich (Steinheim, Germany), and syntheses of SAHA and compound 4 were previously reported.32 All chemicals and solvents were of analytical grade and were used without further purification as obtained from commercial suppliers.
Fluorescence measurements
All microtiter plates were analysed using a Perkin Elmer EnSpire plate reader with excitation at 360 nm and detecting emission at 460 nm. Fluorescence measurements (RFU) were converted to [AMC] concentrations based on an [AMC]-fluorescence standard curve and all data analysis were performed using GraphPad Prism.Fluorescence-based preincubation inhibition experiments
Hydrolase activity of HDAC3-NCoR1 against substrate Ac-LGKac-AMC was evaluated under varying concentrations of trifluoromethyl ketone 4 and preincubation times. HDAC3 (1000 pg μL−1, 10 μL) was added to a solution of trifluoromethyl ketone 4 (30 μL total volume, 16.7 μM–167 pM, 10-fold dilution) at 37 °C (final concentrations 250 pg μL−1 and 12.5 μM–125 pM, respectively) with preincubation times of 2 h, 1 h, 30 min, 15 min and 0 min, then a solution of Ac-LGKac-AMC (10 μL, 50 μM) was added to each well and the plate was incubated for additional 30 min at 37 °C (final concentrations HDAC3: 200 pg μL−1, 4: 10 μM–100 pM, and Ac-LGKac-AMC: 10 μM, respectively). Then a solution of trypsin (50 μL, 0.4 mg mL−1) was added and the reaction mixture was developed for 15 min before analysing fluorescence. The resulting data were analysed to obtain residual enzymatic activity relative to control wells.Fluorescence-based steady-state rate inhibition experiments
Hydrolase activities of HDACs against substrates Ac-LGKac-AMC, Ac-LGKtfa-AMC and Ac-RHKacKac-AMC were evaluated under varying concentrations of SAHA (1), TSA (2), and trifluoromethyl ketone 4. The HDAC enzyme was incubated with the relevant substrate, inhibitor, and trypsin in a total volume of 100 μL of assay buffer (Buffer A, except for HDAC8, where Buffer B was used), using the following concentrations: HDAC1 (2500 pg μL−1), LGKac (20 μM), trypsin (3 ng μL−1); HDAC2 (4000 pg μL−1), LGKac (20 μM), trypsin (3 ng μL−1); HDAC3-NCoR1 (200 pg μL−1), LGKac (20 μM), trypsin (3 ng μL−1); HDAC4 (50 pg μL−1), LGKtfa (50 μM), trypsin (5 ng μL−1); HDAC6 (4000 pg μL−1), LGKac (20 μM), trypsin (3 ng μL−1); HDAC7 (2.5 pg μL−1), LGKtfa (50 μM), trypsin (5 ng μL−1); HDAC8 (5 pg μL−1), LGKtfa (200 μM), trypsin (3 ng μL−1); HDAC8 (400 pg μL−1), RHKacKac (400 μM), trypsin (5 ng μL−1). In situ fluorophore release was monitored immediately by fluorescence readings recorded continuously every 10–15 seconds for 60 min at 25 °C. Using GraphPad Prism, background fluorescence was subtracted and the data were fitted to the relevant equations ([P] = vst, or eqn (2)). Secondary plots were then fitted to the relevant equations (eqn (1), (3) or (4)) to obtain the desired kinetic parameters.Acknowledgements
Financial support from the Lundbeck Foundation (Young Group Leader Fellowship, C. A. O.), the Danish Independent Research Council–Natural Sciences (Steno grant no. 10-080907, C. A. O.), the Danish Independent Research Council–Technical and Production Sciences (Sapere Aude grant no. 12-132328, A. S. M.), and the Carlsberg Foundation (equipment grant, C. A. O.) is gratefully acknowledged.Notes and references
- T. Kouzarides, Cell, 2007, 128, 693–705 CrossRef PubMed.
- M. Biel, V. Wascholowski and A. Giannis, Angew. Chem., Int. Ed., 2005, 44, 3186–3216 CrossRef CAS PubMed.
- M. Haberland, R. L. Montgomery and E. N. Olson, Nat. Rev. Genet., 2009, 10, 32–42 CrossRef CAS PubMed.
- C. Choudhary, C. Kumar, F. Gnad, M. L. Nielsen, M. Rehman, T. C. Walther, J. V. Olsen and M. Mann, Science, 2009, 325, 834–840 CrossRef CAS PubMed.
- M. D. Hirschey, Cell Metab., 2011, 14, 718–719 CrossRef CAS PubMed.
- C. A. Olsen, ChemMedChem, 2014, 9, 434–437 CrossRef CAS PubMed.
- C. A. Olsen, Angew. Chem., Int. Ed., 2012, 51, 3755–3756 CrossRef CAS PubMed.
- C. Choudhary, B. T. Weinert, Y. Nishida, E. Verdin and M. Mann, Nat. Rev. Mol. Cell Biol., 2014, 15, 536–550 CrossRef CAS PubMed.
- S. Zhao, W. Xu, W. Jiang, W. Yu, Y. Lin, T. Zhang, J. Yao, L. Zhou, Y. Zeng, H. Li, Y. Li, J. Shi, W. An, S. M. Hancock, F. He, L. Qin, J. Chin, P. Yang, X. Chen, Q. Lei, Y. Xiong and K. L. Guan, Science, 2010, 327, 1000–1004 CrossRef CAS PubMed.
- Q. Wang, Y. Zhang, C. Yang, H. Xiong, Y. Lin, J. Yao, H. Li, L. Xie, W. Zhao, Y. Yao, Z. B. Ning, R. Zeng, Y. Xiong, K. L. Guan, S. Zhao and G. P. Zhao, Science, 2010, 327, 1004–1007 CrossRef CAS PubMed.
- I. V. Gregoretti, Y. M. Lee and H. V. Goodson, J. Mol. Biol., 2004, 338, 17–31 CrossRef CAS PubMed.
- S. Minucci and P. G. Pelicci, Nat. Rev. Cancer, 2006, 6, 38–51 CrossRef CAS PubMed.
- J. E. Bolden, M. J. Peart and R. W. Johnstone, Nat. Rev. Drug Discovery, 2006, 5, 769–784 CrossRef CAS PubMed.
- P. A. Cole, Nat. Chem. Biol., 2008, 4, 590–597 CrossRef CAS PubMed.
- M. Paris, M. Porcelloni, M. Binaschi and D. Fattori, J. Med. Chem., 2008, 51, 1505–1529 CrossRef CAS PubMed.
- F. F. Wagner, M. Weïwer, M. C. Lewis and E. B. Holson, Neurotherapeutics, 2013, 10, 589–604 CrossRef CAS PubMed.
- P. Bertrand, Eur. J. Med. Chem., 2010, 45, 2095–2116 CrossRef CAS PubMed.
- Y. Chen and S. M. Cohen, ChemMedChem, 2015, 10, 1733–1738 CrossRef CAS PubMed.
- J. A. Day and S. M. Cohen, J. Med. Chem., 2013, 56, 7997–8007 CrossRef CAS PubMed.
- R. Furumai, A. Matsuyama, N. Kobashi, K. H. Lee, M. Nishiyama, H. Nakajima, A. Tanaka, Y. Komatsu, N. Nishino, M. Yoshida and S. Horinouchi, Cancer Res., 2002, 62, 4916–4921 CAS.
- R. R. Frey, C. K. Wada, R. B. Garland, M. L. Curtin, M. R. Michaelides, J. L. Li, L. J. Pease, K. B. Glaser, P. A. Marcotte, J. J. Bouska, S. S. Murphy and S. K. Davidsen, Bioorg. Med. Chem. Lett., 2002, 12, 3443–3447 CrossRef CAS PubMed.
- P. Jones, M. J. Bottomley, A. Carfí, O. Cecchetti, F. Ferrigno, P. Lo Surdo, J. M. Ontoria, M. Rowley, R. Scarpelli, C. Schultz-Fademrecht and C. Steinkühler, Bioorg. Med. Chem. Lett., 2008, 18, 3456–3461 CrossRef CAS PubMed.
- M. J. Bottomley, P. Lo Surdo, P. Di Giovine, A. Cirillo, R. Scarpelli, F. Ferrigno, P. Jones, P. Neddermann, R. de Francesco, C. Steinkühler, P. Gallinari and A. Carfí, J. Biol. Chem., 2008, 283, 26694–26704 CrossRef CAS PubMed.
- M. Ilies, D. P. Dowling, P. M. Lombardi and D. W. Christianson, Bioorg. Med. Chem. Lett., 2011, 21, 5854–5858 CrossRef CAS PubMed.
- A. Saito, T. Yamashita, Y. Mariko, Y. Nosaka, K. Tsuchiya, T. Ando, T. Suzuki, T. Tsuruo and O. Nakanishi, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4592–4597 CrossRef CAS.
- C. J. Chou, D. Herman and J. M. Gottesfeld, J. Biol. Chem., 2008, 283, 35402–35409 CrossRef CAS PubMed.
- J. E. Bradner, N. West, M. L. Grachan, E. F. Greenberg, S. J. Haggarty, T. Warnow and R. Mazitschek, Nat. Chem. Biol., 2010, 6, 238–243 CrossRef CAS PubMed.
- M. S. Finnin, J. R. Donigian, A. Cohen, V. M. Richon, R. A. Rifkind, P. A. Marks, R. Breslow and N. P. Pavletich, Nature, 1999, 401, 188–193 CrossRef CAS PubMed.
- K. N. Allen and R. H. Abeles, Biochemistry, 1989, 28, 8466–8473 CrossRef CAS PubMed.
- T. K. Nielsen, C. Hildmann, D. Riester, D. Wegener, A. Schwienhorst and R. Ficner, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2007, 63, 270–273 CrossRef CAS PubMed.
- E. Muraglia, S. Altamura, D. Branca, O. Cecchetti, F. Ferrigno, M. V. Orsale, M. C. Palumbi, M. Rowley, R. Scarpelli, C. Steinkühler and P. Jones, Bioorg. Med. Chem. Lett., 2008, 18, 6083–6087 CrossRef CAS PubMed.
- A. S. Madsen, H. M. Kristensen, G. Lanz and C. A. Olsen, ChemMedChem, 2014, 9, 614–626 CrossRef CAS PubMed.
- F. F. Wagner, Y. L. Zhang, D. M. Fass, N. Joseph, J. P. Gale, M. Weïwer, P. McCarren, S. L. Fisher, T. Kaya, W. N. Zhao, S. A. Reis, K. M. Hennig, M. Thomas, B. C. Lemercier, M. C. Lewis, J. S. Guan, M. P. Moyer, E. Scolnick, S. J. Haggarty, L. H. Tsai and E. B. Holson, Chem. Sci., 2015, 6, 804–815 RSC.
- J. Zhou, M. Li, N. Chen, S. Wang, H. B. Luo, Y. Zhang and R. Wu, ACS Chem. Biol., 2015, 10, 687–692 CrossRef CAS PubMed.
- K. Brady and R. H. Abeles, Biochemistry, 1990, 29, 7608–7617 CrossRef CAS PubMed.
- M. H. Gelb, J. P. Svaren and R. H. Abeles, Biochemistry, 1985, 24, 1813–1817 CrossRef CAS PubMed.
- A. S. Madsen and C. A. Olsen, J. Med. Chem., 2012, 55, 5582–5590 CrossRef CAS PubMed.
- J. S. Villadsen, B. Kitir, K. Wich, T. Friis, A. S. Madsen and C. A. Olsen, Med. Chem. Commun., 2014, 5, 1849–1855 RSC.
- A. S. Madsen and C. A. Olsen, Angew. Chem., Int. Ed., 2012, 51, 9083–9087 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |