DOI:
10.1039/C6MB00235H
(Paper)
Mol. BioSyst., 2016,
12, 3576-3588
Spectroscopic evidence for the role of a site of the di-iron catalytic center of ferritins in tuning the kinetics of Fe(II) oxidation†
Received
31st March 2016
, Accepted 21st September 2016
First published on 21st September 2016
Abstract
Ferritin is a nanocage protein made of 24 subunits. Its major role is to manage intracellular concentrations of free Fe(II) and Fe(III) ions, which is pivotal for iron homeostasis across all domains of life. This function of the protein is regulated by a conserved di-iron catalytic center and has been the subject of extensive studies over the past 50 years. Yet, it has not been fully understood how Fe(II) is oxidized in the di-iron catalytic center and it is not known why eukaryotic and microbial ferritins oxidize Fe(II) with different kinetics. In an attempt to obtain a new insight into the mechanism of Fe(II) oxidation and understand the origin of the observed differences in the catalysis of Fe(II) oxidation among ferritins we studied and compared the mechanism of Fe(II) oxidation in the eukaryotic human H-type ferritin (HuHF) and the archaeal ferritin from Pyrococcus furiosus (PfFtn). The results show that the spectroscopic characteristics of the intermediate of Fe(II) oxidation and the Fe(III)-products are the same in these two ferritins supporting the proposal of unity in the mechanism of Fe(II) oxidation among eukaryotic and microbial ferritins. Moreover, we observed that a site in the di-iron catalytic center controls the distribution of Fe(II) among subunits of HuHF and PfFtn differently. This observation explains the reported differences between HuHF and PfFtn in the kinetics of Fe(II) oxidation and the amount of O2 consumed per Fe(II) oxidized. These results provide a fresh understanding of the mechanism of Fe(II) oxidation by ferritins.
Introduction
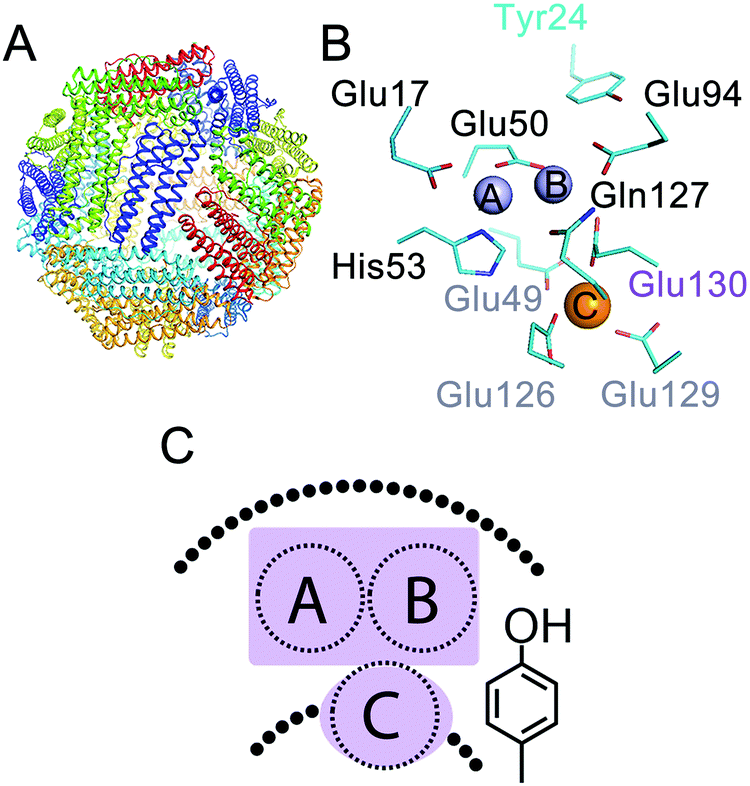
The 24-meric ferritin (Fig. 1A) has a nanocage-like structure, which has found a wide range of applications1–3 in nanotechnology, biocatalysis, and medicine. The major physiological role of ferritin is to manage intracellular concentrations of free Fe(II) and Fe(III) ions. This key function of proteins depends on the oxidation of Fe(II) to Fe(III) in the di-iron center of the catalytically active subunits. This unique di-iron center is known as the ferroxidase center (sites A and B in Fig. 1B). A third transient site, known as site C, has been identified as a gateway to the ferroxidase center in eukaryotic,4–6 bacterial,7,8 and archaeal ferritins1,4 (Fig. 1B and C). The overall mechanism of Fe(III) storage in ferritins can be defined as: (i) Fe(II) entry and access to the ferroxidase center, (ii) Fe(II) oxidation at the ferroxidase center, and (iii) Fe(III) storage in the central cavity. The Fe(II) ions reach the ferroxidase center through the protein shell.5,9–12 Oxidation of Fe(II) occurs in the ferroxidase center and at site C.1,13 The mechanism of Fe(II) oxidation is not fully understood. Previous studies have led to the proposal of different models for the mechanism of Fe(II) oxidation in eukaryotic and microbial ferritins.14–16 While for eukaryotic human H-type ferritin (HuHF)17 and bullfrog M-type ferritin (BfMF)18 it is proposed that under single turnover conditions, i.e. addition of Fe(II) per subunit ≤2, two Fe(II) are simultaneously oxidized in each ferroxidase center, for human mitochondrial ferritin it is proposed that less ferroxidase centers are active and Fe(II) might be oxidized by the Fe(III) mineral core.19 For BfMF18 and BfHF20 similar Mössbauer data obtained during the catalytic reaction have been interpreted differently to reflect different mechanisms of Fe(II) oxidation. It has been proposed that in BfMF Fe(II) is oxidized via a peroxodiferric intermediate, while in BfHF Fe(II) is oxidized via a tyrosine radical. On the other hand it is believed that in E.coli ferritin A (EcFtnA) three Fe(II) are simultaneously oxidized in sites A, B, and C.21 On the basis of these data the diversity view has emerged claiming that the mechanism of Fe(II) oxidation and Fe(III) storage varies among ferritins.14,15,22 In contrast our studies of the hyperthermophilic archaeal ferritin from Pyrococcus furiosus (PfFtn) and HuHF in comparison showed that in eukaryotic and microbial ferritins Fe(III) stays metastably in the ferroxidase center and is displaced by the incoming Fe(II).4 This displacement of Fe(III) by Fe(II) was proposed to be the basis of a common mechanism of Fe(III)-storage among ferritins.1,4,23 Based on these data and a re-evaluation of previous studies on other ferritins we put forward the proposal of unity in the biochemistry of ferritins.1 We proposed that although variations in the amino acid sequences of ferritins exist the chemistry of Fe(II) oxidation and Fe(III) storage is the same among eukaryotic and microbial ferritins.1
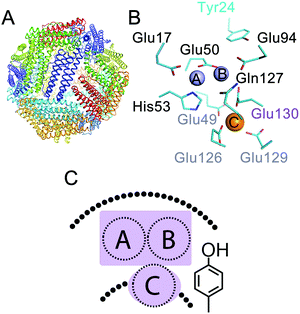 |
| | Fig. 1 Three Fe(II) binding sites exist in ferritins. (A) The conserved nanocage structure of ferritin. (B) The catalytic center in ferritin consists of two sites, i.e. sites A and B, in the middle of the subunit, which form the di-iron ferroxidase center, and a third nearby site named site C. The numbering of the amino acid residues is from Pyrococcus furiosus ferritin (PfFtn, PDB 2JD7). An amino acid residue in the coordination environment of site B and site C that varies among ferritins is numbered in purple. (C) A cartoon showing the ferroxidase center and site C together with the highly conserved tyrosine in the vicinity of site B. | |
In our previous studies using HuHF and PfFtn we observed that the kinetics of Fe(II) oxidation was different,13 but the progress curves of Fe(II) oxidation could be simulated using a common model.13 These observations prompted us to further investigate the intermediates of Fe(II) oxidation in these two ferritins. We applied freeze quench electron paramagnetic resonance (EPR) and Mössbauer spectroscopy together with UV-visible stopped-flow spectroscopy. HuHF and PfFtn were compared because they consist of 24 catalytically active subunits and because they are from two distinct domains of life and should serve as good models to test the diversity view against the unity view. The results strongly suggest that Fe(II) oxidation in both HuHF and PfFtn proceeds via the same peroxodiferric intermediate and results in the same Fe(III) products in support of the proposal of unity in the biochemistry of ferritins. Our data further provide a new insight into the initial step of catalysis of Fe(II)-oxidation, i.e. Fe(II) binding to the catalytic sites, and shed light on a possible explanation for the observed differences in the kinetics of Fe(II) oxidation among eukaryotic and microbial ferritins.
Experimental procedure
Details of chemicals, protein expression and purification, UV-visible stopped-flow experiments, and statistical analysis of Mössbauer data are included in the ESI.†
Choosing the time points for freeze quench to trap the Fe(II) oxidation reaction intermediates of ferritin
In ferritin Fe(II) is the substrate and Fe(III) is the product and during catalysis of Fe(II) oxidation different species such as Fe(II) substrates, Fe(III) intermediates, and Fe(III) products can coexist. As a consequence simulation of Mössbauer data to characterize Fe(III) intermediates will be complex if large amounts of different Fe(III) products are present. To decrease this complexity and to obtain a new insight into various different intermediates, the reaction was quenched at three time points. (i) Before the addition of molecular oxygen (t = 0) at which all the iron should be Fe(II). (ii) A time point after the addition of molecular oxygen at which the absorbance of the peroxodiferric intermediate was close to its maximum in PfFtn and HuHF (see below). The same time point was chosen for HuHF and PfFtn for proper comparison of the intermediates; (iii) at a time point when all of the Fe(II) was converted to Fe(III) products and no further change in the absorbance spectrum from 300 to 700 nm was observed.
Preparation of EPR and Mössbauer samples before the addition of dioxygen (t = 0 s)
The 57Fe(II) or NATFe(II) (natural abundance Fe(II)) solution was prepared in acidic Milli-Q water, i.e. 1 ≤ pH ≤ 2. PfFtn or HuHF was prepared in 1 M MOPS buffer, 200 mM NaCl, pH 7.0. This concentration of buffer was chosen to minimize any change in the pH after mixing protein with the acidic Fe(II) solution in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The final concentration of buffer was 500 mM MOPS, 100 mM NaCl, pH 7.0. To prepare the samples before the reaction of Fe(II)-bound ferritin with dioxygen (samples labelled t = 0 s) anaerobic solutions of Fe(II) and ferritin were mixed (1:1 ratio) in an anaerobic glove box (Coy Laboratories). 250 μl or 500 μl of the solution was then transferred to an EPR tube or a customized Mössbauer sample tube in the glove box, EPR and Mössbauer tubes were tightly closed. Subsequently, they were transferred outside the glove box and immediately frozen in liquid nitrogen.
:1 ratio. The final concentration of buffer was 500 mM MOPS, 100 mM NaCl, pH 7.0. To prepare the samples before the reaction of Fe(II)-bound ferritin with dioxygen (samples labelled t = 0 s) anaerobic solutions of Fe(II) and ferritin were mixed (1:1 ratio) in an anaerobic glove box (Coy Laboratories). 250 μl or 500 μl of the solution was then transferred to an EPR tube or a customized Mössbauer sample tube in the glove box, EPR and Mössbauer tubes were tightly closed. Subsequently, they were transferred outside the glove box and immediately frozen in liquid nitrogen.
Preparation of EPR and Mössbauer samples 0.7 s quenched after reaction with dioxygen
PfFtn or HuHF was in 1 M MOPS buffer, 200 mM NaCl, pH 7.0. The Fe(II) solution should have a pH between 1 and 2 to prevent autoxidation of Fe(II) under dioxygen saturation conditions (ESI,† Fig. S1). Ferritin and Fe(II) solutions were kept in different gas tight bottles and were purged with pure dioxygen gas for circa 10 minutes to reach oxygen saturation conditions. The solutions were then immediately used for rapid freeze quench experiments. Freeze-quench samples were prepared by connecting an in-house build T-mixer cell to the stopped-flow instrument as explained previously.13 One syringe of the stopped-flow instrument was filled with ferritin and the other syringe was filled with Fe(II) solution, each syringe circa 300 μl. The solutions were then rapidly mixed through the T-mixer cell by applying 9 bar pressure behind each syringe. This setup was used because the time scale of the reaction in PfFtn at room temperature is much longer than the millisecond time scale usually associated with rapid freeze-quench techniques. To apply this setup to quench the reaction of PfFtn and HuHF with circa 2 Fe(II) per ferritin subunit, using stopped-flow spectroscopy we determined the optimum temperature at which the absorbance of the peroxodiferric intermediate reached its maximum circa 0.7 s after mixing. This time was chosen because it was the dead time of mixing and freezing for our freeze quench setup, as determined using the myoglobin–azide reaction.13 The optimum temperature for HuHF was 10 °C and for PfFtn it was 47 °C. Increasing the temperature to higher values for PfFtn was not possible due to instrumental limitations. The outflow from the mixer was directly injected into customized EPR or Mössbauer tubes, which were cooled with and kept in liquid nitrogen, by using an extension tubing of 10 cm length. This time is quoted in the text as the shortest quenching time of the reaction for EPR or Mössbauer spectroscopy. To quench the reaction after a long time, circa 1–5 minutes, the solutions were injected into room temperature EPR or Mössbauer tubes. The samples were frozen by immersing in liquid nitrogen 300 s (PfFtn) after incubation at 47 °C, or 60 s (HuHF) after incubation at 10 °C. For Mössbauer spectroscopy the final concentrations of PfFtn and HuHF, after 1:1 mixing with 57Fe(II) solution, were 45 μM (24-mer) and 55 μM (24-mer) respectively. The volume of the Mössbauer samples was either circa 500 μl or circa 250 μl. For EPR spectroscopy the final concentration of PfFtn was 45 μM (24-mer) or 4.4 μM (24-mer), and that of HuHF was 55 μM (24-mer) or 5.5 μM (24-mer). For EPR and Mössbauer samples the final concentration of NATFe or 57Fe was set to achieve a total loading of 50 Fe(II) per ferritin 24-mer, this was done to make sure that two Fe(II) per ferritin subunits were added. The solubility of dioxygen at 10 °C is circa 1.71 mM and at circa 47 °C it is about 0.96 mM. Because under single-turnover conditions in PfFtn the stoichiometry of Fe(II) oxidized per dioxygen is circa 3 and in HuHF it is circa 2.5,13 enough dioxygen for a single turnover of enzyme must be present under our experimental conditions. PfFtn and Fe(II) solutions were preheated to 47 °C for 1 minute before 1:1 mixing, HuHF and Fe(II) solutions were cooled at 10 °C for 1 minute before 1:1 mixing. The pressure of the stopped-flow N2 gas, which is used for shooting the protein and Fe(II) solutions for rapid mixing, was 9 bar.
Electron paramagnetic resonance (EPR) spectroscopy
X-band EPR measurements were performed using a Bruker ECS-106 EPR spectrometer. EPR conditions were: microwave power 0.127–201 mW; modulation frequency 100 kHz; modulation amplitude 12.7 or 4.02 Gauss; temperature 6.4–30 K. EPR spectra were analyzed using programs described in ref. 24.
Mössbauer spectroscopy
Mössbauer spectra were recorded on a conventional spectrometer with alternating constant acceleration of the γ-source. The minimum experimental line width was 0.24 mm s−1 (full width at half-height). The sample temperature was maintained constant either in an Oxford Instruments Variox or an Oxford Instruments Mössbauer-Spectromag cryostat with a split-pair magnet system. Measurements were performed at 80 K. The γ-source (57Co/Rh, 1.8 GBq) was kept at room temperature. By using a re-entrant bore tube the γ-source could be positioned inside the gap of the magnet coils at a position of the zero field. Isomer shifts are quoted relative to iron metal at 300 K.
Results
Fe(II) distribution among three sites is different in HuHF and PfFtn
The first step in the catalysis of Fe(II) oxidation is binding of the Fe(II) ions to the metal ion binding sites in each subunit. As discussed in the introduction three Fe(II) binding sites exist in different eukaryotic and microbial ferritins1,6, i.e. sites A and B of the ferroxidase center and site C close to this center. We have shown previously that Fe(II) distributes among these sites.4 However, we could not determine the Fe(II) occupation of each site to define the amount of different types of Fe(II)-occupied subunits under single-turnover conditions, i.e. addition of circa 2 Fe(II) per ferritin subunit. This knowledge is essential for understanding the mechanism of Fe(II) oxidation. To determine the Fe(II) occupation of each site before the addition of dioxygen we used Mössbauer spectroscopy and combined the results with knowledge of the binding affinity of each site for Fe(II), which we had determined in a previous study using detailed isothermal titration calorimetry experiments under anaerobic conditions4 (ESI,† Table S1). Two Fe(II) ions per ferritin subunit were added to apo-HuHF or apo-PfFtn under anaerobic conditions. Simulation of the Mössbauer spectra required a model of three distinct Fe(II) doublets (Fig. 2A and ESI,† Fig. S2, S3). We attribute these doublets to the three individual sites, i.e. sites A, B, and C (Table 1), in agreement with the observation of three sites with different coordination environments using X-ray crystallography in various ferritins1 including PfFtn25 and HuHF.6 These observations are inconsistent with the possibility that only one or two sites might exist. Furthermore, the hypothesis that two of the Fe(II) doublets might be assigned to a single site with alternative coordination ligands can also be ruled out based on our Mössbauer data. The sum of the amount of any combination of two different doublets exceeds the total number of site A, or B, or C present in a ferritin 24-mer. For example the second and the third doublets in PfFtn together account for circa 60% of the Fe(II)-added. This means circa 29 Fe(II) per ferritin 24-mer. Because there are only 24 sites A, or B, or C per ferritin 24-mer available, the second and the third doublets in PfFtn cannot be assigned to the same site with alternative coordination ligands. The Mössbauer parameters of the first doublet in HuHF and PfFtn are very close (Table 1). Because the Mössbauer parameters of Fe(II) in the absence of dioxygen are mainly affected by its amino acid coordinating residues, the coordination environments of the Fe(II) associated with the doublet in HuHF and PfFtn should be the same. The available structural data1 show exactly the same coordination environment for site A in PfFtn and HuHF (Fig. 2B), but not for sites B and C. Consequently, we attribute the first Fe(II) doublet to the Fe(II) in site A of the ferroxidase center. In PfFtn the second (purple trace in Fig. 2A) and the third (orange trace in Fig. 2A) Fe(II) doublets have 40% and 19% abundance respectively (Table 1). In PfFtn as we reported previously4 the affinity of site B for Fe(II), i.e. (5.5 ± 1.0) × 104 M−1, is 50-fold higher than that of site C, i.e. (1.0 ± 0.3) × 103 M−1 (ESI,† Table S1). Therefore, in PfFtn the doublet with 40% abundance is attributed to site B and the doublet with 19% abundance is attributed to site C. In HuHF the abundances of the second (purple trace in Fig. 2A) and the third (orange trace in Fig. 2A) Fe(II) doublets are the same within the experimental error (Table 1). This is consistent with the observation that sites B and C in HuHF have the same affinity for Fe(II) ions (ESI,† Table S1).4 The exact assignment of the second and the third Fe(II) doublets in HuHF to sites B and C was not possible.
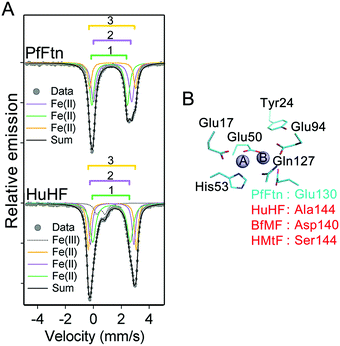 |
| | Fig. 2 The difference in the coordination environment of site B among ferritins results in differences in the amounts of four possible Fe(II)-occupied subunit types. (A) Mössbauer spectrum of Fe(II) in PfFtn and HuHF before addition of dioxygen. In human H-type ferritin (HuHF) a small amount of Fe(III) (less than 9%) is observed which was due to oxidation of Fe(II) before addition to ferritin. The simulation results are not biased by this low amount of ‘dirty’ Fe(III). Both in HuHF and PfFtn three distinct Fe(II) doublets are observed which are assigned to Fe(II) in sites A, B, and C. Measurements were performed at 80 K. (B) Coordination environment of site A is highly conserved and a residue in the coordination environment of site B, which is also nearby site C, varies among ferritins. The structure shows the amino acid residues in the coordination environment of the ferroxidase center of PfFtn. The amino acids that are conserved among ferritins are numbered in black. An amino acid residue in the coordination environment of site B, which varies among ferritins, is numbered in red. Site C is not shown for clarity. | |
Table 1 The amount of Fe(II) in site B and C varies among HuHF and PfFtn
| Protein |
Time (s) |
Doublet |
Oxidation state |
% |
Mössbauer parameters |
Site |
| Fe(II) |
δ (mm s−1) |
ΔEQ (mm s−1) |
| In HuHF at t = 0 s less than 9% of the 57Fe(II) was observed as Fe(III) (gray line in Fig. 2a), which we attribute to dirty Fe(III) possibly due to the presence of Fe(III) in Fe(II) solution before addition to HuHF (ESI Fig. S1). Circa 2 Fe(II) per ferritin subunit were added to PfFtn (45 μM 24-mer) or HuHF (55 μM 24-mer). Measurements were performed under exactly the same conditions. In HuHF Fe(II) is equally distributed among sites B and C and the exact assignment of the second and the third Fe(II) doublet to sites B and C was not possible at this stage. |
| HuHF |
0 |
1 |
Fe(II) |
39(2) |
1.30(2) |
2.70(2) |
A |
| 2 |
Fe(II) |
27(1) |
1.35(1) |
3.44(2) |
B and C |
| 3 |
Fe(II) |
25(1) |
1.34(2) |
3.12(2) |
|
|
| PfFtn |
0 |
1 |
Fe(II) |
41(2) |
1.38(1) |
2.73(2) |
A |
| 2 |
Fe(II) |
40(1) |
1.17(1) |
2.54(1) |
B |
| 3 |
Fe(II) |
19(1) |
1.39(1) |
3.27(1) |
C |
Mössbauer spectroscopy reveals different forms of Fe(II)-filled subunits
For the distribution of Fe(II) among three binding sites, statistically seven Fe(II)-occupation scenarios for subunits can be imagined: subunits with Fe(II)-occupied site A only (AIIB0C0), or site B only (A0BIIC0), or site C only (A0B0CII), subunits with Fe(II)-occupied sites A and B (AIIBIIC0), subunits with Fe(II)-occupied sites A and C (AIIB0CII), subunits with Fe(II)-occupied sites B and C (A0BIICII), and subunits with Fe(II)-occupied sites A, B, and C (AIIBIICII). Site A has the highest affinity for Fe(II) as determined for different ferritins4,26 (ESI,† Table S1). Thus, site A should first be occupied with Fe(II). The occupation of site A will be followed by Fe(II) binding to sites B and C, possibly in a cooperative fashion. Therefore, among the above seven Fe(II)-occupation scenarios four predominate (Fig. 3): (AIIBIIC0) subunits with Fe(II)-occupied sites A and B but empty site C; (AIIBIICII) subunits with Fe(II)-occupied sites A, B, and C; (AIIB0CII) subunits with Fe(II)-occupied sites A and C but empty site B; and (AIIB0C0) subunits with Fe(II)-occupied site A only. To estimate the percentage of each subunit type per ferritin 24-mer using the results of Mössbauer spectroscopy we define three variables:| | 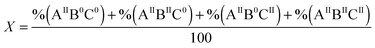 | (1) |
| | 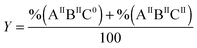 | (2) |
| | 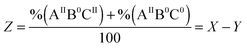 | (3) |
in which X is the sum of the percentages of all subunit types divided by 100, Y is the sum of the percentages of subunits with sites A and B occupied divided by 100, and Z is the percentages of subunits with site B empty divided by 100. As we discussed above, site A is first occupied with Fe(II) and subsequently sites B and C are filled. Thus, ‘X’ or ‘Y’ is a factor of the amount of Fe(II) added per subunit and the percentage of Fe(II) assigned to site A or B respectively. Accordingly we may write:| |  | (4) |
| |  | (5) |
In which “n” is the amount of Fe(II) added per ferritin 24-mer for a single turnover experiment. In our experiments “n” was 50 Fe(II) per ferritin 24-mer. %Fe(II) in site A or B is the percentage of the Fe(II) doublet assigned to site A or B based on the results of Mössbauer spectroscopy for samples before the addition of dioxygen (Table 1). X and Y are calculated using eqn (4) and (5), and subsequently the percentage of four different Fe(II)-occupied subunit types (Fig. 3) was found using the following equations (see the ESI† for details):| | 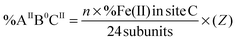 | (6) |
| | 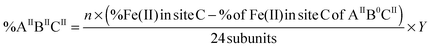 | (7) |
| | | %AIIBIIC0 = (Y × 100) − %AIIBIICII | (8) |
| | | %AIIB0C0 = (Z × 100) − %AIIB0CII | (9) |
in which %Fe(II) in site B or C is obtained from the results of Mössbauer spectroscopy for samples before the addition of dioxygen. Using eqn (6)–(9) we found that the percentage of (AIIBIIC0) subunits in PfFtn and HuHF is circa 52% and 42%, respectively, and that of (AIIBIICII) subunits in PfFtn and HuHF is 32% and 14%, respectively (Fig. 3). The percentages of (AIIB0CII) and (AIIB0C0) subunits in PfFtn are circa 1% each, while in HuHF they are circa 13% and 12% respectively (Fig. 3). Because in some subunits, i.e. (AIIBIICII) subunits, three sites are occupied upon addition of circa 2 Fe(II) per subunit and because the percentage of (AIIBIICII) subunits is more than that of (AIIB0C0) subunits, in total only circa 80–90% of the subunits is observed to be occupied. Moreover, it should be noted that although we could not specifically assign the second and the third Fe(II) doublets in HuHF to sites B and C, because their amounts are the same within experimental error results obtained using our statistical model are valid for HuHF. Our observations regarding the distribution of Fe(II) are consistent with a possible positive cooperativity among subunits and among three binding sites, i.e. binding of Fe(II) to site A in one subunit induces binding of Fe(II) to site A in a nearby subunit and to sites B and C. Indeed kinetics of Fe(II) oxidation have shown positive cooperativity in eukaryotic and microbial ferritins due to a yet to be identified mechanism.13,27
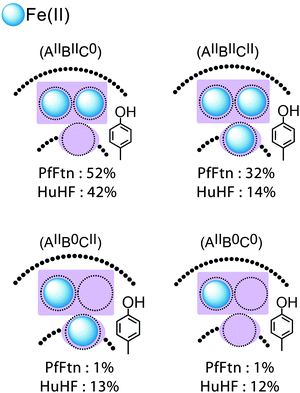 |
| | Fig. 3 The amounts of four Fe(II)-occupied subunit types is different between HuHF and PfFtn. Based on the results of Mössbauer spectroscopy four possibilities for the distribution of Fe(II) among binding sites exist: subunits with sites A and B filled but site C empty (AIIBIIC0 subunits), subunits with sites A, B, and C filled (AIIBIICII subunits), subunits with sites A and C filled (AIIB0CII subunits), and subunits with site A only filled (AIIB0C0 subunits). The percentage of each subunit type varies between HuHF and PfFtn. The major difference between PfFtn and HuHF is in the percentages of (AIIB0CII), (AIIB0C0), and (AIIBIICII) subunits. The percentage of (AIIB0CII) and (AIIB0C0) subunits in PfFtn is negligible. | |
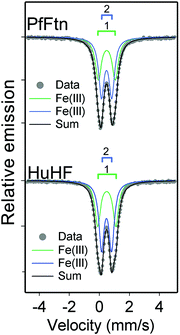
The same peroxodiferric intermediate is formed in HuHF and PfFtn
An intermediate with visible absorbance between 500 and 800 nm and centered at a different wavelength in different ferritins1,7,19,28–30 has been reported during the catalysis of Fe(II) oxidation. For example the progress curves of this intermediate in HuHF (650 nm) and PfFtn (620 nm) are shown in Fig. 4A and B respectively. We applied freeze quench EPR and Mössbauer spectroscopy to obtain molecular insight into the origin of this intermediate in these ferritins. The reaction of HuHF or PfFtn containing circa 2 57Fe(II) per ferritin subunit was quenched 0.7 s after the addition of dioxygen to compare the intermediates at the same freezing time. This time was chosen because the absorbance of the Fe(III) intermediate species reached its maximum in HuHF (Fig. 4A) and was close to maximum in PfFtn (Fig. 4B) (Methods). Simulation of the Mössbauer spectrum of PfFtn suggested the presence of one Fe(II) and two Fe(III) doublets (Fig. 4C and ESI,† Fig. S4), and that of HuHF suggested the presence of two Fe(II) and three Fe(III) doublets (Fig. 4C and ESI,† Fig. S5). The ratio of the two major Fe(III) doublets in HuHF and PfFtn (green and purple traces in Fig. 4C) was constrained to 1:1 abundance (Table 2). This was done because EPR spectroscopy implied that the majority of the Fe(III) ions should be in a spin-coupled diferric intermediate with the S = 0 ground state (EPR silent): EPR spectroscopy showed only negligible spin concentration of the total Fe(II) added as a mononuclear Fe(III) species or a [Fe(II)–Fe(III)] mixed valence cluster4 (ESI,† Table S2). The Mössbauer parameters of the diferric intermediate in HuHF are similar to those of the diferric intermediate in PfFtn (Table 2). This implies that the molecular structure of the diferric intermediate in HuHF and PfFtn is the same. These parameters are compared to those of the various peroxodiferric intermediate species in model compounds31–37 and in dioxygen activating enzymes38–41 (Table 3). From Table 3 one can observe that the Mössbauer parameters assigned to the μ-1,2-peroxodiferric binding mode span over a wide range, but for the majority of cases, at least one of the reported values for the ΔEQ is above 1.4 (mm s−1) (Table 3). On the other hand in the cases in which the peroxo species is assigned to the η2-O2 binding mode a ΔEQ of less than 0.8 (mm s−1) is reported. Similar to the η2-O2 binding mode of the peroxo, in PfFtn and HuHF one of the ΔEQ of the peroxodiferric intermediate is less than 0.8 (mm s−1) (Table 3). Because EPR spectroscopy showed that two Fe(III) in the ferroxidase center are antiferromagnetically coupled, we propose that the peroxodiferric intermediate in HuHF and PfFtn has a μ-η1:η2 core structure. Further investigations by e.g. resonance Raman or EXAFS spectroscopy may be used to corroborate this proposal. It should be noted that the Mössbauer parameters we found in HuHF are different from those reported previously.42 Previous Mössbauer studies with HuHF42 were performed at pH ≤ 6.5, a pH value at which Fe(II) binding to site A of the ferroxidase center is known to be disrupted.26 Fe(II) binding under anaerobic conditions to sites A, B, and C in HuHF has been observed by isothermal titration calorimetry4 or X-ray crystallography10 at pH ≥ 7.
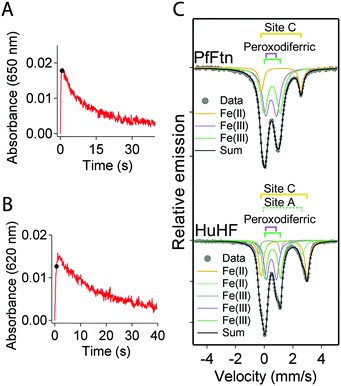 |
| | Fig. 4 The binding mode of dioxygen in the peroxodiferric intermediate is the same in PfFtn and HuHF. (A) Progress curves for the formation and decay of the peroxodiferric intermediate were recorded at 650 nm for HuHF (2.2 μM 24-mer) or (B) at 620 nm for PfFtn (4.5 μM 24-mer). Measurements with HuHF were performed at 10 °C and those with PfFtn were performed at 47 °C. The black sphere shows the quenching time (t = 0.7 s) for preparing the Mössbauer samples. (C) Mössbauer spectra of PfFtn and HuHF 0.7 s after the addition of dioxygen. In HuHF besides the two major Fe(III) doublets attributed to the peroxodiferric intermediate a minor Fe(III) doublet (<6%) was observed (dashed grey line). The black line (Sum) is the superposition of the simulated subspectra. Measurements were performed at 80 K. | |
Table 2 The same peroxodiferric intermediate is formed in PfFtn and HuHF
| Protein |
Time (s) |
Doublet |
Oxidation state |
% |
Mössbauer parameters |
Site |
|
δ (mm s−1) |
ΔEQ (mm s−1) |
| Measurements were performed under exactly the same conditions. Circa 2 Fe(II) per ferritin subunit were added. In HuHF a minor Fe(III) doublet (<6%) was observed (Fig. 3). The Mössbauer parameters of this doublet were different from those of dirty Fe(III) observed in sample before addition of dioxygen: δ (mm s−1) = 0.38 (1) and ΔEQ (mm s−1) = 1.52 (1). This minor Fe(III) species might be the mononuclear Fe(III) observed by EPR spectroscopy (ESI Table S2), whose origin is unknown. The Fe(III) doublets that form the peroxodiferric intermediate. |
| HuHF |
0.7 |
1 |
Fe(III)a |
29(1) |
0.50(2) |
0.70(2) |
A and B |
| 2 |
Fe(III)a |
29(1) |
0.58(2) |
1.10(2) |
| 3 |
Fe(II) |
12(2) |
1.32(1) |
2.70(2) |
A |
| 4 |
Fe(II) |
25(2) |
1.42(2) |
3.14(2) |
C |
|
|
| PfFtn |
0.7 |
1 |
Fe(III)a |
42(1) |
0.49(1) |
0.76(1) |
A and B |
| 2 |
Fe(III)a |
42(1) |
0.56(2) |
1.12(1) |
| 3 |
Fe(II) |
16(2) |
1.20(1) |
2.77(1) |
C |
Table 3 Comparison of the Mössbauer parameters of the peroxodiferric intermediate in HuHF and PfFtn with those reported for the peroxo species in model compounds and other proteins
| |
|
δ (mm s−1) |
ΔEQ (mm s−1) |
Binding mode of dioxygen |
Ref. |
|
Pyrococcus furiosus ferritin (PfFtn); human H-type ferritin (HuHF); methane monooxygenase (MMO); ribonucleotide reductase (RNR); arylamine oxygenase (CmII); human deoxyhypusine hydroxylase (hDOHH); 1, [Fe2(Ph-bimp)(C6H5COO)(O2)]2+; 2, [[Fe2O3(6-Me3-TPA)2](ClO4)3]−; 3, [Fe2(μ-O2)(μ-O2CCH2Ph)2(HB(pz′)3)2]; 4, [Fe2(LPh4)-(RCO2)(O2)]2+ (R = Ph3C (oxy−1)); 5, [Fe(III)(TMC)(O2)]+; 6, [Fe2(O2)(Ar)4(py)2]; 7, [Fe(EDTA)O2]3−. The binding modes proposed for dioxygen in RNR and complexes 1, 2, 3, 4, and 6 are based on detailed spectroscopic studies. The binding mode proposed for complex 5 is a suggestion due to the considerable difference between the Mössbauer parameters of this complex and those reported for complexes with μ-1,2-peroxo bonding mode. For BfMF inconsistent Mössbauer parameters have been obtained from simulation of exactly the same Mössbauer spectra. Based on these inconsistent data a μ-1,2-peroxo binding mode has been proposed. Signs unknown. Postulated. |
| PfFtn |
Fe(III) |
0.49(1) |
0.76(1) |
μ-η1:η2c |
This work |
| Fe(III) |
0.56(2) |
1.12(1) |
| HuHF |
Fe(III) |
0.50(2) |
0.70(2) |
μ-η1:η2c |
This work |
| Fe(III) |
0.58(2) |
1.10(2) |
| BfMFa |
Fe(III) |
0.62 |
1.08 |
— |
29
|
| BfMFa |
Fe(III) |
0.65 |
1.05 |
— |
18
|
| Fe(III) |
0.55 |
1.06 |
| MMO |
Fe(III) |
0.66 |
1.51 |
— |
39
|
| Fe(III) |
| RNR |
Fe(III) |
0.63 |
1.74 |
μ-1,2-Peroxo |
40
|
| Fe(III) |
| CmII |
Fe(III) |
0.61 |
−0.23b |
μ-η1:η2 |
38
|
| Fe(III) |
0.54 |
−0.68b |
| hDOHH |
Fe(III) |
0.55 |
1.16 |
(μ-Hydroxo) (μ-1,2-peroxo) |
41
|
| Fe(III) |
0.58 |
0.88 |
| 1 |
Fe(III) |
0.58 |
0.74 |
cis-μ-1,2-Peroxo |
31
|
|
|
Fe(III) |
0.65 |
1.70 |
| 2 |
Fe(III) |
0.54 |
1.68 |
(μ-Oxo) (μ-1,2-peroxo) |
32
|
| Fe(III) |
| 3 |
Fe(III) |
0.66 |
1.40 |
μ-1,2-Peroxo |
33
|
| Fe(III) |
| 4 |
Fe(III) |
0.57 |
1.44 |
μ-1,2-Peroxo |
34
|
| Fe(III) |
| 5 |
Fe(III) |
0.58 |
−0.92 |
η2-O2 |
35
|
| 6 |
Fe(III) |
0.52(2) |
0.71(2) |
η2-O2 |
36
|
| Fe(III) |
0.65(2) |
1.27(3) |
| 7 |
Fe(III) |
0.65 |
0.72 |
Side-on (η2-O2) |
37
|
Only in (AIIBIIC0) and (AIIBIICII) subunits two Fe(II) can be simultaneously oxidized
It has been previously proposed that in eukaryotic ferritins two Fe(II) together are simultaneously oxidized in each ferroxidase center to form the peroxodiferric intermediate,17,18,43 while in bacterial ferritins three Fe(II), two Fe(II) in the ferroxidase center together with the Fe(II) at site C, are simultaneously oxidized.21,44 These proposals predict that under single-turnover conditions, when the absorbance of the peroxodiferric intermediate reaches its maximum, i.e. 0.7 s in our experiments, all of the Fe(II) added should have been converted to the peroxodiferric intermediate or to products. Our Mössbauer data and those reported previously for BfMF29 and BfHF20 are inconsistent with this proposal. We analysed the Mössbauer data of the Fe(II) doublets before the addition of dioxygen (Table 1) and those of the Fe(II)/Fe(III) doublets 0.7 s after the addition of dioxygen (Table 2). As discussed above the results of Mössbauer spectroscopy before the addition of dioxygen revealed the amounts of different forms of Fe(II)-occupied subunits for PfFtn and HuHF (Fig. 3). In PfFtn and HuHF 0.7 s after the addition of dioxygen the amount of Fe(III) observed as the peroxodiferric intermediate was circa 84% and 58% (Table 2), which represent circa 84% of subunits in PfFtn and 58% of subunits in HuHF. Comparison of these values with the percentages of (AIIBIIC0) and (AIIBIICII) subunits in Fig. 5 shows that they are within experimental error the same as the sum of the percentages of (AIIBIIC0) and (AIIBIICII) subunits in PfFtn (84%) and in HuHF (56%) respectively. These data suggest that both in PfFtn and in HuHF the Fe(II) ions at sites A and B of the (AIIBIIC0) and (AIIBIICII) subunits were oxidized concurrently within 0.7 s to form the peroxodiferric intermediate, but the Fe(II) ions at site C of the (AIIBIICII) subunits or sites A and C of the (AIIB0C0) and (AIIB0CII) subunits were not oxidized rapidly (Fig. 5). Consistently, in PfFtn one Fe(II) doublet (16%) was observed (Table 2) whose amount was within experimental error close to the amount of the Fe(II) doublet attributed to site C (19%) under anaerobic conditions (Table 1). However, the Mössbauer parameters of the Fe(II) doublet attributed to site C before (Table 1) and after (Table 2) the addition of dioxygen were different. The reason for this difference is not known but may suggest a change in the coordination environment of site C in PfFtn upon Fe(II) oxidation in the ferroxidase center. In HuHF 0.7 s after the addition of dioxygen two Fe(II) doublets were observed (Table 2). The Mössbauer parameters of the first Fe(II) doublet (12%) (Table 2) are the same as the Fe(II) doublet attributed to site A before the addition of dioxygen (Table 1). The amount of this doublet (12%) is consistent with the oxidation of Fe(II) in sites A and B, and the formation of the peroxodiferric intermediate in the ferroxidase center: in HuHF before the addition of dioxygen the amount of Fe(II) in site B was only 25–27% of the Fe(II) added. As a result upon addition of dioxygen only 25–27% of the 39% Fe(II) in site A could rapidly oxidize to form the peroxodiferric intermediate. Circa 12% of the Fe(II) in site A could not be oxidized rapidly. The second Fe(II) doublet in HuHF (Table 2) should be the Fe(II) in site C, since this Fe(II) has not entered the ferroxidase center and cannot be oxidized rapidly together with the Fe(II) in site A of the ferroxidase center. In summary, the data for PfFtn and HuHF together demonstrate that only in (AIIBIIC0) and (AIIBIICII) subunits two Fe(II) are oxidized simultaneously in the ferroxidase center. In subunits in which site B is not occupied, Fe(II) in site A cannot be oxidized (Fig. 5). We speculate that site B might be the initial dioxygen binding site. This suggestion is in line with a previous site directed mutagenesis study of HuHF in which differences between sites A and B of the ferroxidase center were observed.45 Replacement of a glutamate residue of each site resulted in a different response to Fe(II) oxidation. Based on this observation it has been proposed that differences exist between sites A and B, and that site B is possibly the initial dioxygen binding site.45
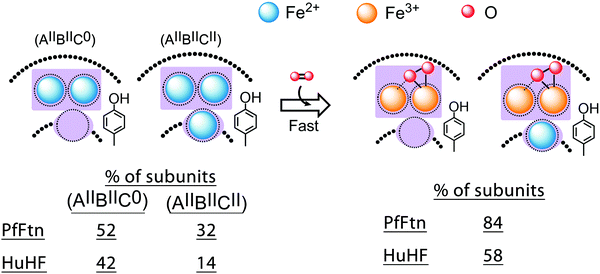 |
| | Fig. 5 Under single-turnover conditions two Fe(II) are not simultaneously oxidized in each subunit of HuHF and PfFtn. A cartoon showing the oxidation of Fe(II) in (AIIBIICII) and (AIIBIIC0) subunits via the peroxodiferric intermediate. The total percentage of (AIIBIICII) and (AIIBIIC0) subunits observed by Mössbauer spectroscopy is within experimental error equal to the percentage of the subunits with the peroxodiferric intermediate. This suggest that only in (AIIBIICII) and (AIIBIIC0) subunits two Fe(II) are simultaneously oxidized in the ferroxidase center to form the peroxodiferric intermediate. | |
Site B tunes the kinetics of Fe(II) oxidation
Progress curves of Fe(III) formation, which are typically measured between 300 and 350 nm, have been recorded for various ferritins using stopped-flow spectroscopy.6,7,17,21 Even though previous Mössbauer data showed that when the peroxodiferric intermediate has its maximum absorbance not all the Fe(II) ions are oxidized,18,20,29 the progress curves have been interpreted as the formation of the peroxodiferric in each subunit as a sudden increase in the absorbance followed by spontaneous transfer of the Fe(III) product to the internal cavity of ferritin observed as a gradual increase of the absorbance in a slower phase.17,18,20,29 UV-visible spectroscopy by itself does not provide direct information on the nature of Fe(II) and Fe(III), e.g. whether the Fe(III) species are intermediates or products. To properly interpret the stopped-flow UV-visible data (Fig. 6) in terms of the formation of different Fe(III) species we used our Mössbauer data. The recorded progress curves are consistent with those reported previously for HuHF13,17,30 or PfFtn.13,30 The data were analyzed based on the amount of doublets assigned to the Fe(II) substrate and the peroxodiferric intermediate observed 0.7 s after the addition of dioxygen (Table 2). Under single-turnover conditions, a two-exponential equation (eqn (10)) was required to obtain a fit to the data using global fit analysis:| | 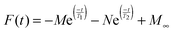 | (10) |
in which M and N are the pre-exponential amplitude factor (the absorbance of each exponential phase), T1 and T2 are time constants, and M∞ is the absorbance at infinite time. The values of M, N, T1, T2, and M∞ for PfFtn and HuHF are given in Table 4. In PfFtn and HuHF the ratio of the M to M∞ was circa 80% and 50%, respectively. This suggests that in PfFtn circa 80% and in HuHF circa 50% of the Fe(II) added were rapidly oxidized in the first phase. This is consistent with the observation of circa 84% and circa 58% Fe(III) as the peroxodiferric intermediate in PfFtn and in HuHF respectively (Table 2). Thus, the fast phase should present the rapid formation of the peroxodiferric intermediate in the (AIIBIIC0) and (AIIBIICII) subunits and not the Fe(III) products. Moreover, the ratio of N to M∞ in PfFtn and HuHF was circa 20% and 50% respectively. These ratios represent the percentages of Fe(II) not oxidized in the first phase but oxidized in the second slow phase plus a possible small change in the absorbance due to conversion of the peroxodiferric intermediate to the Fe(III) products. They are close to the percentages of Fe(II) observed by Mössbauer spectroscopy in PfFtn (16%) and in HuHF (37%) 0.7 s after the addition of dioxygen (Table 2). Therefore, the Fe(II) that was not oxidized rapidly in the first phase was oxidized at a slower rate in the second phase. These data demonstrate that the kinetics of Fe(II) oxidation is defined by the amount of the peroxodiferric intermediate that can rapidly form as a result of the presence of Fe(II) in site B of the ferroxidase center.
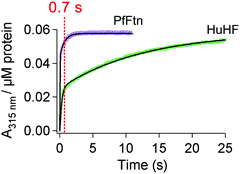 |
| | Fig. 6 Stopped-flow UV-visible spectroscopy shows differences in the kinetics of Fe(II) oxidation. Progress curves of Fe(III)-species formation were recorded at 315 nm. Circa 2 Fe(II) per ferritin subunit were added to PfFtn (4.5 μM 24-mer) or HuHF (2.2 μM 24-mer). Measurements with HuHF were performed at 10 °C and those with PfFtn were performed at 47 °C. The solid black line shows the fit obtained using a two exponential equation (eqn (1)). The red dashed line at 0.7 s shows quenching time for the Mössbauer measurements in Fig. 3. | |
Table 4 Different kinetic parameters obtained for Fe(II) oxidation in PfFtn and HuHF
| Protein |
M
|
N
|
T
1 (s) |
T
2 (s) |
M
∞
|
| The kinetic parameters were obtained from a global analysis of the progress curves of Fe(III) formation in Fig. 6 using eqn (10). The M, N, and M∞ are dimensionless. |
| HuHF |
0.027 ± 0.001 |
0.031 ± 0.002 |
0.27 ± 0.01 |
12.5 ± 0.1 |
0.058 ± 0.001 |
| PfFtn |
0.045 ± 0.001 |
0.012 ± 0.001 |
0.03 ± 0.001 |
0.7 ± 0.02 |
0.058 ± 0.001 |
The Fe(III)-dimer in the ferroxidase center is the major product of the Fe(II) oxidation
After complete oxidation of Fe(II), i.e. after 300 s in PfFtn and 60 s in HuHF, we recorded the presence of different Fe(III)-product species in ferritins. The Mössbauer spectra of PfFtn and HuHF could be simulated using a model of two Fe(III) doublets (Fig. 7 and ESI,† Fig. S6, S7). The Mössbauer parameters of these doublets were different from those of the peroxodiferric intermediate. The first doublet in PfFtn and HuHF accounts for circa 42% of the Fe(III) (Table 5), which is the same as the amount of Fe(II) in site A before the addition of dioxygen (Table 1). Therefore, this doublet is assigned to Fe(III) in site A. The second doublet accounts for 58% Fe(III) (Table 5), which is the same as the sum of the Fe(II) in sites B and C before the addition of dioxygen (Table 1). The Mössbauer parameters of the Fe(III) products in ferritin are similar to those reported for oxo or hydroxo bridged di-iron complexes.46 This is consistent with the results of EPR spectroscopy. Only circa 2–5% of the total Fe(II) added showed up as an EPR detectable (g = 4.3) mononuclear Fe(III) species (ESI,† Table S2). Because 42% of the Fe(III) ions was assigned to site A, at least 42% of the Fe(III) ions should have been in site B to form the antiferromagnetically coupled Fe(III)–O(H)–Fe(III) unit in the ferroxidase center, which is EPR silent. This interpretation is consistent with our previous observation that after complete oxidation of Fe(II) the majority of the ferroxidase centers remain occupied with two Fe(III), and the Fe(III) ions are displaced by new incoming Fe(II) ions.4 Two fates for the remaining 16% of Fe(III) can be considered: some of the Fe(III) stayed in site C and was observed as mononuclear Fe(III) and some moved to the internal cavity to form the Fe(III)-mineral core. Further detailed low temperature high-field Mössbauer measurements are required to study the nature of the mineral core in each ferritin.
 |
| | Fig. 7 Fe(III) products in PfFtn and HuHF are the same. Mössbauer spectra of PfFtn and HuHF after complete oxidation of Fe(II). Mössbauer spectrum of PfFtn was recorded 300 s after addition of circa 2 Fe(II) per ferritin subunit and that of HuHF was recorded 60 s after addition of circa 2 Fe(II) per subunit. Measurements were performed at 80 K. | |
Table 5 The same Fe(III) products are formed in the ferroxidase center of PfFtn and HuHF
| Protein |
Time (s) |
Species |
% |
Mössbauer parameters |
Site |
|
δ (mm s−1) |
ΔEQ (mm s−1) |
| Measurements were performed under exactly the same conditions. Circa 42% of the second Fe(III) doublet in HuHF and PfFtn should be the Fe(III) in site B, because EPR spectroscopy shows negligible amount of Fe(III) monomer. From the remaining amount of the second Fe(III) doublet (circa 16%) some is possibly in site C and is observed as mononuclear Fe(III), and some forms the Fe(III) mineral core. |
| HuHF |
60 |
Fe(III) |
42(2) |
0.49(1) |
1.21(2) |
A |
| Fe(III) |
58(1) |
0.48(1) |
0.67(1) |
B, C, and mineral corea |
|
|
| PfFtn |
300 |
Fe(III) |
42(2) |
0.49(1) |
1.14(1) |
A |
| Fe(III) |
58(2) |
0.48(1) |
0.69(1) |
B, C, and mineral corea |
Discussion
Because oxidation of Fe(II) by ferritin is vital for the iron homeostasis machinery of most life forms, this reaction has been studied intensively for more than half a century using ferritins from different organisms. Although the quaternary structure of ferritins is highly conserved, differences exist in the amino acid residues essential for the functioning of protein. A notable variation among ferritins is in one of the amino acids in the coordination environment of site B of the ferroxidase center (Fig. 2B). As a consequence, studies of the kinetics of Fe(II) oxidation with various ferritins have resulted in the suggestion of core differences and sometimes mutually inconsistent proposals regarding the mechanism of Fe(II) oxidation in eukaryotic, bacterial, and archaeal ferritins. Some of these differences are listed below and have been discussed in more detail previously:1 (i) measurement of the amount of dioxygen consumed for oxidation of two Fe(II) per ferritin subunit led to the report of differences in eukaryotic and microbial ferritins. For eukaryotic HuHF a stoichiometry of circa 0.45–0.5 O2 consumed per Fe(II) oxidized has been reported47,48 while for E.coli ferritin A (EcFtnA) a stoichiometry of circa 0.35 O2 consumed per Fe(II) oxidized has been observed.21 These differences have been interpreted in terms of different mechanisms of Fe(II) oxidation in HuHF and PfFtn.15,16 In HuHF it has been proposed that two Fe(II) are simultaneously oxidized but in EcFtnA three Fe(II) are simultaneously oxidized. (ii) It has been observed that the UV-visible absorbance of the peroxodiferric intermediate at 650 nm in human mitochondrial ferritin (MtFtn) is less than that in human H-type ferritin (HuHF).19 From this observation it has been concluded that in MtFtn less ferroxidase centers are active and that in this ferritin the Fe(III)-mineral core in the internal cavity of protein catalyzes the oxidation of Fe(II).19 (iii) Similar Mössbauer data for bullfrog M ferritin29 (BfMF) and bullfrog H ferritin (BfHF)20 were simulated differently (for detail see ref. 1). In BfMF it has been proposed that two Fe(II) are rapidly oxidized in each ferroxidase center via a μ-1,2-peroxodiferric intermediate, which resembles the peroxodiferric intermediate P in soluble methane monooxygenase. Subsequently, the μ-1,2-peroxodiferric intermediate decays slowly to Fe(III) products, which spontaneously move to the core.17,18,29,42,43,49,50 However, in bullfrog H-type ferritin (BfHF) it has been proposed that Fe(II) is oxidized via a tyrosine radical intermediate and not a peroxodiferric intermediate.20,51 As a result the Mössbauer spectra collected for BfHF during the formation of the intermediates were simulated to show the formation of different Fe(III) products instead of the peroxodiferric intermediate.20 (iv) Measurement of the progress curves of Fe(II) oxidation for E.coli ferritin A (EcFtnA) led to the conclusion that in this ferritin two Fe(II) in the ferroxidase center and an Fe(II) in site C, are oxidized concertedly.37,46 A recent study using EcFtnA led to the conclusion that in this ferritin Fe(II) in site C is oxidized by hydrogen peroxide generated in the ferroxidase center.14 In HuHF using stopped-flow UV-visible spectroscopy progress curves of Fe(III) formation and the peroxodiferrric intermediate were measured.17 In contrast to EcFtnA, the data for HuHF were interpreted as the oxidation of two Fe(II) in each ferroxidase center observed as a sudden jump in the absorbance between 300 and 400 nm and the absorbance of the peroxodifferic intermediate at 650 nm. A subsequent gradual increase in the absorbance at 300–400 nm and a decrease in the absorbance of the peroxodifferic intermediate at 650 nm was interpreted as the release of Fe(III) to the core. Consequently, the UV-visible spectra were simulated using a model to explain Fe(II) oxidation and Fe(III) release. (v) After the complete oxidation of Fe(II), Mössbauer measurements were used to study the formation of Fe(III) products in different ferritins. The results obtained for EcFtnA44 have been interpreted as the formation of the Fe(III) dimer (circa 60%) and some Fe(III) monomer (circa 30%), while the data obtained for HuHF52 have been interpreted as the formation of the Fe(III) dimer as the main product (circa 70%) and some Fe(III) mineral core (circa 30%). (vi) In bacteria a variant of ferritin, named bacterioferritin, is found, which has a very similar structure to that of ferritin except that it has a heme group between pairs of subunits53 with a role in iron release.54 While studies with E.coli bacterioferritin have led to the conclusion that in bacterioferritins the Fe(III) mineralization process is different from that in eukaryotic and microbial ferritins and proceeds via a diiron cofactor site,15,55 studies with a bacterioferritin isolated from Desulfovibrio vulgaris Hildenborough (DvHBfr) have led to the proposal of an Fe(III) mineralization mechanism that is similar to the proposed Fe(III) mineralization process for vertebrate H-type ferritin;56 the ferroxidase center is a substrate site and not a stable cofactor center. Based on the data and interpretations discussed above the diversity view has emerged: the mechanism of Fe(II) oxidation and storage is different among eukaryotic and microbial ferritins.15,22 For example, in eukaryotic ferritin two Fe(II) are simultaneously oxidized in each ferroxidase center and in bacterial ferritin three Fe(II) are simultaneously oxidized in the ferroxidase center and site C.
In contrast to this diversity view our recent studies using HuHF and PfFtn have led to the emergence of the unifying view of a single mechanism of Fe(II) oxidation and storage by ferritins and bacterioferritins.1 For PfFtn we initially suggested that the Fe(III) in the ferroxidase center is a stable cofactor site57 similar to the cofactor site of dioxygen activating enzymes such as soluble methane monooxygenase or similar to the proposed diiron cofactor site in E. coli bacterioferritin. Our subsequent studies using HuHF and PfFtn in comparison showed that in PfFtn and HuHF the Fe(III) is not a stable cofactor site.4 Fe(III) remains metastably in the ferroxidase center. Upon the arrival of the new Fe(II) ions, Fe(III) is sequentially displaced by Fe(II) and moves to the internal cavity.4 We further observed that although the kinetics of Fe(II) oxidation in HuHF and PfFtn was different,13 the progress curves of Fe(II) oxidation could be simulated using a common model.13 Mutagenesis studies of PfFtn compared to those reported for HuHF suggested a role for the highly conserved tyrosine in the vicinity of site B.13 We proposed that this tyrosine acts as a molecular capacitor for the oxidation of Fe(II) in site C via the peroxodiferric intermediate in the ferroxidase center.13 These data suggested a common mechanism of Fe(II) oxidation and Fe(III) mineralization among eukaryotic and microbial ferritins. To understand the origin of the observed differences in the reported kinetics of Fe(II) oxidation among eukaryotic and microbial ferritins and to check if they reflect different mechanisms of Fe(II) oxidation among ferritins we studied and compared the mechanisms of Fe(II) oxidation in HuHF and PfFtn. These two ferritins are from two different domains of life and should serve as a proper model to test the unity view against the diversity view and to understand differences among eukaryotic and microbial ferritins. We focused on the molecular details of the mechanism of Fe(II) oxidation by dioxygen at three stages of the reaction under single-turnover conditions: binding of Fe(II) ions to sites A, B, and C prior to the addition of dioxygen, the formation of Fe(II)/Fe(III) intermediates after the addition of dioxygen, and finally the appearance of Fe(III) products. The results of anaerobic Fe(II) binding measured by Mössbauer spectroscopy revealed the amount of Fe(II) present in each site, and subsequently the amount of four possible Fe(II)-occupied subunit types (Fig. 3 and 8): (AIIBIIC0), (AIIBIICII), (AIIB0CII), and (AIIB0C0) subunits. The major difference between PfFtn and HuHF was the relative amount of each Fe(II)-occupied subunit type. This difference is interpreted to originate from the difference in the affinity of site B in these ferritins for the Fe(II) ion.
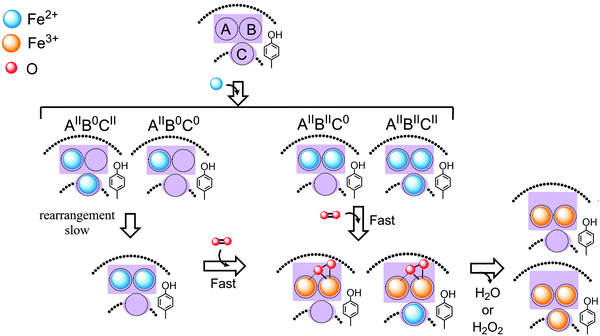 |
| | Fig. 8 A model describing a common mechanism of Fe(II) oxidation for HuHF and PfFtn and the origin of differences observed in the kinetics of Fe(II) oxidation by these ferritins. Upon the addition of Fe(II) to HuHF and PfFtn different subunit types form: subunits with Fe(II)-occupied sites A and B but empty site C (AIIBIIC0); subunits with Fe(II)-occupied sites A, B, and C (AIIBIICII); subunits with Fe(II)-occupied site A and C but empty site B (AIIB0CII); and subunits with Fe(II)-occupied site A only (AIIB0C0). The Fe(II) in sites A and B of the (AIIBIICII) and (AIIBIIC0) subunits is oxidized rapidly via the peroxodiferric intermediate, which presumably has a μ-η1:η2 structure. In these subunits the Fe(II) in site C is possibly oxidized via the peroxodiferric intermediate in the ferroxidase center as proposed previously.13 In (AIIB0CII) and (AIIB0C0) subunits, whose site B is empty, Fe(II) is first rearranged to fill sites A and B. The kinetic of this rearrangement process is the rate limiting step in oxidation of Fe(II) in (AIIB0CII) and (AIIB0C0) subunits. The model shows a single turnover in the ferroxidase center after addition of Fe(II) to apo-ferritin, i.e. ferritin with no Fe(II) or Fe(III) bound, in the presence of molecular oxygen. For subsequent turnovers Fe(III) present in the ferroxidase center is displaced by incoming Fe(II). | |
In the next step we analysed the Fe(II)/Fe(III) intermediates during the catalysis of Fe(II) oxidation. The Mössbauer parameters that we found for the peroxodiferric intermediate were compared to those reported for different peroxodiferric species in other proteins and model compounds. We observed that the values of the quadrupole splitting (ΔEQ) in HuHF and PfFtn (Table 3) are not close to those assigned to the μ-1,2-peroxodiferric binding mode in most of the di-iron cofactor enzymes and model compounds. However, the values of ΔEQ for the peroxodiferric intermediate in PfFtn and HuHF were close to those reported for the η2-O2 binding mode of dioxygen to Fe(III) in model compounds (Table 3).38 Because EPR spectroscopy showed that the majority of Fe(III) ions are antiferromagnetically coupled we propose a μ-η1:η2 binding mode for the peroxodiferric intermediate in ferritins similar to that proposed for arylamine oxygenase.38 For BfMF resonance Raman spectroscopy has been used to determine the molecular structure of the peroxodiferric intermediate. An O–O stretching frequency ν(O–O) = 851 (cm−1) was reported.38 The O–O stretching frequencies (ν(O–O)) typically reported for the μ-1,2-peroxodiferric binding mode span a wide range (830–925 cm−1)38 whose minimum is close to the value reported for the η2-O2 binding mode (ν(O–O) = 822 cm−1).1 Because a wide range of ν(O–O) might be expected for different binding modes of the peroxodiferric species, it appears to us that based on the ν(O–O) alone the exact assignment of the binding mode of the peroxodiferric intermediate in BfMF is not possible and resonance Raman data should be used in combination with Mössbauer data. Because of the available inconsistencies in the reported Mössbauer data for BfMF (Table 3) we cannot conclude that the peroxodiferric intermediate in BfMF has a μ-1,2-peroxo structure. Further experiments using different spectroscopic techniques are required to obtain a better understanding of the molecular structure of the peroxodiferric intermediate in BfMF and other ferritins.
Comparison of the Mössbauer data before the addition of dioxygen and 0.7 s after the addition of dioxygen revealed that only in the (AIIBIIC0) and (AIIBIICII) subunits the Fe(II) in sites A and B could be oxidized rapidly to form the peroxodiferric intermediate (Fig. 8). Thus, the rapid increase in the absorbance at 310 nm in HuHF and PfFtn (Fig. 4) is indeed due to the formation of the peroxodiferric intermediate and not Fe(III) products. The slower phase of the progress curves of Fe(III) formation at 310 nm (Fig. 4), which occurs after 0.7 s, represents the slow oxidation of Fe(II) in site C of the (AIIBIICII) subunits and that of Fe(II) in (AIIB0C0) and (AIIB0CII) subunits. In PfFtn less than 16% of the total Fe(II) added is oxidized slowly and in HuHF circa 37% of the total Fe(II) added is oxidized slowly (Table 2). The difference in the kinetics of Fe(II) oxidation between HuHF and PfFtn originates from the amount of (AIIB0CII), (AIIB0C0), and (AIIBIICII) subunits. In PfFtn the percentages of (AIIB0CII) and (AIIB0C0) subunits were negligible and almost all of the Fe(II) in site C was next to fully occupied ferroxidase centers, i.e. (AIIBIICII) subunits. The Fe(II) in site C of (AIIBIICII) subunits is proposed to be oxidized presumably by the peroxodiferric intermediate,13 and in this mechanism the conserved tyrosine provides a fourth electron for the complete reduction of molecular oxygen to water.13 In contrast in HuHF the percentage of (AIIBIICII) subunits was less than half of that in PfFtn and instead the percentage of (AIIB0CII) and (AIIB0C0) subunits together was 25%. The Fe(II) in (AIIB0CII) and (AIIB0C0) subunits cannot be oxidized rapidly via the peroxodiferric intermediate. In these subunits the Fe(II) should be oxidized via other mechanisms. A reasonable possibility would be the re-organization of Fe(II) to sites A and B (Fig. 8) and subsequent oxidation of Fe(II) via the peroxodiferric intermediate. This is because EPR spectroscopy indicated more than 95% of the Fe(III) to be in antiferromagnetically coupled species. If Fe(II) in site A and site C of the (AIIB0CII) and (AIIB0C0) subunits would have been oxidized separately via other mechanisms at least 12% mononuclear Fe(III) should have been observed by EPR, because the Fe(III) in site A cannot spontaneously move to the internal cavity and the Fe(III) ions in sites A and C are too far away to be coupled by exchange. The proposal that the Fe(II) ions in (AIIB0CII) and (AIIB0C0) subunits first rearrange to sites A and B, for oxidation to occur via the peroxodiferric intermediate is also in line with the previous observations by us4 and others5,10 that site C in different eukaryotic and microbial ferritins is a transient Fe(II) binding site.
In summary, we demonstrated that in PfFtn and HuHF a difference in the occupation of site B with Fe(II) exists, but the same peroxodiferric intermediate forms upon the addition of dioxygen, which decays to a major Fe(III)-dimer product. While the exact molecular structure of the peroxodiferric intermediate remains to be determined, the data support the proposal of unity in the biochemistry of ferritins, and they provide a possible explanation for the observed differences among ferritins in the reaction rates, the amount of Fe(II) oxidized per molecular oxygen, and the formation of different Fe(III) products besides the major Fe(III)-dimer. We propose that because of the variation in an amino acid residue of site B, variation in the affinity of this site for Fe(II) among ferritins exists. As a consequence the amount of (AIIBIICII), (AIIBIIC0), (AIIB0CII), and (AIIB0C0) subunits formed upon addition of Fe(II) will vary. In ferritins with higher percentages of (AIIBIICII) and (AIIBIIC0) subunits, more Fe(II) will be oxidized at a fast rate via the peroxodiferric intermediate because Fe(II) in site B is required for catalysis. This will result in different reaction rates as we observed here for HuHF and PfFtn. A higher percentage of (AIIBIICII) subunits means more Fe(II) will be oxidized in site C together with the Fe(II) in sites A and B to form two water molecules and as a result the amount of Fe(II) oxidized per dioxygen consumed will be different in PfFtn and HuHF as we reported previously.13 Moreover, differences in the relative number of (AIIBIICII), (AIIBIIC0), (AIIB0CII), and (AIIB0C0) subunits among ferritins can lead to the formation of minor Fe(III) products such as the Fe(III)-monomer, the Fe(III)-trimer, and the Fe(III) mineral core, next to the main Fe(III)-dimer product in the ferroxidase center. The validity of this proposal to other microbial and eukaryotic ferritins remains to be evaluated. It is conceivable that the variation observed in the kinetics of Fe(II) oxidation among ferritins might be relevant to the specific requirement of the iron homeostasis machinery of each organism for managing the intracellular concentrations of free Fe(II) and Fe(III) ions.
Competing financial interest
The authors declare no competing financial interest.
Acknowledgements
This work was supported in part by an EMBO Long Term Fellowship (2015-157) to KHE. We thank Marc Strampraad for his assistance with the freeze-quench setup. During the preparation of this work, Dr Simon de Vries whose work on the development of rapid freeze-quench techniques has inspired our present research and who has been a long-time friend passed away unexpectedly. We dedicate this work to his memory.
References
- K. H. Ebrahimi, P.-L. Hagedoorn and W. R. Hagen, Chem. Rev., 2015, 115, 295–326 CrossRef PubMed.
- G. n. Jutz, P. van Rijn, B. Santos Miranda and A. Böker, Chem. Rev., 2015, 115, 1653–1701 CrossRef CAS PubMed.
- M. Uchida, S. Kang, C. Reichhardt, K. Harlen and T. Douglas, Biochim. Biophys. Acta, 2010, 1800, 834–845 CrossRef CAS PubMed.
- K. H. Ebrahimi, E. Bill, P.-L. Hagedoorn and W. R. Hagen, Nat. Chem. Biol., 2012, 8, 941–948 CrossRef PubMed.
- R. K. Behera and E. C. Theil, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7925–7930 CrossRef CAS PubMed.
- C. Pozzi, F. Di Pisa, C. Bernacchioni, S. Ciambellotti, P. Turano and S. Mangani, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 1909–1920 CAS.
- F. Bou-Abdallah, H. Yang, A. Awomolo, B. Cooper, M. Woodhall, S. Andrews and N. Chasteen, Biochemistry, 2014, 53, 483–495 CrossRef CAS PubMed.
- A. S. Pereira, C. G. Timóteo, M. r. Guilherme, F. Folgosa, S. G. Naik, A. r. G. Duarte, B. H. Huynh and P. Tavares, J. Am. Chem. Soc., 2012, 134, 10822–10832 CrossRef CAS PubMed.
- K. H. Ebrahimi, P.-L. Hagedoorn and W. R. Hagen, J. Biol. Chem., 2015, 290, 26801–26810 CrossRef CAS PubMed.
- C. Pozzi, F. Di Pisa, D. Lalli, C. Rosa, E. Theil, P. Turano and S. Mangani, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 941–953 CAS.
- R. K. Watt, R. J. Hilton and D. M. Graff, Biochim. Biophys. Acta, 2010, 1800, 745–759 CrossRef CAS PubMed.
- T. Douglas and D. R. Ripoll, Protein Sci., 1998, 7, 1083–1091 CrossRef CAS PubMed.
- K. H. Ebrahimi, P. L. Hagedoorn and W. R. Hagen, ChemBioChem, 2013, 14, 1123–1133 CrossRef CAS PubMed.
- N. E. Le Brun, A. Crow, M. E. Murphy, A. G. Mauk and G. R. Moore, Biochim. Biophys. Acta, 2010, 1800, 732–744 CrossRef CAS PubMed.
- J. M. Bradley, G. R. Moore and N. E. Le Brun, J. Biol. Inorg. Chem., 2014, 19, 775–785 CrossRef CAS PubMed.
- F. Bou-Abdallah, Biochim. Biophys. Acta, 2010, 1800, 719–731 CrossRef CAS PubMed.
- F. Bou-Abdallah, G. Zhao, H. R. Mayne, P. Arosio and N. D. Chasteen, J. Am. Chem. Soc., 2005, 127, 3885–3893 CrossRef CAS PubMed.
- G. N. Jameson, W. Jin, C. Krebs, A. S. Perreira, P. Tavares, X. Liu, E. C. Theil and B. H. Huynh, Biochemistry, 2002, 41, 13435–13443 CrossRef CAS PubMed.
- F. Bou-Abdallah, P. Santambrogio, S. Levi, P. Arosio and N. D. Chasteen, J. Mol. Biol., 2005, 347, 543–554 CrossRef CAS PubMed.
- A. S. Pereira, P. Tavares, S. G. Lloyd, D. Danger, D. E. Edmondson, E. C. Theil and B. H. Huynh, Biochemistry, 1997, 36, 7917–7927 CrossRef CAS PubMed.
- A. Treffry, Z. Zhao, M. A. Quail, J. R. Guest and P. M. Harrison, FEBS Lett., 1998, 432, 213–218 CrossRef CAS PubMed.
- J. M. Bradley, N. E. Le Brun and G. R. Moore, J. Biol. Inorg. Chem., 2016, 1–16 Search PubMed.
- R. K. Watt, ChemBioChem, 2013, 14, 415–419 CrossRef CAS PubMed.
-
W. R. Hagen, Biomolecular EPR spectroscopy, CRC press, 2008 Search PubMed.
- J. Tatur, W. R. Hagen and P. M. Matias, J. Biol. Inorg. Chem., 2007, 12, 615–630 CrossRef CAS PubMed.
- F. Bou-Abdallah, P. Arosio, P. Santambrogio, X. Yang, C. Janus-Chandler and N. D. Chasteen, Biochemistry, 2002, 41, 11184–11191 CrossRef CAS PubMed.
- X. Liu and E. C. Theil, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8557–8562 CrossRef CAS PubMed.
- A. Treffry, Z. Zhao, M. Quail, J. Guest and P. Harrison, Biochemistry, 1995, 34, 15204–15213 CrossRef CAS PubMed.
- A. S. Pereira, W. Small, C. Krebs, P. Tavares, D. E. Edmondson, E. C. Theil and B. H. Huynh, Biochemistry, 1998, 37, 9871–9876 CrossRef CAS PubMed.
- K. H. Ebrahimi, P.-L. Hagedoorn and W. R. Hagen, FEBS Lett., 2013, 587, 220–225 CrossRef PubMed.
- T. Ookubo, H. Sugimoto, T. Nagayama, H. Masuda, T. Sato, K. Tanaka, Y. Maeda, H. Okawa, Y. Hayashi and A. Uehara, J. Am. Chem. Soc., 1996, 118, 701–702 CrossRef CAS.
- Y. Dong, Y. Zang, L. Shu, E. C. Wilkinson, L. Que, K. Kauffmann and E. Münck, J. Am. Chem. Soc., 1997, 119, 12683–12684 CrossRef CAS.
- K. Kim and S. J. Lippard, J. Am. Chem. Soc., 1996, 118, 4914–4915 CrossRef CAS.
- M. Yamashita, H. Furutachi, T. Tosha, S. Fujinami, W. Saito, Y. Maeda, K. Takahashi, K. Tanaka, T. Kitagawa and M. Suzuki, J. Am. Chem. Soc., 2007, 129, 2–3 CrossRef CAS PubMed.
- F. Li, K. K. Meier, M. A. Cranswick, M. Chakrabarti, K. M. Van Heuvelen, E. Münck and L. Que, J. Am. Chem. Soc., 2011, 133, 7256–7259 CrossRef CAS PubMed.
- F. A. Chavez, R. Y. Ho, M. Pink, V. G. Young Jr, S. V. Kryatov, E. V. Rybak-Akimova, H. Andres, E. Münck, L. Que Jr and W. B. Tolman, Angew. Chem., 2002, 114, 157–160 CrossRef.
- O. Horner, C. Jeandey, J. L. Oddou, P. Bonville and J. M. Latour, Eur. J. Inorg. Chem., 2002, 1186–1189 CrossRef CAS.
- T. M. Makris, V. V. Vu, K. K. Meier, A. J. Komor, B. S. Rivard, E. Münck, L. Que and J. D. Lipscomb, J. Am. Chem. Soc., 2015, 137, 1608–1617 CrossRef CAS PubMed.
- K. E. Liu, A. M. Valentine, D. Wang, B. H. Huynh, D. E. Edmondson, A. Salifoglou and S. J. Lippard, J. Am. Chem. Soc., 1995, 117, 10174–10185 CrossRef CAS.
- D. Yun, R. García-Serres, B. M. Chicalese, Y. H. An, B. H. Huynh and J. M. Bollinger, Biochemistry, 2007, 46, 1925–1932 CrossRef CAS PubMed.
- V. V. Vu, J. P. Emerson, M. Martinho, Y. S. Kim, E. Münck, M. H. Park and L. Que, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14814–14819 CrossRef CAS PubMed.
- F. Bou-Abdallah, G. Papaefthymiou, D. Scheswohl, S. Stanga, P. Arosio and N. Chasteen, Biochem. J., 2002, 364, 57–63 CrossRef CAS PubMed.
- J. Hwang, C. Krebs, B. H. Huynh, D. E. Edmondson, E. C. Theil and J. E. Penner-Hahn, Science, 2000, 287, 122–125 CrossRef CAS PubMed.
- E. R. Bauminger, A. Treffry, M. A. Quail, Z. Zhao, I. Nowik and P. M. Harrison, Biochemistry, 1999, 38, 7791–7802 CrossRef CAS PubMed.
- A. Treffry, Z. Zhao, M. A. Quail, J. R. Guest and P. M. Harrison, Biochemistry, 1997, 36, 432–441 CrossRef CAS PubMed.
- D. M. Kurtz Jr, Chem. Rev., 1990, 90, 585–606 CrossRef.
- G. Zhao, F. Bou-Abdallah, P. Arosio, S. Levi, C. Janus-Chandler and N. D. Chasteen, Biochemistry, 2003, 42, 3142–3150 CrossRef CAS PubMed.
- B. Xu and N. Chasteen, J. Biol. Chem., 1991, 266, 19965–19970 CAS.
- P. Moënne-Loccoz, C. Krebs, K. Herlihy, D. E. Edmondson, E. C. Theil, B. H. Huynh and T. M. Loehr, Biochemistry, 1999, 38, 5290–5295 CrossRef PubMed.
- J. K. Schwartz, X. S. Liu, T. Tosha, E. C. Theil and E. I. Solomon, J. Am. Chem. Soc., 2008, 130, 9441–9450 CrossRef CAS PubMed.
- G. S. Waldo and E. C. Theil, Biochemistry, 1993, 32, 13262–13269 CrossRef CAS PubMed.
- E. Bauminger, P. Harrison, D. Hechel, N. Hodson, I. Nowik, A. Treffry and S. Yewdall, Biochem. J., 1993, 296, 709–719 CrossRef CAS PubMed.
- M. A. Carrondo, EMBO J., 2003, 22, 1959–1968 CrossRef CAS PubMed.
- H. Yao, Y. Wang, S. Lovell, R. Kumar, A. M. Ruvinsky, K. P. Battaile, I. A. Vakser and M. Rivera, J. Am. Chem. Soc., 2012, 134, 13470–13481 CrossRef CAS PubMed.
- Y. Kwak, J. K. Schwartz, V. W. Huang, E. Boice, D. M. Kurtz Jr and E. I. Solomon, Biochemistry, 2015, 54, 7010–7018 CrossRef CAS PubMed.
- C. G. Timóteo, M. Guilherme, D. Penas, F. Folgosa, P. Tavares and A. S. Pereira, Biochem. J., 2012, 446, 125–133 CrossRef PubMed.
- K. H. Ebrahimi, P.-L. Hagedoorn, J. A. Jongejan and W. R. Hagen, J. Biol. Inorg. Chem., 2009, 14, 1265–1274 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mb00235h |
| ‡ Present address: Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: kourosh.honarmandebrahimi@chem.ox.ac.uk; Tel: +44(0)1865 272649 |
|
| This journal is © The Royal Society of Chemistry 2016 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Eckhard
Bill
b,
Peter-Leon
Hagedoorn
a and
Wilfred R.
Hagen
a
*a,
Eckhard
Bill
b,
Peter-Leon
Hagedoorn
a and
Wilfred R.
Hagen
a