
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymerase chain reaction in microfluidic devices
Christian D.
Ahrberg
a,
Andreas
Manz
bc and
Bong Geun
Chung
*a
aDepartment of Mechanical Engineering, Sogang University, Seoul, Korea. E-mail: bchung@sogang.ac.kr
bMicrofluidics group, KIST-Europe, Saarbrücken, Germany
cMechanotronics Department, Universität des Saarlandes, Saarbrücken, Germany
First published on 6th September 2016
Abstract
The invention of the polymerase chain reaction (PCR) has caused a revolution in molecular biology, giving access to a method of amplifying deoxyribonucleic acid (DNA) molecules across several orders of magnitude. Since the first application of PCR in a microfluidic device was developed in 1998, an increasing number of researchers have continued the development of microfluidic PCR systems. In this review, we introduce recent developments in microfluidic-based space and time domain devices as well as discuss various designs integrated with multiple functions for sample preparation and detection. The development of isothermal nucleic acid amplification and digital PCR microfluidic devices within the last five years is also highlighted. Furthermore, we introduce various commercial microfluidic PCR devices.
 Christian D. Ahrberg | Christian D. Ahrberg obtained his Bachelor and Master Degree in Chemical and Bioengineering from ETH Zürich. After graduating in 2012 he worked at Bayer Technology Services in Leverkusen before starting his PhD under supervision of Andreas Manz at the Mechatronics department of Saarland University in cooperation with KIST Europe, during which he worked on a portable real-time PCR device. Since graduating in 2016, currently he is a postdoctoral researcher at the BioNano Technology Lab in Sogang University, Seoul, Korea. |
 Andreas Manz | Andreas Manz is currently professor at the Saarland University, and principal investigator at KIST Europe, Saarbrücken Germany. He graduated 1986 with PhD from Chemistry ETH Zurich Switzerland, was group leader at Ciba-Geigy Ltd. (Novartis) Basel Switzerland 1988–1995, professor at Imperial College London UK 1995–2004, head of the ISAS (Leibniz-Institute of Analytical Sciences) and professor at the Technical University Dortmund Germany 2003–2008. He was founder and chairman of the RSC journal Lab on a Chip 2001–2008, and scientific advisor to Caliper Technologies Inc. (now part of Perkin Elmer) 1995–2000. |
 Bong Geun Chung | Bong Geun Chung is currently Associate Professor at Department of Mechanical Engineering in Sogang University, Seoul, Korea. He received his Bachelor and Master degrees from Hanyang University, Seoul, Korea and his Ph.D. degree from University of California Irvine, USA in 2007. He was an Instructor at Department of Medicine in Harvard Medical School, Boston, USA. His research interests are in the areas of Lab on a Chip, nanomedicine, and tissue engineering. He is now a director of the BioNano Technology Lab in Sogang University, Seoul, Korea. |
Introduction
Since polymerase chain reaction (PCR) was invented by Kary Mullis over 30 years ago,1 it has become a valuable tool for molecular biology as it can exponentially amplify deoxyribonucleic acid (DNA). Several million copies of DNA can be generated from a single molecule by only few cycles of PCR. A PCR cycle typically consists of three distinct steps, denaturation, annealing, and extension. In the denaturation step (≈95 °C), double stranded DNA denaturates into two single strands. Afterwards the temperature is decreased for the annealing step (≈56 °C). Primers, which are short, complementary sequences of DNA, can anneal to the single stranded target DNA. Furthermore, a polymerase attaches to the complex; the polymerase is often derived from thermophile organisms like Thermus aquaticus.2 To increase activity of the polymerase, the temperature is raised for the extension step, typically to around 72 °C. The polymerase gains activity, synthesizing a second complementary strand of DNA from free nucleotides in solution. Therefore, an exponential increase of double stranded DNA concentration is obtained by repeating three temperature steps. The method was extended to reverse transcription PCR.3 Starting with a ribonucleic acid (RNA) molecule, a reverse transcription step is performed to obtain DNA complementary to the RNA strand before PCR. A further modification of PCR introduced fluorescent dyes into the reaction mixture, allowing the real-time monitoring of reaction progress.4 This extension of the method allowed the determination of initial copies of DNA in the reaction mixture through standard curves. The method is referred to as quantitative real-time PCR (qrtPCR). Today, a number of PCR applications (e.g., diagnostics and medicine,5–7 agricultural sciences,8 and food sciences9) can be found.Microfluidic devices offer small thermal mass, low thermal inertia, and rapid heat transfer. They present a number of advantages compared to macroscopic equivalents. The small volumes reduce sample and reagent consumption, leading to inexpensive operation of the systems. However, volumes in most cases are still large enough for bulk kinetics to apply. Furthermore, different functions can easily be integrated in microfluidic devices.10 Diffusive mass transport and heat conduction scale with the characteristic length of the system. While in macroscale systems diffusion and heat conduction are slow for efficient transport, the smaller scales of microfluidic devices allow for utilization of these effects. Furthermore, these effects are easier to control than convective heat and mass transport used on larger scales instead. This allows for a better control of processes in microfluidic devices. A reduction in size also leads to a higher surface to volume ratio. This means that the exchange areas for mass and heat transport are larger, allowing for faster transport. On the other hand, surface and wall effects become more important compared to larger devices. As PCR requires many heating and cooling steps, total analysis time can be significantly reduced by miniaturization. Examples of PCR conducted in microfluidic chambers are among the first examples of PCR in microfluidic devices.11,12 Soon after ligase chain reaction, a method closely related to PCR was shown in the same devices.13 In 1998, the first flow through PCR in a microfluidic device was demonstrated by Kopp et al.14 A sample was flown through a microfluidic channel, repeatedly passing the three temperature zones for PCR. A 10 μL sample could perform 20 cycles of PCR within 90 seconds. Numerous applications of PCR in microfluidic devices have been proposed.15,16 Through the low sample and power consumption, these microfluidic devices are especially interesting for point-of-care applications. The possibility to combine sample preparation, PCR, and detection in the same automated microfluidic device also promises reduction of handling time as well as prevention of sample contamination. This review introduces and discusses various designs of PCR in microfluidic device developed within the last five years. First, space domain PCR microfluidic devices are discussed before moving on to their counterpart time domain PCR microfluidic devices. Second, isothermal nucleic acid amplification and digital PCR in microfluidic devices are discussed (Table 1).
| Method | Design & system | Approach | Ref. |
|---|---|---|---|
| Space domain PCR | Serpentine channel designs | Disposable polyimide chip | 17 |
| 30 cycles of PCR | |||
| CNC fabricated polycarbonate device | 18 | ||
| 30 cycles of PCR | |||
| Steel capillary coated with NOA61 | 19 | ||
| Reducing back pressure | |||
| Metal–polymer composite material | 20 | ||
| Improved temperature stability | |||
| Replacing active pumps with capillary forces | 21 | ||
| Model of capillary forces in channel for PCR | |||
| Reducing the number of heaters | 22 | ||
| Central heater, edges of chip cooled by heat pipes | |||
| Reducing the number of heaters 3D device heated at the bottom | 23 | ||
| Radial designs | Radial design | 24 | |
| Central heater, edges actively cooled | |||
| Radial design | 25, 26 | ||
| Heated by focusing sunlight on center of device | |||
| Capillary wrapped around cylinder | 27 | ||
| Cylinder with different temperature zones | |||
| Centrifugal microfluidic device | 28 | ||
| Mounted on dual shaft centrifuge | |||
| Digital and oscillating designs | Oscillating droplet | 30 | |
| Four parallel channels for multiplexing | |||
| Oscillating droplet | 31 | ||
| Nested PCR to increase specificity | |||
| Digital microfluidic device | 32 | ||
| Droplet moved between temperature zones | |||
| Digital microfluidic device | 33 | ||
| Real-time optical detection | |||
| Digital microfluidic device | 34 | ||
| Method for solvent replenishing | |||
| Heating/cooling systems | Capillaries in water bath | 35 | |
| Forced convection inside capillaries | |||
| Sample moved between water bath using servo motor | 38, 39 | ||
| Time domain PCR | Heating/cooling systems | Infrared heating through tungsten lamp | 40, 41 |
| Fan for active cooling | |||
| Infrared laser for heating | 42 | ||
| Silicon heating chip | 44, 45 | ||
| Portable real-time PCR device | |||
| Silicon heating chip | 46 | ||
| Melting curve analysis, multiplexing | |||
| Sample chamber on top of heat exchanger | 47, 48 | ||
| Heating and cooling through medium | |||
| Silica capillaries with thin film heaters | 49 | ||
| Cooling by fan | |||
| Capillary with bypass channel | 50 | ||
| Heating and cooling by Peltier elements | |||
| Integrated devices | Lysis and PCR in same chamber | 54 | |
| Removal of inhibitors through diffusion | |||
| Chemical lysis followed by DNA extraction with magnetic particles | 55 | ||
| Retention of DNA on silica gel membrane | 57 | ||
| Release by master mix and amplification | |||
| FTA membrane at entrance of amplification chamber retaining DNA | 58 | ||
| Dielectrophoresis to capture pathogens on slip chip | 60 | ||
| Followed by washing and PCR | |||
| Separation of amplicons by CE after PCR | 61, 62 | ||
| PCR in amplification chamber connected to CE | 63 | ||
| Real-time detection of amplicons | |||
| Centrifugal microfluidic designs | Centrifugal device with chambers | 64, 65 | |
| Counting of bacteria through Poisson distribution | |||
| PCR centrifugal device | 66, 67 | ||
| Ice-valves next to PCR chamber | |||
| Centrifugal device dividing sample in 8 sub-samples | 67, 68 | ||
| Centrifugal device subdividing sample | 69, 70 | ||
| Positive, negative control, and internal standards | |||
| LabDisc device with vapor diffusion barrier | 71 | ||
| Preventing bubble formation in amplification chamber during PCR | |||
| LabDisc device including sample preparation | 72 | ||
| Arrays | Printed array of sample droplets in oil | 73 | |
| Dispensing of single cells onto hydrophilic spots for PCR | 74 | ||
| Compartmentalization of sample by centrifugal device | 75 | ||
| Detection of amplicons by printed DNA probe array | |||
| Single cell devices | Printing of single cells into individual reaction volumes | 76 | |
| Trapping of single cells followed by lysis and RNA extraction | 77 | ||
| Encapsulation of single cells into droplets | 78 | ||
| Droplet sorting to recover genome of target species | |||
| PDMS valve to create 96 × 96 array of single cells | 79, 80 | ||
| 300 cell traps separated by valves | 81 | ||
| PCR in chambers adjacent to cell traps | |||
| Isothermal nucleic acid amplification | Implementation of LAMP on microfluidic devices | Continuous flow LAMP in droplets for DNA and microRNA | 84, 85 |
| Multiplexed LAMP in device with 8 amplification | 86 | ||
| LAMP in agarose beads | 87 | ||
| Extraction of RNA using magnetic beads followed by reverse transcription LAMP | 88 | ||
| Centrifugal device with real-time detection of LAMP reaction | 89 | ||
| Centrifugal device for multiplexed detection of 10 different species by LAMP | 90 | ||
| Centrifugal LAMP device including sample preparation and detection of products | 91 | ||
| LAMP for point-of-care devices | LAMP in capillary heated by two pocket warmers for portable format | 92 | |
| Disposable polycarbonate cassette for LAMP | 93 | ||
| LAMP followed by electrochemical detection | 94 | ||
| Ten reaction chambers with different primers for multiplexed LAMP, detection by naked eye | 95 | ||
| Digital PCR | Multiplexed digital PCR | Duplexed digital PCR in device containing 38 panels each with 770 partitions | 104 |
| Duplex digital PCR in droplets utilizing TaqMan-probes with different emission wavelength | 105, 106 | ||
| Various designs | Cross-junction to generate water in oil emulsions for droplet digital PCR | 107 | |
| Creating sample array through PDMS membrane | 108 | ||
| Megapixel digital PCR device | 109, 111, 112 | ||
| Creating droplets at T-junction | 110 | ||
| Creating sample array with negative pressure vapor proof-layer to stop sample evaporation | 113 | ||
| Digital PCR in agarose beads | 114 | ||
| Digital PCR to count nanoparticles tagged with DNA | 115 | ||
| Integrated systems | Centrifugal device dividing samples into reaction volumes | 116 | |
| Centrifugal device using step emulsification for creating droplets | 117–119 | ||
| Device combining sample preparation and digital PCR | 140, 141 | ||
| Slip chip for digital PCR | 120–122 | ||
| Commercial PCR devices | Integrated devices | Centrifugal system with sample preparation and detection | 123 |
| Automated systems including sample preparation, amplification and detection using prefilled reagent strips or blister-films | 124–126 | ||
| Integrated, automated systems utilizing cartridges | 127, 128 | ||
| Microfluidic silicon chip, with hybridization array for detection | 129 | ||
| Digital PCR devices | Based on an array of chambers | 133 | |
| Droplet based | 134, 135 | ||
| Integrated fluidic circuits for sample portioning | 136 | ||
Space domain PCR
When the sample is moved through a microchannel featuring different temperatures along its length, the temperature is dependent on the position in the channel. Thus, this group of devices is referred to as space domain PCR device. Sample preparation can easily be integrated in the microfluidic device. The duration of the thermal steps of PCR and the number of cycles are predefined by the microfluidic design. The operation of space domain PCR devices requires movement of the sample along the channel.Serpentine channel designs
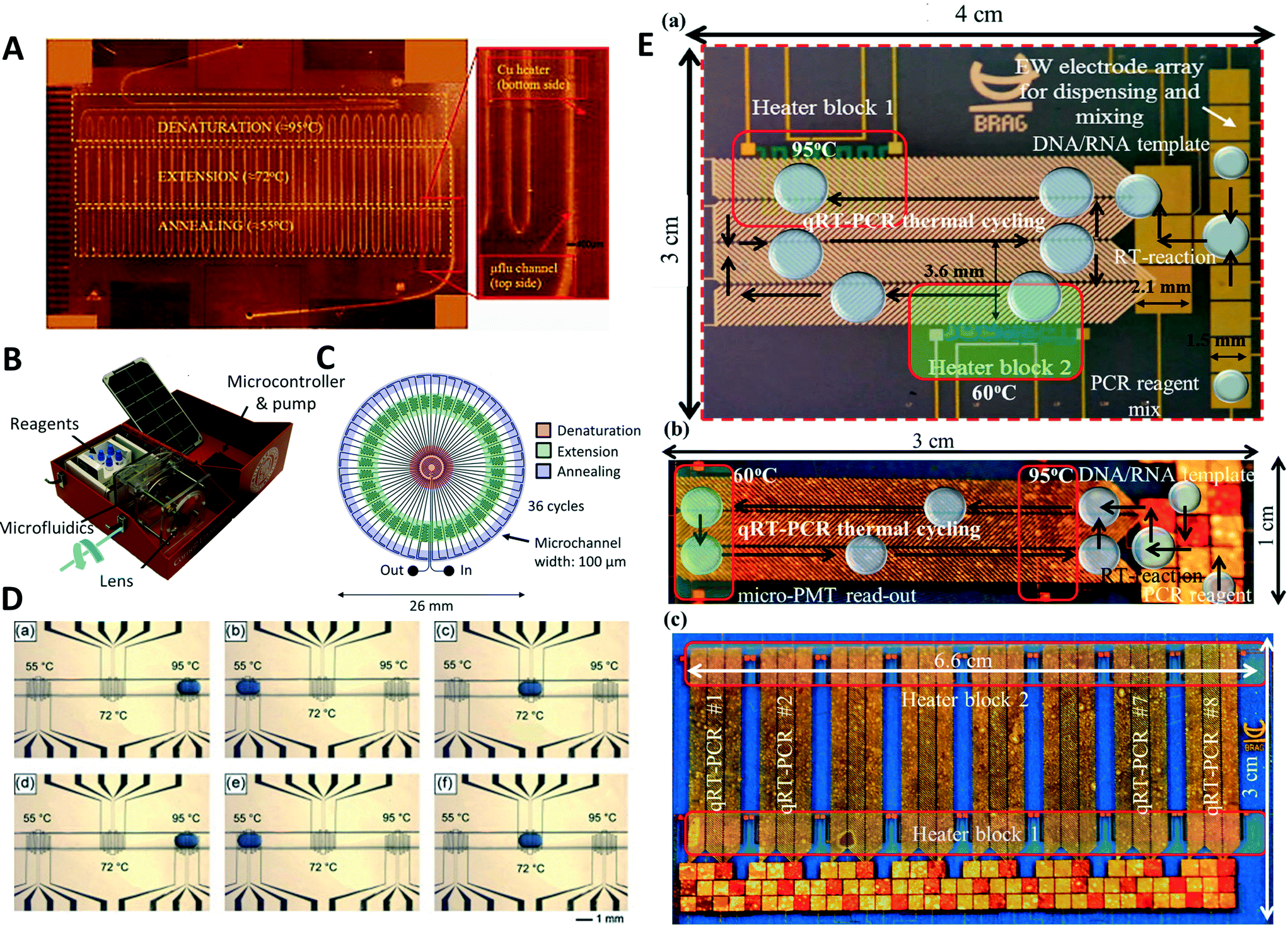
Since the first application of a microfluidic PCR device, a variety of devices improving and evolving the initial design have been developed. A polyimide device with three integrated resistive copper heaters has been presented (Fig. 1A).17 The microfluidic device was designed for 30 cycles of PCR with a time ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 for denaturation, annealing, and extension step. Due to the low cost of the plastic device, it can be disposed after the experiment preventing cross-contamination that could arise through adsorption of DNA onto the channel walls. In another study, a similar design featuring 30 cycles of PCR was created in a polycarbonate substrate using a computer numerical controlled (CNC) vertical milling machine.18 The temperatures were created using two copper heating blocks for denaturation and a combined annealing/extension step. Wu et al. reduced the flow resist of these devices by coating a 0.5 mm inner diameter steel capillary with Norland Optical Adhesive 61 (NOA61).19 A smooth, hydrophobic inner surface was used to reduce the back pressure. While the previous examples experimented with different substrate materials reducing device costs and optimizing flow resistance, other researchers optimized the heaters or simplified device design by changing sample pumping methods and reducing the number of heaters. Harandi et al. optimized the heating of the sample using a metal filled composite material.20 By employing copper comb structures inside of a polyimide matrix, temperature control with a precision of 1 °C could be achieved. In 2014, Tachibana et al. suggested a method to reduce the complexity of devices by replacing active pumps by capillary forces.21 The authors created a numerical model describing how the capillary force periodically changes. They developed a microfluidic device that could perform 50 cycles of PCR in 14 minutes. Capillary forces transported the sample through the microfluidic device, eliminating the need for an active pump.
:1:2 for denaturation, annealing, and extension step. Due to the low cost of the plastic device, it can be disposed after the experiment preventing cross-contamination that could arise through adsorption of DNA onto the channel walls. In another study, a similar design featuring 30 cycles of PCR was created in a polycarbonate substrate using a computer numerical controlled (CNC) vertical milling machine.18 The temperatures were created using two copper heating blocks for denaturation and a combined annealing/extension step. Wu et al. reduced the flow resist of these devices by coating a 0.5 mm inner diameter steel capillary with Norland Optical Adhesive 61 (NOA61).19 A smooth, hydrophobic inner surface was used to reduce the back pressure. While the previous examples experimented with different substrate materials reducing device costs and optimizing flow resistance, other researchers optimized the heaters or simplified device design by changing sample pumping methods and reducing the number of heaters. Harandi et al. optimized the heating of the sample using a metal filled composite material.20 By employing copper comb structures inside of a polyimide matrix, temperature control with a precision of 1 °C could be achieved. In 2014, Tachibana et al. suggested a method to reduce the complexity of devices by replacing active pumps by capillary forces.21 The authors created a numerical model describing how the capillary force periodically changes. They developed a microfluidic device that could perform 50 cycles of PCR in 14 minutes. Capillary forces transported the sample through the microfluidic device, eliminating the need for an active pump.
 | ||
| Fig. 1 Space domain PCR devices. (A) A serpentine-based microfluidic PCR device containing three copper heaters (reprinted with permission from ref. 17, Copyright 2014 Elsevier). (B and C) A portable PCR device powered by solar energy with the radial design featuring a central denaturation zone. Data are transferred wireless to a tablet computer for analysis (reprinted with permission from ref. 26, Copyright 2016 PLoS One). (D) Example for an oscillating droplet inside of a microfluidic channel. The sample droplet is moved between three different heaters for denaturation (a), annealing (b), and extension (c), a second cycle of PCR (d–f). For nested PCR, the droplet is removed from the channel and mixed with the reagents for the nested reaction before being reintroduced to the channel (reprinted with permission from ref. 31, Copyright 2011 Elsevier). (E) Photographs of a spiral droplet-dielectrophoresis digital microfluidic device for PCR (a), the continuous, bi-directional droplet actuation scheme (b), and the eight-plex microfluidic device (c) (reprinted with permission from ref. 33, Copyright 2015 MDPI AG). | ||
Other publications reduce the number of heaters. One example uses a single heater mounted in the middle of the device to create an denaturation temperature, while the edges of the device are cooled using heat pipes cooled with a fan to create an annealing zone.22 Another example used a three-dimensional (3D) poly(dimethylsiloxane) (PDMS) device on a single heater.23 The PDMS microfluidic device consisted of a channel meandering between the top layer and bottom layer of the microfluidic device containing the heater. The heater was set to denaturation temperature creating a denaturation zone at the bottom of the microfluidic device, while the top layer acted as an annealing/extension zone. Most devices are close to the original serpentine channel PCR device. While some advances make operation of these devices simpler and more robust, others might reduce the requirement of equipments. However, implementation of these simplifications often restricts the accessible operating conditions.
Radial designs
The number of heaters can be reduced by creating symmetric devices. Schaerli et al. created a droplet PCR device with a circular geometry.24 The center of the device was heated by a single copper rod to create a denaturation zone. Channels transported sample to the edge of the radial device where temperatures are suitable for annealing and extension due to the natural thermal gradient. A Peltier element was applied to the edge of the design to adjust the thermal gradients. A similar design was recently proposed utilizing the sun as a heat source, making the device independent of external power sources.25 The authors used a radial device and focused the sun light on its center using a lens. An absorber layer on the device converted the sun light into heat, generating temperature zones for PCR. A portable PCR device was constructed by addition of a solar powered syringe pump and fluorescence measurements (Fig. 1B and C).26 The portable device was used for the detection of Kaposi's saracoma in pseudo-biopsy samples with a sensitivity of 83% and specificity of 70%.In a different approach to radial designs, a capillary was wrapped around a cylinder machined out of three parts with a 1 mm gap in-between them.27 Each part contained its own cartridge heater and provided denaturation, annealing, or extension temperature. The number of PCR cycles could be varied by changing the number of loops of the capillary around the cylinder. Furthermore, the system consisted of another heated cylinder before the amplification cylinder to perform a reverse transcription step and a fluorescence detection unit. Miao et al. used a radial centrifugal microfluidic PCR system.28 The device employed a circular PCR containing four microfluidic chambers for denaturation, annealing, extension, and standby. The heaters were mounted underneath each chamber to provide the necessary temperature. The microfluidic device was mounted on a dual-shaft centrifugal device to allow for bidirectional pumping of the fluid using centrifugal forces. While only the number of PCR cycles can be modified in the capillary-based approach, the cycle number and duration of the single temperature steps can be changed by the centrifugal system.
Digital and oscillating microfluidic designs
Electrowetting-mediated droplets are used in digital microfluidic devices. Cycle number and temperature step durations of PCR can be controlled by software manipulation using digital microfluidics. Oscillatory microfluidics is a simplification of digital microfluidics and samples are moved back and forth in a channel through a syringe pump. A first example of oscillating or bidirectional flow PCR was shown in 2007.29 Zhang et al. used an array of independent parallel channels passing through three temperature zones to perform multiplexed PCR.30 The device used a syringe pump to generate the oscillation motion of sample between three temperature zones for the detection of different foodborne bacterial pathogens. In a similar manner, Sciancalepore et al. used a single straight channel with a sample oscillating to perform nested PCR, increasing the specificity of the reaction (Fig. 1D).31 Droplets looping between three different heaters were employed for a digital microfluidic PCR device.32 Optimization algorithms and simulations were used to increase the performance of the thermal looping and reduce sample cross-contamination. In a further use of digital microfluidics, a droplet was shuttled between two temperature zones for denaturation and annealing.33 In Fig. 1E, a photograph of the device can be seen illustrating the movement of the droplets through the different temperature zones. Furthermore, it is illustrated how several devices can be operated in parallel using the same two heating blocks. Normal PCR and reverse transcription PCR were performed using this microfluidic setup. Through the addition of a fluorescence detection unit on top of the digital microfluidic device, end point detection of amplification and quantitative PCR could be performed. A further application added a geometry to replenish solvents to counter evaporation in air-matrix digital microfluidic applications of PCR.34 A hole was drilled in the bottom layer of the digital microfluidic device. If the sample droplet was moved on top of the hole, solvent could be replenished using a syringe pump. In oscillatory and digital microfluidic devices, the number of cycles and duration of temperature steps can be changed unlike other space domain PCR devices.Heating and cooling systems
To regulate the temperature, a small water bath, heated by a tea candle, was used to perform PCR in a low power and portable manner.35 The water bath contained a holder for 20 μL capillaries for the sample. Due to the temperature gradient from the bottom to the top of the capillary, convective flow arose and the samples were cycled between different temperature zones. This demonstration is similar in function to internal convection devices shown previously, however the water bath allows a significant simplification of the heating system.36,37 In a further operation mode using water baths, PCR samples are kept in vials for conventional PCR (≈20 μL) and moved between different water bath for heating and cooling of the sample. A first example for these types of devices used 17 μL glass vials for the PCR sample which was transported between 3–4 water baths using a sample holder on a pan/tilt servo motor.38 The first water bath was used for performing a reverse transcription step when starting from RNA. The second and third were kept at temperatures for a denaturation and annealing/extension step. The last, optional bath remained at room temperature and was used to perform faster cooling steps. They were able to achieve 40 cycles of PCR in 5.5 minutes. By using smaller samples and a similar setup, Farrar et al. managed to perform 35 cycles of PCR in 14.7 seconds.39 Next to the sample capillary, a temperature sensor was placed to trigger the stepper motor moving the sample into the next bath. Additionally, a fluorescence detection unit was placed in the room temperature water bath, allowing real-time monitoring of PCR. These combinations of small sample volumes and large water bathes for heating and cooling allow for fast cycle times. Water baths need significantly more energy to heat up compared to other heating equipments. Furthermore, integration with sample preparation or post amplification analysis is not possible.Time domain PCR
In a second class of devices, samples are stationary and the temperature is changed through active heating and cooling. The sample temperature is dependent on the time; hence it is referred to as time domain PCR device. As the temperature profile is not dependent on the channel design, changes to the PCR thermal cycling can be made by modification of the heating and cooling protocols. As no movement of the sample is required, there is no requirement for pumps. A disadvantage of this method of operation is that devices cannot be operated continuously limiting sample throughput.Different methods of heating and cooling
Time domain devices are reliant on performing a suitable temperature profile on a stationary sample. Active heating and cooling steps are required. Thus, more elaborate systems are required than used for most space domain PCR devices. To solve this problem, many different approaches (e.g., fast heating or accurate temperature control) can be considered. A popular choice for heating is infrared light as the sample can be heated directly using this method without convection or conduction steps. One example used a tungsten lamp for infrared heating and a fan for forced convective cooling, achieving heating and cooling rates of about 10 °C s−1.40 In a similar approach, a tungsten lamp with a 780 nm long pass filter was used for heating.41 Two modulated lasers with different frequencies were employed for the excitation of two dyes, EvaGreen and ROX. Emission was collected by a photomultiplier tube (PMT) in a color-blind manner and was demodulated by frequency analysis, showing that two amplicons could be quantified simultaneously. Pak et al. used a 1450 nm infrared laser for heating a 1.4 μL sample inside of a microfluidic chamber.42 Through the use of a laser instead of a tungsten lamp, heating rates up to 60 °C s−1 could be obtained. The small thermal masses of microfluidic systems allow fast heating rates. Plasmonic photothermal light-to-heat conversion was used in another demonstration by irradiating a thin gold layer inside of microfluidic PCR chambers with LEDs.43 Photon–electron–phonon coupling in the gold membrane allowed heating the PCR solution up to 150 °C within 3 minutes with heating rates of 12.79 °C s−1. In a further example, utilizing the small thermal masses, sample droplets (∼200 nL) were placed on a hydrophobic glass surface and covered with small mineral oil droplets (∼1 μL) to prevent evaporation.44,45 The samples were heated using a silicon device with gold resistive heaters and temperature sensors to heat four samples in parallel. Combined with an optical detection unit and an external 12 V power supply, the system formed a portable real-time PCR device. The small temperature gradients inside of the sample and fast heating rates were also utilized to perform fast melting curve analysis after each cycle of PCR,46 showing that two targets could be amplified and quantified simultaneously using only a single intercalating dye.Active heating and cooling can be accomplished by mounting a heat exchanger beneath the sample chamber for temperature control. To change from heating to cooling, the hot heating medium can be replaced by cooling medium through a valve. For example, Wheeler et al. used water as a medium flowing through a porous matrix as a heat exchanger.47 5 μL sample could be heated and cooled with rates up to 45 °C s−1, resulting in reaction times of under 3 minutes for 30 cycles. A similar approach used a mixture of propylene glycol and water flown through a second chamber on top of the 25 μL sample chamber.48 Melting curve analysis was conducted after amplification by applying a temperature gradient to the reservoir containing the cooling medium. Capillaries were also employed to conduct time domain PCR. Sundberg et al. flowed 50 nL PCR samples into one of eight parallel fused silica capillaries coated with thin film heaters.49 Active cooling was provided by a fan, resulting in heating rates of 50 °C s−1 and cooling rates of 20 °C s−1. Once PCR was completed after 12.5 minutes, flow could be continued and samples were collected from the end of the capillary. Another approach prepared a 14 μL sample in a chamber with a bypass channel.50 The sample chamber would be filled continuously, once the flow resistance increases significantly and sample flows through the bypass capillary. Heating and cooling were actively performed using two Peltier elements placed on each side of the sample chamber. The different methods of heating and cooling show different characteristics. While some offer fast heating and cooling rates, others have a higher precision or are able to handle larger sample volumes. The choice of heating method should be made depending on applications and requirements.
Different approaches to PCR
In time domain PCR, the sample solution does not need to be moved and primers can be immobilized by binding them to a surface. This allows for a combination of PCR and microarray technology.51 The authors conducted a nested PCR inside of a microfluidic chamber. Once the first PCR was completed, a nested PCR was carried out with some of the primers covalently attached to the glass surface. After complete reaction, the chamber was washed and filled with an aqueous solution containing an intercalating dye. From the pattern of fluorescence signals, the type of avian flu RNA could be identified. Jung et al. used optically encoded, hydrophilic porous polyethylene glycol microparticles to immobilize forward primers of several different micro RNAs (Fig. 2C).52 Reverse transcription qrtPCR was performed in a channel. The fluorescence signal could be demultiplexed by decoding the code on the particles, leading to the parallel quantification of different micro RNAs. Qin et al. developed a microfluidic device from NOA81.53 The device consisted of a microfluidic chamber on top of a Peltier element for heating and cooling. As NOA81 inhibits the PCR reaction, the authors coated the device with bovine serum albumin to prevent absorption of DNA and polymerase during the reaction.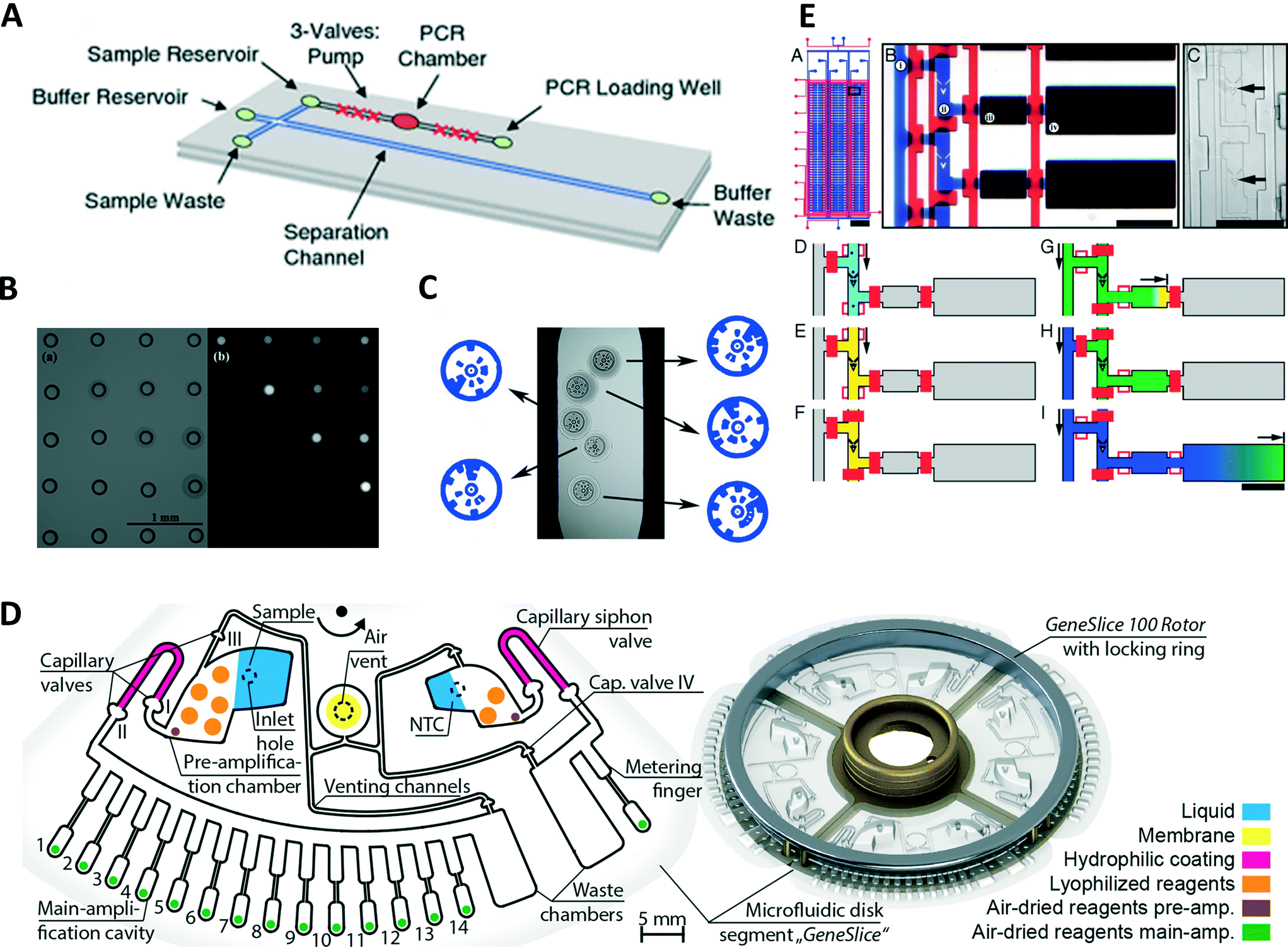 | ||
| Fig. 2 Time domain PCR devices. (A) Device capable of performing PCR from whole blood samples. Adjacent to the PCR chamber is microfluidic valves which can be used for pumping. After amplification targets can be separated using capillary electrophoresis on the same device (reprinted with permission from ref. 62, Copyright 2016 Springer). (B) Bright field image of a contact printed array of droplets (left) and fluorescence image of the same array after selective amplification of droplets using an infrared laser. This technique allows applying individual temperature profiles to each droplet as well as only reducing the testing to selected droplets (reprinted with permission from ref. 73, Copyright 2009 Royal Society of Chemistry). (C) Bright field image illustrating the barcoded hydrogel particles with primers for different microRNAs bound to them in microfluidic PCR device (reprinted with permission from ref. 52, Copyright 2016 Nature Publishing Group). (D) Example of a schematic of centrifugal microfluidic device for nested PCR (left). Image of the assembled centrifugal device fitting a commercial thermocycler (right) (reprinted with permission from ref. 70, Copyright 2015 PLoS One). (E) Device for the analysis of single cells via reverse transcription qrtPCR (a–c). Individual cells are trapped in cell traps (e) before a washing and cell lysis step (d–f). Next samples are flown into a reverse transcription chamber using active valves (g and h). Lastly qrtPCR is carried out in a chamber (i) (reprinted with permission from ref. 81, Copyright 2011 National Academy of Science). | ||
Integrated devices
While some applications focus on heating methods or modifications of PCR process, a number of publications consider the combination of time domain PCR, sample preparation, and post PCR analysis. Ma et al. performed a lysis step in a chamber connected to two side chambers.54 Once cells were lysed, they replaced the lysing buffer in the reaction chamber with PCR solution by diffusion from the side chambers. In parallel, they utilized the difference in diffusivity between long DNA strands and PCR inhibitors to remove the inhibitors from the solution in the same step. DNA was extracted from Helicobacter pylori by thermolysis in a chamber and bound to magnetic particles.55 The magnetic beads remained in the chamber during several washing steps and a subsequent filling of the chamber with PCR master mix. After PCR, successful amplification was confirmed by fluorescence measurements using the intercalating dye SYBR green I. DNA was extracted with magnetic beads, before releasing the DNA into a microfluidic amplification chamber.56 After amplification, targets were analyzed using capillary electrophoresis on the same device. Membranes can also be used to extract DNA from cell lysate. Cell lysate from human blood was prepared using a commercial product before introducing the lysate into a microfluidic device.57 DNA was then extracted from the lysate through a silica gel membrane and several washing steps before the DNA solution was flown into another chamber for PCR. Real-time detection allowed for the extraction of amplification curves for quantification followed by melting curve analysis. A membrane integrated to the entrance of the PCR chamber of a microfluidic device was used for DNA extraction.58 After lysis, DNA retained in the membrane was washed and dried before filling the chamber with PCR master mix releasing the retained DNA. An aluminium oxide membrane was used to extract and concentrate DNA from saliva samples on a microfluidic device.59 Samples added to the device and flown through the membrane, afterwards reaction mix was added to the wells and real-time PCR performed.Dielectrophoresis was used to capture pathogens from diluted whole blood into individual grooves on a slip chip.60 Once the pathogens were separated, a washing step was carried out to remove PCR inhibitors by flushing with water. The grooves were split from the feeding channel through the slip chip and cells were lysed through heating. The slip chip consisted of four parallel sample processing channels, allowing for the parallel detection of four different pathogens. Electrophoresis is the motion of charged particles relative to a fluid caused by an electric field. As particle velocities depend on particle net charge and size, it can be used for separation. Thus, electrophoresis is commonly applied for post amplification analysis in the form of slab gel electrophoresis. Miniaturization of slab gel electrophoresis is often performed in capillary electrophoresis (CE) which can achieve higher separation performance due to the higher area-to-volume ratio countering Joule heating. As CE is a powerful method of post amplification analysis, various integrated time domain PCR CE devices have recently been developed. Le Roux et al. used a PCR device with an infrared laser for heating to perform PCR.61 After amplification samples injected into a microfluidic device, amplicons were separated by amplicon length. This combination of methods was used to quantify short tandem repeats. Similarly, Manage et al. used a CE microfluidic device after amplification to separate and identify amplicons (Fig. 2A).62 As starting PCR from whole blood in this case, the CE separation step is necessary to perform fluorescence detection after the amplification reaction. This approach has the further advantages that intercalating dyes can be used and targets can be identified by length. The integration of valve pumps into the design eliminates the need for external pumps. An interesting variant of PCR combined a PCR chamber with a set of CE channels.63 After each cycle of PCR, a small sample could be drawn from the sample chamber and analyzed by CE. This allows for real-time detection of PCR products as well as their quantification. As a separation is performed in each detection step, this device is capable of monitoring several amplification processes in parallel with the utilization of a single intercalating dye.
Centrifugal microfluidic devices
In centrifugal microfluidics, fluids are distributed using centrifugal force and the Coriolis effect. The microfluidic devices are designed in the format of compact discs (CD) due to the large availability of CD-based centrifugal microfluidic device, which could be a potential candidate for point-of-care applications. A silicon/glass sandwich disk containing 24 channels and 313 microchambers with volumes of 1.5 nL was used to count Salmonella enterica.64 A solution containing the cells was placed in an entrance port and distributed over the microchambers using centrifugal force, leading to a Poisson distribution of bacteria in the microwells. PCR was initiated by thermocycling the entire disc. After PCR, the microwells showing amplification were counted to determine the initial numbers of cells. The same device was used for reverse transcription PCR.65 Direct reverse transcription of RNA was possible by using a robust DNA polymerase immune to inhibition. The introduction of ice valves next to the PCR chamber opened access to other device materials like polycarbonate.66 The liquid was frozen next to the PCR chamber, forming a plug sealing the chamber to prevent sample evaporation.The “LabDisk” format developed at the University of Freiburg in Germany offers a range of different functions integrated on a disc ranging from sample preparation to analysis after amplification. The lab disc contains four units, showing that a sample can divide into eight sub-volumes (Fig. 2D).67,68 Air-dried reagents for the PCR reaction are dissolved by the sample inside of the chambers. Thermocycling is conducted in a commercial thermocycler. This design was later modified to accommodate a positive control, no template control as well as standards.69 The modifications were done without compromising the ability of the disc to fit a commercial thermocycler and without the addition of a significant number of pipetting steps. This design was later used for forensic animal family identification.70 In a further modification, making the design compatible with the “LabDisk Player”, a vapor diffusion barrier added to the system to limit the pressure in the system that can arise through boiling of the PCR solution.71 The vapor diffusion barrier was created by adding a pneumatic chamber filled with air closer to the center of rotation than PCR chamber. In case of gas formation in the PCR chamber, the pressure is relived through the pneumatic chamber preventing undesired pumping of the solution. This “LabDisk” was extended with DNA extraction capabilities, allowing sample preparation steps to be carried out with the device.72 Bacteria samples from culture could be pipetted into the inlets of the device. An automated program on the “LabDisk” player centrifuged the device at the right frequencies to carry out cell lysis, DNA extraction and washing, multiplexed PCR and detection. This allowed a complete sample to answer assay to be carried out with minimal requirements for manual handling of the device. The development of these centrifugal devices is particularly interesting as they provide integrated devices. Only few manual handling steps are required using these devices.
Arrays
As samples are spatially fixed in space domain PCR devices, they can be encoded by their spatial position in arrays for multiplexing applications. For example, samples can be printed in a water-in-oil emulsion on a Petri dish with sample volumes in the nanoliter range (Fig. 2B).73 The sample array was created using contact printing in a custom device utilizing standard pipette tips. Single droplets can be heated by a focused infrared laser. Fast passive cooling is achieved through the surrounding oil. In another example, solution containing cells was dispensed by a pipetting robot on surface patterned with hydrophilic spots.74 The droplets were covered with mineral oil to prevent evaporation. Cells captured in droplets were lysed before adding reagents for reverse transcription by the pipetting robot. Xu et al. first conducted PCR in a device in which the sample was compartmentalized using a centrifugal device.75 After amplification, targets could be identified with an array consisting of printed DNA probes for different targets on aldehyde-modified glass slides.Single cell devices
A number of diseases are generated through abnormalities in a small number of cells. Microfluidics offers great opportunities for solving this task as cells can be compartmentalized into small volumes. Microfluidic devices combine the isolation of cells with PCR to analyze the single cells. Single cells were printed into commercial PCR tubes.76 A microfluidic inkjet printing-inspired technique was utilized in combination with a vacuum shutter to ensure that droplets containing a cell were dispersed in the tubes. In a different approach, single cells were captured in a flow through cell trapping unit.77 A chemical lysis step was carried out and the released messenger RNA captured by magnetic oligo(dT)25 beads. After moving the beads to a PCR chamber, reverse transcription PCR was carried out. The effects of methyl methanesulfonate on a single human cancer cell could be tested. Lim et al. encapsulated cells into individual droplets.78 The droplets were collected in a vial and PCR was carried out in a bench-top PCR device using TaqMan probes. Droplets were inserted into a droplet sorting device and droplets showing positive amplification were separated to recover the entire genome of the cells. Cells can also be isolated by pneumatically actuated valves in microfluidic systems. The system allows the simultaneous capturing and reverse transcription PCR in a 96 × 96 array.79,80 A further valve-based design contained 300 cell traps in individual chambers separated by valves.81 The reverse transcription chamber could be used for a chemical cell lysis step, when a “one pot” reverse transcription PCR was carried out in the PCR chamber. In Fig. 2E, the arrangement of cell traps and reaction chambers were used for reverse transcription PCR. The design of chambers was optimized to obtain a homogeneous distribution of cells, while preventing mechanical damages to them. Furthermore, background noise and effects of PCR inhibitors were reduced and the device was used to measure the expression rates of microRNA in individual leukemia cells.Isothermal nucleic acid amplification
An elimination of the temperature steps during PCR leads to a significant simplification of PCR by removing the requirements of heating and cooling systems. This can be achieved by isothermal nucleic acid amplification methods. Different methods, like nucleic acid sequence-based amplification and self-sustained sequence replication, are available for amplifying DNA at a single temperature. Another method is multiple displacement amplification, which has been demonstrated in microfluidic devices.82 However, these methods often lack specificity, because they are conducted at relatively low temperatures (≈40 °C). In 2000, Notomi et al. suggested loop-mediated isothermal amplification (LAMP), conducted at temperatures around 65 °C.83 Four sets of primers and a DNA polymerase are employed. An inner primer complementary to the sense and antisense strand of the target DNA creates a double strand of DNA. The newly synthesized strand is released through strand replacement initiated by the outer primer. The released strand now serves as a target for the second set of inner and outer primer, resulting in a stem-loop structure. In following reactions, an inner primer initiates strand replacement polymerization, yielding the original stem-loop and a second stem loop. As a single temperature step is only required, devices are simpler and power consumption is lower. This makes LAMP an interesting option for point-of-care applications when combined with low sample volumes in microfluidic devices.Implementations of LAMP on microfluidic devices
Isothermal amplification methods have been used in combination with microfluidic devices. Applications in continuous flow in droplets can be found for the detection of DNA and microRNA.84 A further application used droplets produced in a continuous manner.85 The device was able to generate droplets, incubate the droplets at the temperature required for the LAMP reaction, and perform a fluorescence readout of droplets. A device with eight different sample chambers connected to a common central sample inlet was used for multiplexed LAMP reaction.86 The device could be used to determine either three different human influenza A viruses or eight different swine flu viruses. Real-time detection of reaction progress was obtained through measurements of the redux current. Similar to solid phase PCR conducted in time domain devices, LAMP was carried out in agarose beads (Fig. 3B).87 To prepare the beads, droplets were created from a solution containing agarose and the necessary reagents for LAMP. The droplets were collected for gelation initiated by cooling. Once the droplets had gellated into beads, the LAMP reaction was initiated by heating to 43 °C. Amplification was detected by monitoring fluorescence in a real-time manner using a camera equipped with matching fluorescence filters.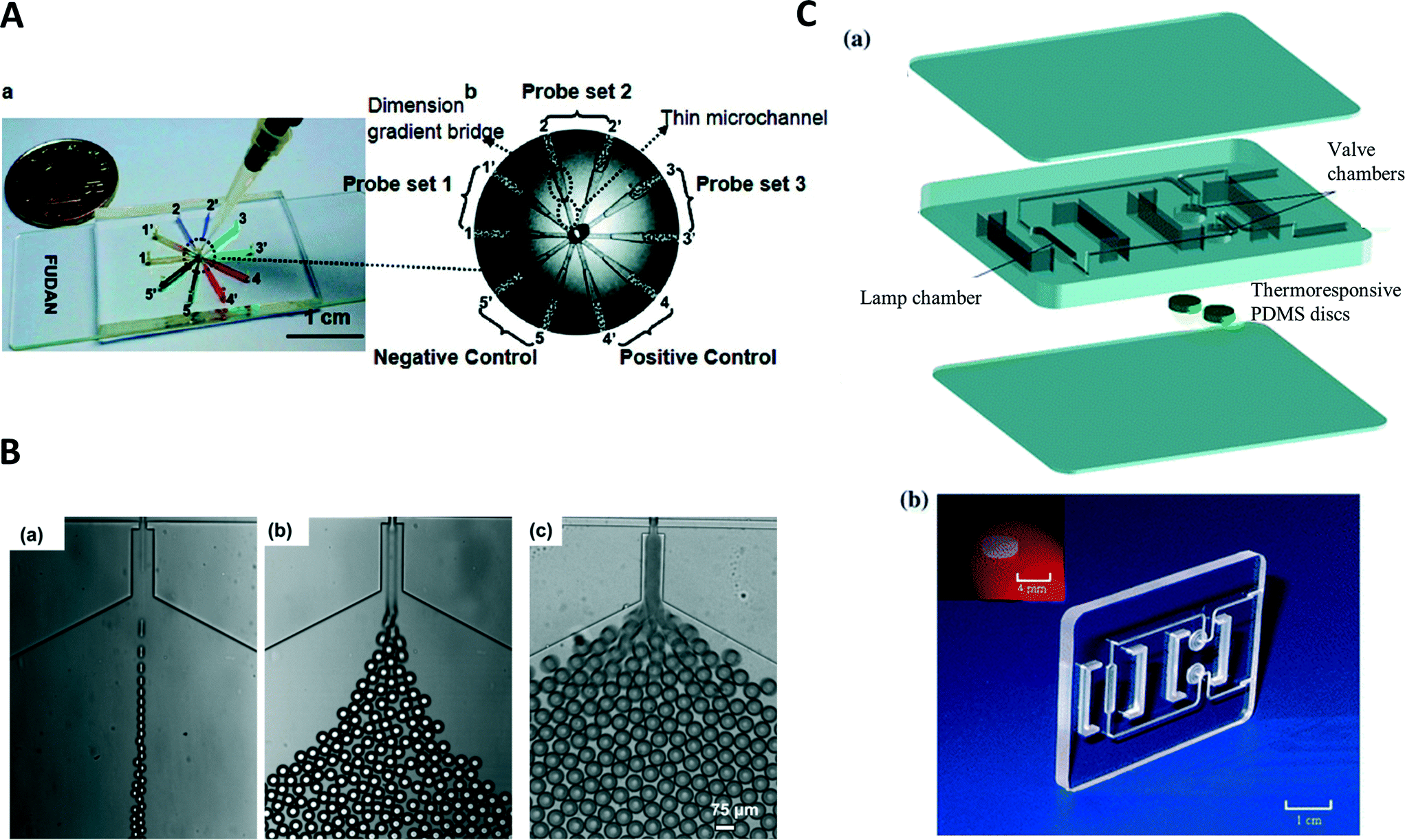 | ||
| Fig. 3 (A) Microfluidic device for the parallel amplification of several genes. (a) Photograph of the device, the channels are filled with different dyes for visualization. (b) Schematic of the device illustrating placement of the probes for the different genes (reprinted with permission from ref. 95, Copyright 2010 American Chemical Society). (B) Generation of agarose precursor droplets with different flow conditions resulting in different droplet sizes. Beads are produced from these droplets and used for LAMP amplification in the solid agarose medium (reprinted with permission from ref. 87, Copyright 2012 AIP Publishing LLC). (C) Disposable polycarbonate cassette for LAMP reactions in point-of-care applications, exploded view of the PCR cassette (top) and photograph of the assembled cassette (bottom) including an image of the thermoresponsive PDMS disc (inset) (reprinted with permission from ref. 93, Copyright 2011 Springer). | ||
Integrated device are also proposed for this type of amplification. Wang et al. suggested a device combining extraction of RNA and reverse transcription LAMP reaction.88 The samples are introduced to the device and RNA is extracted using magnetic beads. After washing steps, RNA transports to a LAMP chamber where a reverse transcription step is carried out. Afterwards products were analyzed using slab-gel electrophoresis of devices. The use of radial device design and centrifugal devices is also a popular choice for LAMP reactions. A radial device with four units containing five reaction chambers was designed.89 Once the device was filled, it was transferred to a combined heating and fluorescence detection unit. The radial symmetry of the device was utilized as the device could be rotated to measure fluorescence in the different reaction chambers using the confocal detection unit. Through this setup, real-time amplification curves could be extracted for the LAMP reaction. Other radial device designs utilize centrifugal forces to reduce the number of required handling steps. One disc-shaped device consisted of 24 reaction wells in which primers for the multiplexed detection of ten different pathogenic bacteria were pipetted and dried.90 A solution containing the extracted DNA from fishes out of aquaculture was pipetted into the inlet of the device and divided between the reaction wells using centrifugal forces. LAMP reaction and real-time detection were performed on a device suitable for the temperature manipulation and fluorescence measurement of these disks. Sayad et al. used an integrated CD device for the detection of Salmonella.91 The disc contained the steps required for reagent preparation, LAMP, and detection of amplification products. All of these steps could be automatically performed by a custom-made computer system.
LAMP for point-of-care devices
A simple point-of-care device was constructed by filling samples for LAMP amplification into a glass capillary.92 The capillary was sealed on both ends using epoxy glue and was sandwiched between two pocket warmers to obtain an amplification temperature. Amplification was detected by naked eye using a dye and a portable UV light. This allows for performing a LAMP reaction completely independent of external power supplies. Liu et al. produced a disposable polycarbonate cassette with a size of 46 mm × 36 mm × 3.7 mm (Fig. 3C).93 It contained two chambers for facilitating an isothermal amplification reaction and could be filled with only few pipetting steps due to thermoresponsive valves. An electrochemical DNA sensor was used by Hsieh et al. to detect the presence of a target DNA.94 The reaction is performed before the electrochemical detection. Ten chambers connected to a central inlet were used for a multiplexed LAMP reaction.95 Each chamber was functionalized with probes for a different target, allowing the multiplexed detection of ten different targets (Fig. 3A). After one hour of incubation at 63 °C, successful amplification could be detected by the naked eye through the precipitation of magnesium phosphate in the chambers, making this simple detection device suitable for point-of-care applications.Digital PCR
Digital PCR is an advanced refinement of PCR. While in PCR the process is carried out in a bulk solution, digital PCR divides the sample into hundreds to thousands of sub-samples. The sample is partitioned and sub-samples contain less than one molecule of DNA on average with the molecule distribution following a Poisson distribution. After a digital PCR runs the number of partitions, showing that amplification can be counted and the starting number of molecules is estimated through the Poisson distribution. Digital PCR has the advantages over conventional qrtPCR that absolute quantification does not require the measurement of a standard curve. Furthermore, digital PCR offers advantages when working with minor differences in starting molecule count or target DNAs that are rare in the sample. A requirement for digital PCR is the partitioning of the sample in sufficiently small sub-samples. Microfluidics offers several options for performing this task, either by creating droplets or microwells, which can be filled through a variety of techniques.Applications
Droplet-based digital PCR has been used for several applications. Zhu et al. used droplet-based digital PCR for the detection of HER2 and CEP17.96 The same samples were measured using alternative techniques, such as immunochemistry and fluorescence in situ hybridization to test patients with breast and gastric carcinomas. A high concordance of droplet digital PCR with the standard methods showed a high sensitivity. Another study compared the ability of digital PCR to amplify rare targets compared to qrtPCR.97 In this study, KRAS mutation was detected in patient samples. While digital PCR was able to correctly identify 11 out of 14, adenomas qrtPCR only identified 9, demonstrating that the advantage of digital PCR has over conventional PCR in handling rare targets. These advantages were utilized to measure DNA contaminations of protein drugs produced by yeast cells.98 For these experiments, the protein drug directly added to the digital PCR droplets. Thermal cycling carried out with primers for yeast, which produced the drug in this case. The higher amounts of sample could be tested in comparison to qrtPCR methods and the experiment protocol could be simplified.Floren et al. used digital PCR to identify several different species.99 Additionally they were able to determine from what tissue samples were taken due to minute differences in the amount of mitochondrial DNA, not detectable by qrtPCR. DNA from soil samples was quantified by Hoshino et al. using droplet digital PCR and standard qrtPCR.100 While qrtPCR was prone to underestimating the DNA content of the samples due to inhibition, digital PCR showed to be more robust. While for some samples the inhibition effects could be overcome by dilution prior to amplification, other samples still displayed an underestimation of DNA content by qrtPCR compared to digital PCR. The robustness of digital PCR against inhibition was also confirmed by Sedlak et al. who looked into quantification of inhibition prone samples by qrtPCR and digital PCR.101 The isothermal nucleic acid amplification is generally affected by inefficiencies in the reaction compared to normal PCR. Experiments were carried out to evaluate the suitability of isothermal amplification for digital PCR.102 It found that the reaction speed of the isothermal reaction did not correlate with the digital efficiency. Furthermore, no correlation between reaction speed and specificity was found in the experiments. Digital PCR can also be used to gain into early cycles of PCR.103 The ogive shape of digital PCR assays was decoded and information about the amplification reaction could be gained, especially about early cycle amplification.
Multiplexed digital PCR
A device with 48 individual panels, each panel containing 770 partitions, was used for duplexed digital PCR.104 The device was used to detect genetically modified organisms by measuring the amount of modified genome in a sample. In another approach to droplet-duplexed digital PCR, Escherichia coli and Listeria monocytogenes were detected in parallel (Fig. 4B).105 Droplets contained primers and TaqMan probes for both bacteria. To differentiate between the two species, colored reporters were used for the TaqMan probes. After amplification in a droplet array, two fluorescence images of the droplet array were taken. From the separate images, the number of amplified targets could be determined for each target. While the previous two methods employ spatially separated samples on the same device or different fluorescence wavelength for demultiplexing the results, Zhong et al. used a different approach.106 In this assay, two different fluorophores were used to differentiate five different targets. To allow five different targets in the multiplexed reaction, linear combinations of the two dyes are formed using different brightness for the probes. This creates a unique fluorescent signature for each target. The same technique can also be used to run duplexed digital PCR using only one fluorescent reporter with different fluorescence intensities.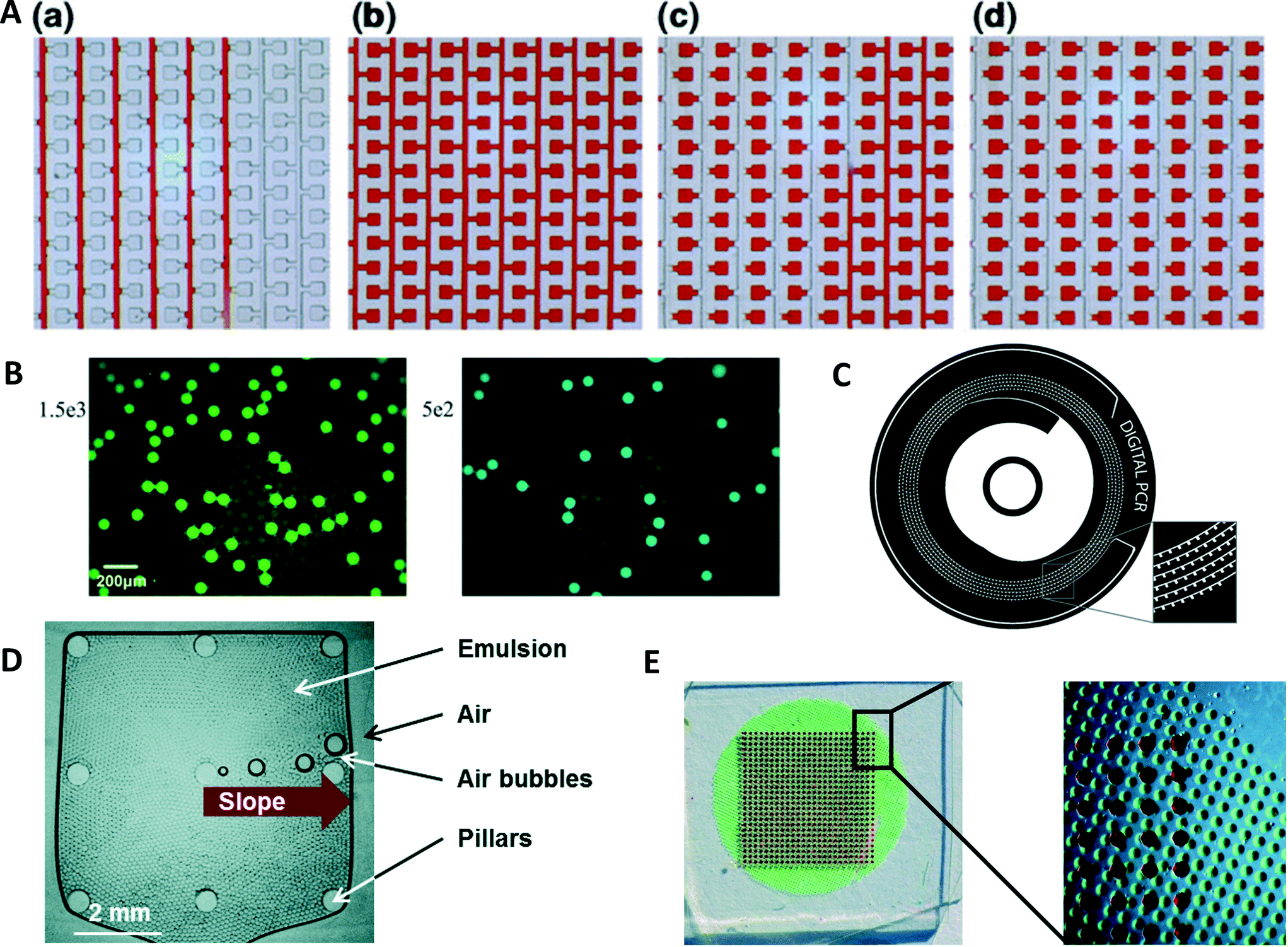 | ||
| Fig. 4 (A) A digital PCR device with a serpentine channel design with reaction chambers branching of the main channel. (a) In a first step, sample (replaced here with red dye) is flown through the degassed PDMS device. Once the reaction chambers are filled with sample (b) oil is flown through the channels (c) separating the sample into individual volumes (d) (reprinted with permission from ref. 112, Copyright 2015 Springer). (B) Multiplexed droplet digital PCR of E. coli and L. monocytogenes. Two different TaqMan probes are used, labelled with FAM E. coli (left) and VIC for L. monocytogenes (right) (reprinted with permission from ref. 105, Copyright 2015 Elsevier). (C) Centrifugal microfluidic device for the compartmentalization of sample. The sample is introduced in the central inlet chamber and then divided into the individual chambers by centrifugal force. Cross talk is prevented by filling the spiral channel with oil before amplification (reprinted with permission from ref. 116, Copyright 2014 Springer). (D) Photograph of an amplification chamber in a centrifugal droplet digital PCR device. A pyramid structure in the center of the chamber guides air bubbles developing during thermal cycling to the edge of the chamber (reprinted with permission from ref. 118, Copyright 2016 Royal Society of Chemistry). (E) Image of a device featuring an array of reaction chambers for amplification (filled with red dye). Below the array there is a vapor proof-layer preventing sample evaporation from the PDMS device (filled with green dye for visualization) (reprinted with permission from ref. 113, Copyright 2015 Royal Society of Chemistry). | ||
Designs
Several different options have been suggested for the generation of reaction compartments using different microfluidic techniques. A cross-junction has been used to generate homogenous water-in-oil droplets in a high-frequency manner.107 The droplets contained microRNA from lung cancer tissues and were incubated in a chamber, holding approximately 2 × 104 sample droplets. The density of sample volumes per area was increased in microwell assays.108 A device featuring 82000 wells with a volume of 36 fL only occupied an area of 2 mm × 2 mm, giving a well density of 20000 wells mm−2. After removing the excess sample solution, wells were sealed by applying an overpressure to a second chamber above the microwell structure. A PDMS membrane was bent to close the wells preventing possible cross-contamination. Heating and cooling for PCR was performed by a Peltier heater. A megapixel digital PCR device was created with a density of 440000 sample wells cm−2.109 The PDMS device consisted of channels from which several dead end chambers branched off. To fill the chambers, the sample solution was flown into the device under pressure, causing the air in the chambers to diffuse out through the PDMS. The channels were separated by flowing an immiscible oil through the channel to isolate the chambers. A microarray scanner was used for image analysis after the PCR run. A microfluidic device with a T-junction was used to generate droplets.110 The device consisted of two inlets, one for an oil phase and another one for the sample and a reservoir to collect the droplets. A flow was created from the air in the reservoir diffusing into the degassed PDMS creating a negative pressure. Droplet dispensing frequencies of up to 12 Hz were achieved and droplet size was dependent on the channel size.
Thompson et al. proposed a device with a serpentine channel with 1020 reaction chambers.111 The device was filled by applying a vacuum to the outlet, while pipetting sample solution in the inlet. Once chambers were filled, an oil phase was flown through the channel to separate the chambers. A similar device had several parallel channels with 1040 chambers branching of them with volumes of 2.08 nL (Fig. 4A).112 A fluorinated oil phase is filled into the channel. A device containing 650 chambers with volumes of 6.28 nL was filled by negative pressure in a chamber separated from the reaction chambers by a thin layer of PDMS (Fig. 4E).113 Once chambers were filled, they were capped with silicon oil and an adhesive cover layer. To stop evaporation of sample during digital PCR, the vapor proof-layer consisting of an array of chambers containing water was placed instead of the negative pressure chamber. During PCR, a vapor proof-layer keeps the PDMS saturated with water to minimize sample evaporation which is can be a major problem when working with small sample volumes. Agarose solution, forming nanoliter-sized beads, was used to perform digital PCR.114 While forward primers were in free solution, the reverse primers conjugated to the agarose. As the agarose melts during PCR, the efficiency of the reaction is not influenced by the conjugation of the primers to the agarose matrix. Digital PCR in microliter wells was used to count nanoparticles.115 The nanoparticles were tagged with DNA and diluted to a concentration of less than one particle per microliter. The digital PCR could be performed and the number of particles was counted by determining the number of positive microwells and the Poisson statistics.
Integrated systems
The function of microfluidic PCR devices has been integrated into the design to simplify sample preparation and integrate detection of amplification products. A spinning disc for the enrichment and quantification of rare DNA variants was suggested by Sundberg et al. (Fig. 4C).116 The disc consisted of a central loading chamber in which the sample and oil phase were injected. It demonstrated that compartments were generated through chambers rather than droplets on a centrifugal platform. The sample was separated over 1000 wells during rotation of the disc and crosstalk was eliminated through the oil phase. The digital PCR was termed quasi-digital as all chambers contained a number of copies of wild type DNA which was not the target of amplification. In another example of a spinning disc device, monodispersed droplets were created by centrifugal step emulsification.117 The droplets could be created using a chamber filled with oil in which the aqueous sample injected through a nozzle over a step. Liquids were moved by centrifugal force which was created by the “LabDisc Player”. The player heated the sample to perform recombinase polymerase amplification, another method of isothermal nucleic acid amplification. Detection of successful amplification was also carried out by the “LabDisc Player” after amplification. To carry out conventional PCR protocols with the device, a method had to be developed to remove air bubbles during thermal cycling. Thus, the disk was extended with a pyramid structure in the middle of the amplification chamber and a capillary channel leading back to the sample inlet (Fig. 4D).118 When gas bubbles developed, they were guided towards the edge of the amplification chamber through the pyramid structure. Once they arrived at the edge, they were removed from the chamber through the capillary channel enabling the conduction of thermal cycling digital PCR. This development allowed for the effective removal of gas bubbles, which could lead to issues in the analysis and droplet stability. This design was later modified to become independent of the “LabDisc Player”.119 The smaller device contained the same features as the disk device was miniaturized to fit a mini-centrifuge. Isothermal digital PCR was conducted in an in situ cycler and transferred to a microarray scanner for fluorescence scanning.Slip chips consist of two plates in contact with each other.120 While the bottom plate contains reagent wells, the top plate acts as a cover. Furthermore, the bottom plate features ducts forming a fluidic path with wells in the top plate. A first reagent can be preloaded into the wells of the top device, while a second reagent can be filled into the wells of the bottom plate. Once the chambers in the top plate are filled, the top plate can be moved to align with the wells in the bottom plate. This slip chip technology was used to compartmentalize a sample for digital PCR to detect HIV RNA.121 In further work, the robustness of digital isothermal amplification was compared against qrtPCR and was found favorable for point-of-care applications.122 Thus, a fluorescence readout unit was developed for the slip chip using a smartphone-camera equipped with an objective lens and fluorescence filters. Digital PCR offers great advantages over conventional qrtPCR, such as robustness against inhibitors and quantification without the requirement for standard curves. Due to these advantages, digital PCR could be able to replace qrtPCR in a near future.
Commercial PCR devices
The microfluidic PCR devices integrated into commercial thermal cyclers and real-time thermal cyclers. Furthermore, digital PCR instruments were introduced to the market, which requires microfluidics for sample partition.Integrated systems
A number of integrated, commercial thermal cyclers have been developed in the last years. They employ a variety of microfluidic techniques to integrate sample preparation, amplification and detection. Furthermore, integrated systems can allow for a test to be conducted in a doctor's office instead of requiring an external laboratory for testing. The GenePoc™ system uses a centrifugal platform for sample preparation as well as PCR on the same device.123 The system is capable of testing eight different samples in parallel, while testing for up to 12 different targets per sample. Similarly, the cobas® Liat PCR system from Roche Diagnostics uses prefilled reagent strips to perform sample extraction, PCR, and subsequent detection.124 Currently, three different assays are available for the system that can test for either Group A Streptococcus DNA or RNA of influenza A/B. The device can test one sample per run in 20 minutes due to the small sample volumes. In these small and low throughput systems, microfluidics is used to prepare samples for PCR followed by integrated amplification and detection on the same devices. The cross-contamination can be prevented and tests can be conducted easily. This allows medical doctors to conduct test, without requiring a specialised laboratory and trained laboratory staff. Other systems are aimed at high-throughput applications. The main advantage of microfluidics is that sample preparation can be automated to reduce the handling times. Two different approaches are most prominent. Reagents can be provided in reagent strips, like in the case of the BD MAX™ system.125 Another option is to provide reagents in blister films joined by channels as done in the FilmArray® system from bioMérieux.126 Alternatively, cartridges can be provided for different assays as done in the case of Idylla™ system from Biocartis127 or the GeneXpert® system from Cepheid.128 All of these systems are able to run multiplexed PCR assays including automated sample preparation as well as detection.The VerePLEX™ Biosystem from Veredus Laboratories does not offer automated sample preparation.129 The system uses silicon-based microfluidic devices with a volume of 25 μL for end point PCR and is capable of performing up to 50 different tests on a single sample in a single reaction. After amplification, samples are hybridized on a microarray of up to 400 individual spots, allowing for multiplexed detection. The advantages of miniaturization are used to increase the number of detectable amplifications products per PCR run from up to eight in conventional TaqMan probe assays to up to 50. A study was carried out evaluating the performance of the cobas® Liat system for the detection of Influenza A and B viruses from throat swabs.130 The study found a high sensitivity of 99.2% for Influenza A and 100% for Influenza B. Specificity for both targets was 100% in the study. A panel for testing 21 common viral and bacterial respiratory pathogenes was developed for the FilmArray® platform.131 The panel performed automated nested, multiplexed PCR on cultured organisms as well as nasal aspirants collected from patients, showing an analytical and clinical sensitivity as well as specificity comparable to other diagnostic platforms. A detection and identification of human influenza viruses was performed on the VerePLEX™ system.132
Digital PCR systems
The microfluidics can achieve a division of samples into several thousand sub-volumes in the pico to nanoliter range. Three different microfluidic approaches can be found in commercial digital PCR devices. The QuantStudio® 3D Digital PCR system by Thermo Fisher Scientific uses an array of reaction chambers with volumes of 809 picoliter.133 The array is filled using a doctor blading method, before thermalcycling is performed on a Peltier system. After amplification, arrays are analyzed on a separated array reader to measure fluorescence on two different wavelengths. Other methods use droplets, such as QX200™ Droplet Digital™ PCR system from Bio-Rad Laboratories134 or the RainDrop® system from RainDance™ Technologies.135 Both systems generate droplets on a microfluidic device. Droplets are collected in a vial and thermal cycling is performed on a conventional PCR machine. After amplification, droplets were introduced into a second microfluidic device to measure the fluorescence intensity of droplets. Both systems are capable of measuring two different colors of fluorescence probes, enabling multiplexing. While the QX200™ system can only differentiate two targets, the RainDance™ system can differentiate up to ten targets by applying different concentrations of probes for targets. The Biomark HD system from Fluidigm® uses a system of integrated fluidic circuits (IFCs).136 Reaction compartments are generated by separating the sample into sub-volumes using microfluidic valves. Samples are divided in 9180 partitions, each with a volume of 6 nL. The device has five fluorescence channels for detection. These commercial digital PCR devices are used for a variety of tasks. The droplet-based microfluidics from Bio-Rad was used for the differentiation of twelve different genetically modified maize lines,137 while the droplet digital PCR device from RainDance™ was employed to detect and quantify of three cancer mutations.138 The Fluidigm system was used for testing the suitability of digital RNA quantification.139Conclusions and perspectives
Since the first PCR in microfluidic devices was demonstrated, a number of techniques have been developed. They aim to improve the efficiency, reduce the complexity of devices, and integrate other functions, such as sample preparation and detection. However, a number of time and space domain PCR devices do not address the issue of providing simple user interfaces. While microfluidic devices offer great advantages over conventional bench-top systems, typical PCR users do not have access to microfluidic techniques, often lacking equipment or skill required for the operation. This provides a significant barrier for PCR users, preventing them from adapting microfluidic techniques. Additionally, the use of microfluidic devices can be difficult for users who do not have access to fabrication facilities. Only a few examples, like the “LabDisk”, went the additional step of not only designing the microfluidic device but also methods facilitating simple use of the microfluidic devices. Future research, especially in time and space domain PCR devices, should focus on making the microfluidic methods easily available for users. Through the origin of LAMP, technical requirements for devices were reduced. Furthermore, isothermal nucleic acid amplification methods show a higher robustness against temperature variations and inhibition compared to conventional PCR. This makes isothermal nucleic acid amplification an interesting candidate for point-of-care applications. However, like conventional PCR methods, access for end users should be as simple as possible. The higher requirements for primer design and advances in conventional PCR devices provide a further obstacle for isothermal devices. The microfluidic-based digital PCR has become a valuable alternative to conventional qrtPCR and has the potential to revolutionize the field. Digital PCR allows absolute quantification without the requirement for standard curves. Unlike conventional PCR, rare targets can be amplified and quantified with high precision and inhibition has less severe effects. Digital PCR depends on a compartmentalization of DNA molecules into individual volumes from bulk solution. Although digital PCR is already widely used, the impact of the technology could be extended and the technique might replace conventional qrtPCR. To achieve this, research should be conducted to allow for as simple handling of digital PCR devices as for qrtPCR devices. Furthermore, automatic, high-throughput, and multiplexed digital PCR has to gain the same ease of use as currently offered by qrtPCR systems.The commercial integrated PCR systems show that the automation possibilities and small fluidic handling are required. Thus, integrated microfluidic PCR devices have great advantages for high-throughput applications. Through the simple operation and high robustness against cross-contamination, integrated PCR devices allow for tests to be conducted in a medical doctor's office, eliminating the need for external laboratories. Although tests can be conducted in 20 minutes with the cobas® Liat PCR system, current applications do not seem to profit from further reductions in reaction time. Although assay costs could be decreased by a reduction of sample volumes, the cost advantages are not significant enough for most applications. Legal barriers can provide another hurdle for microfluidic systems; the commercial devices introduced here have less than 50 FDA-approved tests. This also prevents a wider application of portable point-of-care systems, in combination with the assay costs which are relatively high for developing countries. As microfluidics is the only way of creating digital PCR devices, a bigger variety of techniques is adapted in commercial digital PCR devices. Systems like RainDrop® or QX200™ require three different devices to perform the analysis. Utilizing the advantages of microfluidics has to offer integration of different functions and automation. Simplification of digital PCR processes and reduction of required equipment numbers could help to get more attention from end users.
Alternative methods of DNA and RNA detection, like DNA arrays and next generation sequencing that have been developed in the last years, offer another interesting perspective for development of microfluidic-based PCR. While PCR can detect single molecules of DNA or RNA in a rapid manner, its specificity is determined by the primer design. Furthermore, the detection of single nucleotide polymorphisms or repeats with PCR is difficult. Detection is significantly easier using DNA arrays. With sequencing methods, like pyrosequencing, the entire sequence of the target can be found. However, both DNA arrays and sequencing methods often need a minimum amount of DNA or RNA and thus require a sample preparation steps by PCR. Therefore, an integration of PCR and DNA arrays or sequencing methods on the same microfluidic device is an interesting option for future research.
Acknowledgements
This work was also supported by BioNano Health-Guard Research Center funded by the Ministry of Science, ICT & Future Planning (MSIP) of Korea as Global Frontier Project (grant number H-GUARD_2014M3A6B2060503), Republic of Korea. This research was supported by the National Research Foundation (NRF) of Korea grant funded by the MSIP (Grant number 2016R1A6A1A03012845). This work was also supported by the NRF of Korea grant funded by the MSIP (Grant number 2015R1A2A1A15054236).References
- K. Mullis, F. Faloona, S. Scharf, R. Saiki, G. Horn and H. Erlich, Cold Spring Harbor Symp. Quant. Biol., 1986, 51, 263–273 CrossRef CAS PubMed.
- R. Saiki, D. Gelfand, S. Stoffel, S. Scharf, R. Higuchi, G. Horn, K. Mullis and H. Erlich, Science, 1988, 239, 487–491 CAS.
- H. M. Temin, Virology, 1963, 20, 577–582 CrossRef CAS PubMed.
- F. F. Chehab and Y. W. Kan, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 9178–9182 CrossRef CAS.
- F. Osman, E. Hodzic, A. Omanska-Klusek, T. Olineka and A. Rowhani, J. Virol. Methods, 2013, 194, 138–145 CrossRef CAS PubMed.
- Y. Kim, Y. Choi, B.-Y. Jeon, H. Jin, S.-N. Cho and H. Lee, Yonsei Med. J., 2013, 54, 1220–1226 CrossRef CAS PubMed.
- J. F. Mehrabadi, P. Morsali, H. R. Nejad and A. A. Imani Fooladi, J. Infect. Public Health, 2012, 5, 263–267 CrossRef PubMed.
- A. Abd-Elmagid, P. A. Garrido, R. Hunger, J. L. Lyles, M. A. Mansfield, B. K. Gugino, D. L. Smith, H. A. Melouk and C. D. Garzon, J. Microbiol. Methods, 2013, 92, 293–300 CrossRef CAS PubMed.
- M. Safdar and M. F. Abasıyanık, Appl. Biochem. Biotechnol., 2013, 171, 1855–1864 CrossRef CAS PubMed.
- A. Manz, N. Graber and H. M. Widmer, Sens. Actuators, B, 1990, 1, 244–248 CrossRef CAS.
- P. Wilding, M. A. Shoffner and L. J. Kricka, Clin. Chem., 1994, 40, 1815–1818 CAS.
- M. A. Northrup, C. Gonzalez, S. Lehew and R. Hills, Micro Total Analysis Systems: Proceedings of the μTAS '94 Workshop, Springer, 1995, p. 139 Search PubMed.
- J. Cheng, M. A. Shoffner, K. R. Mitchelson, L. J. Kricka and P. Wilding, J. Chromatogr. A, 1996, 732, 151–158 CrossRef CAS PubMed.
- M. U. Kopp, A. J. d. Mello and A. Manz, Science, 1998, 280, 1046–1048 CrossRef CAS PubMed.
- P. A. Auroux, Y. Koc, A. deMello, A. Manz and P. J. R. Day, Lab Chip, 2004, 4, 534–546 RSC.
- L. Chen, A. Manz and P. J. R. Day, Lab Chip, 2007, 7, 1413–1423 RSC.
- D. Moschou, N. Vourdas, G. Kokkoris, G. Papadakis, J. Parthenios, S. Chatzandroulis and A. Tserepi, Sens. Actuators, B, 2014, 199, 470–478 CrossRef CAS.
- M. L. Ha and N. Y. Lee, Food Control, 2015, 57, 238–245 CrossRef CAS.
- J. Wu, W. Guo, C. Wang, K. Yu, Y. Ma, T. Chen and Y. Li, Cell Biochem. Biophys., 2015, 72, 605–610 CrossRef CAS PubMed.
- A. Harandi and T. Farquhar, J. Micromech. Microeng., 2014, 24, 115009 CrossRef.
- H. Tachibana, M. Saito, K. Tsuji, K. Yamanaka, L. Q. Hoa and E. Tamiya, Sens. Actuators, B, 2015, 206, 303–310 CrossRef CAS.
- J. J. Chen, M. H. Liao, K. T. Li and C. M. Shen, Biomicrofluidics, 2015, 9, 014107 CrossRef PubMed.
- W. Wu and N. Y. Lee, Anal. Bioanal. Chem., 2011, 400, 2053–2060 CrossRef CAS PubMed.
- Y. Schaerli, R. C. Wootton, T. Robinson, V. Stein, C. Dunsby, M. A. A. Neil, P. M. W. French, A. J. deMello, C. Abell and F. Hollfelder, Anal. Chem., 2009, 81, 302–306 CrossRef CAS PubMed.
- L. Jiang, M. Mancuso, Z. Lu, G. Akar, E. Cesarman and D. Erickson, Sci. Rep., 2014, 4, 4137 Search PubMed.
- R. Snodgrass, A. Gardner, L. Jiang, C. Fu, E. Cesarman and D. Erickson, PLoS One, 2016, 11, e0147636 Search PubMed.
- Y. Li, C. Zhang and D. Xing, Microfluid. Nanofluid., 2010, 10, 367–380 CrossRef.
- B. Miao, N. Peng, L. Li, Z. Li, F. Hu, Z. Zhang and C. Wang, Sensors, 2015, 15, 27954 CrossRef CAS PubMed.
- L. Chen, J. West, P.-A. Auroux, A. Manz and P. J. R. Day, Anal. Chem., 2007, 79, 9185–9190 CrossRef CAS PubMed.
- C. Zhang, H. Wang and D. Xing, Biomed. Microdevices, 2011, 13, 885–897 CrossRef PubMed.
- A. G. Sciancalepore, A. Polini, E. Mele, S. Girardo, R. Cingolani and D. Pisignano, Biosens. Bioelectron., 2011, 26, 2711–2715 CrossRef CAS PubMed.
- Z. Li, T.-Y. Ho and K. Chakrabarty, ACM Transactions on Design Automation of Electronic Systems, 2016, 21, 1–27 CrossRef.
- R. Prakash, K. Pabbaraju, S. Wong, A. Wong, R. Tellier and K. Kaler, Micromachines, 2015, 6, 63 CrossRef.
- M. J. Jebrail, R. F. Renzi, A. Sinha, J. Van De Vreugde, C. Gondhalekar, C. Ambriz, R. J. Meagher and S. S. Branda, Lab Chip, 2015, 15, 151–158 RSC.
- Y.-F. Hsieh, E. Yonezawa, L.-S. Kuo, S.-H. Yeh, P.-J. Chen and P.-H. Chen, Appl. Phys. Lett., 2013, 102, 173701 CrossRef.
- M. Krishnan, V. M. Ugaz and M. A. Burns, Science, 2002, 298, 793 CrossRef PubMed.
- N. Agrawal, Y. A. Hassan and V. M. Ugaz, Angew. Chem., Int. Ed., 2007, 46, 4316–4319 CrossRef CAS PubMed.
- K. Chan, P.-Y. Wong, P. Yu, J. Hardick, K.-Y. Wong, S. A. Wilson, T. Wu, Z. Hui, C. Gaydos and S. S. Wong, PLoS One, 2016, 11, e0149150 Search PubMed.
- J. S. Farrar and C. T. Wittwer, Clin. Chem., 2015, 61, 145–153 Search PubMed.
- Y. Ouyang, G. R. M. Duarte, B. L. Poe, P. S. Riehl, F. M. dos Santos, C. C. G. Martin-Didonet, E. Carrilho and J. P. Landers, Anal. Chim. Acta, 2015, 901, 59–67 CrossRef CAS PubMed.
- A. M. Schrell and M. G. Roper, Analyst, 2014, 139, 2695–2701 RSC.
- N. Pak, D. C. Saunders, C. R. Phaneuf and C. R. Forest, Biomed. Microdevices, 2012, 14, 427–433 CrossRef CAS PubMed.
- J. H. Son, B. Cho, S. Hong, S. H. Lee, O. Hoxha, A. J. Haack and L. P. Lee, Light: Sci. Appl., 2015, 4, e280 CrossRef CAS.
- C. D. Ahrberg, B. R. Ilic, A. Manz and P. Neuzil, Lab Chip, 2016, 16, 586–592 RSC.
- C. D. Ahrberg, A. Manz and P. Neužil, Anal. Chem., 2016, 88, 4803–4807 CrossRef CAS PubMed.
- C. D. Ahrberg, A. Manz and P. Neuzil, Sci. Rep., 2015, 5, 11479 CrossRef PubMed.
- E. K. Wheeler, C. A. Hara, J. Frank, J. Deotte, S. B. Hall, W. Benett, C. Spadaccini and N. R. Beer, Analyst, 2011, 136, 3707–3712 RSC.
- T. Houssin, J. Cramer, R. Grojsman, L. Bellahsene, G. Colas, H. Moulet, W. Minnella, C. Pannetier, M. Leberre, A. Plecis and Y. Chen, Lab Chip, 2016, 16, 1401–1411 RSC.
- S. O. Sundberg, C. T. Wittwer, R. M. Howell, J. Huuskonen, R. J. Pryor, J. S. Farrar, H. M. Stiles, R. A. Palais and I. T. Knight, Clin. Chem., 2014, 60, 1306–1313 CAS.
- C. Hurth, J. Yang, M. Barrett, C. Brooks, A. Nordquist, S. Smith and F. Zenhausern, Biomed. Microdevices, 2014, 16, 905–914 CrossRef CAS PubMed.
- Y. Sun, R. Dhumpa, D. D. Bang, J. Hogberg, K. Handberg and A. Wolff, Lab Chip, 2011, 11, 1457–1463 RSC.
- S. Jung, J. Kim, D. J. Lee, E. H. Oh, H. Lim, K. P. Kim, N. Choi, T. S. Kim and S. K. Kim, Sci. Rep., 2016, 6, 22975 CrossRef CAS PubMed.
- K. Qin, X. Lv, Q. Xing, R. Li and Y. Deng, Anal. Methods, 2016, 8, 2584–2591 RSC.
- S. Ma, D. N. Loufakis, Z. Cao, Y. Chang, L. E. K. Achenie and C. Lu, Lab Chip, 2014, 14, 2905–2909 RSC.
- C.-Y. Chao, C.-H. Wang, Y.-J. Che, C.-Y. Kao, J.-J. Wu and G.-B. Lee, Biosens. Bioelectron., 2016, 78, 281–289 CrossRef CAS PubMed.
- P. Liu, X. Li, S. A. Greenspoon, J. R. Scherer and R. A. Mathies, Lab Chip, 2011, 11, 1041–1048 RSC.
- S.-M. Zhao, L. Zhu, C.-C. Zhu, Y. Li, H.-D. Wang, L. Zhang, D.-W. Du, G.-Q. Deng, A. Wang and Y. Liu, Fenxi Huaxue, 2014, 42, 1393–1399 CAS.
- X. Qiu and M. G. Mauk, Microsyst. Technol., 2014, 21, 841–850 CrossRef.
- E. A. Oblath, W. H. Henley, J. P. Alarie and J. M. Ramsey, Lab Chip, 2013, 13, 1325–1332 RSC.
- D. Cai, M. Xiao, P. Xu, Y. Xu and W. Du, Lab Chip, 2014, 14, 3917–3924 RSC.
- D. Le Roux, B. E. Root, C. R. Reedy, J. A. Hickey, O. N. Scott, J. M. Bienvenue, J. P. Landers, L. Chassagne and P. de Mazancourt, Anal. Chem., 2014, 86, 8192–8199 CrossRef CAS PubMed.
- D. P. Manage, Y. C. Morrissey, A. J. Stickel, J. Lauzon, A. Atrazhev, J. P. Acker and L. M. Pilarski, Microfluid. Nanofluid., 2010, 10, 697–702 CrossRef.
- Y. Liu, C. Li, Z. Li, S. D. Chan, D. Eto, W. Wu, J. P. Zhang, R.-L. Chien, H. G. Wada, M. Greenstein and S. Satomura, Electrophoresis, 2016, 37, 545–552 CrossRef CAS PubMed.
- S. Furutani, H. Nagai, Y. Takamura and I. Kubo, Anal. Bioanal. Chem., 2010, 398, 2997–3004 CrossRef CAS PubMed.
- S. Furutani, H. Nagai, Y. Takamura, Y. Aoyama and I. Kubo, Analyst, 2012, 137, 2951–2957 RSC.
- M. Amasia, M. Cozzens and M. J. Madou, Sens. Actuators, B, 2012, 161, 1191–1197 CrossRef CAS.
- O. Strohmeier, S. Laßmann, B. Riedel, D. Mark, G. Roth, M. Werner, R. Zengerle and F. Stetten, Microchim. Acta, 2013, 181, 1681–1688 CrossRef.
- M. Keller, S. Wadle, N. Paust, L. Dreesen, C. Nuese, O. Strohmeier, R. Zengerle and F. von Stetten, RSC Adv., 2015, 5, 89603–89611 RSC.
- O. Strohmeier, N. Marquart, D. Mark, G. Roth, R. Zengerle and F. von Stetten, Anal. Methods, 2014, 6, 2038–2046 RSC.
- M. Keller, J. Naue, R. Zengerle, F. von Stetten and U. Schmidt, PLoS One, 2015, 10, e0131845 Search PubMed.
- G. Czilwik, I. Schwarz, M. Keller, S. Wadle, S. Zehnle, F. von Stetten, D. Mark, R. Zengerle and N. Paust, Lab Chip, 2015, 15, 1084–1091 RSC.
- G. Czilwik, T. Messinger, O. Strohmeier, S. Wadle, F. von Stetten, N. Paust, G. Roth, R. Zengerle, P. Saarinen, J. Niittymaki, K. McAllister, O. Sheils, J. O'Leary and D. Mark, Lab Chip, 2015, 15, 3749–3759 RSC.
- H. Kim, S. Vishniakou and G. W. Faris, Lab Chip, 2009, 9, 1230–1235 RSC.
- Y. Zhu, Y.-X. Zhang, W.-W. Liu, Y. Ma, Q. Fang and B. Yao, Sci. Rep., 2015, 5, 9551 CrossRef PubMed.
- Y. Xu, H. Yan, Y. Zhang, K. Jiang, Y. Lu, Y. Ren, H. Wang, S. Wang and W. Xing, Lab Chip, 2015, 15, 2826–2834 RSC.
- F. Stumpf, J. Schoendube, A. Gross, C. Rath, S. Niekrawietz, P. Koltay and G. Roth, Biosens. Bioelectron., 2015, 69, 301–306 CrossRef CAS PubMed.
- H. Sun, T. Olsen, J. Zhu, J. Tao, B. Ponnaiya, S. A. Amundson, D. J. Brenner and Q. Lin, RSC Adv., 2015, 5, 4886–4893 RSC.
- S. W. Lim, T. M. Tran and A. R. Abate, PLoS One, 2015, 10, e0113549 Search PubMed.
- V. Sanchez-Freire, A. D. Ebert, T. Kalisky, S. R. Quake and J. C. Wu, Nat. Protoc., 2012, 7, 829–838 CrossRef CAS PubMed.
- K. H. Narsinh, N. Sun, V. Sanchez-Freire, A. S. Lee, P. Almeida, S. Hu, T. Jan, K. D. Wilson, D. Leong, J. Rosenberg, M. Yao, R. C. Robbins and J. C. Wu, J. Clin. Invest., 2011, 121, 1217–1221 CAS.
- A. K. White, M. VanInsberghe, O. I. Petriv, M. Hamidi, D. Sikorski, M. A. Marra, J. Piret, S. Aparicio and C. L. Hansen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13999–14004 CrossRef CAS PubMed.
- L. Chen, A. Manz and P. J. R. Day, Anal. Biochem., 2008, 372, 128–130 CrossRef CAS PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, e63 CrossRef CAS PubMed.
- M. C. Giuffrida, L. M. Zanoli, R. D'Agata, A. Finotti, R. Gambari and G. Spoto, Anal. Bioanal. Chem., 2015, 407, 1533–1543 CrossRef CAS PubMed.
- T. D. Rane, L. Chen, H. C. Zec and T.-H. Wang, Lab Chip, 2015, 15, 776–782 RSC.
- J. Luo, X. Fang, D. Ye, H. Li, H. Chen, S. Zhang and J. Kong, Biosens. Bioelectron., 2014, 60, 84–91 CrossRef CAS PubMed.
- L. Desbois, A. Padirac, S. Kaneda, A. J. Genot, Y. Rondelez, D. Hober, D. Collard and T. Fujii, Biomicrofluidics, 2012, 6, 044101 CrossRef PubMed.
- C.-H. Wang, K.-Y. Lien, T.-Y. Wang, T.-Y. Chen and G.-B. Lee, Biosens. Bioelectron., 2011, 26, 2045–2052 CrossRef CAS PubMed.
- T. Wang, Y. Zhang, G. Huang, C. Wang, L. Xie, L. Ma, Z. Li, X. Luo, H. Tian, Q. Li, X. Li, Z. Lv and X. Bao, Sci. China: Chem., 2012, 55, 508–514 CrossRef CAS.
- Q.-J. Zhou, L. Wang, J. Chen, R.-N. Wang, Y.-H. Shi, C.-H. Li, D.-M. Zhang, X.-J. Yan and Y.-J. Zhang, J. Microbiol. Methods, 2014, 104, 26–35 CrossRef CAS PubMed.
- A. A. Sayad, F. Ibrahim, S. M. Uddin, K. X. Pei, M. S. Mohktar, M. Madou and K. L. Thong, Sens. Actuators, B, 2016, 227, 600–609 CrossRef CAS.
- Y. Zhang, L. Zhang, J. Sun, Y. Liu, X. Ma, S. Cui, L. Ma, J. J. Xi and X. Jiang, Anal. Chem., 2014, 86, 7057–7062 CrossRef CAS PubMed.
- C. Liu, M. G. Mauk and H. H. Bau, Microfluid. Nanofluid., 2011, 11, 209–220 CrossRef CAS.
- K. Hsieh, B. S. Ferguson, M. Eisenstein, K. W. Plaxco and H. T. Soh, Acc. Chem. Res., 2015, 48, 911–920 CrossRef CAS PubMed.
- X. Fang, H. Chen, S. Yu, X. Jiang and J. Kong, Anal. Chem., 2011, 83, 690–695 CrossRef CAS PubMed.
- Y. Zhu, D. Lu, M. E. Lira, Q. Xu, Y. Du, J. Xiong, M. Mao, H. C. Chung and G. Zheng, Exp. Mol. Pathol., 2016, 100, 287–293 CrossRef CAS PubMed.
- D. Azuara, M. M. Ginesta, M. Gausachs, F. Rodriguez-Moranta, J. Fabregat, J. Busquets, N. Pelaez, J. Boadas, S. Galter, V. Moreno, J. Costa, J. de Oca and G. Capellá, Clin. Chem., 2012, 58, 1332–1341 CAS.
- M. Hussain, R. Fantuzzo, S. Mercorelli and C. Cullen, J. Pharm. Biomed. Anal., 2016, 123, 128–131 CrossRef CAS PubMed.
- C. Floren, I. Wiedemann, B. Brenig, E. Schütz and J. Beck, Food Chem., 2015, 173, 1054–1058 CrossRef CAS PubMed.
- T. Hoshino and F. Inagaki, Syst. Appl. Microbiol., 2012, 35, 390–395 CrossRef CAS PubMed.
- R. H. Sedlak, J. Kuypers and K. R. Jerome, Diagn. Microbiol. Infect. Dis., 2014, 80, 285–286 CrossRef CAS PubMed.
- E. M. Khorosheva, M. A. Karymov, D. A. Selck and R. F. Ismagilov, Nucleic Acids Res., 2016, 44, e10 CrossRef PubMed.
- D. L. Duewer, M. C. Kline and E. L. Romsos, Anal. Bioanal. Chem., 2015, 407, 9061–9069 CrossRef CAS PubMed.
- P. Zhu, W. Fu, C. Wang, Z. Du, K. Huang, S. Zhu and W. Xu, Anal. Chim. Acta, 2016, 916, 60–66 CrossRef CAS PubMed.
- X. Bian, F. Jing, G. Li, X. Fan, C. Jia, H. Zhou, Q. Jin and J. Zhao, Biosens. Bioelectron., 2015, 74, 770–777 CrossRef CAS PubMed.
- Q. Zhong, S. Bhattacharya, S. Kotsopoulos, J. Olson, V. Taly, A. D. Griffiths, D. R. Link and J. W. Larson, Lab Chip, 2011, 11, 2167–2174 RSC.
- P. Wang, F. Jing, G. Li, Z. Wu, Z. Cheng, J. Zhang, H. Zhang, C. Jia, Q. Jin, H. Mao and J. Zhao, Biosens. Bioelectron., 2015, 74, 836–842 CrossRef CAS PubMed.
- Y. Men, Y. Fu, Z. Chen, P. A. Sims, W. J. Greenleaf and Y. Huang, Anal. Chem., 2012, 84, 4262–4266 CrossRef CAS PubMed.
- K. A. Heyries, C. Tropini, M. VanInsberghe, C. Doolin, O. I. Petriv, A. Singhal, K. Leung, C. B. Hughesman and C. L. Hansen, Nat. Methods, 2011, 8, 649–651 CrossRef CAS PubMed.
- H. Tanaka, S. Yamamoto, A. Nakamura, Y. Nakashoji, N. Okura, N. Nakamoto, K. Tsukagoshi and M. Hashimoto, Anal. Chem., 2015, 87, 4134–4143 CrossRef CAS PubMed.
- A. M. Thompson, A. Gansen, A. L. Paguirigan, J. E. Kreutz, J. P. Radich and D. T. Chiu, Anal. Chem., 2014, 86, 12308–12314 CrossRef CAS PubMed.
- Q. Song, Y. Gao, Q. Zhu, Q. Tian, B. Yu, B. Song, Y. Xu, M. Yuan, C. Ma, W. Jin, T. Zhang, Y. Mu and Q. Jin, Biomed. Microdevices, 2015, 17, 1–8 CrossRef CAS PubMed.
- Q. Tian, Q. Song, Y. Xu, Q. Zhu, B. Yu, W. Jin, Q. Jin and Y. Mu, Anal. Methods, 2015, 7, 2006–2011 RSC.
- X. Leng, W. Zhang, C. Wang, L. Cui and C. J. Yang, Lab Chip, 2010, 10, 2841–2843 RSC.
- D. Paunescu, C. A. Mora, L. Querci, R. Heckel, M. Puddu, B. Hattendorf, D. Günther and R. N. Grass, ACS Nano, 2015, 9, 9564–9572 CrossRef CAS PubMed.
- S. O. Sundberg, C. T. Wittwer, L. Zhou, R. Palais, Z. Dwight and B. K. Gale, Biomed. Microdevices, 2014, 16, 639–644 CrossRef CAS PubMed.
- F. Schuler, F. Schwemmer, M. Trotter, S. Wadle, R. Zengerle, F. von Stetten and N. Paust, Lab Chip, 2015, 15, 2759–2766 RSC.
- F. Schuler, M. Trotter, M. Geltman, F. Schwemmer, S. Wadle, E. Dominguez-Garrido, M. Lopez, C. Cervera-Acedo, P. Santibanez, F. von Stetten, R. Zengerle and N. Paust, Lab Chip, 2016, 16, 208–216 RSC.
- F. Schuler, C. Siber, S. Hin, S. Wadle, N. Paust, R. Zengerle and F. von Stetten, Anal. Methods, 2016, 8, 2750–2755 RSC.
- W. Du, L. Li, K. P. Nichols and R. F. Ismagilov, Lab Chip, 2009, 9, 2286–2292 RSC.
- B. Sun, F. Shen, S. E. McCalla, J. E. Kreutz, M. A. Karymov and R. F. Ismagilov, Anal. Chem., 2013, 85, 1540–1546 CrossRef CAS PubMed.
- D. A. Selck, M. A. Karymov, B. Sun and R. F. Ismagilov, Anal. Chem., 2013, 85, 11129–11136 CrossRef CAS PubMed.
- GenePOC, http://www.genepoc-diagnostics.com/en/technology/, (accessed 31.08.2016).
- Roche Diagnostics, https://usdiagnostics.roche.com/en/instrument/cobas-liat.html, (accessed 31.08.2016).
- BD Diagnostic Systems, http://moleculardiagnostics.bd.com/product/max/, (accessed 31.08.2016).
- bioMérieux SA, http://www.biomerieux-diagnostics.com/filmarray, (accessed 31.08.2016).
- Biocartis, https://www.biocartis.com/, (accessed 31.08.2016).
- Cepheid, http://www.cepheid.com/us/cepheid-solutions/systems/genexpert-systems/genexpert-i, (accessed 31.08.2016).
- Veredus Laboratories, http://vereduslabs.com/system-and-software/vereplex-biosystem/, (accessed 31.08.2016).
- M. J. Binnicker, M. J. Espy, C. L. Irish and E. A. Vetter, J. Clin. Microbiol., 2015, 53, 2353–2354 CrossRef CAS PubMed.
- M. A. Poritz, A. J. Blaschke, C. L. Byington, L. Meyers, K. Nilsson, D. E. Jones, S. A. Thatcher, T. Robbins, B. Lingenfelter, E. Amiott, A. Herbener, J. Daly, S. F. Dobrowolski, D. H. F. Teng and K. M. Ririe, PLoS One, 2011, 6, e26047 CAS.
- J. Teo, P. D. Pietro, F. S. Biagio, M. Capozzoli, Y.-M. Deng, I. Barr, N. Caldwell, K.-L. Ong, M. Sato, R. Tan and R. Lin, Arch. Virol., 2011, 156, 1371–1378 CrossRef CAS PubMed.
- Thermo Fisher Scientific, https://www.thermofisher.com/de/de/home/life-science/pcr/digital-pcr/quantstudio-3d-digital-pcr-system.html, (accessed 01.09.2016).
- Bio-Rad, http://www.bio-rad.com/en-ch/product/qx200-droplet-digital-pcr-system, (accessed 01.09.2016).
- RainDance Technologies, http://raindancetech.com/digital-pcr-tech/raindrop-digital-pcr-system/, (accessed 01.09.2016).
- Fluidigm, https://www.fluidigm.com/products/biomark-hd-system, (accessed 01.09.2016).
- D. Dobnik, B. Spilsberg, A. Bogožalec Košir, A. Holst-Jensen and J. Žel, Anal. Chem., 2015, 87, 8218–8226 CrossRef CAS PubMed.
- E. Zonta, F. Garlan, N. Pécuchet, K. Perez-Toralla, O. Caen, C. Milbury, A. Didelot, E. Fabre, H. Blons, P. Laurent-Puig and V. Taly, PLoS One, 2016, 11, e0159094 Search PubMed.
- R. Sanders, D. J. Mason, C. A. Foy and J. F. Huggett, PLoS One, 2013, 8, e75296 CAS.
- Q. Tian, B. Yu, Y. Mu, Y. Xu, C. Ma, T. Zhang, W. Jin and Q. Jin, RSC Adv., 2015, 5, 81889–81896 RSC.
- Q. Tian, Y. Mu, Y. Xu, Q. Song, B. Yu, C. Ma, W. Jin and Q. Jin, Anal. Biochem., 2015, 491, 55–57 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |