
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous cell sorting in a flow based on single cell resonance Raman spectra†
David
McIlvenna
a,
Wei E.
Huang
b,
Paul
Davison
c,
Andrew
Glidle
a,
Jon
Cooper
a and
Huabing
Yin
*a
aDivision of Biomedical Engineering, School of Engineering, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: Huabing.yin@glasgow.ac.uk
bDepartment of Engineering Science, University of Oxford, Parks Road, Oxford OX1 3PJ, UK
cKroto Research Institute, Department of Civil and Structural Engineering, North Campus, The University of Sheffield, Broad Lane, Sheffield S3 7HQ, UK
First published on 2nd March 2016
Abstract
Single cell Raman spectroscopy measures a spectral fingerprint of the biochemistry of cells, and provides a powerful method for label-free detection of living cells without the involvement of a chemical labelling strategy. However, as the intrinsic Raman signals of cells are inherently weak, there is a significant challenge in discriminating and isolating cells in a flowing stream. Here we report an integrated Raman-microfluidic system for continuous sorting of a stream of cyanobacteria, Synechocystis sp. PCC6803. These carotenoid-containing microorganisms provide an elegant model system enabling us to determine the sorting accuracy using the subtly different resonance Raman spectra of microorganism cultured in a 12C or 13C carbon source. Central to the implementation of continuous flow sorting is the use of “pressure dividers” that eliminate fluctuations in flow in the detection region. This has enabled us to stabilise the flow profile sufficiently to allow automated operation with synchronisation of Raman acquisition, real-time classification and sorting at flow rates of ca. <100 μm s−1, without the need to “trap” the cells. We demonstrate the flexibility of this approach in sorting mixed cell populations with the ability to achieve 96.3% purity of the selected cells at a speed of 0.5 Hz.
Introduction
Recently, the importance of individual heterogeneity in populations has become increasingly recognised in understanding cell behaviour and signalling in both health and pathology. This has led to the rapid development of single cell technologies,1–4 including a range of methods for cell separation and sorting. Despite fluorescence activated sorting dominating current methods,5 the desire to simplify analytical workflows and not to interfere with the “natural” cell state makes label-free sorting strategies extremely attractive.6 To date, the majority of label-free sorting methods have exploited differences in the physical properties of cells.6–8Single cell Raman spectroscopy (SCRS) effectively measures the biochemical profile of all Raman active components in an individual cell, enabling quantitative and multiplexed studies of cellular functionality without extrinsic and external labelling processes.9–11 These advantages have been illustrated in many applications, with examples including cell phenotype identification (e.g. microbes and tumour cells12–14), monitoring cell differentiation and cell physiological states,15–17 evaluation of biomass stoichiometry of single cells18 and in vivo interrogation of cellular composition.19 Furthermore, combining SCRS with stable isotope probing, enables cell metabolic activity and functions to be correlated,12,20–22 allowing the enrichment of a cell population in a label-free and non-destructive manner, that does not alter cell metabolism or state.23
In a similar way to fluorescence activated cell sorting, Raman activated cell sorting (RACS) can be implemented in single cell, chip based systems, although its implementation in a continuous flow system is hampered by the long acquisition times required for inherently weak Raman signals.1 Consequently the majority of techniques for acquiring single cell Raman spectra involve immobilisation of cells, e.g. by trapping in solution.24,25 The problem is illustrated by the recent approach of using surface enhanced Raman scattering (SERS) probes,26 which although increasing the Raman intensity by around an order of magnitude, itself involves the addition of extra manipulative steps in the workflow.27,28 Other recent approaches including optimisation of optical modules29 and the use of resonance Raman (RR) of active cellular compositions30 have enabled the acquisition of reproducible, intrinsic Raman spectra of cells in around 100 ms29 or even at 1 ms,31 greatly enhancing the feasibility of flow based RACS in the future.
To date, all the reported RACS systems are based on “trap-and-release” methods.32 Among reported RACS systems, and despite their weak trapping-forces and low throughput (only a few cells per minute33), optical tweezers have been the most commonly used method to maintain the cell in position during measurement.24,33–37 Here, photo-damage is highly possible when laser tweezers of visible wavelengths are used.38–40 Recently, work in low conductivity, non-physiological buffers, has shown that the trapping capability of dielectrophoresis in relatively high flow rates (4 mm s−1) has led to an overall improvement of the throughput of RACS.41 However, using these buffers can lead to concerns surrounding cell viability.25,42,43
Here we demonstrate “trap-free” RACS in a flow that allows continuous and automated sorting of individual microbial cells, based on their intrinsic single cell resonance Raman spectra. Key to the development is the on-chip integration of novel microfluidic pressure dividers to eliminate local pressure fluctuations to provide a stable flow field in the detection region. As a result, a mechanical switching system can be used in low flow rates. A notable feature of employing mechanical actuation is that it does not impose any additional constraints on the physical properties of cells and medium, making it a generic platform for a broad range of applications.
In developing this platform, we have made use of signals from carotenoids, one of the most structurally diverse pigments found in bacteria.44 These pigments have strong, characteristic, resonance Raman signals.30 Present in nearly all photosynthetic cells, they have been used as intrinsic biomarker to indicate cells' physiological function.31,45 The capability of sorting carotenoid-containing cells based on a specific function, such as CO2 fixation, will provide an invaluable tool for environmental science such as studies of ocean acidification. Using the strain of photosynthetic bacterium, Synechocystis sp. PCC6803 as a model, we demonstrated the capability of the pressure divider RACS system for the automated isolation of individual bacterial cells based on small Raman shifts linked to CO2 fixation function (at a sorting frequency of ∼0.5 Hz and an achieved purity of 96% target cells in the collection fraction).
Experimental section
Cell culture and preparation
BG-11 medium was prepared using BG-11 solution (Sigma-Aldrich, UK) diluted 50 times with sterilised deionised (DI) water (0.2 μm filtration). The pH of BG-11 medium was adjusted to 7.1 by adding NaOH. Synechocystis sp. PCC6803 cells were cultured for 4 days in a light incubator (30 °C temperature, 30 μmol m−2 s−1 light intensity and 150 rpm shaking) in BG-11 media containing 5 mM of either 12C or 13C-labelled sodium bicarbonate (Sigma Aldrich, UK) as the sole carbon source, giving rise to 12C or 13C-containing cells. Cell growth was monitored by measuring optical density (OD) at 730 nm in a microplate reader (Biotek Synergy HT, Biotek, UK). Mixtures of different ratios of 12C to 13C Synechocystis sp. PCC6803 cultures were then prepared by mixing different volumes of 12C to 13C Synechocystis sp. PCC6803 taking into account different cell concentration obtained from the OD730 readings.Raman signal acquisition
Raman signals were obtained using a Horiba Jobin-Yvon HR800 UV Raman spectrometer fitted with a Horiba Jobin-Yvon Synapse CCD. The system used a Quantum Laser Torus 532 nm laser with an air corrected Leica 50×/0.55NA objective and 1 mm spectrometer entrance pinhole. When measurements were made through a 1 mm thick quartz substrate, this resulted in an effective size of the detection area of around 14 μm diameter (determined by mapping features of known sizes) and approximately 5 mW intensity at the sample. A 600 g per inch grating was used and centred around 1300 cm−1. Labspec 5 software was used in all cases to set up the Raman spectrometer. However, Raman signals were directly readout from the CCD using a custom Labview VI (National Instruments Corp., UK) programme that was developed to synchronise the operation of the CCD and the pressure control system (MFCS-1000, Fluigent, Villejuif, France).Microfluidic device manufacture
A mould for the devices was manufactured using standard lithography techniques with Microchem SU8-2005 resist. After treatment of the mould with trichloro(1H,1H,2H,2H-perfluoro-octyl)silane in a desiccator under vacuum, Sylgard 184 PDMS mixed in a ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 elastomer to curing agent was poured onto it, then cured in an oven. The cured device was peeled from the mould, connection holes punched, then bonded to a glass microscope slide following treatment in oxygen plasma.
:1 elastomer to curing agent was poured onto it, then cured in an oven. The cured device was peeled from the mould, connection holes punched, then bonded to a glass microscope slide following treatment in oxygen plasma.
Device and flow characterization
FS04F 1 μm diameter envy green fluorescent polystyrene beads (Bangs Laboratories, Inc., Indiana, USA) were used to characterise flow. Optical images and videos were acquired using an inverted Olympus lX71 microscope (Olympus, Hamburg, Germany) and Andor CCD (Andor Technology Ltd., Belfast, UK). Image J was used to process Optical images. To measure the speed that beads were moving in the devices, videos were taken with a fixed exposure time of 0.2 s for each frame. These were then converted into a series of individual frames and the lengths of the bright light streaks, caused by the movement of the beads during the exposure, were measured.Results and discussions
The pressure divider concept
The capability to precisely manipulate cells in a continuous flow is central to any method for effective and efficient cell sorting. At the microscale, a characteristic of pressure driven flow is that there is often instability whether this be caused by pressure fluctuations, valve actuations, or environmental vibrations. These effects become prominent in low flow rate regimes, as is required for continuous RACS (e.g. <500 μm s−1) (see Fig. S1†). To address these challenges, we have developed a novel approach that integrates “microfluidic pressure dividers” on chip to protect regional flow stability.The concept of a pressure divider was inspired by the voltage control resistor networks used in electronics (where ‘pressure’ is analogous to ‘voltage’ and ‘hydrodynamic resistance’ replaces ‘electrical resistance’). As illustrated in Fig. 1A, when a large resistance (e.g. Radd) is in series with a small resistance (e.g. Rd), any variation in the pressure applied across both (e.g. ε) is distributed across each individual resistance in proportion to its size. As a consequence, pressure fluctuations in a region of interest (e.g. εd for the detection region) are minimised (i.e. εd ∼ 0 when Radd ≫ Rd; in contrast, εd = ε when Radd = 0). In low flow rate regimes the pressure drop across the detection region (ΔPd) is often close to or less than the pressure fluctuations intrinsic to the commonly available pressure pumps (i.e. ΔPd ≤ ε). By making Radd ≫ Rd, undisturbed delivery of samples to the detection point can be achieved regardless of pressure variations elsewhere in the system such that pressure-switching mechanism becomes feasible.
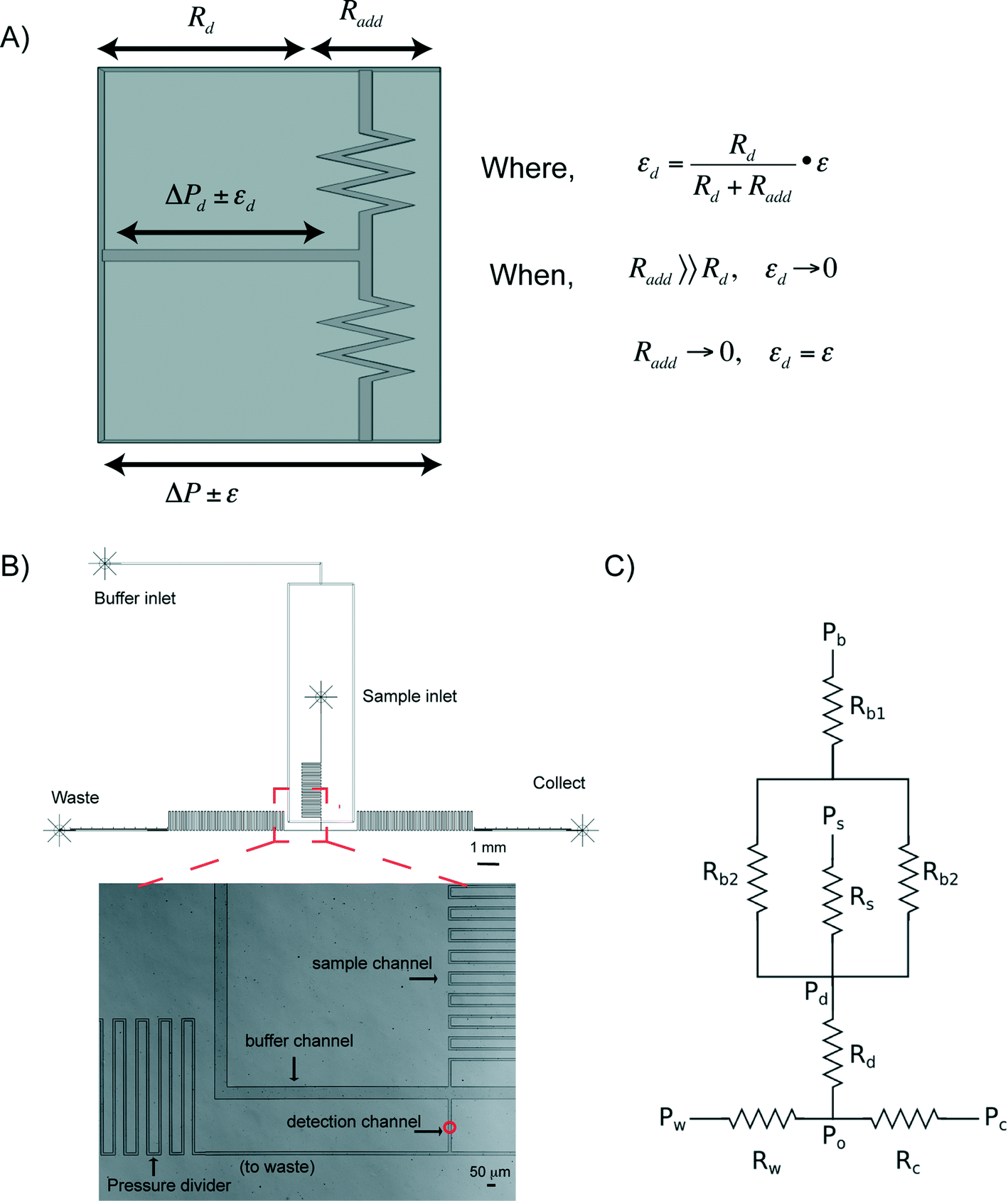 | ||
| Fig. 1 (A) The concept of a pressure divider. (B) The outline of a prototype device and an optical image of the microfluidic channels in the vicinity of the detection chamber. The red circle in the detection channel indicates the location of the focussed laser spot. (C) The schematic of the resistor model used to design the microfluidic device. | ||
Microfluidic device and flow characterization
In addition to the pressure divider concept above, hydrodynamic focusing was used to focus cells to the detection point, and subsequently for sorting. A schematic of a prototype, Fig. 1B shows three regions, namely sample and buffer channels, a detection channel, and two sorting channels (leading to either waste or collection outlets). Within the detection channel, the sample stream is focused on the x–y plane by the buffer flows. Since microfluidic channels are of tens of hundreds of microns, laminar flow dominates (Reynolds number ≪ 1). The microfluidic system can be designed based upon an electronic circuit, Fig. 1C, in which, for a rectangular channel with a height:width ratio of less than 0.7, the fluidic resistance R across it can be described in eqn (1) (Table 1).46 Using Ohm's law and Kirchhoff's current law to describe flow balances,47 the pressure at each junction can be derived, allowing simulation of operational conditions (Table 1).
| Parametersa | Expressionb | Eqn |
|---|---|---|
| a Subscripts indicate the channels as illustrated in Fig. 1B: d – detection channel; s – sample channel; b – buffer channel, where Rb = (Rb1 + Rb2)/2; c – channel between junction point O and the collection outlet; w – channel between the junction point O and the waster outlet. b Where w, h and L in eqn (1) are the width, height and length of the channel respectively, ΔP is the pressure drop across the channel length, Q is the volumetric flow rate (volume/time) and μ is the fluid density. | ||
| R b, Rd, Rs, Rwa (resistance of each channel) |
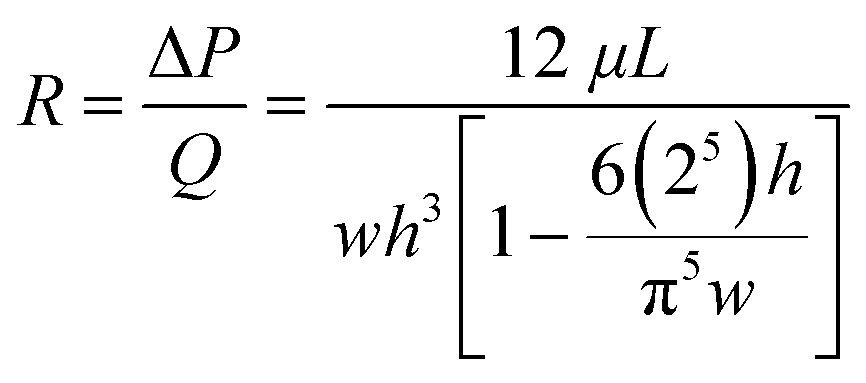
|
(1) |
| P o (pressure at junction O) | P c = Po | (2) |
| P c pressure at collection outlet | P c = Pd − QdRd | (3) |
| P w pressure at waste outlet | P w = Po − QdRw | (4) |
| P d pressure at the start of detection channel |
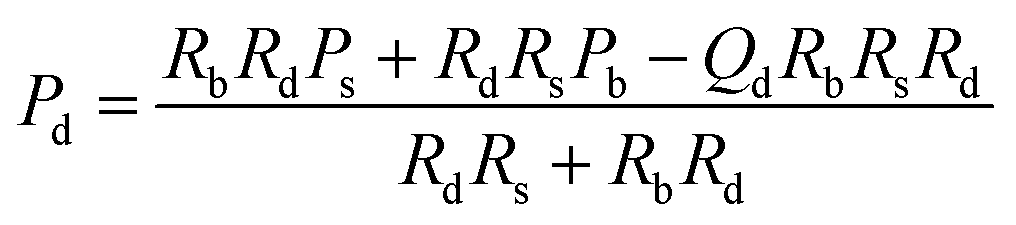
|
(5) |
Using these principles, the system was designed so that a Raman signal integration time of 100 ms could be employed;23,29,48,49 this dictated that the velocity in the detection chamber should be ∼100 μm s−1 for an effective detection area of around 14 μm diameter (as detailed in Experimental section). In pressure-driven flow, the influence of the Poiseuille effect on sample focusing and actual sample velocity has to be considered. By modelling the flow profiles in the channel of a candidate design, the position of a sample cell in the flow stream can be estimated (ESI† Fig. S2). These simulations show that for the channels with the ratio of height:width <0.7, the average velocity is close to 50% of the maximum velocity in the centre of the channel. Based on these and similar simulations, a series of devices were designed, an example of which has dimensions shown in Table 2.
| Channels | Length (mm) | Width (μm) | Height (μm) |
|---|---|---|---|
| Sample channel | 37.5 | 15 | 7.2 |
| Buffer channel | 11 | 90 | 7.2 |
| Detection channel | 0.2 | 20 | 7.2 |
| Output channel | 73 | 15 | 7.2 |
Although low flow rates in the detection region are necessary for long Raman acquisition, it is preferable for the flow in areas outside the detection region to be faster (e.g. to reduce sample sedimentation). This can be achieved by simply varying the geometrical cross sections of the channels. To evaluate the performance of devices in controlling sample position and to measure the velocity in the detection region, 1 μm sized fluorescence beads were used. In the x–y plane, individual beads were well focused and confined to a 1–2 μm wide region in the centre of the detection channel, as illustrated in Fig. S3A and B† (indicated by the arrow). In Fig. S3C† the speeds of individual beads in the detection channel are plotted for different inlet/outlet pressure differentials. Stable bead speeds below 300 μm s−1 were obtained when using a Fluigent pump which had pressure fluctuations (pressure ‘noise’) of ∼0.7 mbar. This shows the distinct advantages of employing an integrated pressure divider network. Observations of particles flowing in the detection region showed that the average velocities of beads were close to the theoretical maximum (Fig. S2†). Based on the flow profile simulations (Fig. S2†), it was concluded that all the beads lie within 3 μm distance of the channel midline for a 7.2 μm height channel, suggesting tight focusing of cells in 3D.
Having established that a stable flow stream, we optimised the conditions for switching the flow to either outlet through simulation. For any given volumetric flow rate Qd in the detection channel (e.g. 52 μL h−1 for a velocity of 100 μm s−1), the output pressures (Pw, Pc) can be changed in such a way that Po was unchanged (Fig. 1B), and therefore the flow could be switched to either output without disruption to the flow profile in the detection channel. The expressions for the pressure at strategic points in the network for the situation where all of the flow is directed to the waste output (i.e. Pc = Po) are shown in Table 1. In practice, to ensure that there was no flow from one outlet channel to another, a small proportion of the detection channel flow was directed to the outlet that was not selected by the pressure switch. This required the pressure difference between the outlets to be slightly less than that calculated using eqn (1)–(5) in Table 1.
As shown in Fig. 2 and Video S1,† beads can be reliably directed to alternate outlets every 0.5 seconds. The beads (labelled as 1 and 2) in the outlet channels continue to move towards their selected outlet regardless of which outlet is currently selected, removing the possibility of samples from the waste channel being moved into the collection channel caused by the switching process. The images between times 1.92 s and 2.63 s in Video S1† demonstrate that the velocities of the beads in the detection channel (labelled as 3, 4, and 5) were not affected by the pressure switching process. The excellent agreement between the simulation and experimental results laid the foundation for automated operation.
 | ||
| Fig. 2 Time-lapse images of switching fluorescent beads between the output channels. Switching frequency was at 2 Hz. | ||
Real-time spectra analysis for accurate classification
Essential for reliable and fast Raman based cell sorting is the capability of fast and accurate classification of cells in situ. However, differences in Raman spectra between target and non-target cells are often subtle.50 This, in combination with the inherent weakness of Raman signals and background interference, impose significant barriers to accurate on-the-fly identification of cells. To overcome these, real time classification of cells was carried out via programmed, multi-parameter analysis of Raman spectra. To reduce processing time, Raman spectra were directly read out from the CCD chip and indexed by pixel number.Using a carotenoid containing photosynthetic microorganism, Synechocystis sp. PCC6803 as a model system together with a stable isotope substrate (13C-bicarbonate) we can detect the red shifts in carotenoid bands that are indicative of active dissolved-CO2-fixing cells, denoted as 13C-cells (in contrast, cells grown in normal medium are denoted as 12C-cells).31Synechocystis sp. PCC6803 are 1–2 μm spherical cells and were found not to adhere to the surface of the microfluidic device. Fig. 3A shows the single cell Raman spectra of dried 12C- and 13C-cells and Fig. 3B similar cells moving at ∼100 μm s−1 in the detection region. Comparison of the spectra of dried cells (Fig. 3A) shows that the slight shifts of the υ1 (1155 cm−1) and υ2 (1516 cm−1) bands are clearly discernable. For cells moving in the flow stream, additional peaks are found in the spectra that arise from the PDMS chip (indicated by arrows in Fig. 3B). These can interfere with discriminating between cell types and had to be taken into account when developing the on-the-fly signal processing routine outlined below.
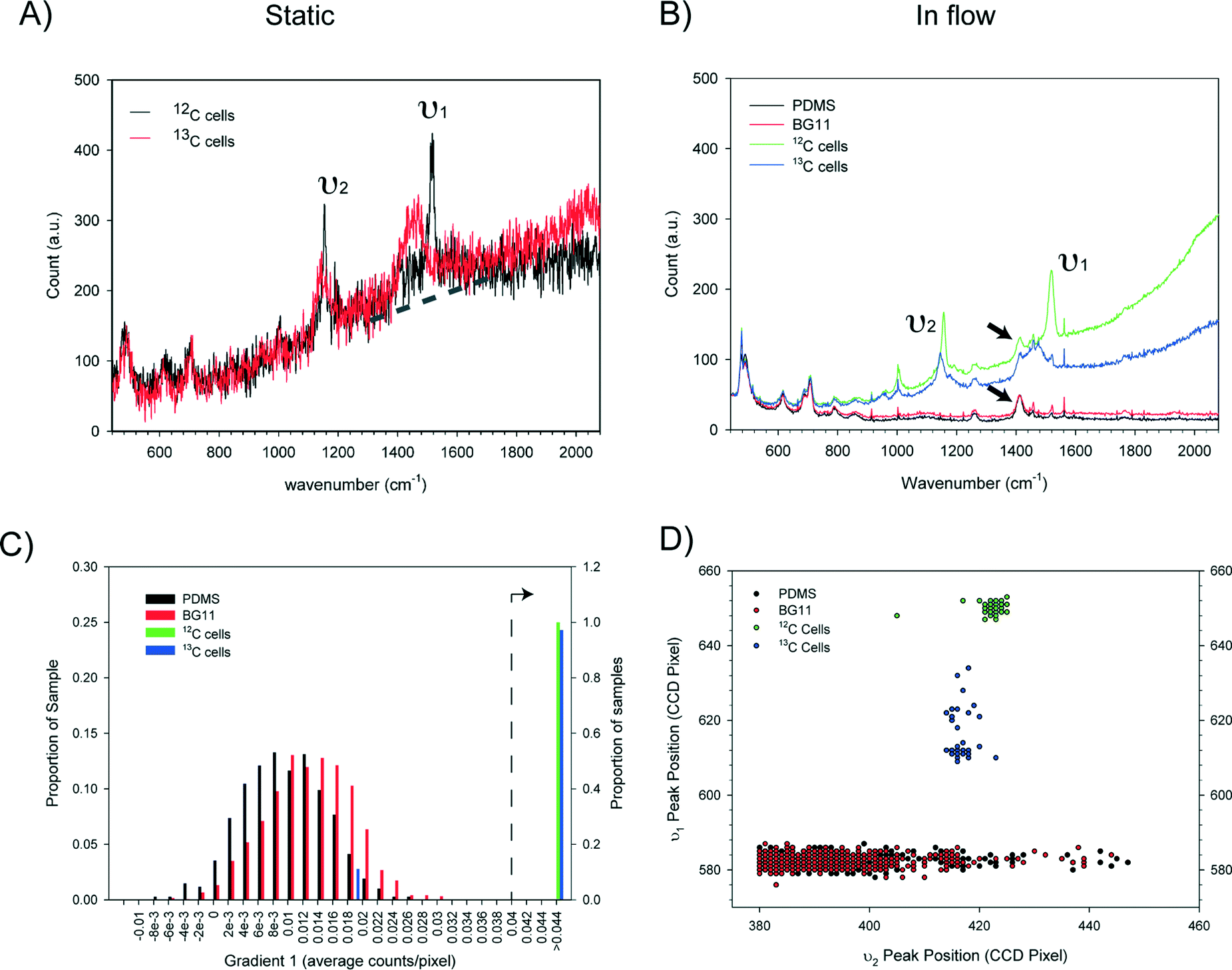 | ||
| Fig. 3 Criteria for cell classification. (A) Single cell Raman spectra of 12C- and 13C-containing Synechocystis PCC6803 cells dried on a glass side (100 ms integration, single spectrum). The dotted line is indicative of the sloping baseline due to cell autofluorescence. (B) Representative Raman spectra of single 12C and 13C-cells moving at 100 μm s−1 on chip (green and blue traces), the PDMS chip alone (black trace), and the PDMS chip with BG11 medium (red trace). For clarity, each spectrum represents an average of five spectra collected using a 50 ms acquisition time and a low pass filter. (C) Histograms showing how the value of the baseline gradient indicated in (A) differs for the four groups of spectra shown in (B) (for clarity, gradients below 0.04 are plotted using the left hand Y-axis and gradients above 0.04 use the right hand Y-axis). (D) υ1 and υ2 peak positions of the four groups, showing characteristic, tight clustering of 12C- and 13C-cells and the lack of correlation between υ1 and υ2 peaks in PDMS or BG-11 spectra. The υ1 and υ2 peak positions were determined by finding the pixel with the maximum intensity within a specified range. | ||
Notwithstanding the strong Raman peaks from PDMS chip, we were able to use multiple parameters derived from Raman spectra of cells (i.e. the υ1 and υ2 signatures, and the baseline gradient due to the cell's autofluorescence – indicated by the dotted line in Fig. 3A) in conjunction with noise filtering methods to formulate a classification criteria. Statistical analysis of spectra from cell populations (Fig. 3C and D) allowed the thresholds of each parameter to be set for the on-the-fly classification. For example, from Fig. 3B, it is clear that the baseline gradient in a cell spectrum is much higher than that in a spectrum of either PDMS or BG11 medium (i.e. when a spectrum was acquired at a time when no cells were passing though the detection zone). The values of this baseline gradient for a series of spectra collected from different types of sample are presented in Fig. 3C. It is apparent that the baseline gradients in cell spectra are generally greater than 0.04 counts per pixel. In addition, processing these spectra showed that the υ1 and υ2 peak positions for the 12C and 13C cells fell into two clusters whereas there was no correlation for PDMS or BG-11 spectra (Fig. 3D). Based on this analysis, in the on-the-fly classification, first a lower threshold of 0.0375 was set on the gradient parameter to identify that the spectrum has come from a cell. Then, the cell type was identified from the position of the υ1 and υ2 peaks using the criteria given in Table 3.
| Type | υ 1 peak position (CCD pixel) | υ 2 peak position υ2, (CCD pixel) | Gradient 1 (counts per pixel) |
|---|---|---|---|
| a Positions on the CCD, values correspond to row numbers on a pixelated CCD chip. | |||
| 12C cells | 644 ≤ υ1 ≤ 659 | 414 ≤ υ2 ≤ 430 | G 1 ≥ 0.0375 |
| 13C cells | 600 ≤ υ1 ≤ 643 | 400 ≤ υ2 ≤ 450 | G 1 ≥ 0.0375 |
Integration for automated, programmable cell sorting
The outline of the whole sorting process is illustrated in Fig. 4. To achieve automated and reliable operation, hardware/software integration was developed using Labview that gave reliable synchronization of Raman acquisition, real-time signal processing and sorting. The workflow of the software and its interface are shown in Fig. 5(I) and (II). The labels A–E indicate each function and its corresponding interface in the sorting process. Apart from the initial cell-loading step, the whole process was run automatically based on pre-defined classification criteria and operational parameters (Fig. 5A–C in II). It should be noted that this program can be easily modified to accommodate any selection criteria required for other applications. During the sorting, Raman spectra are continuously acquired. If a spectrum meets the sorting criteria, the programme triggers the pump (i.e. the MFCS-1000 system) to switch the output pressures to direct the flow to the collection channel. The collection channel is then opened for a pre-programmed delay time (i.e. the switching time, Fig. 5) before the flow is directed back to the waste outlet.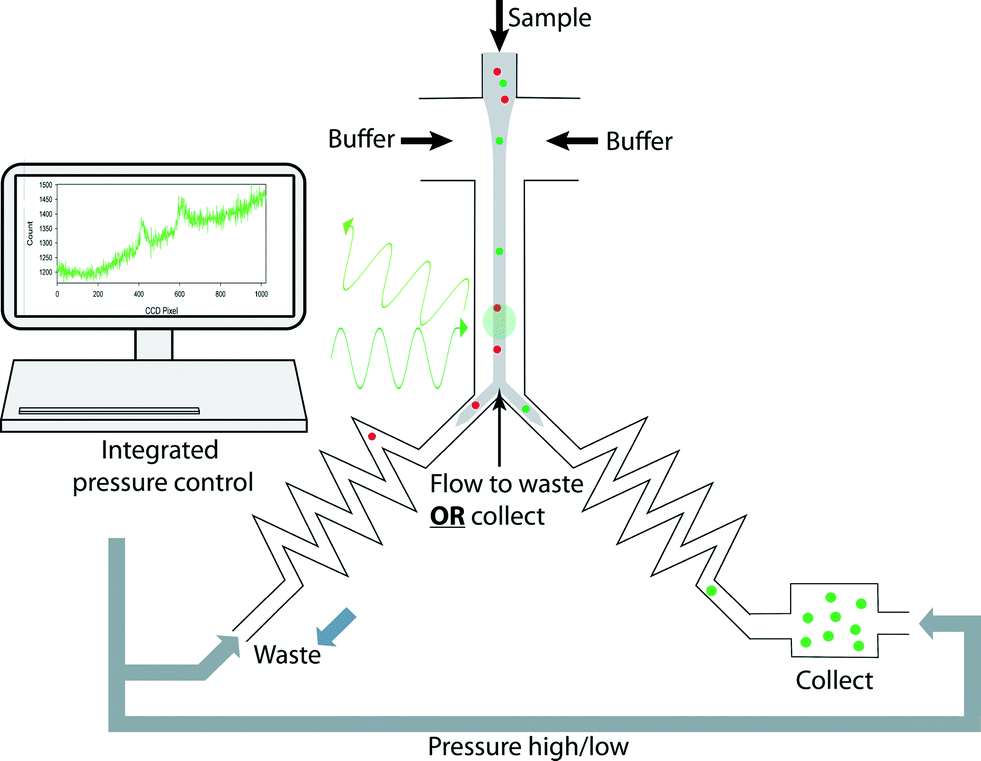 | ||
| Fig. 4 Schematic drawing of the experimental design. A stream of cells were hydrodynamically focused in the detection channel for continuous Raman acquisition; on-the-fly classification was carried out to identify target cells and was immediately followed by alternating the pressures applied to the waste and collection channels, to direct the target cells to the collection chamber. Integrated software was developed to synchronize and automate all the operations. | ||
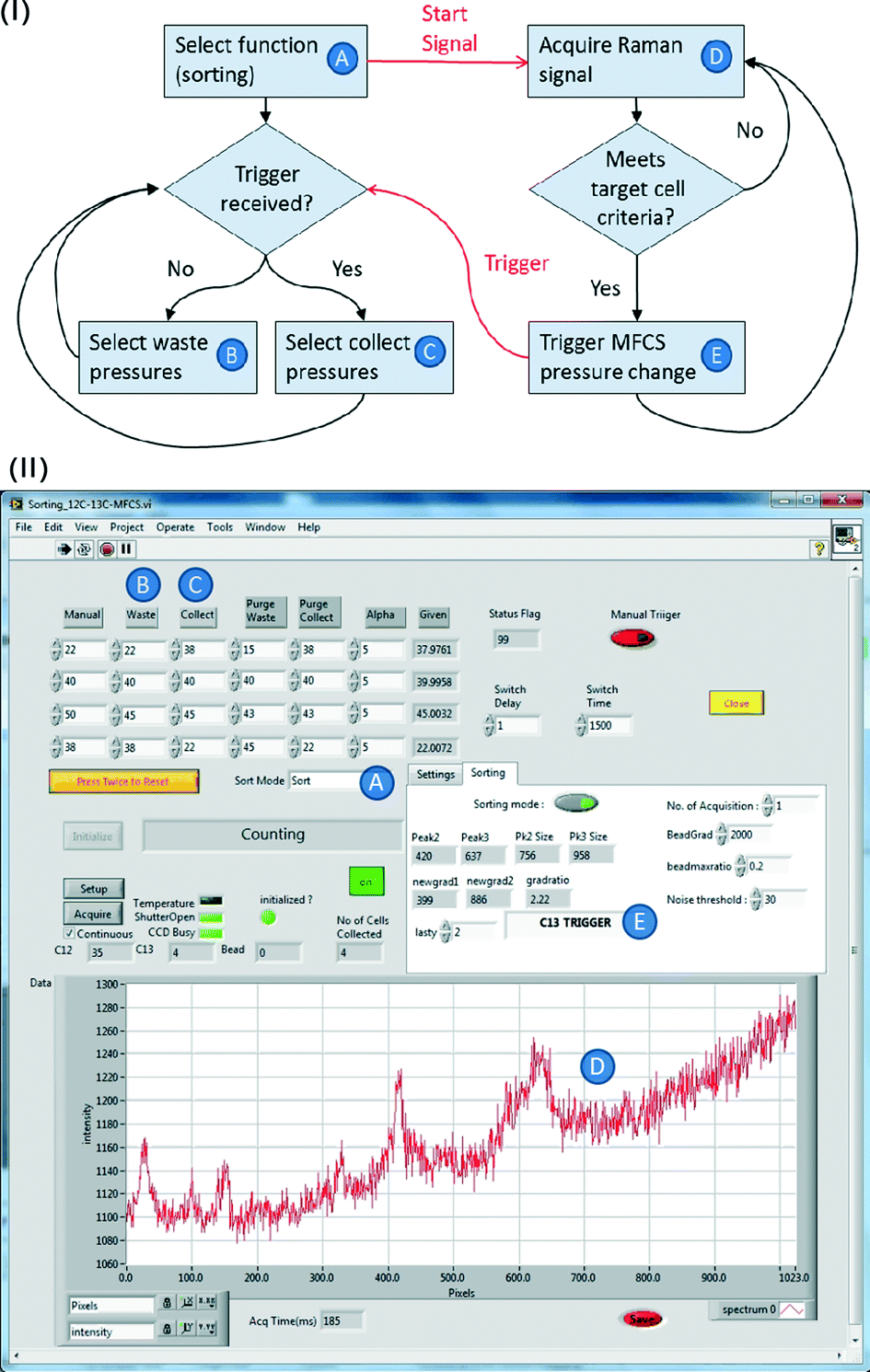 | ||
| Fig. 5 (I) A flow chart of the integration software. (II) A screenshot of the software running panel. Letters A to E correlate the functions of each step in (I) with settings shown in panel (II). The panel shows the detection of a target 13C cell and the resulting changing of the software to change the pressure to the collecting setting, to redirect the target cell to the collection channel. | ||
Direct processing of CCD indexed signals enhanced the speed of spectra analysis in real-time, and consequently enhanced the throughput of sorting (it also provides for greater flexibility in programming to classify the signals using any criteria, and thereby enhancing the capability for the differentiation of complex samples on-line, which would otherwise require off-line spectra analysis26,48). Notwithstanding this, there is a hardware based delay before the switching actuation can occur. In our system, this actuation delay consists of ∼120 ms for the analogue to digital conversion (ADC) time on the CCD, and ∼200 ms for the physical switching of the pump to be effective. This restricts the overall sorting rate to around 2 Hz. In future a more responsive switching system, implementation of faster ADC settings and reduction in the mechanical compliance of the system will further improve this performance.
To evaluate the efficiency of sorting, isolation of 13C cells or 12C cells from mixtures containing both cells at different ratios was carried out using a Raman acquisition time of 50 ms. To avoid cells being undetected during the “dead” ADC delay time (i.e. 120 ms), the total cell density was diluted (OD < 0.3) to give a sufficient gap between adjacent individual cells in the focussed flow stream. As shown in Fig. 6 and Video S2,† cells were focused tightly into a single line and passed the detection point in succession, with adjacent cells spaced by more than 0.2 seconds.
 | ||
| Fig. 6 Time-lapse images showing continuous delivery of individual cells to the detection point. The gap between cells 1 and 2 is approximately 0.4 seconds. | ||
When high sorting purity was required, a programmable delay of 1500 ms was set to allow reliable physical switching (i.e. ∼0.5 Hz sorting speed). The fully automated cell sorting is shown in Video S3,† where single cell Raman spectra and the total number of collected target cells were updated in real-time on the user-friendly interface. To evaluate the sorting accuracy, after sorting, Raman spectra of all the cells in the collection channel (Fig. 7A) were collected for cell type classification. To avoid ambiguity associated with manual inspection, correlation coefficients were calculated between the spectrum of each cell and reference spectra for both 12C and 13C cell types. This provided a post-sorting means to determine the numbers of each cell type in the collection channel and thus the efficiency and accuracy of the sorting method (further details are provided in ESI† section 4). Using this method it was found that isolation of 13C cells from a mixture of 13% 13C cells and 87% 12C gave an average of 75.9% 13C cells in the collection channel, which is around a six fold enrichment (Fig. 7B, total collected cell number = 79, Table S2, Fig. S4†). Similarly, isolation of 12C cells from 12.5 ± 3.5% 12C cells in mixed populations (three independent experiments) gave an average of 82.24 ± 4.1% 12C in the collection (total collected cell number = 289).
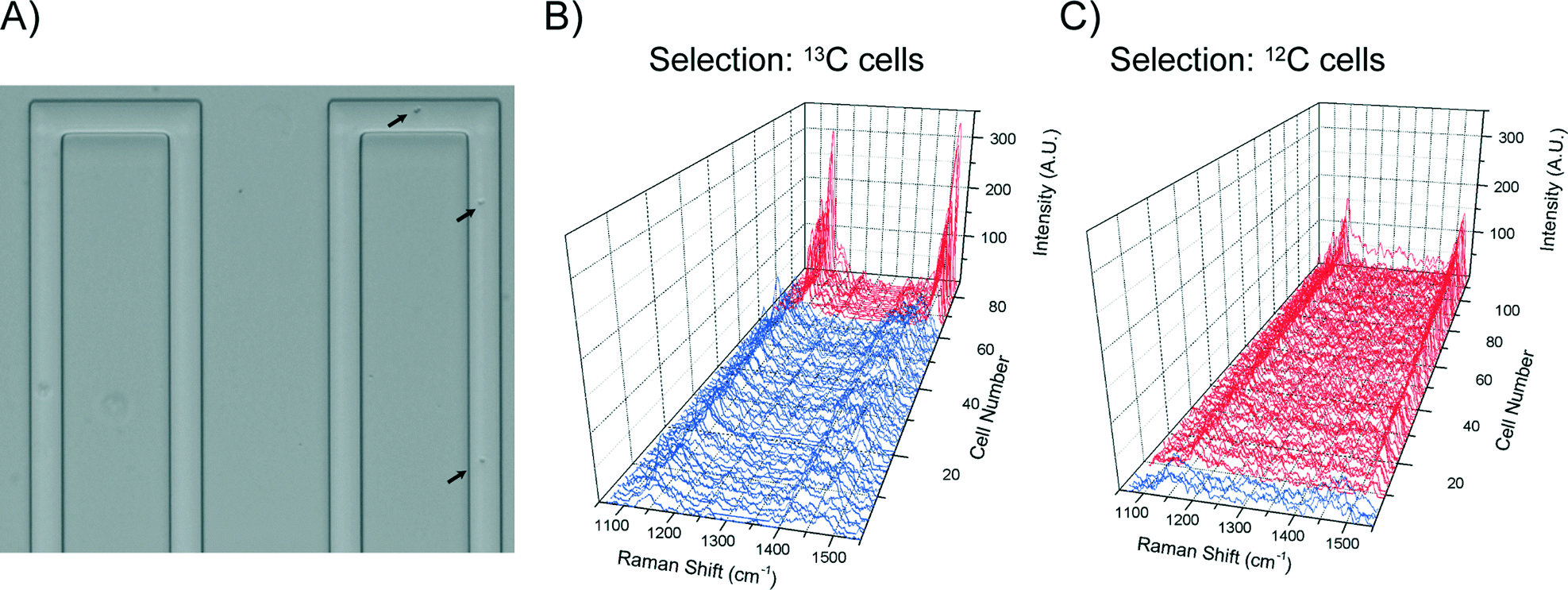 | ||
| Fig. 7 Sorting efficiency. (A) Optical image of sorted cells in the collection channel. Arrows indicate cells. (B) Raman spectra of 79 cells in the collection channel sorted from an initial 13% 13C-cells in the mixture. The purity of the selected 13C-cells is 75.9%. (C) Raman spectra of 109 cells in the collection channel sorted from an initial 45% 12C-cells in the mixture. The purity of the selected 12C-cells was 96.3%. | ||
Close observations of the sorting process suggested that the less than perfect enrichment was mainly due to the occurrence of the majority cell type in the immediate vicinity of a targeted cell at the point at which the sorting occurs. Statistically, the frequency of such occurrence is in inverse relationship to the initial concentration of the targeted cells in a population. Therefore, a simple way to achieve higher purity is to increase initial concentration of the targeted cells. As expected, isolation of 12C from a mixture of 45% 12C cells and 55% 13C cells gave a purity of 96.3% 12C cells in the collection (total collected cell number = 109, Fig. 7C). Together, these results suggest that a two-stage enrichment can be an effective way to obtain high purity of targeted cells that are initially of low abundance in a population.
Conclusions
We have developed an automated RACS system, which demonstrates the capability of continuous and “trap-free” cell sorting in flow based on intrinsic Raman signals. High accuracy sorting of 96.3% was achieved, as a result of reliable synchronisation of Raman signal acquisition, real-time identification and cell sorting. Currently, the sorting speed of 2 Hz is mainly due to the actuation delay of the external components used (pump and CCD), providing scope for further improvement in throughput. Importantly, because this approach utilises simple hydrodynamic focusing and a switch mechanism, it removes any dependence on the physical properties of the cells or medium involved in a “trapping” method, and will offer great advantages to isolate cells from a complex community and in their native biological fluid.Acknowledgements
We thank partial support from EPSRC (EP/H04986X/1 and EP/J009121/1). We gratefully acknowledge the technical team of the James Watt Nanofabrication Centre (JWNC) at University of Glasgow for the support in fabricating the devices.References
- N. Navin, J. Kendall, J. Troge, P. Andrews, L. Rodgers, J. McIndoo, K. Cook, A. Stepansky, D. Levy, D. Esposito, L. Muthuswamy, A. Krasnitz, W. R. McCombie, J. Hicks and M. Wigler, Nature, 2011, 472, 90–94 CrossRef CAS PubMed.
- B. F. Brehm-Stecher and E. A. Johnson, Microbiol. Mol. Biol. Rev., 2004, 68, 538–549 CrossRef CAS PubMed.
- H. B. Yin and D. Marshall, Curr. Opin. Biotechnol., 2012, 23, 110–119 CrossRef CAS PubMed.
- M. E. Lidstrom and M. C. Konopka, Nat. Chem. Biol., 2010, 6, 705–712 CrossRef CAS PubMed.
- S. Muller and G. Nebe-von-Caron, FEMS Microbiol. Rev., 2010, 34, 554–587 CrossRef PubMed.
- D. R. Gossett, W. M. Weaver, A. J. Mach, S. C. Hur, H. T. K. Tse, W. Lee, H. Amini and D. Di Carlo, Anal. Bioanal. Chem., 2010, 397, 3249–3267 CrossRef CAS PubMed.
- C. W. Shields, C. D. Reyes and G. P. Lopez, Lab Chip, 2015, 15, 1230–1249 RSC.
- L. Q. Ren, Y. C. Chen, P. Li, Z. M. Mao, P. H. Huang, J. Rufo, F. Guo, L. Wang, J. P. McCoy, S. J. Levine and T. J. Huang, Lab Chip, 2015, 15, 3870–3879 RSC.
- P. Rosch, M. Harz, M. Schmitt, K. D. Peschke, O. Ronneberger, H. Burkhardt, H. W. Motzkus, M. Lankers, S. Hofer, H. Thiele and J. Popp, Appl. Environ. Microbiol., 2005, 71, 1626–1637 CrossRef PubMed.
- H. J. van Manen, Y. M. Kraan, D. Roos and C. Otto, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10159–10164 CrossRef CAS PubMed.
- M. Harz, P. Rosch and J. Popp, Cytometry, Part A, 2009, 75, 104–113 CrossRef CAS PubMed.
- W. E. Huang, R. I. Griffiths, I. P. Thompson, M. J. Bailey and A. S. Whiteley, Anal. Chem., 2004, 76, 4452–4458 CrossRef CAS PubMed.
- M. Harz, M. Kiehntopf, S. Stockel, P. Rosch, E. Straube, T. Deufel and J. Popp, J. Biophotonics, 2009, 2, 70–80 CrossRef CAS PubMed.
- S. Dochow, C. Krafft, U. Neugebauer, T. Bocklitz, T. Henkel, G. Mayer, J. Albert and J. Popp, Lab Chip, 2011, 11, 1484–1490 RSC.
- T. Ichimura, L. D. Chiu, K. Fujita, S. Kawata, T. M. Watanabe, T. Yanagida and H. Fujita, PLoS One, 2014, 9, 8 Search PubMed.
- N. M. S. Sirimuthu, C. D. Syme and J. M. Cooper, Anal. Chem., 2010, 82, 7369–7373 CrossRef CAS PubMed.
- X. L. Zhang, H. B. Yin, J. M. Cooper and S. J. Haswell, Anal. Bioanal. Chem., 2008, 390, 833–840 CrossRef CAS PubMed.
- E. K. Hall, G. A. Singer, M. Polzl, I. Hammerle, C. Schwarz, H. Daims, F. Maixner and T. J. Battin, ISME J., 2011, 5, 196–208 CrossRef PubMed.
- H. W. Wu, J. V. Volponi, A. E. Oliver, A. N. Parikh, B. A. Simmons and S. Singh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3809–3814 CrossRef CAS PubMed.
- W. E. Huang, K. Stoecker, R. Griffiths, L. Newbold, H. Daims, A. S. Whiteley and M. Wagner, Environ. Microbiol., 2007, 9, 1878–1889 CrossRef CAS PubMed.
- W. E. Huang, A. Ferguson, A. C. Singer, K. Lawson, I. P. Thompson, R. M. Kalin, M. J. Larkin, M. J. Bailey and A. S. Whiteley, Appl. Environ. Microbiol., 2009, 75, 234–241 CrossRef CAS PubMed.
- D. Berry, E. Mader, T. K. Lee, D. Woebken, Y. Wang, D. Zhu, M. Palatinszky, A. Schintimeister, M. C. Schmid, B. T. Hanson, N. Shterzer, I. Mizrahi, I. Rauch, T. Decker, T. Bocklitz, J. Popp, C. M. Gibson, P. W. Fowler, W. E. Huang and M. Wagner, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E194–E203 CrossRef CAS PubMed.
- M. Q. Li, J. Xu, M. Romero-Gonzalez, S. A. Banwart and W. E. Huang, Curr. Opin. Biotechnol., 2012, 23, 56–63 CrossRef CAS PubMed.
- C. Xie, J. Mace, M. A. Dinno, Y. Q. Li, W. Tang, R. J. Newton and P. J. Gemperline, Anal. Chem., 2005, 77, 4390–4397 CrossRef CAS PubMed.
- U. C. Schroder, A. Ramoji, U. Glaser, S. Sachse, C. Leiterer, A. Csaki, U. Hubner, W. Fritzsche, W. Pfister, M. Bauer, J. Popp and U. Neugebauer, Anal. Chem., 2013, 85, 10717–10724 CrossRef PubMed.
- D. A. Watson, L. O. Brown, D. R. Gaskill, M. Naivar, S. W. Graves, S. K. Doorn and J. P. Nolan, Cytometry, Part A, 2008, 73, 119–128 CrossRef PubMed.
- R. M. Jarvis and R. Goodacre, Anal. Chem., 2004, 76, 40–47 CrossRef CAS PubMed.
- R. M. Jarvis, N. Law, L. T. Shadi, P. O'Brien, J. R. Lloyd and R. Goodacre, Anal. Chem., 2008, 80, 6741–6746 CrossRef CAS PubMed.
- F. C. Pascut, H. T. Goh, V. George, C. Denning and I. Notingher, J. Biomed. Opt., 2011, 16, 4 CrossRef PubMed.
- B. Robert, Photosynth. Res., 2009, 101, 147–155 CrossRef CAS PubMed.
- M. Q. Li, D. P. Canniffe, P. J. Jackson, P. A. Davison, S. FitzGerald, M. J. Dickman, J. G. Burgess, C. N. Hunter and W. E. Huang, ISME J., 2012, 6, 875–885 CrossRef CAS PubMed.
- Q. Zhang, P. Zhang, H. Gou, C. Mou, W. E. Huang, M. Yang, J. Xu and B. Ma, Analyst, 2015, 140, 6163–6174 RSC.
- Z. Pilat, J. Jezek, J. Kanka and P. Zemanek, in Imaging, Manipulation, and Analysis of Biomolecules, Cells, and Tissues Xii, ed. D. L. Farkas, D. V. Nicolau and R. C. Leif, Spie-Int Soc Optical Engineering, Bellingham, 2014, vol. 8947 Search PubMed.
- C. G. Xie, M. A. Dinno and Y. Q. Li, Opt. Lett., 2002, 27, 249–251 CrossRef PubMed.
- A. Y. Lau, L. P. Lee and J. W. Chan, Lab Chip, 2008, 8, 1116–1120 RSC.
- W. E. Huang, A. D. Ward and A. S. Whiteley, Environ. Microbiol. Rep., 2009, 1, 44–49 CrossRef CAS PubMed.
- X. L. Wang, S. X. Chen, M. Kong, Z. K. Wang, K. D. Costa, R. A. Li and D. Sun, Lab Chip, 2011, 11, 3656–3662 RSC.
- Z. Pilat, J. Jezek, M. Sery, M. Trtilek, L. Nedbal and P. Zemanek, J. Photochem. Photobiol., B, 2013, 121, 27–31 CrossRef CAS PubMed.
- G. J. Puppels, J. H. F. Olminkhof, G. M. J. Segersnolten, C. Otto, F. F. M. Demul and J. Greve, Exp. Cell Res., 1991, 195, 361–367 CrossRef CAS PubMed.
- I. Notingher, S. Verrier, H. Romanska, A. E. Bishop, J. M. Polak and L. L. Hench, Spectroscopy, 2002, 16, 43–51 CrossRef CAS.
- P. R. Zhang, L. H. Ren, X. Zhang, Y. F. Shan, Y. Wang, Y. T. Ji, H. B. Yin, W. E. Huang, J. Xu and B. Ma, Anal. Chem., 2015, 87, 2282–2289 CrossRef CAS PubMed.
- F. F. Becker, X. B. Wang, Y. Huang, R. Pethig, J. Vykoukal and P. R. C. Gascoyne, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 860–864 CrossRef CAS.
- R. Pethig, Biomicrofluidics, 2010, 4, 022811 CrossRef PubMed.
- S. Takaichi, in The purple phototrophic bacteria, ed. C. N. Hunter, Springer Science, 2008 Search PubMed.
- D. A. Bryant and N. U. Frigaard, Trends Microbiol., 2006, 14, 488–496 CrossRef CAS PubMed.
- H. A. Stone, A. D. Stroock and A. Ajdari, Annu. Rev. Fluid Mech., 2004, 36, 381–411 CrossRef.
- M. S. Ferry, I. A. Razinkov and J. Hasty, in Methods in Enzymology, Vol 497: Synthetic Biology, Methods for Part/Device Characterization and Chassis Engineering, Pt A, ed. C. Voigt, Elsevier Academic Press Inc, San Diego, 2011, vol. 497, pp. 295–372 Search PubMed.
- M. Li, P. C. Ashok, K. Dholakia and W. E. Huang, J. Phys. Chem. A, 2012, 116, 6560–6563 CrossRef CAS PubMed.
- Y. Wang, Y. T. Ji, E. S. Wharfe, R. S. Meadows, P. March, R. Goodacre, J. Xu and W. E. Huang, Anal. Chem., 2013, 85, 10697–10701 CrossRef CAS PubMed.
- W. E. Huang, M. Q. Li, R. M. Jarvis, R. Goodacre and S. A. Banwart, in Advances in Applied Microbiology, ed. A. I. Laskin, S. Sariaslani and G. M. Gadd, Elsevier Academic Press Inc, San Diego, 2010, vol. 70, pp. 153–186 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Videos S1–S3, Fig. S1–S4 and Table S1. See DOI: 10.1039/c6lc00251j |
| This journal is © The Royal Society of Chemistry 2016 |