
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntibiograms in five pipetting steps: precise dilution assays in sub-microliter volumes with a conventional pipette†
Ladislav
Derzsi‡
ab,
Tomasz S.
Kaminski‡
a and
Piotr
Garstecki
*a
aInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: garst@ichf.edu.pl
bUniversity of Padova, Department of Physics and Astronomy, Via Marzolo 8, 35131 Padova, Italy
First published on 12th January 2016
Abstract
We demonstrate a standalone microfluidic chip that allows us to carry out commonly executed antibiotic susceptibility assays in an array of nanoliter droplets. We eliminated the need for automation in performing an exemplary complicated liquid handling assay on a chip. Operations on droplets are hard-wired into the microfluidic chip. The liquid handling protocol can be executed with a simple and commonly available source of flow such as an automatic manual pipette. The system passively prepares a series of dilutions of a chemical compound and mixes them with portions of the sample. The precision of metering, merging, mixing, and splitting of discrete portions of liquid samples is rooted in the passive capillary action in microfluidic traps and not in the precision of dosing with a pipette. We show an exemplary use of the device in the determination of the minimum inhibitory concentration (MIC) of ampicillin against E. coli ATCC 25922.
Here we demonstrate a lab-on-a-chip device that does not require automation and can be operated with a standard automatic pipette. The microfluidic chip passively automates a common laboratory liquid handling protocol of preparation of a series of dilutions and merging them with a constant factor. The system reduces the number of manual pipetting steps to a minimum required just to deliver the sample and reagents. The technique offers precision in handling nanoliter volumes while not requiring neither skill nor precision in using a common pipetting tool to operate the microfluidic chip.
A vast majority of chemical, biochemical and microbial assays use liquids. A large fraction of these tests are commonly executed manually via multiple pipetting steps. For example an antimicrobial assay that determines the MIC (minimum inhibitory concentration) of an antibiotic against a given bacterial sample is performed by growing bacteria in a range of dilutions of an antibiotic. The preparation of this assay requires a large number of pipetting steps, equal to a multiple of the number of dilutions.1,2 For 11 dilutions, the technician must perform at least 33 manual pipetting actions (Fig. 1a).
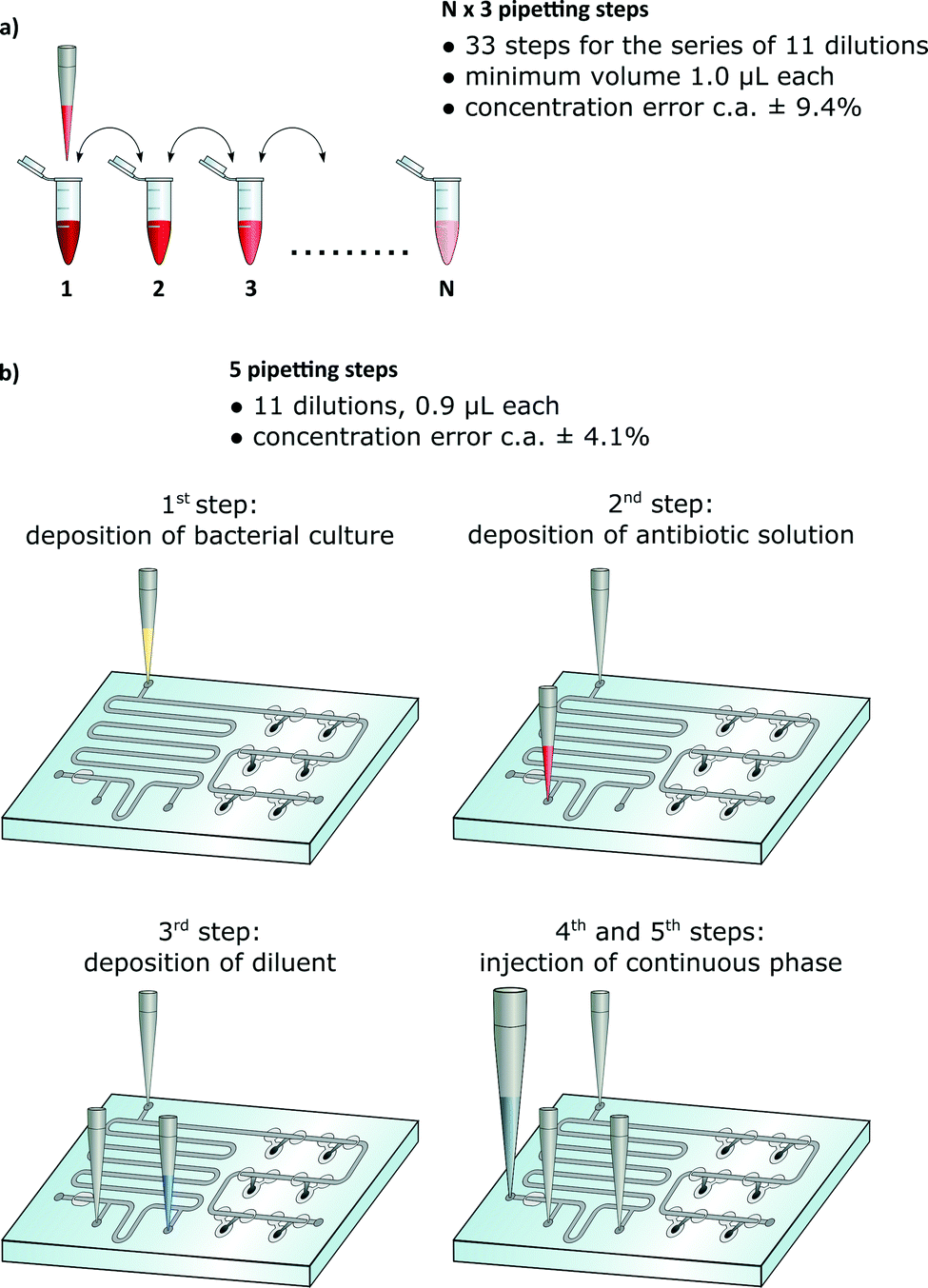 | ||
| Fig. 1 The comparison of the classical way of testing the antibiotic susceptibility with a protocol in a passive droplet microfluidic device. a) A ‘golden standard’ method requires 3N pipetting steps: aliquoting of pure broth in each tube, dilution of antibiotic, and addition of liquid bacterial culture. b) The microfluidic device requires 5 pipetting steps, regardless of the total number of dilutions. | ||
Many sites, both in the rich and developing countries, do not have access to commercial automated systems. A viable question arises whether it is possible to design disposable microfluidic chips that would minimize the number of manual operations while maintaining at least the precision and flexibility of standard protocols (Fig. 1b).
Here we show an example of such a solution that not only dramatically reduces the amount of labor needed to execute a common assay but also allows us to reduce the volume of the reagents and increase the precision of preparation of the dilution series.
Droplet microfluidics
Droplet microfluidics offers a convenient technology for compartmentalization of reagents and samples in arrays of microdroplets.3 This technology has been so far mostly used in automated systems for generation of large screens (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) for digital PCR assays4–7 or in academic demonstrations of digital detection of proteins,8,9 directed evolution of enzymes,10,11 single cell sequencing,12,13 and clonal selection of cells secreting antibodies.14
000) for digital PCR assays4–7 or in academic demonstrations of digital detection of proteins,8,9 directed evolution of enzymes,10,11 single cell sequencing,12,13 and clonal selection of cells secreting antibodies.14
Autonomous microfluidic chips
In recent years, there has been a continuous effort for the development of simple microfluidic devices that could be operated with minimum external instrumentation. Such devices could be used to execute simple assays in point-of-care or in-field applications. The most impressive examples include paper microfluidics,15,16 SlipChip,17,18 pressurized PDMS microchips,19,20 chips equipped with oscillatory peristaltic pumps,21,22 or, among others, passive pumping based on Laplace pressure.23,24 These devices are promising solutions for execution of simple protocols and/or efficient compartmentalization of reagents.However, dilution of reagents is limited to diffusion-based gradients or slow mixing of fluids in the single-phase flow. This limits the flexibility in terms of protocols that can be executed. In the particular example of dilution series, it also limits the span of concentrations that can be addressed. More generally, a broad spectrum of common laboratory protocols require dilution of one component of the reaction (e.g. an antibiotic or an inhibitor) while other components are kept at a constant concentration (e.g. bacterial cells, enzyme). This operation while being labor intensive is simple at the macroscale. In contrast, it remains a challenge for microfluidic systems unless they use sophisticated control of flow.
Passive droplet microfluidics
Droplet microfluidic techniques do offer the flexibility of performing virtually any (including iterative) operations on liquids. One of major challenges, tackled in the past decade, is in designing droplet microfluidic systems that would be easy to use – i.e. that would not require programmable syringe pumps or other automated instrumentation for controlling the flow on chip. A promising direction of research in this respect includes the development of techniques for passive operations on microdroplets. Recent developments within this trend include passive formation of monodisperse droplets25–28 which do not require precise pumps or valves for fluid delivery to the chip. Techniques for subsequent operations on droplets have also been developed. For example droplets can be passively merged in dedicated geometries such as pillar traps,29,30 static arrays31,32 or rails and anchors.33,34 A more advanced system presented the possibility of formation of simple gradients using the technology of hydrodynamic traps.31,32,35,36 The application of these microfluidic dilution systems for laboratory tests is significantly limited due to the fact that all components of the sample were diluted. Many assays require a stable load of selected analytes (e.g. bacteria, enzymes or substrates for reaction), while the concentration of others (inhibitors, antibiotics) should gradually decrease in successive compartments. Components with a fixed concentration can be added to each droplet after formation of a gradient – e.g. by direct injection of the additional laminar stream32 or by using active modules such as picoinjectors.37 However, these strategies increase the complexity of the system, and the process of injection is prone to cross-contamination or uncontrollable break-up of droplets. Most importantly, to date, there are no solutions that would demonstrate the integration of a commonly executed assay in a format that can be operated with a simple automatic pipette.Here we present a system capable of generating gradients of concentration of selected chemical components, merging these with metered portions of undiluted sample and storing the mixtures for subsequent optical analysis.
Results and discussion
We demonstrate a microfluidic chip for standard microbial susceptibility testing. Execution of the test with our chip requires just five pipetting steps: three for the deposition of aqueous solutions: bacterial culture, antibiotic solution and pure broth. The final two operations are required to drive the flow in the chip: the first one for metering equal volumes of bacterial culture and the second for metering the subvolumes of pure broth and for diluting the antibiotic. In the final step, the droplets containing bacteria autonomously merge with droplets comprising gradually decreasing concentration of the antibiotic.The Meter and Store (M&S) module
The functionality of the device is based on the unique geometries of hydrodynamic traps that passively meter, merge, route and lock nanoliter droplets. The basic functional unit of our system is a Meter & Store module (Fig. 2). Each of these units serves as an autonomous droplet handling module that precisely meters the droplet phase into a pre-defined volume, allows for exchange of the trapped droplet in a dilution scheme, and after switching the flow off, stores the metered aliquot in the side chamber.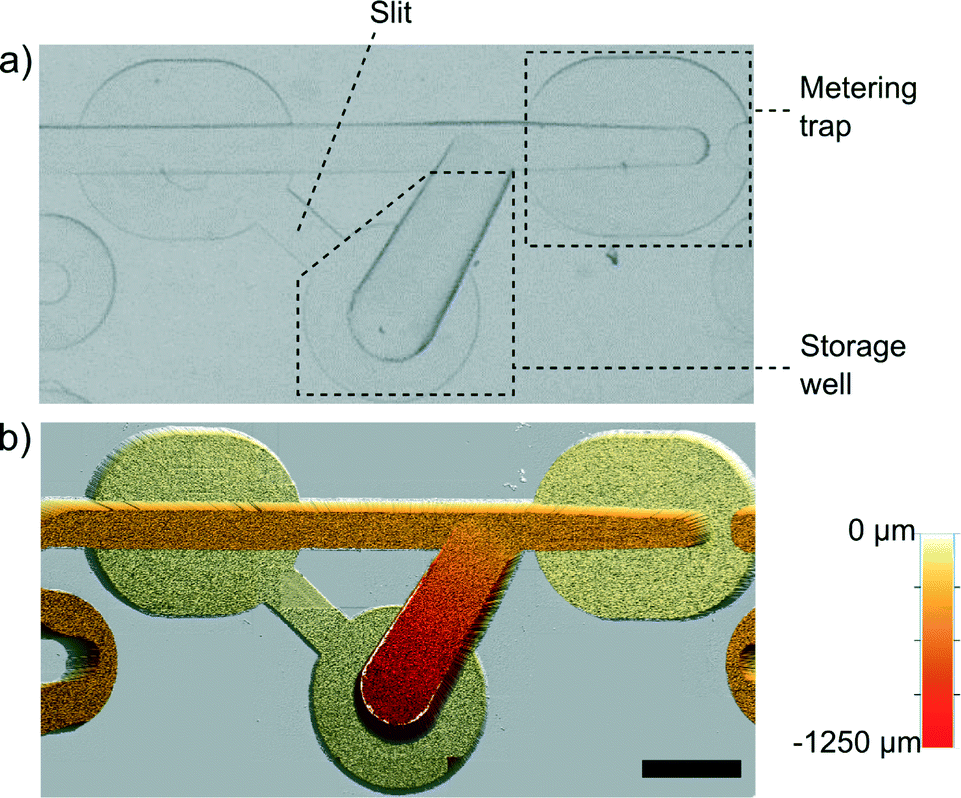 | ||
| Fig. 2 Single unit of the Meter & Store module. a) Snapshot of the module that comprises the metering trap with a narrowed front and the storage chamber connected with the slit to the main channel. b) Image from the optical profilometer (Bruker ContourGT-K) showing the 3D geometry. The scale bar is 1 mm. | ||
The unit comprises a modified metering trap35 that is able to precisely meter a tiny volume of liquid (Fig. 2). Along the metering trap, there are bypasses that are milled to 30% of the depth of the main channel. At the terminus of the trap, we placed a barrier in the main channel that extends upwards to 70% of the height, i.e. of equal height to the bottom of the bypasses. When a droplet longer than the trap reaches the barrier, the rear part of the droplet blocks the entrance to the bypasses. Consequently, the continuous phase cannot flow by the droplet efficiently and it pushes the droplet forward until the rear of the droplet aligns with the entrance to the metering trap. At that moment, continuous liquid begins to flow around the droplet via the bypasses and the droplet itself stops. Subsequently, the Rayleigh–Plateau instability breaks the liquid neck at the front barrier, leaving a precisely metered volume of the droplet locked in the metering trap. The main channel is 500 μm deep and 500 μm wide at the entrance (upstream end of the metering trap). Within the metering trap, we progressively narrowed the main channel to a width of 375 μm at the terminus that is aligned with the barrier. The 25% reduction of the width of the main channel produces a higher curvature of the interface at the front of the droplet than at its rear. When the flow is stopped, the droplet minimizes its surface energy by leaving the trap in the reverse direction and flowing to the storage well within ∼30 s. The storage well (Fig. 2a) is gradually widening and deepening away from the main channel. When the flow is stopped, the droplet metered in the trap starts to flow spontaneously towards the storage well due to the action of the capillary pressure (higher at the front of the droplet than at its rear). The storage chamber has the second connector (marked as ‘slit’ in Fig. 2a) with the main channel. This slit allows for drainage of the side chamber when a droplet flows into it.
Fig. 3 and Video 1 show the process of formation of a gradient in two successive M&S modules. In the first step, a large yellow plug of 0.01% (w/w) solution of tartrazine (Sigma Aldrich, Germany) representing the sample is metered into micro-aliquots of a tightly predefined volume in the successive metering traps (Fig. 3a–c). Next, the flow of oil is halted, and the newly formed droplets of the sample are derailed to the storage wells (Fig. 3d). We use the term sample to refer to the stable component of the assay – e.g. bacterial suspension. In the following step, two plugs are injected into the system. First, a large blue plug of 0.001% (w/w) resazurin (Sigma Aldrich, Germany) represents the diluent, and the second smaller (red) plug of 0.05% (w/w) Congo Red (POCH, Poland) models the reagent – e.g. an antibiotic. As the large blue plug flows through the M&S units, it is portioned into micro-aliquots in the metering traps (Fig. 3e). As we do not stop the flow of oil, these droplets do not flow back into the store chambers but are immediately merged with the reagent plug that has a larger volume than the metering trap (Fig. 3f). The reagent plug merges with the droplet of diluent locked in the metering trap and pushes out a plug that has a volume equal to the initial volume of the reagent yet is composed of both the reagent and the diluent liquids (Fig. 3g). The droplet comprising both solutions flows in the winding channel towards the next M&S unit and its content mixes by convection.38 The operation of dilution is repeated in the successive ten M&S modules (Fig. 3h). In the final step, the flow of the continuous phase is switched-off again, and the droplets of the diluted reagent are derailed to the storage well (Fig. 3i). There, they merge with the already locked sample droplets (Fig. 3j). This initiates the reaction.
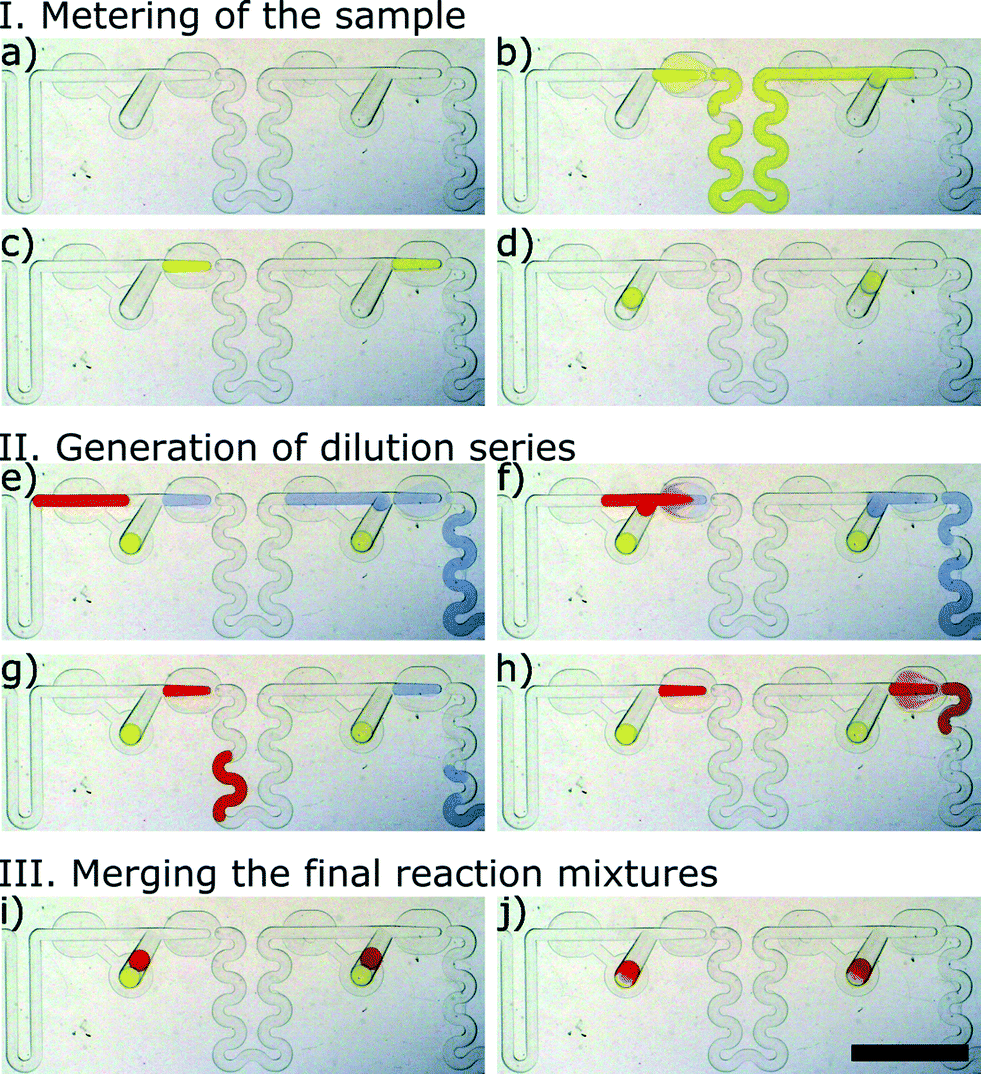 | ||
| Fig. 3 Two subsequent Meter & Store handling modules. Micrographs (a) through (j) illustrate the metering, dilution and storage of microdroplets in a side chamber: (a) empty modules filled with the continuous phase, (b and c) metering of the sub-volume of the first sample – stable component of the assay, (d) derailing of the droplets with the first component to the storage chamber, (e–h) metering the second component (buffer) and dilution of the sample. (i) Derailing of droplets with the diluted sample to the storage well and (j) merging with the first component. The scale bar is 5 mm. | ||
Microfluidic system for antibiotic susceptibility testing
Fig. 4a shows the micrograph of the microfluidic chip. We highlighted three functionally distinct sections: (1) for deposition of the liquid samples, (2) for transport of plugs along a delay channel, and the dilution section (3) for formation of the gradation of concentrations and for storing the mixtures for subsequent analysis. The dilution section comprises 11 serially connected Meter & Store units. The number of dilutions might be higher and adjusted for a particular application. We chose the number of chambers to be 11 because this is a sufficient number of dilutions for an MIC assay and because this number fitted with the volume that can be handled with the particular pipette that we had in the lab. | ||
| Fig. 4 a) The microfluidic chip consisted of 3 parts – module (1) for deposition of a sample to be diluted (e.g. antibiotic solution) and buffer (e.g. pure broth); delay channel (2) with inlet for deposition of an another reagent (e.g. bacterial culture) in which the concentration will be stable in each droplet. The scheme of the chip is presented in Fig. S1.† The main part (3) comprises 11 Meter & Store modules for generation of a gradient. b–d) Snapshots depicting the depositions of the subsequent portions of liquids. The letter ‘T’ indicates the metering trap, and the letter ‘W’ indicates the waste reservoir. The scale bar is 5 mm. | ||
The chip comprises three layers, all fabricated in PDMS (Fig. S2†): a bottom (flat) slab, an intermediate fluidic layer and an upper thick slab with through-holes that serve as wells for oil and prevents injection of bubbles during deposition of samples from the pipette. The fluidic functionality of the chip can be captured by a chip made of a single injection molded part (comprising the wells and ports for deposition of the samples on the top side and the fluidic channels on the bottom) laminated with a plastic foil from the bottom.
The full procedure of preparation of a dilution series with a component at constant concentration
We start the experiment with the system filled with the continuous liquid. We then deposit the sequence of three plugs comprising solutions of different dyes on the chip (Fig. 4b–d and Video 1) using a simple automatic pipette (Brand Transferpette S, volume range 0.5–10 μL, Germany). First, we deposit a large (9 μL) plug (Fig. 4b) representing the sample. Then, we inject the second 4.5 μL plug comprising the reagent (Fig. 4c), and finally a 9 μL plug modeling the buffer (Fig. 4d).During the deposition of the buffer solution, the much higher hydrodynamic resistance towards the dilution section (to the right) forces the reagent plug to flow through the metering trap located on the left side of the deposition module (the trap is indicated by the letter ‘T’ in Fig. 4c–d). This additional metering trap has twice the volume of the metering traps in the dilution section and meters a droplet containing 900 nL of the reagent. The excess reagent liquid is locked away in a deep on-chip waste container (indicated by the letter ‘W’ in Fig. 4c–d).
After deposition of each specimen, each pipette tip is pressed against the bottom of the inlet channel and released from the pipette (Fig. S2†). The diameter of inlets in the PDMS chip are smaller than the outer diameter of the terminus of the pipette tip, allowing us to seal the port.
In the next step, we inject 100 μL of the continuous liquid (solution of fluorocarbon fluids) into the chip as the continuous phase at a constant flow rate of ca. 9 ml h−1. For this purpose, we use an electronic hand dispenser Multipette Xstream with a 0.1 mL Combitip (both from Eppendorf, Germany). As a result, the yellow plug of tartrazine solution is pushed to the dilution section for metering of portions of the sample, while the two other plugs (buffer and reagent) flow along the delay channel. Next, we remove the oil tip from the inlet and this operation generates additional transient back-pressure that speeds up the derailing of the sample droplets into the side chambers. This action takes up to few seconds (Video 2). Next, we inject another 100 μL of the continuous phase into the chip via the same inlet. The flow generated in the second step pushes the buffer and the reagent through the network to generate the gradation of concentration of the reagent in the M&S modules. Finally, the dispenser tip is removed and the droplets with a gradually decreasing concentration of the reagent are derailed to the storage well. They merge with the portions of the sample within a few seconds.
Serial dilution of a fluorescent sample
We prepared the gradient in the same way as described above, but as a sample, we used 10 μM rhodamine 110 (Sigma Aldrich, Germany) dissolved in Mueller-Hinton (MH) broth (BD Biosciences, USA). The pure MH broth served as the sample (not diluted in the experiment) and also as the dilution buffer. Fig. 5 shows the decrease in fluorescence and dilution ratios between 11 successive droplets. The results are averaged between 7 independent executions of the whole experiment. The averaged dilution ratio equals 1.68 ± 0.07 (RSD 4.1%). We observed that mixing inside the droplets is efficient in agreement with previous studies.38,39 The mixing process may be further optimized for specific applications via tuning the geometry and dimensions of the channels. The dilution ratio is not the same as the ratio (equal to 2) of the volumes of the reagent plug (900 nL) and of the droplet of diluent aliquoted in the Meter & Store unit (450 nL). The difference is rooted in the observation that when a plug of reagent (or a mixture of reagent with diluent) flows into a metering trap that locks a droplet of pure diluent, not all of the diluent liquid is flushed forward. The fact that the dilution ratio does not follow straightforward from the ratio of volumes necessitates calibration of any new geometry of the chip. Most importantly, however, the system generates reproducibly the same dilution ratio in all of the M&S units in the series and within a broad spectrum of the rates of flow of the continuous liquid, ranging from 5 μL h−1 to 16 μL h−1.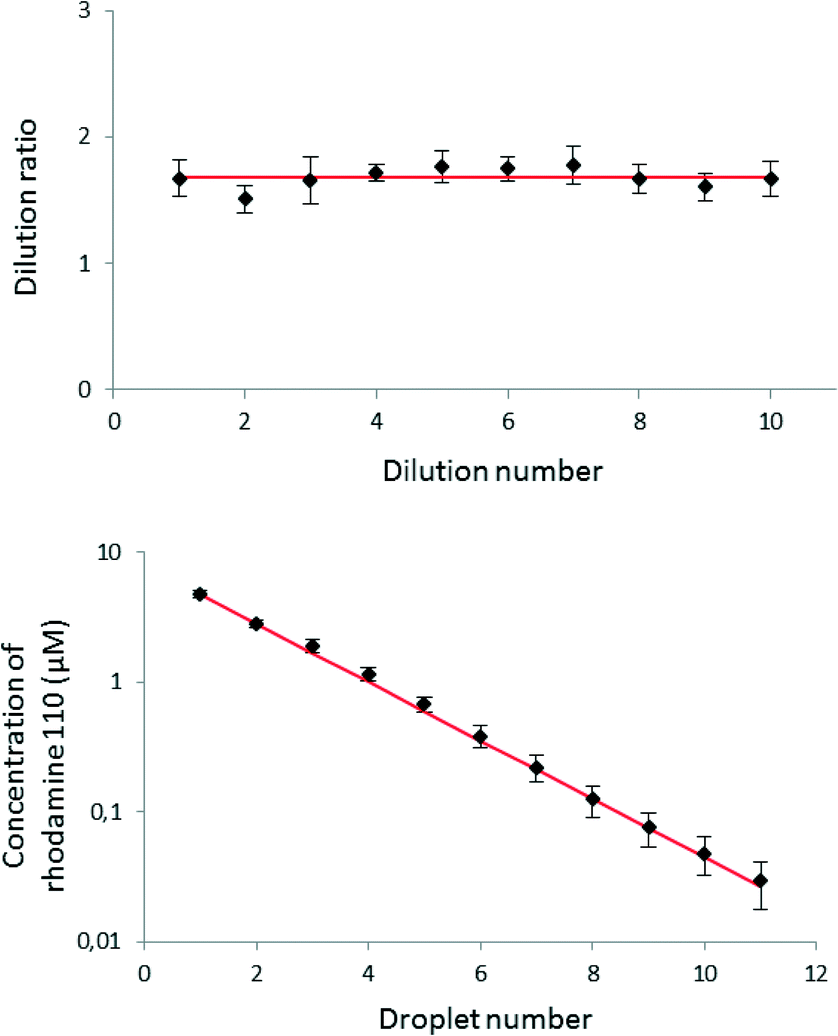 | ||
| Fig. 5 A result of the measurement of the fluorescence intensity of the droplets after the dilutions. The error bars represent standard deviation (n = 7). The red line represents the average dilution ratio (upper plot) and the expected decrease in concentration in successive droplets. Larger values of the relative standard deviation of concentration at the higher dilution ratios may be due to the accumulation of errors during the mixing process in the successive traps. | ||
We compared the level of reproducibility of the microfluidic generation of gradient with the precision and accuracy of a classical automatic pipette. We used the values40 describing technical features of an Eppendorf Research® plus pipette – a comparable device to Brand Transferpette® S used in this study. The pipette prepares 0.5 μL aliquots of liquid with the inaccuracy of ±8.0% and imprecision of ±5.0%. These values can be recalculated to the relative standard deviation of ±9.4%, that is significantly higher than the RSD of dilution in our microfluidic system. As two components (bacterial culture and solution of an antibiotic) are needed for an antibiogram assay, the minimum reaction volume prepared with a pipette is ca. 1.0 ± 0.1 μL.
We can distinguish two sources of variation in the microfluidic system: extrinsic (or user dependent) and intrinsic resulting from imperfections of the dimensions of channels and traps. The main reason of the user-dependent variation is low precision of metering of the sample (Fig. 4d). During this operation, the fluid is pushed by the manual pipette, so the fluctuations of flow rate might be much higher than during the use of an electronic dispenser that drives the oil through the system. The dilution ratio also varied between successive traps on the same chip (RSD between 5.5 and 10.0%). This intrinsic variation might be explained by the limited precision of micromilling – for example depths of the main channel inside the traps differed by up to 3%. Variations in the dimensions of the trap structures might introduce additional errors during gradient formation. Other, more precise method of fabrication should completely alleviate this source of intrinsic error. Fabrication of tilted channels with soft lithography is challenging but possible.26 The structures that we propose are also fully compatible with precision injection molding.
Determination of minimum inhibitory concentration (MIC) of ampicillin
In a model use of our system, we deposited 3 plugs of MH broth – 1st with bacteria (density of 106 CFU mL−1), 2nd with an ampicillin (concentration 200 mg L−1) and 3rd with the pure medium for dilution of the antibiotic. The antibiotic solution and pure medium contained the same concentration of viability indicator – 88 μM of the resazurin sodium salt dye (Sigma Aldrich, Germany) which is converted to highly fluorescent resorufin in the presence of viable bacteria.We generated droplets with the same concentration of bacteria and resazurin but with decreasing concentration of antibiotic in consecutive droplets. Importantly, bacteria contacted only the already-diluted solutions of the antibiotic, just as in a classic antibiotic susceptibility test. The chemical composition of final droplets was the following: 44 μM of the resazurin sodium salt dye, E. coli cells at a density of 5 × 105 CFU mL−1 and ampicillin decreasing from 47 ± 3 mg L−1 (1st droplet) to 0.3 ± 0.1 mg L−1 (11th droplet).
Next, we incubated the reaction mixtures for 4 h at 37 °C and performed fluorescent readout using gel imaging system (Fig. 6). We did not observe influence of evaporation during 4 h of incubation. For longer incubations, additional humidity control might be introduced to prevent excessive evaporation of the samples. We did observe formation of bubbles during incubation at elevated temperatures – a phenomenon which is one of the more important practical challenges in biphasic microfluidic systems, particularly in the case of use of fluorocarbons, that are characterized by high solubility of gases that decreases at elevated temperatures. This restriction was circumvented by performing the incubation outside the chip – e.g. during emulsion PCR4,6 or, in single phase flows, by introduction of bubble traps.41,42 In our chip, bubbles are almost exclusively formed in the main reservoirs of the oil – delay channels (section 1 in Fig. 4a) or deposition module (section 2 in Fig. 4a). Most importantly, the architecture of the side chambers warrants that bubbles do not change the position of droplets locked in the storage wells – neither directly nor by flow of oil caused by expansion of bubbles. Fig. S3† presents exemplary snapshots of the chips after incubation.
 | ||
| Fig. 6 Results of an antibiogram assay. Snapshots from the a) fluorescence gel imager and b) conventional camera depicting conversion of resazurin to resorufin across the droplets locked dilution module after 5 h of incubation at 37 °C. The scale bar is 5 mm. | ||
Fig. 7 illustrates the averaged (n = 5) and normalized intensity of fluorescence for each droplet. These data combined with the dilution ratio from Fig. 5 enabled us to determine the MIC value for ampicillin to be equal to 5.9 ± 0.9 mg L−1. This result is in good agreement with the values obtained by the classical dilution broth method (e.g. the MIC values for ampicillin and E. coli ATCC 25922 in a range of 2.0 and 8.0 mg L−1 reported in the literature).2
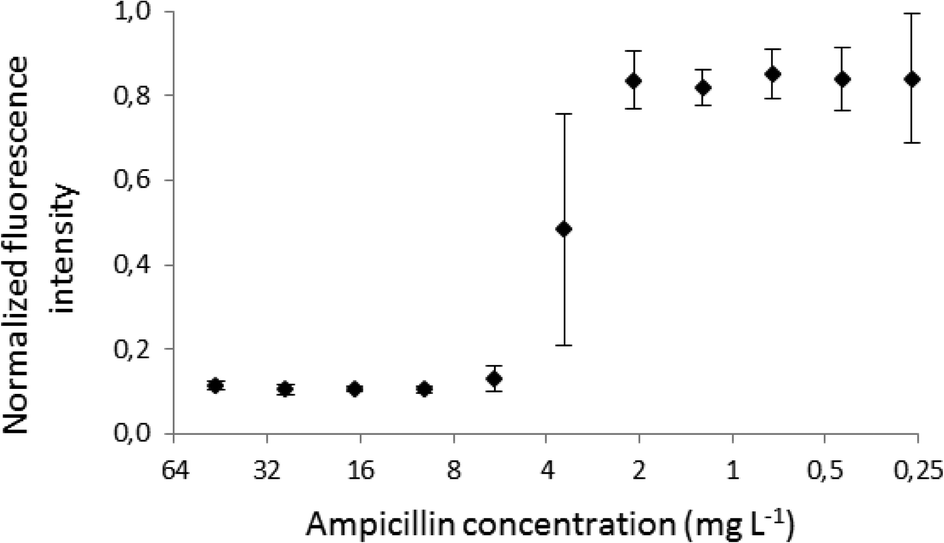 | ||
| Fig. 7 The MIC plot for ampicillin against E. coli ATCC 25922. Each point is an average of 5 experiments, and the error bars show the standard deviation for the set (n = 5). | ||
Discussion
We used a gel imaging system equipped with a fluorescence camera for optical fluorescence readout; however, the detection setup can be simplified, e.g. portable devices such as mobile phones can be used for the fluorescence measurement.43Another challenge is limited precision of droplet cutting that might be improved by more precise fabrication and use of lower flow rates of the continuous phase. The volumes we used here are quite large (the metered droplet is ca. 450 nL), but they were determined by the characteristics of commercial pipettes. We envisage that dedicated pipettes with lower flow rates and relatively large volumes should be developed and used with microfluidic chips such as the one presented here.
We observed that large plugs, of volume greater than 6 μL, broke before they flowed entirely through the metering trap (Videos 1 and 2). This break-up is caused by the slow drainage of the fluid from bypasses to the storage well (Fig. S4,† and Videos 3 and 4) However, the fragmentation of a large plug does not affect significantly the process of formation of a gradation of concentration in subsequent M&S units. This is because smaller droplets merge with the volume metered and locked in the next trap and the system automatically pushes the excess volume forward. Premature break-up of long droplets is not a problem unless the 0.9 μL droplet of reagent leaves the metering trap as a single plug. In the future, systems based on M&S units can be improved towards better precision or faster operations. Both features are associated with the capillary number and dimensions of the metering trap. For instance, a decrease in surfactant concentration or introduction of higher barrier in the trap will provide better precision, but the whole system will work in the range of lower flow velocities.
The system presented here can be complementary for other portable technologies such as SlipChips.17,18 Instead of injection, liquids can be stored in wells, and other methods of flow generation can be implemented, e.g. pumping lids44 or oscillatory pumps.21,22 The throughput of the technology presented here can be improved by parallelization of the modules for gradient generation.
Conclusions
One of the ultimate goals in droplet microfluidics is to construct inexpensive, portable and robust lab-on-chip devices for biological and chemical assays. Such simple-to-use systems will be required to perform multiple liquid handling operations in sequence, including i) preparation of the test sample, ii) generation of a gradation of concentration of reagent taken from stock solution, and subsequently, iii) mixing and incubation of the reaction mixtures. The fluid actuation should ideally be simple and preferably insensitive to fluctuations.Some of these functions have already been demonstrated, as e.g. dilution,31,32,35,36 yet without the necessity of adding a component at a constant concentration and storage for subsequent analysis and without a demonstration of operating the system with a tool as commonly available in the laboratories as an automatic pipette.
In summary, the system that we presented here addresses all the above challenges as it i) precisely meters small portions of the sample, ii) prepares a concentration gradient of the reagent on the same chip, iii) can be operated with a simple micropipette and iv) does not require additional moving elements or external forces to control the transport of droplets. Another advantage is stable locking of droplets after formation of the gradient. Once the simple protocol of five pipetting steps is executed, the chip can be transported, shaken, and incubated, and the droplets can safely stay in their reaction chambers where they are kept there by capillary forces.
The method of gradient generation using passive traps might also find use in other biomedical application such as quantification of DNA, protein crystallization, formation of lipid membranes or cell assays.
Materials and methods
Fluids
We used a mixture of fluorocarbon fluids: 50% (w/w) Novec HFE-7500, 49.6% (w/w) FC-40 (both acquired from 3M, USA) with 0.4% (w/w) 1H,1H,2H,2H-perfluorooctanol (Alfa Aesar, Germany) as a continuous phase.Fabrication of the PDMS three-layer device
The top layer comprised 5 mm in diameter and 10 mm deep round through holes that served as wells for the excess of oil. The middle layer comprised microfluidic channels, and the bottom part of the chip was a flat slab of PDMS. The fabrication of a fluidic layer and a layer with through holes was as follows: in the first step, we fabricated the polycarbonate molds in 5 mm (middle layer) or 10 mm thick (upper layer) plates of polycarbonate (Macroclear, Bayer, Germany) using a CNC milling machine (MSG4025, Ergwind, Poland). Next, we poured PDMS (Sylgard 184, Dow Corning, USA) onto the PC chip and subsequently polymerize the mold at 70 °C for 2 h. In a following step, we silanized the PDMS mold during 3 h under 10 mbar pressure with vapors of tridecafluoro-1,1,2,2,-tetrahydrooctyl)-1-trichlorosilane (United Chemical Technologies, USA). Cleaned PDMS negative masters were used for molding the positive PDMS replicas that we subsequently bonded with each other by exposing three parts to 30 s O2 plasma and placing them together. We modified the microfluidic channels hydrophobically by filing the chip with Novec 1720 (3M, USA) for 10 minutes. Next, we blew out Novec 1720 with compressed air and let the rest of the fluid evaporate at room temperature. In order to preserve modification, we baked the chip at 135 °C for 1 h.Measurement of fluorescence
Rhodamine gradient
We used a confocal microscope Nikon A1-R (Japan) in order to assess the quality of the gradient. The ratio of values of intensity of fluorescence excited at 488 nm wavelength for two successive droplets indicated the dilution ratio. We repeated the measurement for all 11 droplets. In order to collect the values of fluorescence intensities over nearly 3 orders of magnitude, we set the laser power so that the fluorescence intensity of the first droplet is just below the limit of the dynamic range of the detector of the microscope. Then we measure the fluorescence intensities of the sample droplet (the excessive volume trapped in a waste container located behind the large metering trap, Fig. 4c–d) and first 6 droplets. Then we set the laser power so that the fifth droplet in the dilutor has a fluorescence intensity just below the limit of the dynamic range of the microscope. We then measure fluorescence intensities of droplets 5 to 11. Next, we stitch the data from three obtained pictures by proportionality based on the overlapping data for droplets 5 and 6. The values of fluorescence were subtracted by the auto-fluorescence of pure MH medium.Antibiogram
In order to measure the results of the antibiogram assay, we performed the readout of the fluorescence signal using a Gel Doc XR System (Bio-Rad, USA). We excite the fluorescence of the droplets using UV light passing through the XcitaBlue Conversion Screen. The emission filter enables for acquisition of signal in the range of wavelengths between 460 and 700 nm.Microbiology
As tested bacteria, we used the recommended1,2E. coli ATCC 25922 strain. We prepared a stock solution of cells in Luria Bertani medium (BD Biosciences, USA) containing 30% (v/v) glycerol (Chempur, Poland) and froze them at −80 °C. Before the experiment, cells were streaked on LB agar plates and incubated overnight at 37 °C. For the susceptibility testing, we used Mueller-Hinton (MH) broth (BD Biosciences, USA) also recommended by the Clinical and Laboratory Standards Institute.1,2 We picked individual colonies, used them to inoculate MH liquid broth, and cultured the cells at 37 °C overnight with shaking at 200 rpm. Aliquots of the overnight cultures were used to seed fresh liquid media and were grown to a 0.5 unit of McFarland range. Next we dilute the suspension 100× times, and the approximate starting concentration of bacteria inside the plug deposited on a chip was ∼106 CFU mL−1.We prepared antibiotic stocks of ampicillin using deionized water—at a concentration that was 500× more higher than in the plug injected into a chip. Before each experiment, we diluted antibiotic stocks in MH broth to the desired concentration and transferred the samples to the inlets of the device. The concentration of antibiotics was subsequently reduced during the merging of droplets on-chip.
We used resazurin sodium salt (Sigma-Aldrich, Germany) diluted in MH broth at a concentration of 44 μM as an indicator of cell viability. In order to maintain a constant concentration of resazurin in the incubation mixtures, we added the compound to all solutions introduced into the device with the exception of the suspension of bacteria.
Acknowledgements
The project was co-financed by the European Research Council Starting Grant 279647, within the Foundation for Polish Science MISTRZ fellowship (to P.G.), grant VENTURES/2012-10/4 (to T.S.K.) and START fellowship (to T.S.K.). T.S.K. obtained the funds for the preparation of the doctoral dissertation from the National Science Centre within the scholarship on the basis of decision number DEC-2014/12/T/ST4/00649. This project was partially performed in the laboratories funded by NanoFun POIG.02.02.00-00-025/09 and co-financed by the National Science Centre (DEC-2014/12/W/NZ6/00454).References
- I. Wiegand, K. Hilpert and R. E. W. Hancock, Nat. Protoc., 2008, 3, 163–175 CrossRef CAS PubMed
.
- F. R. Cockerill, M. A. Wikler, J. Adler, M. N. Dudley, G. M. Elipoulos, M. J. Ferraro, D. J. Hardy, D. W. Hecht, J. A. Hindler, J. B. Patel, M. Powell, J. M. Swenson, R. B. Thomson, M. M. Traczewski, J. D. Turnidge, M. P. Weinstein and B. L. Zimmer, Performance Standards for Antimicrobial Susceptibility Testing, Twenty-Second Informational Supplement, CLSI document M100-S22, Wayne, PA: Clinical and Laboratory Standards Institute, 2012, vol. 32, no. 3, p. 136 Search PubMed.
- A. B. Theberge, F. Courtois, Y. Schaerli, M. Fischlechner, C. Abell, F. Hollfelder and W. T. S. Huck, Angew. Chem., Int. Ed., 2010, 49, 5846–5868 CrossRef CAS PubMed
- B. J. Hindson, K. D. Ness, D. A. Masquelier, P. Belgrader, N. J. Heredia, A. J. Makarewicz, I. J. Bright, M. Y. Lucero, A. L. Hiddessen, T. C. Legler, T. K. Kitano, M. R. Hodel, J. F. Petersen, P. W. Wyatt, E. R. Steenblock, P. H. Shah, L. J. Bousse, C. B. Troup, J. C. Mellen, D. K. Wittmann, N. G. Erndt, T. H. Cauley, R. T. Koehler, A. P. So, S. Dube, K. A. Rose, L. Montesclaros, S. Wang, D. P. Stumbo, S. P. Hodges, S. Romine, F. P. Milanovich, H. E. White, J. F. Regan, G. A. Karlin-Neumann, C. M. Hindson, S. Saxonov and B. W. Colston, Anal. Chem., 2011, 83, 8604–8610 CrossRef CAS PubMed
- M. M. Kiss, L. Ortoleva-Donnelly, N. R. Beer, J. Warner, C. G. Bailey, B. W. Colston, J. M. Rothberg, D. R. Link and J. H. Leamon, Anal. Chem., 2008, 80, 8975–8981 CrossRef CAS PubMed
- D. Pekin, Y. Skhiri, J.-C. Baret, D. Le Corre, L. Mazutis, C. Ben Salem, F. Millot, A. El Harrak, J. B. Hutchison, J. W. Larson, D. R. Link, P. Laurent-Puig, A. D. Griffiths and V. Taly, Lab Chip, 2011, 11, 2156–2166 RSC
- A. C. Hatch, J. S. Fisher, A. R. Tovar, A. T. Hsieh, R. Lin, S. L. Pentoney, D. L. Yang and A. P. Lee, Lab Chip, 2011, 11, 3838–3845 RSC
- H. N. Joensson, M. L. Samuels, E. R. Brouzes, M. Medkova, M. Uhlen, D. R. Link and H. Andersson-Svahn, Angew. Chem., Int. Ed., 2009, 48, 2518–2521 CrossRef CAS PubMed
- J. Shim, R. T. Ranasinghe, C. A. Smith, S. M. Ibrahim, F. Hollfelder, W. T. S. Huck, D. Klenerman and C. Abell, ACS Nano, 2013, 7, 5955–5964 CrossRef CAS PubMed
- M. Fischlechner, Y. Schaerli, M. F. Mohamed, S. Patil, C. Abell and F. Hollfelder, Nat. Chem., 2014, 6, 791–796 CrossRef CAS PubMed
- J. J. Agresti, E. Antipov, A. R. Abate, K. Ahn, A. C. Rowat, J.-C. Baret, M. Marquez, A. M. Klibanov, A. D. Griffiths and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4004–4009 CrossRef CAS PubMed
- E. Z. Macosko, A. Basu, R. Satija, J. Nemesh, K. Shekhar, M. Goldman, I. Tirosh, A. R. Bialas, N. Kamitaki, E. M. Martersteck, J. J. Trombetta, D. A. Weitz, J. R. Sanes, A. K. Shalek, A. Regev and S. A. McCarroll, Cell, 2015, 161, 1202–1214 CrossRef CAS PubMed
- A. M. Klein, L. Mazutis, I. Akartuna, N. Tallapragada, A. Veres, V. Li, L. Peshkin, D. A. Weitz and M. W. Kirschner, Cell, 2015, 161, 1187–1201 CrossRef CAS PubMed
- B. El Debs, R. Utharala, I. V. Balyasnikova, A. D. Griffiths and C. A. Merten, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11570–11575 CrossRef CAS PubMed
- A. W. Martinez, S. T. Phillips and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19606–19611 CrossRef CAS PubMed
- J. L. Osborn, B. Lutz, E. Fu, P. Kauffman, D. Y. Stevens and P. Yager, Lab Chip, 2010, 10, 2659–2665 RSC
- W. Du, L. Li, K. P. Nichols and R. F. Ismagilov, Lab Chip, 2009, 9, 2286–2292 RSC
- L. Li, W. Du and R. F. Ismagilov, J. Am. Chem. Soc., 2010, 132, 112–119 CrossRef CAS PubMed
- I. K. Dimov, L. Basabe-Desmonts, J. L. Garcia-Cordero, B. M. Ross, A. J. Ricco and L. P. Lee, Lab Chip, 2011, 11, 845–850 RSC
- N. J. Cira, J. Y. Ho, M. E. Dueck and D. B. Weibel, Lab Chip, 2012, 12, 1052–1059 RSC
- P. N. Duncan, T. V. Nguyen and E. E. Hui, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18104–18109 CrossRef CAS PubMed
- M. Rhee and M. A. Burns, Lab Chip, 2009, 9, 3131–3143 RSC
- G. M. Walker and D. J. Beebe, Lab Chip, 2002, 2, 131–134 RSC
- M. W. Toepke, V. V. Abhyankar and D. J. Beebe, Lab Chip, 2007, 7, 1449–1453 RSC
- R. Dangla, E. Fradet, Y. Lopez and C. N. Baroud, J. Phys. D: Appl. Phys., 2013, 46, 114003 CrossRef
- R. Dangla, S. C. Kayi and C. N. Baroud, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 853–858 CrossRef CAS PubMed
- V. van Steijn, P. M. Korczyk, L. Derzsi, A. R. Abate, D. A. Weitz and P. Garstecki, Biomicrofluidics, 2013, 7, 024108 CrossRef PubMed
- N. Mittal, C. Cohen, J. Bibette and N. Bremond, Phys. Fluids, 2014, 26, 082109 CrossRef
- X. Niu, S. Gulati, J. B. Edel and A. J. deMello, Lab Chip, 2008, 8, 1837–1841 RSC
- J. Clausell-Tormos, A. D. Griffiths and C. A. Merten, Lab Chip, 2010, 10, 1302–1307 RSC
- S. S. Bithi, W. S. Wang, M. Sun, J. Blawzdziewicz and S. A. Vanapalli, Biomicrofluidics, 2014, 8, 034118 Search PubMed
- M. Sun and S. A. Vanapalli, Anal. Chem., 2013, 85, 2044–2048 CrossRef CAS PubMed
- P. Abbyad, R. Dangla, A. Alexandrou and C. N. Baroud, Lab Chip, 2011, 11, 813–821 RSC
- E. Fradet, P. Abbyad, M. H. Vos and C. N. Baroud, Lab Chip, 2013, 13, 4326–4330 RSC
- P. M. Korczyk, L. Derzsi, S. Jakiela and P. Garstecki, Lab Chip, 2013, 13, 4096–4102 RSC
- X. Niu, F. Gielen, J. B. Edel and A. J. deMello, Nat. Chem., 2011, 3, 437–442 CrossRef CAS PubMed
- A. R. Abate, T. Hung, P. Mary, J. J. Agresti and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19163–19166 CrossRef CAS PubMed
- H. Song, M. R. Bringer, J. D. Tice, C. J. Gerdts and R. F. Ismagilov, Appl. Phys. Lett., 2003, 83, 4664–4666 CrossRef CAS PubMed
- J. D. Tice, A. D. Lyon and R. F. Ismagilov, Anal. Chim. Acta, 2004, 507, 73–77 CrossRef CAS
-
http://www.eppendorf.com/int/index.php?l=211&pb=307f5fcfc3cfee65&action=products&contentid=1&catalognode=67026&productpage=3
- A. M. Skelley and J. Voldman, Lab Chip, 2008, 8, 1733–1737 RSC
- C. Lochovsky, S. Yasotharan and A. Guenther, Lab Chip, 2012, 12, 595–601 RSC
- H. Zhu, O. Yaglidere, T.-W. Su, D. Tseng and A. Ozcan, Lab Chip, 2011, 11, 315–322 RSC
- S. Begolo, D. V. Zhukov, D. A. Selck, L. Li and R. F. Ismagilov, Lab Chip, 2014, 14, 4616–4628 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5lc01151e |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2016 |