
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a method for the simultaneous detection of Cr(III) and Cr(VI) in exhaled breath condensate samples using μLC-ICP-MS
Elizabeth
Leese
*ab,
Jackie
Morton
a,
Philip H. E.
Gardiner
b and
Vikki A.
Carolan
b
aHealth and Safety Executive, Harpur Hill, Buxton, Derbyshire SK17 9JN, UK. E-mail: Liz.leese@hsl.gsi.gov.uk; Tel: +44 (0)1298218434
bBiomolecular Sciences Research Centre, Sheffield Hallam University, Howard Street, Sheffield S1 1WB, UK
First published on 8th February 2016
Abstract
The method described in this study is a robust and sensitive method for the simultaneous determination of trivalent and hexavalent chromium (Cr(III) and Cr(VI)) in an exhaled breath condensate (EBC) sample and drinking water proficiency testing material sample. The method uses a hyphenated micro liquid chromatography (μLC) system coupled to inductively coupled plasma mass spectrometry (ICP-MS). The optimised method incorporates a pH adjusted EDTA complexation step to stabilise Cr(VI) and Cr(III). Separation was achieved using an anion exchange micro-sized column. The limits of quantification were 0.040 μg L−1 and 0.013 μg L−1 for Cr(III) and Cr(VI) respectively. EBC is the non-invasive collection of condensate from cooled exhaled breath during regular tidal breathing. We propose that EBC may be a suitable alternative biological matrix to give a more comprehensive picture of exposure to Cr(VI) following an inhalation exposure. This study shows that water samples and EBC samples containing Cr(III) and Cr(VI) can be stored in the fridge (∼4 °C) for up to 6 weeks when they have been diluted 10 fold with an EDTA solution, and that EBC samples must not be frozen after collection if the integrity of Cr(VI) is to be maintained. This study has demonstrated a practical approach to collecting EBC samples from workers potentially occupationally exposed to hexavalent chromium and that the integrity of the chromium species will be maintained prior to analysis.
Introduction
Trivalent chromium is biologically important as an essential element for humans, needed for glucose and lipid metabolism.1 Hexavalent chromium however is considered toxic and IARC has classified it as a group 1 agent, defined as carcinogenic to humans.2 Hexavalent chromium is known to cause lung and nasal cancer as well as other health conditions such as skin allergies, skin ulcerations, nasal perforations, respiratory problems, kidney and liver damage.3 Trivalent chromium [+3, Cr(III)] is the most common form of chromium, whilst hexavalent chromium [+6, Cr(VI)] is rarely found naturally in the environment, therefore the majority of hexavalent chromium is a product of industry and manufacturing.4Chromium compounds are occupationally important in many industries such as the production and welding of stainless steel including cutting and finishing, electroplating, smelting, pigments and paints used in aerospace and maritime industries, construction and those using pesticides and tanning agents.4 Exposure to Cr(VI) and its compounds can occur by ingestion or dermal exposure but in some industries the inhalation of fumes and mists is most likely. Upon exposure to Cr(VI), the body naturally reduces Cr(VI) to Cr(III)5,6 before it is excreted in the urine, with studies suggesting occupational exposure to chromium mainly via inhalation results in elimination half-lives of 7 hours and 15–30 days.7 Apart from in bacterial cells, there is no evidence of humans or other biological systems having the ability to re-oxidise Cr(III) back to Cr(VI).8 One approach to assessing occupational exposure uses biological monitoring, involving the measurement of total chromium (without differentiation of Cr(III) and Cr(VI)) in a urine sample. Background unexposed reference ranges and occupational guidance values for total chromium in urine have been established in the UK,9 Germany10 and the US.11 Cr(VI) has the ability to permeate cell membranes meaning Cr(VI) can also be measured in red blood cells (RBC),12,13 determined as Cr(III). Once inside RBCs Cr(VI) is rapidly reduced to Cr(III) as it binds with haemoglobin and other proteins.6,12 Methods for the separate measurements of Cr(III) and Cr(VI) have focused on the assessment of environmental sources of exposure such as natural water,14,15 drinking water,14,15 waste water16 and industrial waste.17
Whilst the measurement of total chromium in urine is a practical bio-monitoring approach, it does not give a comprehensive picture of exposure. It is possible that an alternative biological matrix to measure exposure to Cr(VI) for those exposed through inhalation might be exhaled breath condensate (EBC). EBC is the non-invasive collection of condensate from cooled exhaled breath during regular tidal breathing. The collection of exhaled air will contain mostly water vapour but also droplets of fluid from the respiratory tract,18 meaning EBC measurements will reflect markers and molecules from the mouth, tracheobronchial system and the alveoli.19 EBC is reported to contain both volatile substances in the gaseous phase along with low volatile and non-volatile substances in droplets.20 The non-volatile substances will include salts, proteins, lipids, and environmental contaminants.18–20 Because EBC is relatively free of the interfering solutes found in other biological matrices it represents a suitable biological matrix for elemental analysis to assess inhalation exposure to trace elements. Studies have already shown that occupationally inhaled elements have been detected in EBC for cobalt and tungsten,21,22 iron, nickel and total chromium,23,24 manganese25 and hexavalent chromium (using liquid/liquid solvent extraction and analysis by electrothermal atomic absorption spectrometry).26 There are currently no publications for the simultaneous detection and determination of Cr(III) and Cr(VI) in an EBC sample. Due to the difference in toxicities of Cr(III) and Cr(VI) and the conversion from Cr(VI) to Cr(III) in the human body after exposure, it is important to measure both species of chromium to enable a more comprehensive picture of exposure assessment.
In order to measure Cr(III) and Cr(VI) it is necessary to maintain the integrity of each species in the sample. The stability of both trivalent and hexavalent chromium depends on pH. In acidic solutions, trivalent chromium compounds mainly exist as Cr(H2O)63+ and hydrolyse to a hydroxide in less acidic and alkaline conditions. Hexavalent chromium compounds exist either as a chromate (CrO42−, HCrO4−, H2CrO4) or dichromate (Cr2O72−).3,27 In extremely acidic conditions only, hexavalent chromium exists as H2CrO4; in these acidic conditions hexavalent chromium is a potent oxidant, which after oxidising can also begin to reduce to trivalent chromium compounds. In the pH range of 2–6 hexavalent chromium compounds exist as HCrO4− and HCr2O7−, with HCr2O7− reducing to Cr2O72− at concentrations higher than 10−2 M. Above pH 6, hexavalent chromium only exists as CrO42−.3,8,27,28 Due to this pH dependence and the fact that trivalent chromium compounds are typically cationic in solution and hexavalent chromium compounds are typically anionic in solution, speciation using ion exchange chromatography methodologies is an analytical challenge. These challenges have resulted in analysis either only determining Cr(VI) using colorimetric analysis with diphenylcarbohydrazide (DPC)15 or by determining both Cr(VI) by anion chromatography and total chromium to calculate Cr(III) by subtraction.14 Cr(III) and Cr(VI) have been simultaneously determined by ICP-MS using reverse phase chromatography combined with ion-pairing reagents.27,29 The use of non-traditional HPLC chromatographic methods such as solid phase extraction based fractionation,14,30,31 micro columns (PTFE tubes filled with activated alumina)32–35 and short column capillary electrophoresis36 with subsequent detection by AAS, ICP-AES or ICP-MS have all been employed as simple techniques to separate and determine Cr(III) and Cr(VI). Whilst all these methods have their own advantages and disadvantages (such as interferences, contamination, raised backgrounds, unsuitable limits of detection and time consuming procedures), it has meant that other research has focused on anion exchange chromatography coupled to ICP-MS. Both Chen et al.37 and Pantsar-Kallio and Manninen38 employed a dual column approach to overcome the issue of the opposite charges of the chromium species. Other authors overcame this ionic challenge by using derivatisation, where Cr(III) is complexed with EDTA to change the ionic charge from a cation to an anion complex, enabling separation of the two chromium species using the same sample injection on an anion exchange column.39,40 Suzuki and Serita41 were the first to report this technique using HPLC-AAS, with Chen et al.37 reporting that alternative complexing agents such as diethylenetriaminepentaacetic acid (DTPA) and 2,6-pyridinedicarboxylic acid (PDCA) were not as effective as EDTA. Other publications have reported the successful speciation of Cr(III) and Cr(VI) using newly introduced speciality anion columns with sulfonic and alkyl quaternary ammonium functional groups which boast both anionic and cationic properties, and have been employed by authors concerned with species' integrity related to sample pre-treatment.42–44
The initial aim of this study was to develop a speciation method to simultaneously determine both Cr(III) and Cr(VI) using a small anion exchange speciality column with cationic capabilities using a OneFAST system coupled to an ICP-MS. The method developed was then used to investigate the stability of both chromium species in an EBC sample with the view to establishing a viable technique to employ for workplace monitoring. The objective, being to establish a method to allow the collection and analysis of EBC samples from workers who may be occupationally exposed to hexavalent chromium.
Experimental
Reagents
Chromium speciation compounds potassium dichromate (K2Cr2O7) and chromium(III) chloride (CrCl3) were obtained from Fisher Scientific (Loughborough, UK). Ammonia solution (NH3), UpA nitric acid and UpA hydrochloric acid was supplied by Romil (UK). Butan-1-ol was from Fisher Scientific (Loughborough, UK).All solutions were made using ultrapure deionised water (18.2 MΩhm cm) from a Millipore system (Merck Millipore, Billerica, MA, USA). Single standard stock solutions of 1000 mg L−1 of Cr(III) and Cr(VI) were prepared fresh in water for each day of analysis.
A commercially available proficiency testing material (PTM) from Sigma Aldrich (UK) for Cr(VI) in drinking water (Sample WS13-4 Lot LRAA1427) was used to validate the method. Manufacturer instructions suggest the PTM is diluted 100 fold in water. However for this study the PTM was diluted 1000 fold in water to more accurately reflect the levels of Cr(VI) expected in EBC samples. Previous studies that measured total chromium in EBC24 or Cr(VI) only in EBC45 reported levels less than 10 μg L−1 and 5 μg L−1 of chromium in EBC respectively.
Instrumentation
Separation was achieved using a hyphenated μLC system with an ICP-MS (XSERIES 2, Thermo Fisher Scientific, Hemel Hempstead, UK). The separation of Cr(III) and Cr(VI) was achieved using a 5 cm anion exchange guard column, (Dionex IONPAC AG7 4 mm × 50 mm i.d., 10 μm, Thermo Fisher Scientific). The Dionex AG7 anion exchange column has cationic exchange capability due to the presence of sulfonic and alkyl quaternary ammonium functional groups, and is suitable for a full pH range of 0–14. The micro-flow delivery of both sample and mobile phase was accomplished using an ESI OneFAST system (Elemental Scientific, Warrington, UK), with a six port switching valve and 500 μL sample loop. The ICP-MS was operated in collision cell (CCT) mode using 7% hydrogen in helium (approximately 3.5 mL min−1). The ICP-MS conditions were optimised daily using a 10 μg L−1 tuning solution containing arsenic, chromium, cobalt, indium and uranium (made in 1% (v/v) nitric acid from 1000 mg L−1 stock standards (ICP-MS standards, BDH, Poole, UK)). Table 1 summarises the ICP-MS and OneFAST operating conditions.| XSERIES2 ICP-MS Settings | Parameters |
|---|---|
| RF power | 1400 W |
| Nebuliser gas flow | 0.89–0.90 L min−1 |
| Sample/skimmer cones | Ni/Ni Xt cones |
| Collision gas 7% He+H2 flow rate | 3.5 mL min−1 |
| Isotope monitored | 52Cr |
| Dwell time | 100 ms |
| μLC conditions | Parameters |
|---|---|
| Analytical column | Dionex IONPAC AG7 (4 mm × 50 mm, 10 μm) |
| Mobile phase | 4% v/v ammonia solution & 3.2% v/v nitric acid, pH 1.8–2 |
| Sample injection time | 30 s |
| Flow rate | 1 mL min−1 |
| Total acquisition time | 230 s |
| Sample loop | 500 μL |
| Peristaltic pump tubing | Green/orange 0.38 mm |
| ESI switching valve | P6 – 6 port |
| Valve to column connector | 0.25 ID (green) |
| Column to ICP-MS nebuliser | 0.5 ID (orange) |
| Nebuliser | PFA-ST |
Exhaled breath condensate collection
EBC was collected using a portable TURBO-DECCS condenser (Transportable Unit for Research on Biomarkers Obtained from Disposable Exhaled Condensate Collection Systems) from ItalChill (Parma, Italy). This technique consists of a disposable respiratory system comprising of a mouthpiece connected to a one-way aspiration valve and saliva trap with an EBC sample collection tube at the end inserted into a temperature controlled chilling unit set at −5 °C (see Fig. 1). The collection of exhaled air results in low sample volumes of EBC; 15 minutes of regular tidal breathing will typically provide less than 2 mL of EBC sample. It was necessary to collect large volumes of EBC from a volunteer not occupationally exposed to hexavalent chromium for use in method development as a blank sample and for recovery. In addition to the volunteer supplying the bulk EBC, five further volunteers supplied EBC samples for the optimised method applications. Blank EBC was collected from the volunteer by regular tidal breathing through the mouth piece of the TURBO-DECCS. For the most part a complete seal around the mouthpiece with the mouth is maintained allowing periodic swallowing of any accumulated saliva. | ||
| Fig. 1 TURBO-DECCS condenser consisting of a disposable respiratory system comprising of a mouthpiece connected to a one-way aspiration valve and saliva trap with an EBC sample collection tube at the end inserted into a temperature controlled chilling unit set at −5 °C. | ||
Method development
See Fig. 2 for a flow chart outlining the six stages of the method development. | ||
| Fig. 2 Flow chart showing optimisation stages of the method development. | ||
Sample storage experiment
The optimised method incorporates a derivatisation step, with a 10 fold dilution of the EDTA solution (at a concentration of 0.5 mM adjusted to pH 8 with 10% v/v ammonia solution) with the EBC sample. From the 1000 mg L−1 single standard stock solutions of Cr(III) and Cr(VI) 100 μg L−1 single and mixed species working solutions were prepared daily. Using the 100 μg L−1 mixed species working solution, calibration standards of 0.01, 0.02, 0.05, 0.1, 0.5, 1, 2 and 5 μg L−1 were prepared in the 0.5 mM EDTA solution using a 10 fold dilution to 50 mL. Blank and spiked EBC samples were diluted 10 fold with the EDTA solution to 1.5 mL. The original 1 in 1000 preparation of the proficiency testing material to validate the method was subsequently diluted 10 fold to 50 mL with the EDTA solution. All samples including standards were left to stand at room temperature for a minimum of 1 hour prior to analysis by μLC-ICP-MS.
Results & discussion
Method development
Following initial work with the AG7 ion exchange column, the simultaneous separation of Cr(III) and Cr(VI) was achieved in spiked aqueous samples using an isocratic flow of 4% v/v ammonia solution and 3.2% v/v ultrapure nitric acid, with pH adjustment to between pH 1.8–2 using the ammonia solution. | ||
| Fig. 3 Chromatograms of 5 μg L−1 single standards of Cr(VI) and Cr(III) analysed using an ESI OneFAST system coupled to a Dionex AG7 anion exchange column and ICP-MS using a nitric acid and ammonia mixed mobile phase. (A1–A4) Chromatograms of Cr(III). (B1–B4) Chromatograms of Cr(VI). (A1 & B1) Chromium species stored at laboratory room temperature (∼22 °C). (A2 & B2) Chromium species stored at fridge temperature (4 °C). (A3 & B3) Chromium species stored at freezer temperature (−20 °C) for 24 hour. (A4) Baseline of chromatogram (A2) showing Cr(III) intermediate compound. (B4) Baseline of chromatogram (B3) showing the unknown chromium intermediate compound. | ||
The chromatograms in Fig. 4 show that the complexation reaction of Cr(III) with the EDTA solution was not instantaneous but achieved near full complexation after one hour at room temperature prior to analysis. In many of the studies published where EDTA complexation was used to stabilise or change the charge of Cr(III), concentrations lower than 0.5 mM EDTA were used and heat was also used for the derivatisation reaction to occur. Both Suzuki & Serita41 and Byrdy et al.39 heated the samples in a heat block for 1 hour at 50 °C; Gurleyuk & Wallschlager40 heated their samples for 1 hour at 70 °C and Leist et al.47 heated for 1.5 hours at 70 °C. This approach would not be practical after the collection of EBC samples from workers at a place of work, and samples may not be delivered to the laboratory for 24 hours, meaning immediate heating of samples after collection in a heat block is not a viable option. However, Wolf et al.27 found that the complexation reaction was fully achieved with 0.5 mM of EDTA when the samples were left to stand at room temperature for 30 minutes prior to analysis; having adjusted the pH of the EDTA solution to pH 7.6. The pH of the EDTA solution used in this study without any adjustment was pH 5, which may explain why the complexation reaction shown in chromatogram B of Fig. 4 was not 100% effective.
 | ||
| Fig. 4 Chromatograms of a 5 μg L−1 Cr(III) standard prepared in 0.5 mM EDTA analysed using an ESI OneFAST system coupled to a Dionex AG7 anion exchange column and ICP-MS using a nitric acid and ammonia mixed mobile phase. (A) Analysed immediately after preparation. (B) Analysed after being left to stand for 1 hour. | ||
 | ||
| Fig. 5 Chromatograms showing the effect of pH of the EDTA solution to successfully complex and stabilise Cr(III), analysed using an ESI OneFAST system coupled to a Dionex AG7 anion exchange column and ICP-MS using a nitric acid and ammonia mixed mobile phase. | ||
Single standards of 5 μg L−1 Cr(VI) were prepared in the three pH solutions of EDTA that achieved full complexation of Cr(III); pH 7.6, 8 and 9. Fig. 6 shows that pH 7.6 and 8 had no effect on Cr(VI); however peak splitting was observed at pH 9.
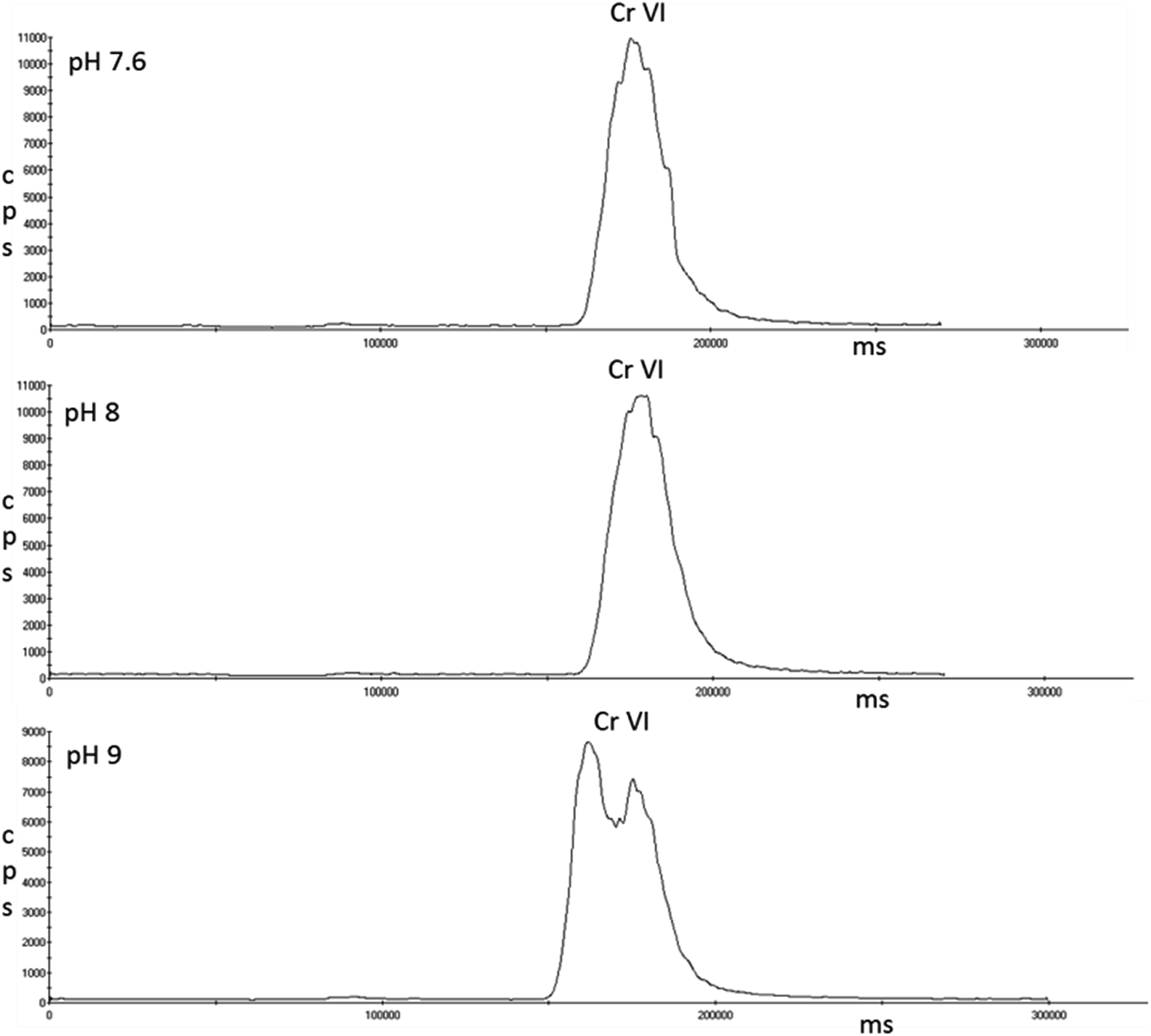 | ||
| Fig. 6 Chromatograms showing the effect of pH of the EDTA solution on Cr(VI) analysed using an ESI OneFAST system coupled to a Dionex AG7 anion exchange column and ICP-MS using a nitric acid and ammonia mixed mobile phase. | ||
 | ||
| Fig. 7 Chromatograms of two Cr(VI) spiked EBC samples, stored at fridge temperature (∼4 °C) for 6 weeks prior to analysis using an ESI OneFAST system coupled to a Dionex AG7 anion exchange column and ICP-MS using a nitric acid and ammonia mixed mobile phase. (A) EDTA pH 7.6. (B) EDTA pH 8. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ms). The calibration range used in this study was 0–5 μg L−1 for both species of chromium. The limit of detection (LOD) was calculated as three times the standard deviation of the blank; this was 0.006 μg L−1 and 0.002 μg L−1 for Cr(III) and Cr(VI) respectively. The limit of quantification (LOQ) was calculated as 10 times the mean background equivalent concentration (BEC); this was 0.040 μg L−1 and 0.013 μg L−1 for Cr(III) and Cr(VI) respectively.
000 ms). The calibration range used in this study was 0–5 μg L−1 for both species of chromium. The limit of detection (LOD) was calculated as three times the standard deviation of the blank; this was 0.006 μg L−1 and 0.002 μg L−1 for Cr(III) and Cr(VI) respectively. The limit of quantification (LOQ) was calculated as 10 times the mean background equivalent concentration (BEC); this was 0.040 μg L−1 and 0.013 μg L−1 for Cr(III) and Cr(VI) respectively.
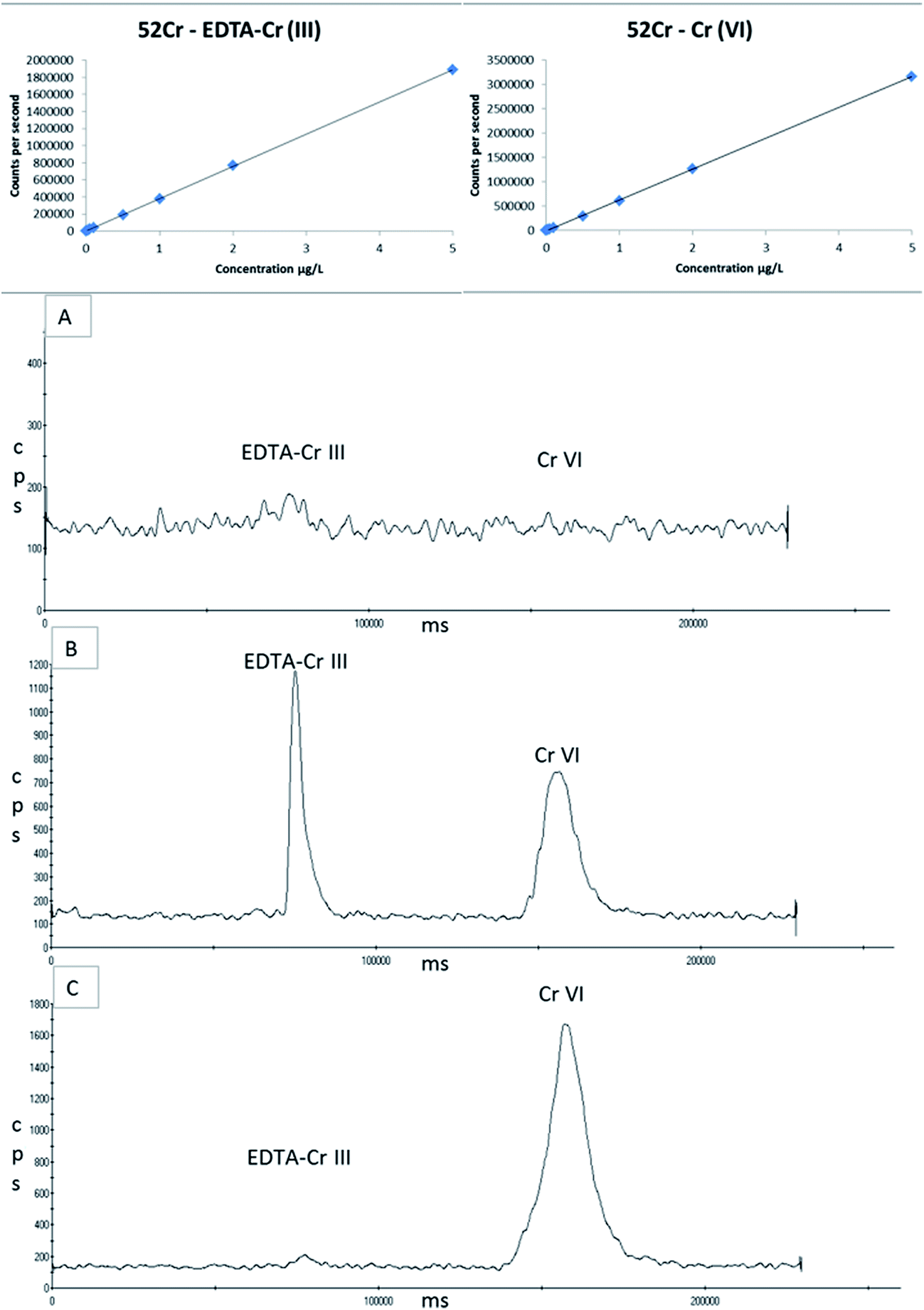 | ||
| Fig. 8 Linear calibration curves of 0–5 μg L−1 for both EDTA–Cr(III) and Cr(VI) using an ESI OneFAST system coupled to a Dionex AG7 anion exchange column and ICP-MS using a nitric acid and ammonia mixed mobile phase. (A) Blank EBC sample. (B) A 2 μg L−1 spiked blank EBC sample with EDTA–Cr(III) and Cr(VI). (C) Commercially available proficiency testing material for Cr(VI) in drinking water. | ||
The “blank” EBC spiked at a concentration of 5 μg L−1, analysed on four separate occasions over the 6 week storage period, gave an average recovery of 91% ± 0.13 for Cr(III) and 95% ± 0.02 for Cr(VI). The proficiency testing material when prepared 1 in 1000 for this study has a target value of 2.81 μg L−1 and an acceptance range of 2.28–3.28 μg L−1. The PTM sample was analysed three times and gave an average within range result of 2.65 ± 0.03 μg L−1 for Cr(VI).
Calibration standards, samples and quality control material are diluted 10 fold with 0.5 mM EDTA solution adjusted to pH 8 with 10% v/v ammonia solution. These samples are left to stand a minimum of 1 hour prior to analysis μLC-ICP-MS, and can be stored refrigerated for up to 6 weeks.
The use of ICP-MS can result in spectroscopic interferences, e.g. with polyatomic interferences 40Ar12C+ and 36Ar16O+ on 52Cr. However the optimised use of the collision cell will avoid most of these interferences. It is also possible that the sample matrix could create additional interferences given that element such as C, O and Cl are abundant in biological samples. The main contributor to a potential high background signal intensity on 52Cr due to 40Ar12C+. Having injected a weak butanol solution with and without the EBC matrix, no peak or increase in background signal intensity was observed. The main chlorine based interference on 52Cr would be the 35Cl16O1H+. To investigate the impact of a chloride interference on the chromium species a weak solution of HCl was injected onto the column. Prior to complexing Cr(III) with EDTA, Cl eluted before Cr(VI) and Cr(III). However, after complexing Cr(III) with EDTA and altering the retention time, when a weak solution of HCl is injected onto the column or added to ‘blank’ EBC, Cl and EDTA–Cr(III) elute at the same time. However, unlike a urine sample, the chloride content in EBC is negligible, as can be seen in the blank EBC in Fig. 8a. Furthermore the recovery of the spiked sample for Cr(III) in the stability study was 91% so no over recovery from an interference is evident. To confirm the absence of a chloride interference, two EBC samples were collected from five volunteers who were not occupationally exposed to chromium. There was no significant peaks obtained at Cr(III) in fact both EDTA–Cr(III) and Cr(VI) were less than the LOQ for all 10 EBC samples analysed.
Conclusion
The method described in this study is a robust and sensitive method for the simultaneous determination of Cr(III) and Cr(VI) in an EBC sample. This speciation method has been validated with spiked samples of blank EBC and a commercially available drinking water proficiency testing material sample for Cr(VI). It has been demonstrated that water samples and EBC samples containing Cr(III) and Cr(VI) can be stored in the fridge (∼4 °C) for up to 6 weeks when they have been diluted 10 fold with the EDTA solution, and that EBC samples must not be frozen after collection if the integrity of Cr(VI) is to be maintained. This study has developed a practical approach to collecting EBC samples in the workplace from workers who are potentially occupationally exposed to hexavalent chromium and that the integrity of the chromium species will be intact prior to analysis by μLC-ICP-MS. The next stage of this study will be to undertake site visits to collect EBC samples from chromium workers and apply the methods developed here to these ‘real’ samples.Acknowledgements
This publication and the work it describes were funded by the Health and Safety Executive (HSE). Its contents, including any opinions and/or conclusions expressed, are those of the authors alone and do not necessarily reflect HSE policy.References
- G. F. Nordberg, B. A. Fowler, M. Nordberg and L. T. Friberg, Handbook on the toxicology of metals, Elsevier Academic Press, London, 3rd edn, 2007 Search PubMed.
- Agents classified by the IARC monographs, volumes 1–113, http://monographs.iarc.fr/ENG/Classification/latest_classif.php (Accessed September 2015).
- R. Rakhunde, L. Deshpande and H. D. Juneja, Crit. Rev. Environ. Sci. Technol., 2012, 42(7), 776–810 CrossRef CAS.
- IARC monographs on the evaluation of carcinogenic risks to humans, Volume 100 C. A review of human carcinogens: arsenic, metals, fibres and dusts. http://monographs.iarc.fr/ENG/Monographs/vol100C/index.php (accessed September 2015).
- D. J. Paustenbach, B. L. Finley, F. S. Mowat and B. D. Kerger, J. Toxicol. Environ. Health, Part A, 2003, 66(14), 1295–1339 CrossRef CAS PubMed.
- F. S. De, D. Serra, A. Camoirano and P. Zanacchi, Biol. Trace Elem. Res., 1989, 21, 179–187 CrossRef.
- P. Hoet, in Handbook of elemental speciation II: Species in the environment, food, medicine and occupational health, ed. R. Cornelis, J. Caruso, H. Crews and K. Heumann, Wiley-Blackwell, 2005, ch. 2.6.2, pp. 136–157 Search PubMed.
- A. Zhitkovich, Chem. Res. Toxicol., 2011, 24(10), 1617–1629 CrossRef CAS PubMed.
- HSL, Guidance on laboratory techniques in occupational medicine, HSE, 12th edn, 2013 Search PubMed.
- DFG German DRF Deutsche Forschungsgemeinschaft, List of MAK and BAT values 2009, Wiley-VCH, Germany, 2012 Search PubMed.
- American Conference of Governmental Industrial Hygienists, TLVs and BEIs: Threshold limit values for chemical substances and physical agents, Signature Publications, USA, 2015 Search PubMed.
- G. E. Corbett, D. G. Dodge, E. O'Flaherty, J. Liang, L. Throop, B. L. Finley and B. D. Kerger, Environ. Res., 1998, 78(1), 7–11 CrossRef CAS PubMed.
- J. P. Goulle, E. Saussereau, J. Grosjean, C. Doche, L. Mahieu, J. M. Thouret, M. Guerbet and C. Lacroix, Forensic Sci. Int., 2012, 217(1–3), 8–12 CrossRef PubMed.
- M. L. Chen, Y. N. Zhao, D. W. Zhang, Y. Tian and J. H. Wang, J. Anal. At. Spectrom., 2010, 25(11), 1688–1694 RSC.
- Y. M. Scindia, A. K. Pandey, A. R. Reddy and S. B. Manohar, Anal. Chim. Acta, 2004, 515(2), 311–321 CrossRef CAS.
- A. S. Stasinakis, N. S. Thomaidis and T. D. Lekkas, Anal. Chim. Acta, 2003, 478(1), 119–127 CrossRef CAS.
- L. Girard and J. Hubert, Talanta, 1996, 43(11), 1965–1974 CrossRef CAS PubMed.
- M. Corradi and A. Mutti, Acta Biol. Med., 2005, 76(2), 20–29 Search PubMed.
- S. A. Kharitonov and P. J. Barnes, Am. J. Respir. Crit. Care Med., 2001, 163(7), 1693–1722 CrossRef CAS PubMed.
- M. Goldoni, M. Corradi, P. Mozzoni, G. Folesani, R. Alinovi, S. Pinelli, R. Andreoli, D. Pigini, R. Tillo, A. Filetti, C. Garavelli and A. Mutti, J. Breath Res., 2013, 7(1), 017101 CrossRef PubMed.
- M. Goldoni, S. Catalani, G. De Palma, P. Manini, O. Acampa, M. Corradi, R. Bergonzi, P. Apostoli and A. Mutti, Environ. Health Perspect., 2004, 112(13), 1293–1298 CrossRef CAS PubMed.
- H. C. Broding, B. Michalke, T. Goen and H. Drexler, Int. Arch. Occup. Environ. Health, 2009, 82(5), 565–573 CrossRef CAS PubMed.
- F. Hoffmeyer, T. Weiss, M. Lehnert, B. Pesch, H. Berresheim, J. Henry, M. Raulf-Heimsoth, C. H. Broding, J. Buenger, V. Harth and T. Bruening, J. Environ. Monit., 2011, 13(1), 212–218 RSC.
- A. Caglieri, M. Goldoni, O. Acampa, R. Andreoli, M. V. Vettori, M. Corradi, P. Apostoli and A. Mutti, Environ. Health Perspect., 2006, 114(4), 542–546 CrossRef CAS PubMed.
- S. Hulo, N. Cherot-Kornobis, M. Howsam, S. Crucq, V. de Broucker, A. Sobaszek and J. L. Edme, Toxicol. Lett., 2014, 226(1), 63–69 CrossRef CAS PubMed.
- M. Goldoni, A. Caglieri, D. Poli, M. V. Vettori, M. Corradi, P. Apostoli and A. Mutti, Anal. Chim. Acta, 2006, 562(2), 229–235 CrossRef CAS PubMed.
- R. E. Wolf, J. M. Morrison and M. B. Goldhaber, J. Anal. At. Spectrom., 2007, 22(9), 1051–1060 RSC.
- J. Scancar and R. Milacic, J. Anal. At. Spectrom., 2014, 29(3), 427–443 RSC.
- K. Neubauer, W. Reuter and P. Perrone, Application Note, Perkin Elmer Life and Analytical Sciences, USA, 2003 Search PubMed.
- C. Puls and A. Limbeck, J. Anal. At. Spectrom., 2009, 24(10), 1434–1440 RSC.
- M. M. L. Guerrero, E. V. Alonso, J. M. C. Pavon, M. T. S. Cordero and A. G. de Torres, J. Anal. At. Spectrom., 2012, 27(4), 682–688 RSC.
- M. J. Marques, A. Morales-Rubio, A. Salvado and M. de la Guardia, Talanta, 2001, 53(6), 1229–1239 CrossRef CAS PubMed.
- J. Prokisch, S. A. Katz, B. Kovacs and Z. Gyori, J. Chromatogr. A, 1997, 774(1–2), 363–371 CrossRef CAS.
- R. P. Monasterio, G. E. Lascalea, L. D. Martinez and R. G. Wuilloud, J. Trace Elem. Med. Biol., 2009, 23(3), 157–166 CAS.
- M. Sperling, S. K. Xu and B. Welz, Anal. Chem., 1992, 64(24), 3101–3108 CrossRef CAS.
- B. H. Li and X. P. Yan, Electrophoresis, 2007, 28(9), 1393–1398 CrossRef CAS PubMed.
- Z. Chen, M. Megharaj and R. Naidu, Talanta, 2007, 72(2), 394–400 CrossRef CAS PubMed.
- M. Pantsar-Kallio and P. Manninen, Anal. Chim. Acta, 1996, 318(3), 335–343 CrossRef CAS.
- F. A. Byrdy, L. K. Olson, N. P. Vela and J. A. Caruso, J. Chromatogr. A, 1995, 712(2), 311–320 CrossRef CAS PubMed.
- H. Gurleyuk and D. Wallschlager, J. Anal. At. Spectrom., 2001, 16(9), 926–930 RSC.
- Y. Suzuki and F. Serita, Ind. Health., 1985, 23(3), 207–220 CrossRef CAS PubMed.
- F. Seby, S. Charles, M. Gagean, H. Garraud and O. F. X. Donard, J. Anal. At. Spectrom., 2003, 18(11), 1386–1390 RSC.
- C. Barnowski, N. Jakubowski, D. Stuewer and J. A. C. Broekaert, J. Anal. At. Spectrom., 1997, 12(10), 1155–1161 RSC.
- L. Xing and D. Beauchemin, J. Anal. At. Spectrom., 2010, 25(7), 1046–1055 RSC.
- M. Goldoni, A. Caglieri, G. De Palma, O. Acampa, P. Gergelova, M. Corradi, P. Apostoli and A. Mutti, J. Environ. Monit., 2010, 12(2), 442–447 RSC.
- M. Gube, J. Ebel, P. Brand, T. Goeen, K. Holzinger, U. Reisgen and T. Kraus, Int. Arch. Occup. Environ. Health, 2010, 83(7), 803–811 CrossRef CAS PubMed.
- M. Leist, R. Leiser and A. Toms, The Application Notebook-Advertising Supplement Mass Spectrometry, Varian Inc., 2006, pp. 29–31 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |