
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bulk sensitive determination of the Fe3+/FeTot-ratio in minerals by Fe L2/3-edge X-ray Raman scattering
Alexander
Nyrow†
*a,
Christian
Sternemann
*a,
John S.
Tse
b,
Christopher
Weis
a,
Christoph J.
Sahle‡
a,
Kolja
Mende
a,
D. C. Florian
Wieland§
a,
Valerio
Cerantola‡
c,
Robert A.
Gordon¶
d,
Georg
Spiekermann
eg,
Tom
Regier
f,
Max
Wilke
g and
Metin
Tolan
a
aFakultät Physik/DELTA, Technische Universität Dortmund, 44227 Dortmund, Germany. E-mail: alexander.nyrow@tu-dortmund.de; christian.sternemann@tu-dortmund.de
bDepartment of Physics and Engineering Physics, University of Saskatchewan, Saskatoon S7N 5E2, Canada
cBayerisches Geoinstitut, Universität Bayreuth, D-95440 Bayreuth, Germany
dPNCSRF, APS Sector 20, Argonne, IL 60439, USA
eDeutsches Elektronen-Synchrotron DESY, D-22607, Hamburg, Germany
fCLS, 44 Innovation Boulevard, Saskatoon S7N 2V3, Canada
gSection 3.3, Deutsches GeoForschungsZentrum, 14473 Potsdam, Germany
First published on 10th December 2015
Abstract
We present the first measurements of the iron L2/3-edge of the compounds FeO, Fe2O3, and Fe3O4 at ambient pressure and of FeCO3 at high pressures of 2.4 and 40 GPa using a diamond anvil cell by X-ray Raman scattering spectroscopy, a bulk sensitive probe of soft X-ray absorption edges making use of hard X-rays. We show that the spectral shape of the Fe L2/3-edge can be analyzed quantitatively to reveal the oxidation state of iron in matter. Consequently, in situ X-ray Raman scattering spectroscopy at the iron L-edge at high pressure and temperature opens exciting perspectives to characterize the local coordination, oxidation, and spin state of iron at high pressure and temperature, conditions that are of relevance for e.g. geological sciences or chemical processing.
1 Introduction
Iron is one of the most common elements of the inner Earth, strongly affecting the macroscopic properties of related minerals, melts, and glasses.1,2 Under pressure–temperature conditions of the deep Earth, the local chemical and physical properties of iron, e.g. oxidation state, local coordination, and spin state, can vary significantly.3–5 Quantitative information about these parameters is used in order to understand the structural changes during geochemical and geophysical processes such as Fe–Mg partitioning, iron melting and density variations for instance associated with the spin crossover in iron. In situ experiments under the relevant thermodynamic conditions provide unique insights into geochemical and geophysical processes.6,7Next to Mössbauer spectroscopy,8,9 the local chemical environment of iron in minerals is often studied by X-ray absorption spectroscopy (XAS) at the Fe K-edge, which is a very sensitive tool to study the oxidation state and local coordination.10–13 The related pre-edge features, which contain information on the Fe 3d states, are difficult to resolve and a high-resolution monochromator setup is needed. So far, XAS studies of the near edge structure in situ at high pressure are rare, and analysis of the main edge is used.14 Moreover, the 3d state can be probed directly by resonant X-ray emission.15 Information on the orbital and spin magnetic moment is obtained using X-ray magnetic circular dichroism16 and X-ray linear magnetic dichroism.17
Fe L2/3-edge spectroscopy (2p → 3d) is also a sensitive method in order to study the local properties of iron and is performed either using soft X-rays18–20 or by electron-energy loss spectroscopy (EELS)21–23 with a transmission electron microscope. Compared to pre-edge studies of the redox state at the Fe K-edge, the variation of the corresponding intensity parameter determined on the Fe L-edge is large, thus offering a potentially higher precision for determining the oxidation state. In other words, if there would be no experimental constraints for measuring either one of the edges, the L-edge should provide results with better precision. Therefore, measurements of the L-edge, if feasible, may be a valuable alternative where changes in the pre-edge of the K-edge are too small to be resolved. XAS and EELS at the L-edges at X-ray energies below 1 keV are used for applications in e.g. geology,3,21,24 physics,19,25–29 biology,30,31 catalysis,32–35 and materials science.18,36 However, these techniques cannot be applied for in situ high pressure experiments owing to the highly absorbing sample environment, e.g. resistively or laser heated diamond anvil cells.
The potential of X-ray Raman scattering spectroscopy can be exploited for bulk sensitive in situ studies of low-energy X-ray absorption edges even for embedded samples.37–43 Recently, in situ high pressure measurements of the Fe M2/3-edge demonstrated the ability to track the pressure induced spin cross-over in FeS and to extract quantitative information about the crystal field splitting of the 3d states in iron.44 The Fe M2/3-edge is significantly modified by weak changes of the oxidation and spin state, but it is less sensitive to the local coordination.45 Recently, magnetic circular dichroism of X-ray Raman scattering was observed at the iron L2/3-edge of pure iron.46 In general, measurements of the Fe L2/3-edge by X-ray Raman scattering spectroscopy are a very good alternative for studies of the oxidation state, the local coordination, and the spin state of iron in compounds under geologically relevant conditions. In this paper we focus on the capability of X-ray Raman scattering spectroscopy to analyze the oxidation state of a sample both at ambient and at high pressure.
2 X-ray Raman scattering
X-ray Raman scattering (XRS), a non-resonant inelastic X-ray scattering technique, provides spectroscopic information on soft X-ray absorption edges similar to XAS and EELS but using high-energy X-rays.47–51 Consequently, the local atomic and electronic structure can be probed bulk sensitively yielding information about e.g. oxidation state, spin state and local coordination. By scanning the energy loss in the vicinity of an electron binding energy, core level excitations can be studied both in the dipole and non-dipole limits.48 The intensity of the scattered radiation is proportional to the dynamic structure factor, which is given for transition metals and rare-earth elements due to the strong interaction between the 3d electron and the hole wave functions by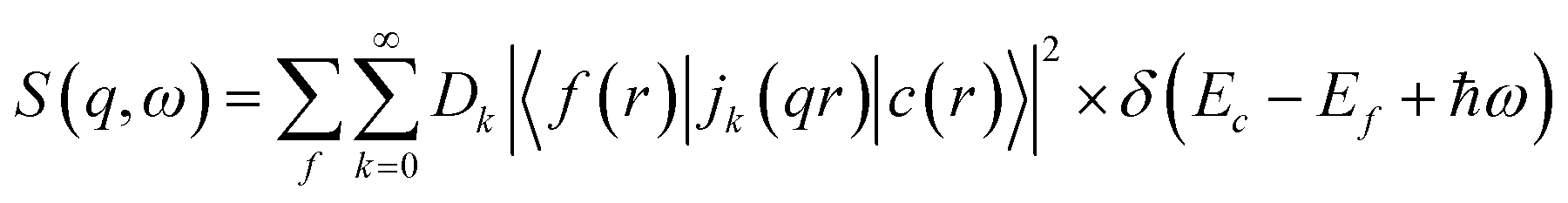 | (1) |
3 Experimental details
The XRS experiment was performed at beamline PNC/XSD 20-ID of the Advanced Photon Source employing the LERIX spectrometer.54 Here, 19 Si(444) analyzer crystals are arranged in a semicircle (radius of 1 m) covering an angular range from 9° to 171°, which results in a momentum transfer range from 0.7 Å−1 to 8.7 Å−1. The energy of the incident X-rays with an overall resolution of 1.0 eV was tuned by using a Si(111) monochromator. In order to measure the Fe L2/3-edge, the energy of incident X-rays was scanned from 8.582 keV to 8.682 keV resulting in an energy loss interval from 670 eV to 770 eV, respectively. Polycrystalline FeO ([6]Fe2+, space group Fm3m, corresponding to the geologically relevant mineral wustite), Fe2O3 ([6]Fe3+, space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c, corresponding to hematite), and Fe3O4 (corresponding to magnetite with a composition of 1/3 [6]Fe2+, 1/3 [6]Fe3+, and 1/3 [4]Fe3+) powders with a trace metal basis of 99.9%, 99.995%, and 99.99%, respectively, were measured without further treatment. The samples were characterized by X-ray diffraction at beamline BL9 of the DELTA synchrotron source.55 Phase purity was found for Fe2O3 and Fe3O4 whereas FeO contained a small amount (<3%) of iron. The spectra were collected in 1.5 hours (FeO) and 4 hours (Fe2O3 and Fe3O4). Due to the small momentum transfer dependence of the shape of the Fe L2/3-edge, the spectra were summed up over the entire q-range. Subsequently, a background described by a polynomial function was fitted to the energy loss values below 700 eV and subtracted from the data. This background subtraction procedure is similar to that typically applied to EELS data.21 Finally, background corrected spectra were normalized to the integrated intensity in the energy loss region from 700 eV to 728 eV. For a general description of the XRS data analysis see ref. 56 and 57.
c, corresponding to hematite), and Fe3O4 (corresponding to magnetite with a composition of 1/3 [6]Fe2+, 1/3 [6]Fe3+, and 1/3 [4]Fe3+) powders with a trace metal basis of 99.9%, 99.995%, and 99.99%, respectively, were measured without further treatment. The samples were characterized by X-ray diffraction at beamline BL9 of the DELTA synchrotron source.55 Phase purity was found for Fe2O3 and Fe3O4 whereas FeO contained a small amount (<3%) of iron. The spectra were collected in 1.5 hours (FeO) and 4 hours (Fe2O3 and Fe3O4). Due to the small momentum transfer dependence of the shape of the Fe L2/3-edge, the spectra were summed up over the entire q-range. Subsequently, a background described by a polynomial function was fitted to the energy loss values below 700 eV and subtracted from the data. This background subtraction procedure is similar to that typically applied to EELS data.21 Finally, background corrected spectra were normalized to the integrated intensity in the energy loss region from 700 eV to 728 eV. For a general description of the XRS data analysis see ref. 56 and 57.
4 Results and discussion
The results of the XRS measurements of the Fe L2/3-edges on FeO, Fe2O3, and Fe3O4 are presented in Fig. 1. The spin–orbit splitting for FeO given by the distance between the inflection points of the Fe L3- and Fe L2-edges can be estimated to be 12.8 ± 0.2 eV, which is in very good agreement with earlier results.58 Significant spectral differences can be observed, if the Fe L2/3-edge of FeO is compared to that of Fe2O3. The energy shift between the inflection points of the Fe L3- (707.1 eV) and Fe L2-edges (720.3 eV) of Fe2O3 results in a spin–orbit coupling of 13.2 ± 0.2 eV, which agrees very well with values obtained by EELS.21,58 The shape of the Fe L2/3-edge of Fe3O4 shows a very broad, asymmetric, and featureless maximum due to the contribution of both FeO- and Fe2O3-like Fe L2/3-edge spectra. | ||
| Fig. 1 Results of the Fe L2/3-edge measurements of FeO (black), α-Fe2O3 (blue), and Fe3O4 (green) at ambient pressure. Moreover, the spectrum of FeCO3 measured in situ at 2.4 GPa (grey) and 40 GPa (red) using a diamond anvil cell is presented. The gray dashed line represents a background modeled by a double arc-tangent function. The spectra are vertically shifted for a better overview. | ||
A comparison of the XRS results with XAS and EELS data, e.g. published in ref. 19, 20 and 59, shows a very good agreement concerning the relative energy position and shape of the Fe L2/3-edges, which is a surprising result considering the origin of the excitation mechanism of the experimental techniques. While only dipole transitions are probed by XAS, a certain momentum transfer contribution can be found in EELS. However, typical EELS measurements are performed at low scattering angles, which restrict electronic excitations to dipole-like transitions. In contrast, XRS spectra are in general strongly dominated by higher order transitions at high scattering angles. Although the momentum transfer dependence of the multiplet spectra is very strong for transition metals,45,52,60 it is hardly observed at the Fe L2/3-edge of FeO, Fe2O3 and Fe3O4. The ability to access non-dipole excitations is governed by the absolute value of the momentum transfer as well as the length scale of the effective radial overlap between the initial and final state wave functions, which is small for the 2p and 3d wave functions of iron. Thus, the momentum transfer range examined in this study is too small to get significant higher order contributions and the statistical accuracy of the spectra can be significantly enhanced due to the fact that the signal collected at different scattering angles by using multi-element spectrometers can be summed up.
The collected spectra can be employed to extract quantitative information about the Fe3+/FeTot-ratio. Therefore, the position and spectral shape of the Fe L2/3-edge can be used as an indicator of the oxidation state and several extraction algorithms are discussed in the literature (see e.g.ref. 23). In order to obtain the Fe3+/FeTot-ratio, Garvie and Buseck61 propose to fit Fe2+ and Fe3+ references to a spectrum taken on a mixed oxidation state sample. This algorithm is only applicable for materials with identical end-members and coordination of iron, which is not the case for Fe3O4. Alternatively, a series of analytical functions, e.g. Gaussian- or Lorentzian-type, can be fitted instead of reference spectra. The position and relative intensities of the series of analytical functions contain information about the Fe3+/FeTot-ratio and local coordination of iron. However, several profiles with a large number of free parameters must be used in order to obtain accurate results.22,62 Another alternative proposed by van Aken et al.21 exploits the intensity ratio between the Fe L3-edge and the Fe L2-edge, which strongly depends on the Fe3+/FeTot-ratio contained in the material. The applicability of this algorithm was demonstrated for different examples of high-spin iron species with a variable oxidation state. Keeping in mind that this procedure can only be applied for high-spin iron species with conserved site symmetry,20 the algorithm will be discussed in detail and applied to the XRS data in order to demonstrate the potential of XRS measurements to obtain quantitative information about the Fe3+/FeTot-ratio in the following.
The Fe L2/3-edge measurements presented in Fig. 1 are further analyzed in order to remove the contribution of the excitations into the continuum states by subtracting a double arc-tangent function with the fixed inflexion points at 708.65 eV and 721.65 eV, respectively.21 The resulting arc-tangent background for FeO, Fe2O3, and Fe3O4 is shown in Fig. 1. Subsequently, background corrected spectra shown in Fig. 2 are integrated in the energy loss ranges from 708.5 eV to 710.5 eV (I(L3)) and from 719.5 eV to 721.5 eV (I(L2)), as suggested by van Aken et al.21 The intensity ratio I(L3)/I(L2) is then correlated with the Fe3+/FeTot-ratio x of well characterized samples via
 | (2) |
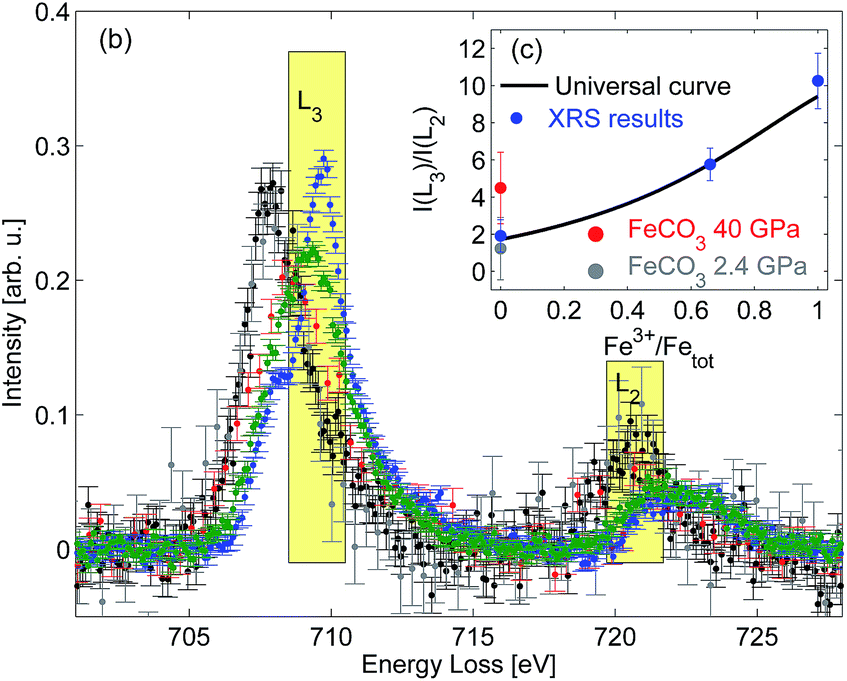 | ||
| Fig. 2 Background corrected XRS spectra with indicated integration ranges; inset: calculated I(L3)/I(L2) fractions as a function of the Fe3+/FeTot ratio in comparison with the calibration curve proposed by van Aken et al.21 | ||
In order to demonstrate the potential of this method to determine the iron oxidation state in situ under extreme conditions we performed a high pressure XRS measurement at beamline ID20 of ESRF (European Synchrotron Radiation Facility) using a multiple analyzer spectrometer.57 An FeCO3 single crystal ([6]Fe2+, space group Rc corresponding to siderite) was loaded in a Böhler–Almax diamond anvil cell using a rhenium gasket and helium as the pressure medium to guarantee quasi-hydrostatic conditions at high pressure. The sample with dimensions of about 15 μm × 25 μm × 25 μm was measured for 12 hours at 2.4 GPa and 8 hours at 40 GPa. The pressure was determined via the ruby fluorescence method.63 The analyzer energy was 12.92 keV and the incident energy was scanned from 13.623 keV to 13.663 keV with an overall energy resolution of 2.1 eV at a momentum transfer of 3.2 ± 0.9 Å−1. The spectra were analyzed as discussed above. The results obtained at 2.4 GPa are presented in Fig. 1 and show a good agreement with the FeO spectral shape despite the different energy resolution. Following the analysis procedure of van Aken et al.21 (see Fig. 2) the extracted Fe3+/FeTot ratio fits to the universal curve within the accuracy of the experiment. The deviation between the L-edge's shape measured at 40 GPa and thus the change of the I(L3)/I(L2) ratio compared to 2.4 GPa is due to the fact that the high spin to low spin transition already started.64 This clearly confirms that the analysis by van Aken is strictly limited to iron compounds in the high spin state. The different energy resolution achieved with XRS and EELS does not affect the results of this analytical procedure. However, both the energy resolution and statistical accuracy can be improved in future experiments, which clearly demonstrates the feasibility of in situ measurements of the Fe L2/3 edge at high pressure to evaluate the redox state.
5 Conclusion
In this paper, bulk sensitive measurements of the Fe L2/3-edge by XRS spectroscopy are presented and their sensitivity to the oxidation state of iron is discussed for the examples of FeO, Fe2O3, and Fe3O4. Relative intensities of the spectral features of the Fe L2/3-edge are used to extract quantitative information about the Fe3+/FeTot-ratio using the calibration curve by van Aken et al.21 This approach is a prospective method to study the oxidation and spin state of iron bulk sensitively in situ under high pressure and high temperature conditions beyond the limits of other experimental techniques. The feasibility of in situ measurements under high pressure conditions using a diamond anvil cell was successfully demonstrated with the example of FeCO3 probed at 2.4 GPa and 40 GPa.Acknowledgements
The authors acknowledge the APS, ESRF, and DELTA for providing synchrotron radiation and beamline support, respectively. AN and KM would like to thank BMBF projects 05K10PEC and 05K13PE2 (within FSP-302) for financial support, respectively. JST and RAG acknowledge the support from NSERC of Canada (MFA, Discover grant).References
- J. Badro, G. Fiquet, F. Guyot, J.-P. Rueff, V. V. Struzhkin, G. Vankó and G. Monaco, Science, 2003, 300, 789–791 CrossRef CAS PubMed.
- T. S. Duffy, Nature, 2008, 451, 269–270 CrossRef CAS PubMed.
- C. McCammon, D. Frost, J. Smyth, H. Laustsen, T. Kawamoto, N. Ross and P. van Aken, Phys. Earth Planet. Inter., 2004, 143144, 157–169 CrossRef.
- M. Wilke and H. Behrens, Contrib. Mineral. Petrol., 1999, 137, 102–114 CrossRef CAS.
- A. Zerr and R. Boehler, Nature, 1994, 371, 506–508 CrossRef CAS.
- I. J. Parkinson and R. J. Arculus, Chem. Geol., 1999, 160, 409–423 CrossRef CAS.
- B. J. Wood and D. Virgo, Geochim. Cosmochim. Acta, 1989, 53, 1277–1291 CrossRef CAS.
- Mössbauer Spectroscopy, ed. D. P. E. Dickson and F. J. Berry, Cambridge University Press, 1986 Search PubMed.
- T. Glaser, Angew. Chem., Int. Ed., 2011, 50, 10019–10020 CrossRef CAS.
- A. J. Berry, G. M. Yaxley, A. B. Woodland and G. J. Foran, Chem. Geol., 2010, 278, 31–37 CrossRef CAS.
- M. Munõz, V. de Andrade, O. Vidal, E. Lewin, S. Pascarelli and J. Susini, Geochem., Geophys., Geosyst., 2006, 7, Q11020 CrossRef.
- M. Wilke, C. Schmidt, F. Farges, V. Malavergne, L. Gautron, A. Simionovici, M. Hahn and P.-E. Petit, Chem. Geol., 2006, 229, 144–161 CrossRef CAS.
- M. Wilke, O. Hahn, A. B. Woodland and K. Rickers, J. Anal. At. Spectrom., 2009, 24, 1364–1372 RSC.
- O. Narygina, M. Mattesini, I. Kantor, S. Pascarelli, X. Wu, G. Aquilanti, C. McCammon and L. Dubrovinsky, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 174115 CrossRef.
- J.-F. Lin, Z. Mao, I. Jarrige, Y. Xiao, P. Chow, T. Okuchi, N. Hiraoka and S. D. Jacobsen, Am. Mineral., 2010, 95, 1125–1131 CrossRef CAS.
- G. Schütz, W. Wagner, W. Wilhelm, P. Kienle, R. Zeller, R. Frahm and G. Materlik, Phys. Rev. Lett., 1987, 58, 737–740 CrossRef PubMed.
- M. M. Schwickert, G. Y. Guo, M. A. Tomaz, W. L. O'Brien and G. R. Harp, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, R4289 CrossRef CAS.
- S. Bonhommeau, N. Pontius, S. Cobo, L. Salmon, F. M. F. de Groot, G. Molnar, A. Bousseksou, H. A. Durr and W. Eberhardt, Phys. Chem. Chem. Phys., 2008, 10, 5882–5889 RSC.
- F. M. F. de Groot, P. Glatzel, U. Bergmann, P. A. van Aken, R. A. Barrea, S. Klemme, M. Hävecker, A. Knop-Gericke, W. M. Heijboer and B. M. Weckhuysen, J. Phys. Chem. B, 2005, 109, 20751–20762 CrossRef CAS PubMed.
- C. Piquer, M. A. Laguna-Marco, A. G. Roca, R. Boada, C. Guglieri and J. Chaboy, J. Phys. Chem. C, 2014, 118, 1332–1346 CAS.
- P. A. van Aken, B. Liebscher and V. J. Styrsa, Phys. Chem. Miner., 1998, 25, 323–327 CrossRef CAS.
- P. A. van Aken and B. Liebscher, Phys. Chem. Miner., 2002, 29, 188–200 CrossRef CAS.
- C. C. Calvert, A. Brown and R. Brydson, J. Electron Spectrosc. Relat. Phenom., 2005, 143, 173–187 CrossRef CAS.
- D. J. Frost, F. Langenhorst and P. A. van Aken, Phys. Chem. Miner., 2001, 28, 455–470 CrossRef CAS.
- J. P. Crocombette, M. Pollak, F. Jollet, N. Thromat and M. Gautier-Soyer, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 3143–3150 CrossRef CAS.
- R. K. Hocking, E. C. Wasinger, F. M. F. de Groot, K. O. Hodgson, B. Hedman and E. I. Solomon, J. Am. Chem. Soc., 2006, 128, 10442–10451 CrossRef CAS PubMed.
- S. Miao, M. Kocher, P. Rez, B. Fultz, R. Yazami and C. C. Ahn, J. Phys. Chem. A, 2007, 111, 4242–4247 CrossRef CAS PubMed.
- T. Kroll, S. Bonhommeau, T. Kachel, H. A. Dürr, J. Werner, G. Behr, A. Koitzsch, R. Hübel, S. Leger, R. Schönfelder, A. K. Ariffin, R. Manzke, F. M. F. de Groot, J. Fink, H. Eschrig, B. Büchner and M. Knupfer, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 220502 CrossRef.
- B. Gilbert, J. E. Katz, J. D. Denlinger, Y. Yin, R. Falcone and G. A. Waychunas, J. Phys. Chem. C, 2010, 114, 21994–22001 CAS.
- H. Wang, G. Peng, L. M. Miller, E. M. Scheuring, S. J. George, M. R. Chance and S. P. Cramer, J. Am. Chem. Soc., 1997, 119, 4921–4928 CrossRef CAS.
- N. Bergmann, S. Bonhommeau, K. M. Lange, S. M. Greil, S. Eisebitt, F. M. F. de Groot, M. Chergui and E. F. Aziz, Phys. Chem. Chem. Phys., 2010, 12, 4827–4832 RSC.
- W. M. Heijboer, A. A. Battiston, A. Knop-Gericke, M. Hävecker, R. Mayer, H. Bluhm, R. Schlögl, B. M. Weckhuysen, D. C. Koningsberger and F. M. F. de Groot, J. Phys. Chem. B, 2003, 107, 13069–13075 CrossRef CAS.
- W. M. Heijboer, A. A. Battiston, A. Knop-Gericke, M. Havecker, H. Bluhm, B. M. Weckhuysen, D. C. Koningsberger and F. M. F. de Groot, Phys. Chem. Chem. Phys., 2003, 5, 4484–4491 RSC.
- W. M. Heijboer, P. Glatzel, K. R. Sawant, R. F. Lobo, U. Bergmann, R. A. Barrea, D. C. Koningsberger, B. M. Weckhuysen and F. M. F. de Groot, J. Phys. Chem. B, 2004, 108, 10002–10011 CrossRef CAS.
- W. Heijboer, D. Koningsberger, B. Weckhuysen and F. M. F. de Groot, Catal. Today, 2005, 110, 228–238 CrossRef CAS.
- J. P. Wang, N. Ji, X. Liu, Y. Xu, C. Sanchez-Hanke, Y. Wu, F. M. F. de Groot, L. F. Allard and E. Lara-Curzio, IEEE Trans. Magn., 2012, 48, 1710–1717 CrossRef CAS.
- J.-F. Lin, H. Fukui, D. Prendergast, T. Okuchi, Y. Q. Cai, N. Hiraoka, C.-S. Yoo, A. Trave, P. Eng, M. Y. Hu and P. Chow, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 012201 CrossRef.
- S. K. Lee, P. J. Eng and H.-k. Mao, Rev. Mineral. Geochem., 2014, 78, 139–174 CrossRef CAS.
- S. K. Lee, J.-F. Lin, Y. Q. Cai, N. Hiraoka, P. J. Eng, T. Okuchi, H.-k. Mao, Y. Meng, M. Y. Hu, P. Chow, J. Shu, B. Li, H. Fukui, B. H. Lee, H. N. Kim and C.-S. Yoo, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7925–7929 CrossRef CAS PubMed.
- C. J. Sahle, C. Sternemann, C. Schmidt, S. Lehtola, S. Jahn, L. Simonelli, S. Huotari, M. Hakala, T. Pylkkänen, A. Nyrow, K. Mende, M. Tolan, K. Hämäläinen and M. Wilke, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6301–6306 CrossRef CAS PubMed.
- C. Sternemann, C. J. Sahle, K. Mende, C. Schmidt, A. Nyrow, L. Simonelli, M. Moretti Sala, M. Tolan and M. Wilke, J. Phys.: Conf. Ser., 2013, 425, 202011 CrossRef.
- J. S. Tse, L. Yang, S. J. Zhang, C. Q. Jin, C. J. Sahle, C. Sternemann, A. Nyrow, V. Giordano, J. Z. Jiang, S. Yamanaka, S. Desgreniers and C. A. Tulk, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 184105 CrossRef.
- J. S. Tse, M. Hanfland, R. Flacau, S. Desgreniers, Z. Li, K. Mende, K. Gilmore, A. Nyrow, M. Moretti Sala and C. Sternemann, J. Phys. Chem. C, 2014, 118, 1161–1166 CAS.
- A. Nyrow, J. S. Tse, N. Hiraoka, S. Desgreniers, T. Büning, K. Mende, M. Tolan, M. Wilke and C. Sternemann, Appl. Phys. Lett., 2014, 104, 262408 CrossRef.
- A. Nyrow, C. Sternemann, M. Wilke, R. Gordon, K. Mende, H. Yavaş, L. Simonelli, N. Hiraoka, C. Sahle, S. Huotari, G. Andreozzi, A. Woodland, M. Tolan and J. S. Tse, Contrib. Mineral. Petrol., 2014, 167, 1012 CrossRef.
- N. Hiraoka, M. Takahashi, W. B. Wu, C. H. Lai, K. D. Tsuei and D. L. Huang, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 241112(R) CrossRef.
- U. Bergmann, P. Glatzel and S. P. Cramer, Microchem. J., 2002, 71, 221–230 CrossRef CAS.
- W. Schülke, Electron Dynamics by Inelastic X-Ray Scattering, Oxford Univ. Press, 2007 Search PubMed.
- K. Hämäläinen and S. Manninen, J. Phys.: Condens. Matter, 2001, 13, 7539 CrossRef.
- M. Krisch and F. Sette, Surf. Rev. Lett., 2002, 09, 969–976 CrossRef CAS.
- C. Sternemann, H. Sternemann, S. Huotari, F. Lehmkühler, M. Tolan and J. S. Tse, J. Anal. At. Spectrom., 2008, 23, 807–813 RSC.
- R. A. Gordon, G. T. Seidler, T. T. Fister, M. W. Haverkort, G. A. Sawatzky, A. Tanaka and T. K. Sham, EPL, 2008, 81, 26004 CrossRef.
- M. W. Haverkort, A. Tanaka, L. H. Tjeng and G. A. Sawatzky, Phys. Rev. Lett., 2007, 99, 257401 CrossRef CAS PubMed.
- T. T. Fister, G. T. Seidler, C. Hamner, J. O. Cross, J. A. Soininen and J. J. Rehr, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 214117 CrossRef.
- C. Krywka, C. Sternemann, M. Paulus, N. Javid, R. Winter, A. Al-Sawalmih, S. Yi, D. Raabe and M. Tolan, J. Synchrotron Radiat., 2007, 14, 391 CrossRef.
- H. Sternemann, C. Sternemann, G. T. Seidler, T. T. Fister, A. Sakko and M. Tolan, J. Synchrotron Radiat., 2008, 15, 162–169 CrossRef CAS PubMed.
- C. J. Sahle, A. Mirone, J. Niskanen, J. Inkinen, M. Krisch and S. Huotari, J. Synchrotron Radiat., 2015, 22, 400–409 CrossRef CAS PubMed.
- C. Colliex, T. Manoubi and C. Ortiz, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 11402–11411 CrossRef CAS.
- J. H. Paterson and O. L. Krivanek, Ultramicroscopy, 1990, 32, 319–325 CrossRef CAS.
- R. A. Gordon, M. W. Haverkort, S. Sen Gupta and G. A. Sawatzky, J. Phys.: Conf. Ser., 2009, 190, 012047 CrossRef.
- L. A. J. Garvie and P. R. Buseck, Nature, 1998, 396, 667–670 CrossRef CAS.
- T. Manoubi, M. Tencé and C. Colliex, Ultramicroscopy, 1989, 28, 49–55 CrossRef CAS.
- H. K. Mao, J. Xu and P. M. Bell, J. Geophys. Res., 1986, 91, 4673–4676 CrossRef CAS.
- V. Cerantola, C. McCammon, I. Kupenko, I. Kantor, C. Marini, M. Wilke, L. Ismailova, N. Solopova, A. Chumakov, S. Pascarelli and L. Dubrovinsky, Am. Mineral., 2015, 100, 2670 CrossRef.
Footnotes |
| † Present address: Max-Planck-Institute for Chemical Energy Conversion, D-45470 Mülheim. |
| ‡ Present address: European Synchrotron Radiation Facility, F-38043 Grenoble Cedex, France. |
| § Present address: Helmholtz-Zentrum Geesthacht, Institut für Werkstoffforschung, D-21502 Geesthacht, Germany. |
| ¶ Present address: Moyie Institute, Port Coquitlam, BC, Canada. |
| This journal is © The Royal Society of Chemistry 2016 |