
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vinculin head–tail interaction defines multiple early mechanisms for cell substrate rigidity sensing†
Zengzhen
Liu‡
,
Philippe
Bun§
,
Nicolas
Audugé
,
Maïté
Coppey-Moisan
and
Nicolas
Borghi
*
Institut Jacques Monod, Unité Mixe de Recherche 7592, Centre National de la Recherche Scientifique, Université Paris-Diderot, 304B Bât. Buffon, 15 rue Hélène Brion, Paris 75013, France. E-mail: nicolas.borghi@ijm.fr; Tel: +33 1 57 27 80 41
First published on 4th May 2016
Abstract
Rigidity sensing is a critical determinant of cell fate and behavior but its molecular mechanisms are poorly understood. Focal adhesions (FAs) are complexes that anchor cells to the matrix. Among their components, vinculin undergoes an auto-inhibitory head–tail interaction that regulates the recruitment of, and interactions with its partners in a force-dependent manner. It is unknown, however, whether this mechanism is involved in substrate rigidity sensing. Here, we use a range of quantitative fluorescence microscopies on live human Mesenchymal Stem Cells to address this question. We identify two distinct rigidity-sensing molecular modules in FAs, one of which involves vinculin and talin, is regulated by vinculin head–tail interaction, and targets cell morphology. Vinculin and talin are recruited independently in a rigidity-dependent manner to FAs where they directly interact in a rigidity-independent stoichiometry at a site proximal to talin head. Vinculin head–tail interaction is required on soft substrates to destabilize vinculin and talin in FAs, and to allow hMSCs branching. Another module involves paxillin and FAK, which soft substrates also destabilize, but independently of vinculin head–tail interaction. This multi-modularity may be key to allow a versatile response to complex biomechanical cues.
Insight, innovation, integrationRigidity sensing is a critical determinant of cell fate and behavior but its molecular mechanisms are poorly understood. Focal adhesions are complexes that anchor cells to the matrix. Among their components, vinculin undergoes an auto-inhibitory head–tail interaction regulating interactions with its partners in a force-dependent manner. Whether this mechanism is involved in rigidity sensing is unknown. Using quantitative fluorescence microscopies on live human Mesenchymal Stem Cells, we identify two distinct rigidity-sensing molecular modules in FAs, one of which involves vinculin and talin, is regulated by vinculin head–tail interaction, and targets cell morphology. Another module involves paxillin and FAK, but independently of vinculin head–tail interaction. This multi-modularity may be key to allow a versatile response to complex biomechanical cues. |
Introduction
Extracellular matrix (ECM) rigidity is a critical determinant of cell fate and behavior. In culture, stem cells differentiate into lineages of tissues whose rigidity matches that of the culture substrates.1,2 Fibroblast cell lines assemble the ECM, spread and migrate as a function of substrate rigidity.3,4 Thus, rigidity sensing likely plays a major role in multicellular development, homeostasis and regeneration. Moreover, abnormal tissue rigidity and cell response to rigidity are signatures of diseases such as cancer.5 The molecular mechanisms of rigidity-sensing are, however, poorly understood.Focal adhesions (FAs) are macromolecular complexes that transmit cell traction forces to the cell substrate. FAs were early found to respond to substrate rigidity,4,6 and a number of their components respond to mechanical forces in situ or in vitro.7–9 Notably, vinculin recruitment to FAs increases with cell contractility, external forces7,10–12 and substrate rigidity.4,13 Moreover, a recent study has shown the requirement of vinculin for proper rigidity-dependent differentiation of human Mesenchymal Stem Cells (hMSCs).14
Interestingly, vinculin comprises a head and a tail domain that bind a number of FA and actin-regulating proteins and membrane lipids,15 but these interactions are competed by a head–tail homo-interaction.16,17 These functional, as well as structural studies18–20 support a model by which the release of vinculin head–tail interaction in FAs involves at least two binding partners, and possibly tension-dependent conformational changes. Indeed, vinculin undergoes conformation opening in FAs,21 and is also under mechanical tension.22 Talin, in turn, exhibits a number of vinculin-head binding sites (VBS) that recruit vinculin in vitro upon actomyosin-generated stretch,23 and is stretched in cells.24 Importantly, a recent study demonstrated in cells that talin tension depends on rigidity, vinculin association and actomyosin activity.25
In summary, the general picture is that in which vinculin act as a cell contractility-dependent master regulator of FA protein recruitment through modulation of its head–tail interaction, and is involved in a positive feedback between talin stretching and actomyosin contractility as a function of substrate rigidity. It is unknown, however, whether vinculin head–tail interaction is involved in rigidity sensing, whether talin interacts with variable numbers of vinculin proteins as a function of rigidity, and the overall effect of these events on FA growth.
To address these issues, we investigate the molecular mechanisms by which substrate rigidity affects hMSCs morphology, FA proteins recruitment, mobility and interactions, and their modulation by a vinculin interaction mutant (T12) that bears four point-mutations in the head-binding region of the tail.17 From higher-than-bone (glass, >50 GPa) to brain tissue-like (0.2 kPa) rigidity, we show that vinculin and talin are recruited independently in a rigidity-dependent manner to FAs, where they directly interact in a rigidity-independent stoichiometry in a site proximal to talin head. Impairment of vinculin head–tail interaction is not sufficient alone to allow vinculin–talin interaction but it stabilizes vinculin and talin in FAs so that their amount is no longer rigidity-dependent. In addition, the T12 mutant prevents hMSCs branching of soft substrates. In contrast, paxillin and FAK amount and turnover in FAs remain rigidity-sensitive independently of vinculin head–tail interaction.
Material & methods
Cells
Three different batches of human bone marrow stromal cells (hMSCs) were purchased from StemCell (MSC-001F) in 1.8 mL vials. We did not detect major batch variability in the results. Cells were cultured in MesenCult® MSC Basal Medium (Human; Catalog #05401) completed with Mesenchymal Stem Cell Stimulatory Supplements (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, Human; Catalog #05402), Antibiotics (1:500, LONZA, MycoZap™ Plus-PR, Catalog # VZA-2021) and were used at passages 3–5 for all experiments.
:10, Human; Catalog #05402), Antibiotics (1:500, LONZA, MycoZap™ Plus-PR, Catalog # VZA-2021) and were used at passages 3–5 for all experiments.
Constructs
Plasmids pEGFP-Talin1, pEGFP-paxillin, pmCherry-FAK-HA, pEGFP and pmCherry were obtained from Addgene. pEGFP-T12, pEGFP-vinculin, pmCherry-vinculin, pmCherry-paxillin and pEGFP FAK were generous gifts from Susan W. Craig, Tova Volberg, Clare M. Waterman, Michael W. Davidson and Jun-Lin Guan. The pEGFP-mCherry tamdem plasmid was described elsewhere.26 The mCherry form of T12 were generated by removing the GFP cassette by BglII and HindIII digestion followed by ligation of the product of a PCR on the pmCherry plasmid. Cells were transfected by electroporation using the Amaxa Nucleofector II and the hMSC kit (LONZA, Catalog # VPE-1001) according to manufacturer's directions, and used 24 h after transfection.Elastic substrates
Collagen-coupled polyacrylamide gels were prepared on round cover slips (32 mm) according to a previously established protocol.27 Briefly, 40% acrylamide and 2% bis-acrylamide stock solutions were mixed in various proportions with TEMED, APS and water, and allowed to polymerize for 15 min between silanized and glutaraldehydized glass coverslips. The gel was then covered with sulfosuccinimidyl-6-[4′-azido-2′-nitrophenylamino]hexanoate (Sulfo-SANPAH, Pierce) in DMSO. After being exposed to UV light for 6 min twice, the polyacrylamide sheet was washed with Hepes buffer twice and incubated with a solution of type I collagen (0.2 mg mL−1, Sigma) overnight at 4 °C. Cells were seeded at a density of 104 cm−2. In these conditions, cells are able to adhere to the pre-coated collagen as well as the matrix proteins, including fibronectin, they produce and harvest from the medium, as previously shown.28,29 Because of this, and that we use saturating collagen conditions, cells are able to respond to substrate rigidity through integrin pathways previously identified,29 and no variation in initial collagen density is expected to significantly contribute to the observations.To avoid intercellular mechanical interactions through the deformable substrate, we only considered in all experiments individual cells without neighbors in the observation field. Thus, as many observation fields as cells were captured in all experiments (see figure legends). Reported rigidities are those indicated in the original protocol.
Cell morphometrics
Cell morphology during the first 24 h after seeding were performed in phase-contrast (10×) on an environment-controlled (37 °C, 5% CO2) Leica microscope equipped with a CoolSNAP HQ (Photometrics) CCD camera. Cell morphology and morphodynamics quantitative analyses were performed on cells expressing fluorescent proteins allowed to spread on gels for 24 h before capturing time-lapses (1 frame per min during 30–60 min) on an environment-controlled Zeiss LSM710 confocal-microscope (40× water objective).Cell shape was extracted with ImageJ using standard background smoothing and contour segmentation procedures within custom-written macros. Cell morphology was quantified by its circularity (100 × 4π × area/perimeter2): the more circular, the less branched. Cell morphodynamics were quantified by the area A of the non-overlapping cell surface S (protrusions and retractions) between two consecutive time points (A(S(t + 1)XORS(t))) normalized to the average cell surface area A(S), similar to previously done.30
FAs protein recruitment patches morphometrics
Cells expressing fluorescent proteins were observed 24 h after seeding on an upright wide-field Zeiss 700 microscope equipped with an AxioCam MRm CCD camera and a 63× water-immersion objective.FA protein recruitment patches were segmented in ImageJ with an intensity-based mask. Patches were sorted in two bins based on their surface area (below or above 0.2 μm2).
FA protein turnover
FRAP (Fluorescence Recovery After Photobleaching) experiments were carried out on a Leica DMI6000 microscope equipped with a Yokogawa CSU22 spinning-disk head, a Leica Plan APO 63×/1.2 NA objective and an EMCCD camera (Photometrics QuantEM). A FRAP Head (Roper scientific) equipped with a 473 nm diode laser (100 mW) was used to perform point bleaching (100% laser power). Pre-bleach and post-bleach image acquisitions were performed with a 491 nm diode laser (50 mW) at 5% laser power.Data were fitted with a single-exponential recovery model F = F0 + Fτ(1 − exp(−t/τ)), where F0 is the fluorescence that recovers before the first time point, Fτ the fluorescence of the mobile fraction characterized by the turnover time τ.
FA protein mobilities and interactions
FCS (Fluorescence Fluctuation Correlation Spectroscopy), FCCS (Fluorescence Fluctuation Cross-Correlation Spectroscopy) and FRET/FLIM (Förster Resonance Energy Transfer by Fluorescence Lifetime Imaging Microscopy) experiments were performed on the confocal time-resolved microscope MicroTime 200 (Picoquant). Briefly, a 470 nm pulsed diode laser (Picoquant) and a 561 continuous diode-pumped solid-state laser (Coherent) are focused through a 60×, NA 1.3 water immersion objective on an inverted Olympus microscope. The beam waist, ω0, is determined using the point spread function of the confocal system (260 nm in the green channel and 320 nm in the red channel). For subsequent data analysis, ω0 is fixed to 260 nm (the smallest volume) and the structural parameter, z0/ω0 to 4 (corresponding to a volume of 0.4 fL).The fluorescence emission passes through a double dichroic mirror (DM 470/571 nm) is focused on a pinhole. A second dichroic mirror (DM 550 nm) splits the fluorescence toward emission filters 525 ± 25 nm and 593 ± 20 nm in front of two avalanche photodiodes (SPADs, Perkin Elmer). Data acquisition and analysis use the TimeHarp 300 PC board and Symphotime software (PicoQuant). Time-Tagged Time-Resolved (TTTR) Single photon counts are acquired during 60 seconds to perform one measurement.For FCS, auto-correlation functions G(τ) were analysed with a two species 3D diffusion model to recover slow and fast apparent diffusion coefficients:

FCCS analysis was performed after fluorescence lifetime filtering between channels. This approach efficiently corrects for spectral bleed-through and enable concentration- and laser power-independent cross-correlation amplitude measurement.31 Therefore, it allows unambiguous discrimination of true positive results from false positive cross-correlation systematically observed on non-filtered data. Presence of a complex involving the proteins of interest was considered positive when the cross-correlation curve could be well fitted with a 1-species 3D diffusion model yielding a physically sound diffusion coefficient. The positive control was a tandem dimer of GFP and mCherry proteins and the negative control co-expressed GFP and mCherry. This approach typically allows to measure interacting fractions as low as 10% of the total amount of either protein. As FCS, we focused on dimmer regions of interest.
FLIM-FRET analysis was performed by fitting the fluorescence decay of protein-tagged GFP with a single-exponential decay model N = N0exp(−t/τD) to yield an average fluorescence lifetime τD (or τDA) of protein-tagged GFP in the absence (or the presence) of the mCherry-tagged second protein of interest. We controlled that fluorescence lifetime differences between conditions were not due to variable expression ratios of GFP- and mCherry-tagged proteins.
Results
Vinculin head–tail interaction is required for rigidity-dependent hMSCs branching
hMSCs spread differently on substrates of different rigidities. In less than 24 h, hMSCs adopted a branched morphology with small cell bodies and long and thin branches on soft substrates, large cell bodies and short branches on rigid substrates, and an intermediate morphology on intermediate rigidities (Fig. 1A). | ||
| Fig. 1 Vinculin conformation change is required for rigidity-dependence of cell morphology. (A) hMSC on 0.2 kPa, 11 kPa and 34 kPa substrates during the first 24 h after plating. Scale bar = 10 μm. (B) Cell surface after intensity-based threshold for WT and T12 vinculin-GFP expressing cells on glass and 0.2 kPa. Scale bar = 10 μm. (C) hMSC circularity for cells expressing GFP-tagged talin, FAK, paxillin, WT vinculin, T12 vinculin mutant or no fluorescent protein (ctrl) on rigid (34 kPa) and soft (0.2 kPa) substrates. Mean ± SEM. ANOVA and uncorrected Fisher's LSD test; n = 3–5 cells per condition. Shown p-values are for any pair of same GFP-tagged protein conditions between rigid and soft substrates. No significant difference of circularity between rigidities for T12 only. | ||
To quantify cell branching as a function of substrate rigidity, we measured cell circularity on rigid and soft substrates (Fig. 1B and C). Cell circularity was lower on soft than on rigid substrates, regardless of whether or not cells exogenously expressed GFP-tagged WT FA proteins (Fig. 1C). In contrast, cells expressing the vinculin T12 mutant, impaired for head–tail interaction, exhibited no circularity decrease on soft substrates (Fig. 1C). Due to transient expression, GFP-tagged proteins levels span over ranges larger within than between rigidity conditions, and may not account for differences in results between conditions. Therefore, rigidity-dependent hMSCs branching requires vinculin head–tail interaction.
To test whether substrate rigidity also affected hMSCs morphodynamics, we quantified cell protrusions and retractions (Fig. S1, ESI†). In cells expressing GFP-tagged WT vinculin, cell protrusions and retractions surface area did not exhibit any significant trend as a function of substrate rigidity. Therefore, we hypothesized they do not depend on vinculin head–tail interaction. Indeed, cells expressing T12 vinculin exhibited rigidity-independent morphodynamics indistinguishable from that of WT vinculin cells (Fig. S1, ESI†).
Thus, substrate rigidity specifically affects hMSCs branching in a manner dependent on vinculin head–tail interaction.
FA protein recruitment is rigidity-dependent and differentially relies on vinculin head–tail interaction
Since hMSCs branching depends on substrate rigidity, we sought to assess the effect of substrate rigidity on FA protein recruitment. In these experiments, each FP-tagged protein reports its own recruitment, and not necessarily that of the other FA proteins. Therefore, we use hereafter the term ‘patch’, rather than FA, to define the surface to which the corresponding protein is recruited. Since cells exhibited size variability, we measured the size distribution of recruitment patches, rather than their total area or number, for a range of FA proteins and a range of substrate rigidities (Fig. 2 and Fig. S2, ESI†). The proportion of recruitment patches of vinculin, talin, paxillin and FAK larger than 0.2 μm2, the plateau size of nascent FAs,32 fell from about 60% to 20–30% as the substrate rigidity decreased (Fig. 2B–E). Therefore, protein amount in FAs is rigidity-dependent. In contrast, expression of the T12 mutant prevented the proportion of large T12 patches to fall below 50% on soft substrates, so that the size distribution of T12 patches remained high and independent of substrate rigidity (Fig. 2B). Therefore, the rigidity-dependence of vinculin recruitment to the cell–ECM interface requires vinculin head–tail interaction.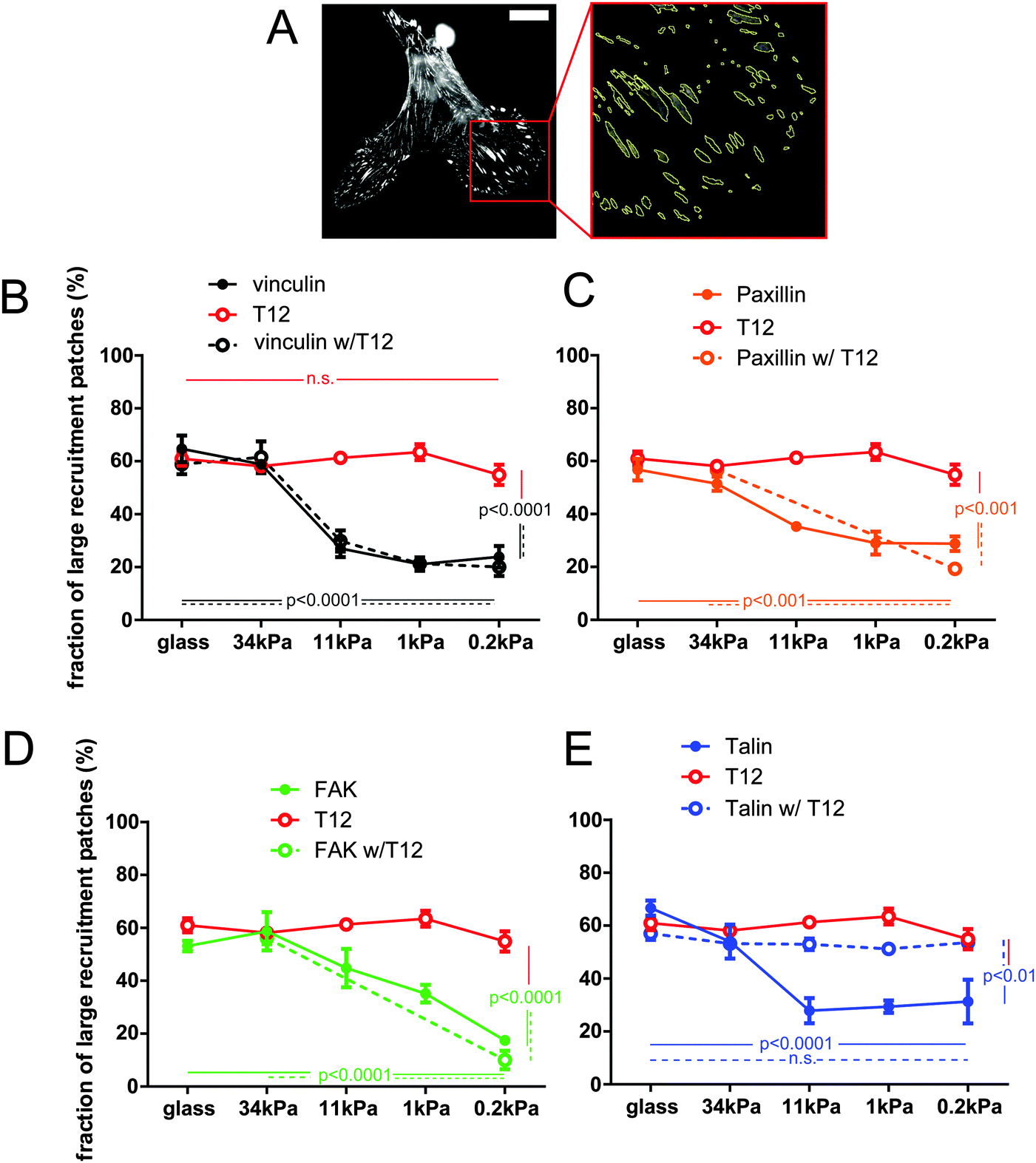 | ||
| Fig. 2 FA protein steady-state recruitment to the cell–ECM interface is rigidity-dependent. Vinculin conformation change is required for the rigidity-dependence of its recruitment and that of Talin, but not that of paxillin and FAK. (A) Left: hMSC expressing vinculin-GFP. Scale bar = 5 μm. Right: Zoom in of the cell lamellum with automatically contoured recruitment patches. (B–E) Fraction (per cell) of large (>0.2 μm2) recruitment patches of FA proteins with and without coexpression of the T12 vinculin mutant for a range of substrate rigidities. (B) vinculin and T12. (C) Paxillin. (D) FAK. (E) Talin. Mean ± SEM. ANOVA and Tukey's HSD test; n = 3–5 cells per condition (more than 40 patches per cell). Shown p-values are between extreme rigidities and WT vs. mutant conditions at lowest rigidity. | ||
To assess the role of vinculin head–tail interaction in the recruitment of other FA proteins as a function of substrate rigidity, we measured the sizes of FA protein recruitment patches in cells co-expressing the T12 mutant. Patches of T12 and of the other FA protein generally colocalized but their sizes were not necessarily the same (Fig. S2B, ESI†). Indeed, the size distribution of mCherry-tagged WT vinculin, paxillin and FAK patches followed the same rigidity-dependence whether or not the cells co-expressed the GFP-T12 mutant (Fig. 2B–D). Thus, T12 mutants do not affect the recruitment of WT vinculin, paxillin or FAK as a function of substrate rigidity. Of note, that cells recruit both WT and T12 vinculin in FAs on rigid substrates shows that T12 is expressed at levels that do not compete with WT vinculin. In contrast, expression of the mCherry-T12 mutant significantly affected GFP-talin recruitment as a function of rigidity (Fig. S2B, ESI†). In those cells, the size distribution of talin recruitment patches did not significantly decrease as substrate rigidity decreased (Fig. 2E). In other words, soft substrates tend to deplete FAs of WT vinculin, paxillin, and FAK but not of T12 and talin in the presence of T12. Therefore, vinculin head–tail interaction appears to control the rigidity-dependence of talin recruitment to FAs, but not that of paxillin or FAK.
FA proteins mobilities are rigidity-dependent and that of vinculin and talin depend on vinculin head–tail interaction
Since substrate rigidity affects FA protein recruitment, we sought to assess its effect on FA proteins mobility. To assess FAs proteins mobility within the sec timescale, we performed FRAP on recruitment patches. We focused on recruitment patches at the periphery of the cell–ECM interface. Fluorescence recovery evidenced a mobile fraction faster than our time resolution (F0) and a slower fraction (Fτ) characterized by a turnover time τ (see Methods). GFP-vinculin, talin and paxillin all exhibited turnover times in the same 50 s range, whereas FAK exhibited shorter turnover times (15 s). Remarkably, all turnover times exhibited a significant decrease with substrate rigidity decrease (Fig. 3). In addition, slow mobile fractions Fτ of vinculin, paxillin and FAK exhibited a significant decrease, compensated by a significant increase in fast mobile fraction F0 of vinculin and FAK. Therefore, all FA proteins tested exhibited an overall higher mobility on soft substrates. | ||
| Fig. 3 FA proteins turnover is rigidity-dependent, and that of vinculin and talin depends on vinculin head–tail interaction. (A) Typical fluorescence recovery profile on FAK-GFP. The red circle indicates the frapped region. Bar = 10 μm. The corresponding normalized fluorescence intensity (red curve) is fitted with a single exponential model (black curve) that defines a turnover time τ, and slow (Fτ) and fast (F0) mobile fractions. (B–E) FRAP turnover time and mobile fractions of (B) vinculin and T12, (C) talin and talin coexpressed with T12, (D) paxillin and (E) FAK as a function of substrate rigidity. Mean ± SEM. One-way (D and E) or two-way (B and C) ANOVA and Tukey's HSD test, n = 10–25 per condition. Shown p-values are between extreme rigidities, and between WT and mutant conditions at lowest rigidity when ‘vinculin mutant-rigidity’ statistical interaction was significant, or between all rigidities-averaged values of WT and mutant conditions when ‘vinculin mutant-rigidity’ statistical interaction was not significant. | ||
While FRAP is well suited to study protein mobility in the sec and min timescale, it misses faster phenomena. To access the ms timescale, we thus performed FCS in the dimmer regions of recruitment patches (see Methods). GFP-vinculin, talin and paxillin all exhibited mobilities best fitted with two apparent diffusion coefficients each: a high coefficient in the 20 μm2 s−1 range, close to that of a freely diffusing protein, and a low coefficient in the 1 μm2 s−1 range, which magnitude reveals a protein mobility hindered by transient interactions with immobile ligands. Slow apparent diffusion coefficients of all tested proteins increased when substrate rigidity decreased (Fig. S3, ESI†). Therefore, ms-scale and s-scale mobilities of FA proteins are similarly rigidity-dependent.
To assess the role of vinculin head–tail interaction on FA protein mobilities as a function of substrate rigidity, we first monitored by FRAP the turnover of the mCherry-T12 mutant as well as that of GFP-talin in cells co-expressing the mCherry-T12 mutant. The mCherry-T12 mutant turnover was about an order of magnitude slower than that of GFP-WT vinculin and did not speed up with decreased substrate rigidity. In addition, the slow mobile fraction of T12 was much larger than that of WT vinculin, at the expense of the fast mobile fraction, and both mobile fractions were no longer rigidity-dependent (Fig. 3B). Similarly, apparent diffusion coefficients of T12 mutant were lower than that of WT vinculin and insensitive to substrate rigidity (Fig. S3B, ESI†). Therefore, vinculin mobility in general, and its rigidity-dependence in particular depend on vinculin head–tail interaction.
Moreover, although GFP-talin turnover time in mCherry-T12 mutant cells was rigidity-dependent, it was overall larger than in WT cells. Interestingly, the T12 mutant decreased the slow mobile fraction of talin, especially on soft substrates, but to the benefit of immobile fraction rather than the fast mobile fraction (Fig. 3C). At the msec timescale, T12 expression abolished the rigidity-dependence of the slow apparent diffusion coefficient of talin (Fig. S3C, ESI†). Therefore, vinculin head–tail interaction regulates talin mobility dependence on substrate rigidity.
FA proteins are recruited to FA individually, regardless of vinculin head–tail interaction
Since talin amount in FAs depends on vinculin head–tail interaction, and their mobilities in FAs were similar, we wondered whether vinculin and talin were recruited as a complex. To test this, we performed lifetime-filtered FCCS in the cytoplasm and in dimmer regions of recruitment patches of cells expressing GFP-talin and mCherry-WT vinculin or GFP-talin and the mCherry-T12 mutant. Note that as FCS, FCCS provides information on mobile proteins that are therefore mostly not yet or no longer involved in FAs. Based on the ability to fit the cross-correlation curves with a 3D diffusion model yielding a physically sound diffusion coefficient, we were unable to detect fluorescence cross-correlation between talin and vinculin, in the cytoplasm and in dimmer regions of recruitment patches, whether or not vinculin was WT or T12 (Table 1 and Fig. S3, ESI†). Therefore, talin does not appear to form a complex with vinculin in the cytoplasm prior its recruitment or after its release from FAs, even when vinculin head–tail interaction is impaired. In addition we tested whether complexes involving multiple vinculins could form in the cytoplasm and found no cross-correlation between WT or T12 vinculin proteins (Table 1). Therefore, no such complexes appear to significantly form in the cytoplasm, whether vinculin head–tail interaction is impaired or not. Similarly, we tested whether paxillin and FAK formed complexes with vinculin or talin in the cytoplasm and found no evidence for such complexes, even with the T12 vinculin mutant (Table S1, ESI†).| GFP-mCherry tandem | GFP-Talin | GFP-Vinculin | GFP-T12 | |
|---|---|---|---|---|
| GFP-mCherry tandem | Yes (Fig. S3E, ESI) | |||
| mCherry-Vinculin | No (Fig. S3G, ESI) | No | ||
| mCherry-T12 | No | No |
Then, we sought to assess whether vinculin and talin directly interacted in FAs. To do so, we performed FRET-FLIM on hMSCs expressing either mCherry-WT or T12 vinculin, and GFP-talin where mCherry and GFP are bound to talin-binding vinculin head and talin head, respectively. As expected from FCCS results, we observed in the cytoplasm similar GFP-talin fluorescence lifetimes whether or not mCherry-tagged vinculin WT or T12 mutant was co-expressed (Fig. 4A). In contrast, GFP-talin fluorescence lifetime in recruitment patches was significantly lower when mCherry-tagged vinculin WT or T12 mutant were expressed (Fig. 4A). Fluorescence lifetime differences were not due to differences in relative expression of donor and acceptor (Fig. S4, ESI†). Therefore, talin and vinculin directly interact in recruitment patches but not in the cytoplasm, and the interaction occurs in proximity of talin head. In addition, GFP-talin fluorescence lifetime appeared independent of substrate rigidity or the expression of the T12 mutant (Fig. 4A). Therefore, the stoichiometry of talin and vinculin bound in proximity of talin head is independent of substrate rigidity or the possibility of head–tail interaction in vinculin.
 | ||
| Fig. 4 Vinculin and talin directly interact in FAs but not in the cytoplasm. (A) GFP fluorescence lifetime of talin-GFP alone or coexpressed with WT vinculin-mCherry or T12-mCherry, in the cytoplasm (left) and in FAs (right), on glass (G), 34 and 1 kPa. Mean ± SEM. ANOVA and Tukey's HSD test, n = 12 cells per condition on average. Shown p-values are for any pair of control, talin w/WT vinculin and T12 vinculin conditions at same rigidity, and any pair of rigidity conditions at same vinculin form expressed. (B) Working model of rigidity sensing by FA proteins in hMSCs. See Discussion for details. | ||
Discussion
In their pioneering study, Engler et al. had observed that hMSCs morphologies and expression of lineage specification markers were under substrate rigidity control.2 Here, we confirm that hMSCs branching, an early morphological feature of differentiation on soft substrate, depends on substrates rigidity as early as 24 h after plating (Fig. 1). Since FAs are presumably the most upstream complexes of rigidity sensing pathways and FA protein vinculin is required for proper rigidity-dependent hMSC differentiation,14 we focused our study on the impact of substrate rigidity on FA proteins and the role of vinculin in hMSC rigidity sensing.In a recent study, Yamashita et al. have investigated in MEF cells the impact of substrate rigidity on FA proteins.13 In those cells, recruitment of vinculin was rigidity-dependent. Specifically, the total area, number, and intensity of vinculin recruitment patches insoluble in a cytoskeleton stabilization buffer were all higher on rigid substrates than on soft, and, somewhat surprisingly, higher than that of total (soluble + insoluble) vinculin recruitment patches on rigid substrates. Here, we observed in hMSCs that total vinculin recruitment patches were larger on rigid substrates (Fig. 2). Thus, while our conditions and results are not identical, they both are consistent with an increased recruitment of vinculin in FAs by rigid substrates. In addition, we show that recruitment patches of paxillin, talin and FAK are also rigidity-dependent, as they are larger on rigid substrates, similarly to vinculin (Fig. 2). Therefore, substrate rigidity appears to modulate FA protein recruitment in a fashion that retain their overall stoichiometry.
Previous studies have evidenced a key role for vinculin head–tail interaction in regulating cell adhesion and migration. Expression of the T12 mutant, impaired for head–tail interaction, increased cell spread area in vinculin-null MEF cells,33 and impaired the directional persistence of membrane protrusions in mouse melanoma cells.34 Here, we show that in hMSCs, T12 expression does not affect cell morphodynamics, which is moreover rigidity-independent (Fig. S1, ESI†). In contrast, T12 expression does impair increased cell branching on soft substrates (Fig. 1). Therefore, vinculin head–tail interaction is specifically required for the rigidity-sensitivity of cell branching in hMSCs. Importantly, rigidity sensing is significantly abolished by T12 expression even though cells also express endogenous WT vinculin.
The role of vinculin head–tail interaction in cell morphology is thought to result from its function in the recruitment of vinculin and other FA proteins to the cell–substrate interface. Indeed, expression of the T12 mutant in vinculin-null and NIH 3T3 cells induced more numerous vinculin recruitment patches that also recruited paxillin and talin.17,35 Moreover, T12 recruitment, unlike that of WT vinculin, persisted even after myosin activity disruption in vinculin-null cells,34 while vinculin closed conformation was observed to precede vinculin release from disassembling FAs.21 All together, these results indicate that vinculin head–tail interaction is required for the regulation of contractility-dependent vinculin recruitment to, and release from the cell–substrate interface. Here, we find that the T12 mutant forms larger recruitment patches than that of WT vinculin in hMSCs on soft substrates but not on rigid substrates, so that T12 recruitment patches size does not depend on substrate rigidity (Fig. 2). This result suggests that soft substrates favor vinculin conformation closing and head–tail interaction. Therefore, substrate rigidity appears to recapitulate the effect of intracellular contractility on vinculin recruitment, a result consistent with the correlation between substrate rigidity and cell traction forces in fibroblasts and epithelial cells,36,37 and that vinculin head–tail interaction limits the range of cell traction forces.33 Nevertheless, it is noteworthy that there is no one-to-one relationship between vinculin conformation and its contractility-dependent molecular tension22 or between vinculin molecular tension and cell traction forces.38
Additionally, we show that recruitment patch sizes of WT vinculin, paxillin and FAK but not talin are still rigidity-dependent in hMSCs also expressing the T12 mutant (Fig. 2). In contrast, a previous study has shown that paxillin, FAK, talin and other FA proteins still colocalize with the T12 mutant at the cell–substrate interface in cells impaired for actomyosin contractility.34 All together, these results reveal that paxillin and FAK recruitment appears to require both rigidity- and contractility-dependent vinculin recruitment and, independently of vinculin, a rigid substrate. This result also indicates that cell morphology rigidity response depends on vinculin head–tail interaction (Fig. 1) but not on paxillin and FAK recruitment as a function of rigidity (Fig. 4B). As a consequence, rigidity sensing exhibits an early signaling divergence toward cell morphology through talin and vinculin head–tail interaction on one hand, and toward other targets through FAK and paxillin on the other hand.
Protein amounts in FAs are generally likely to reflect recruitment kinetics: the more recruited the protein, the less mobile. Indeed, vinculin, talin, paxillin and FAK, which all exhibited smaller recruitment patches on softer substrates (Fig. 2), concomitantly underwent increased turnover, and most exhibited increased fast mobile fractions and slow apparent diffusion coefficients, and decreased slow mobile fractions (Fig. 3 and Fig. S3, ESI†). Noteworthy, Dumbauld et al. have observed that vinculin turnover in FAs correlates with the magnitude of cell traction forces,33 further supporting the similar roles of substrate rigidity and cell contractility on vinculin recruitment. Consistently, Yamashita et al. have recently found that in FAs of MEF cells, vinculin exhibits on soft substrates a smaller immobile fraction than on rigid substrates. However, they observed that paxillin rather exhibits on rigid substrates more mobile features than on soft substrates.13 All together, these results suggest a fairly robust conservation of the rigidity-dependence of vinculin recruitment through cell lineages, while other FA proteins such as paxillin may exhibit cell-type specific behaviors. These differences further support a model involving multiple rigidity-sensing modules within FAs.
Previous studies have shown that the T12 mutant expressed in vinculin-null or NIH 3T3 cells on rigid substrates exhibits in FAs a much slower turnover and smaller mobile fraction than WT vinculin, and also slowed down the turnover of talin, but not that of paxillin, in vinculin-null cells.17,35 Here, we consistently find that the T12 mutant has a much slower turnover and decreased fast mobile fraction and apparent diffusion coefficients compared to WT vinculin (Fig. 3 and Fig. S3, ESI†). Together with increased amounts in FAs on soft substrates compared to WT vinculin (Fig. 2), this further confirms the correlation between lower mobility and higher protein amount in FAs. In addition, we find that decreased substrate rigidity does not increase mobility features of the T12 mutant. Consistently, Dumbauld et al. have shown that T12 mutant turnover does not correlate with the magnitude of cell traction forces.33 Finally, we find that talin mobility features are generally lower or less rigidity-dependent in cells expressing T12 (Fig. 3 and Fig. S3, ESI†), consistent with larger amounts in FAs on soft substrates when the T12 mutant is expressed (Fig. 2). All together, these results supports that substrate rigidity, similarly to cell contractility, controls vinculin and talin turnover in FAs through a modulation of vinculin head–tail interaction.
Indeed, vinculin head domain interacts with talin in vitro, but vinculin head–tail binding competes with this interaction.39 Consistently, full length vinculin does not form a complex with talin in coimmunoprecipitates from NIH 3T3 cells.35 The T12 mutant from HEK283 cell lysates, however, is able to form a complex with the talin rod (residues 397–2541).17 Therefore, the release of vinculin head–tail interaction may allow full-length vinculin–talin interaction. Here, our FLIM-FRET experiments bring direct evidence that full length vinculin and talin do interact directly in proximity of talin head in FAs of live hMSCs (Table 1 and Fig. 4A). As recently proposed,40,41 we show by FLIM-FRET and FCCS that talin and vinculin do not interact in the cytoplasm (Table 1 and Fig. 4A). Additionally, we also show that the T12 mutant does not interact with talin in the cytoplasm (Table 1 and Fig. 4A). Thus, the mere release of vinculin head–tail interaction is not sufficient to allow interaction between the full-length proteins in cells, possibly because vinculin conformation change also requires tension. This is also consistent with a talin pre-stretch requirement, in agreement with a previous study42 but in contrast with another.9 The lack of cytoplasmic complex between the T12 mutant and vinculin putative direct-binding partner paxillin (Table S1, ESI†) also supports this model and, together with the consistent lack of cytoplasmic complex between WT vinculin and paxillin (Table S1, ESI†), contradicts recently observed vinculin–paxillin cytoplasmic complexes.40 The reason for this latter discrepancy possibly lies in our filtered FCCS method that efficiently discards false positive correlation systematically observed in unfiltered FCCS experiments. Altogether, our results do not support that multi-molecular FA protein modules are pre-assembled in the cytoplasm.
A recent study has shown that full length talin under actomyosin-generated tension recruits full length vinculin in vitro.23 In addition, the more stretched the talin rod, the higher the number of vinculin heads it binds in vitro.9 Since in fibroblast FAs talin stretching occurs24 and talin tension is rigidity-dependent,25 an attractive model would be that talin increasingly stretches and thereby interacts with an increasing number of vinculin proteins as rigidity increases. Talin displays five VBS in the three alpha-helix bundles most proximal to its head (R1 to R3), the farthest of which (VBS1 in R3) binding vinculin under the weakest stretching force.42 Therefore, additional vinculin–talin interactions upon increased stretching should occur closer to talin head than R3, namely in R1 and R2. Nevertheless, our FRET results support that the stoichiometry of the interaction between vinculin and talin in proximity of its head is rigidity- and T12-independent (Fig. 4A). It is therefore unlikely that the VBS most proximal to talin head in R1 and R2 be increasingly occupied with vinculin upon increased rigidity. Whether only VBS1 or additional VBS in R1 and R2 are constitutively occupied regardless of substrate rigidity is, however, unknown. Additionally, we show that the amounts and mobilities in FAs of T12 and talin in the presence of T12 are rigidity-independent (Fig. 2, 3 and Fig. S3, ESI†). Therefore, our results are more consistent with a model where vinculin and talin recruitment and direct interaction in proximity of talin head in FAs involve a rigidity-dependent release of vinculin head–tail interaction and conformation change, and a rigidity-independent, vinculin-binding-sufficient talin constitutive stretching within our rigidity range. Symmetrically, vinculin head–tail interaction, rather than talin relaxation, may trigger vinculin and talin dissociation and concomitant release from FAs. In other words, while vinculin head and talin interact in a ratchet-like fashion upon tension,42 vinculin tail loosens the ratchet as a function of rigidity. Noteworthy, this model is not inconsistent with talin tension dependence on substrate rigidity. Indeed, talin tension substantially changes below 1 kPa but increases only modestly in comparison at higher rigidities.25 Conversely, most vinculin head–tail interaction-dependent recruitment of vinculin and talin beyond the size of nascent FAs occurs at rigidities higher than about 10 kPa (Fig. 2). Consequently, low rigidities may be sufficient for talin tension to allow maximum occupation of VBS near talin head. Finally, rigidity sensing at FAs may rely on talin stretching at very low rigidities and vinculin head–tail interaction at higher rigidities. Conserved stoichiometry between talin and vinculin bound near talin head may be useful to ensure balanced protein recruitment during FA growth beyond nascent FA size.
Conclusion
In summary, our results provide insights into the earliest molecular mechanisms of rigidity sensing and the regulation of FAs in general. We evidence that from harder than bone to as soft as brain, substrate rigidity modulates cell morphology through the rigidity-dependent recruitment of vinculin and talin. Cell branching appears to result from the destabilization of a direct interaction between open vinculin and talin in proximity of its head, due to vinculin head–tail interaction, but independently of changes in talin stretching. In contrast, substrate rigidity modulates FAK and paxillin amounts and turnover in FAs independently of vinculin and talin stabilization, unlike cell contractility. The early divergence in rigidity sensing signals through distinct molecular modules may be essential to finely regulate the multiple targets of rigidity independently from each other, through the regulation by other cues of module-specific downstream effectors.Author contributions
ZL performed the experiments. ZL, PB, NA and NB analyzed the data. PB and NA contributed new tools/reagents. NA, MC-M and NB conceived and designed the experiments. NB wrote the manuscript.Conflict of interest
The authors declare no conflict of interest.Acknowledgements
We thank the members of the laboratory for insightful discussions and of the ImagoSeine core facility of the Institut Jacques Monod (member of IBiSA and the France-BioImaging (ANR-10-INBS-04) infrastructure) for technical assistance: Xavier Baudin for help with FRAP experiments, Guan Wang for elastic substrate fabrication, and with Philippe Girard, Vincent Contremoulins and Junjun Liu for image analysis. We thank Susan W. Craig (John Hopkins University), Tova Volberg (Weizmann Institute of Science), Clare M. Waterman (National Institutes of Health), Michael W. Davidson (Florida State University) and Jun-Lin Guan (University of Michigan) for the generous gift of constructs. This material is based in part upon work supported by the Centre national de la recherche scientifique (CNRS), the Agence nationale de la recherche (ANR) grant ANR-10-BLAN-1515. ZL was supported by the China Scholarship Concil (CSC grant #2008630113) and ANR-10-INSB-04 grant.References
- A. J. Engler, M. A. Griffin, S. Sen, C. G. Bönnemann, H. L. Sweeney and D. E. Discher, Myotubes differentiate optimally on substrates with tissue-like stiffness: pathological implications for soft or stiff microenvironments, J. Cell Biol., 2004, 166, 877–887 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Matrix elasticity directs stem cell lineage specification, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- N. L. Halliday and J. J. Tomasek, Mechanical properties of the extracellular matrix influence fibronectin fibril assembly in vitro, Exp. Cell Res., 1995, 217, 109–117 CrossRef CAS PubMed.
- R. J. Pelham and Y.-L. Wang, Cell locomotion and focal adhesions are regulated by substrate flexibility, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 13661–13665 CrossRef CAS.
- D. R. Welch, T. Sakamaki, R. Pioquinto, T. O. Leonard, S. F. Goldberg, Q. Hon, R. L. Erikson, M. Rieber, M. S. Rieber, D. J. Hicks, J. V. Bonventre and A. Alessandrini, Transfection of Constitutively Active Mitogen-activated Protein/Extracellular Signal-regulated Kinase Kinase Confers Tumorigenic and Metastatic Potentials to NIH3T3 Cells, Cancer Res., 2000, 60, 1552–1556 CAS.
- D. Choquet, D. P. Felsenfeld and M. P. Sheetz, Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages, Cell, 1997, 88, 39–48 CrossRef CAS PubMed.
- D. Riveline, E. Zamir, N. Q. Balaban, U. S. Schwarz, T. Ishizaki, S. Narumiya, Z. Kam, B. Geiger and A. D. Bershadsky, Focal Contacts as Mechanosensors: Externally Applied Local Mechanical Force Induces Growth of Focal Contacts by an mDia1-dependent and Rock-independent Mechanism, Cell, 2001, 153, 1175–1185 CrossRef CAS.
- Y. Sawada, M. Tamada, B. J. Dubin-Thaler, O. Cherniavskaya, R. Sakai, S. Tanaka and M. P. Sheetz, Force Sensing by Mechanical Extension of the Src Family Kinase Substrate p130Cas, Cell, 2006, 127, 1015–1026 CrossRef CAS PubMed.
- A. del Rio, R. Perez-jimenez, R. Liu, P. Roca-Cusachs, J. M. Fernandez, M. P. Sheetz and A. Rio, Stretching single talin rod molecules activates vinculin binding, Science, 2009, 323, 638–641 CrossRef CAS PubMed.
- H. Hirata, H. Tatsumi, C. T. Lim and M. Sokabe, Force-dependent vinculin binding to talin in live cells: a crucial step in anchoring the actin cytoskeleton to focal adhesions, Am. J. Physiol.: Cell Physiol., 2014, 306, C607–C620 CrossRef CAS PubMed.
- C. G. Galbraith, K. M. Yamada and M. P. Sheetz, The relationship between force and focal complex development, J. Cell Biol., 2002, 159, 695–705 CrossRef CAS PubMed.
- A. M. Pasapera, I. C. Schneider, E. Rericha, D. D. Schlaepfer and C. M. Waterman, Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation, J. Cell Biol., 2010, 188, 877–890 CrossRef CAS PubMed.
- H. Yamashita, T. Ichikawa, D. Matsuyama, Y. Kimura, K. Ueda, S. W. Craig, I. Harada and N. Kioka, Interaction of the vinculin proline-rich linker region with vinexin α in sensing extracellular matrix stiffness, J. Cell Sci., 2014, 127, 1875–1886 CrossRef CAS PubMed.
- A. W. Holle, X. Tang, D. Vijayraghavan, L. G. Vincent, A. Fuhrmann, Y. S. Choi, J. C. Del Álamo and A. J. Engler, In situ mechanotransduction via vinculin regulates stem cell differentiation, Stem Cells, 2013, 31, 2467–2477 CrossRef CAS PubMed.
- A. Carisey and C. Ballestrem, Vinculin, an adapter protein in control of cell adhesion signalling, Eur. J. Cell Biol., 2011, 90, 157–163 CrossRef CAS PubMed.
- R. P. Johnson and S. W. Craig, An intramolecular association between the head and tail domains of vinculin modulates talin binding, J. Biol. Chem., 1994, 269, 12611–12619 CAS.
- D. M. Cohen, H. Chen, R. P. Johnson, B. Choudhury and S. W. Craig, Two distinct head–tail interfaces cooperate to suppress activation of vinculin by talin, J. Biol. Chem., 2005, 280, 17109–17117 CrossRef CAS PubMed.
- C. Bakolitsa, D. M. Cohen, L. A. Bankston, A. A. Bobkov, G. W. Cadwell, L. Jennings, D. R. Critchley, S. W. Craig and R. C. Liddington, Structural basis for vinculin activation at sites of cell adhesion, Nature, 2004, 430, 583–586 CrossRef CAS PubMed.
- R. A. Borgon, C. Vonrhein, G. Bricogne, P. R. J. Bois and T. Izard, Crystal structure of human vinculin, Structure, 2004, 12, 1189–1197 CrossRef CAS PubMed.
- T. Izard, G. Evans, R. A. Borgon, C. L. Rush, G. Bricogne and P. R. J. Bois, Vinculin activation by talin through helical bundle conversion, Nature, 2004, 427, 171–175 CrossRef CAS PubMed.
- H. Chen, D. M. Cohen, D. M. Choudhury, N. Kioka and S. W. Craig, Spatial distribution and functional significance of activated vinculin in living cells, J. Cell Biol., 2005, 169, 459–470 CrossRef CAS PubMed.
- C. Grashoff, B. D. Hoffman, M. D. Brenner, R. Zhou, M. Parsons, M. T. Yang, M. A. McLean, S. G. Sligar, C. S. Chen and T. Ha, et al., Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics, Nature, 2010, 466, 263–266 CrossRef CAS PubMed.
- C. Ciobanasu, B. Faivre and C. Le Clainche, Actomyosin-dependent formation of the mechanosensitive talin-vinculin complex reinforces actin anchoring, Nat. Commun., 2014, 5, 3095 Search PubMed.
- F. Margadant, L. L. Chew, X. Hu, H. Yu, N. Bate, X. Zhang and M. Sheetz, Mechanotransduction In Vivo by Repeated Talin Stretch-Relaxation Events Depends upon Vinculin, PLoS Biol., 2011, 9, e1001223 CAS.
- K. Austen, P. Ringer, A. Mehlich, A. Chrostek-Grashoff, C. Kluger, C. Klingner, B. Sabass, R. Zent, M. Rief and C. Grashoff, Extracellular rigidity sensing by talin isoform-specific mechanical linkages, Nat. Cell Biol., 2015, 17, 1597–1606 CrossRef CAS PubMed.
- M. Tramier, M. Zahid, J.-C. Mevel, M.-J. Masse and M. Coppey-Moisan, Sensitivity of CFP/YFP and GFP/mCherry pairs to donor photobleaching on FRET determination by fluorescence lifetime imaging microscopy in living cells, Microsc. Res. Tech., 2006, 69, 933–939 CrossRef CAS PubMed.
- J. R. Tse and A. J. Engler, Preparation of hydrogel substrates with tunable mechanical properties, Curr. Protoc. Cell Biol., 2010, ch. 10, unit 10.16 Search PubMed.
- E. G. Hayman, Distribution of fetal bovine serum fibronectin and endogenous rat cell fibronectin in extracellular matrix, J. Cell Biol., 1979, 83, 255–259 CrossRef CAS PubMed.
- B. Li, C. Moshfegh, Z. Lin, J. Albuschies and V. Vogel, Mesenchymal stem cells exploit extracellular matrix as mechanotransducer, Sci. Rep., 2013, 3, 2425 Search PubMed.
- N. Borghi, M. Lowndes, V. Maruthamuthu, M. L. Gardel and W. J. Nelson, Regulation of cell motile behavior by crosstalk between cadherin- and integrin-mediated adhesions, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13324–13329 CrossRef CAS PubMed.
- S. Padilla-Parra, N. Audugé, M. Coppey-Moisan and M. Tramier, Dual-color fluorescence lifetime correlation spectroscopy to quantify protein-protein interactions in live cell, Microsc. Res. Tech., 2011, 74, 788–793 CrossRef CAS PubMed.
- C. K. Choi, M. Vicente-Manzanares, J. Zareno, L. A. Whitmore, A. Mogilner and A. R. Horwitz, Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner, Nat. Cell Biol., 2008, 10, 1039–1050 CrossRef CAS PubMed.
- D. W. Dumbauld, T. T. Lee, A. Singh, J. Scrimgeour, C. A. Gersbach, E. A. Zamir, J. Fu, C. S. Chen, J. E. Curtis, S. W. Craig and A. J. Garcia, How vinculin regulates force transmission, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9788–9793 CrossRef CAS PubMed.
- A. Carisey, R. Tsang, A. M. Greiner, N. Nijenhuis, N. Heath, A. Nazgiewicz, R. Kemkemer, B. Derby, J. Spatz and C. Ballestrem, Vinculin regulates the recruitment and release of core focal adhesion proteins in a force-dependent manner, Curr. Biol., 2013, 23, 271–281 CrossRef CAS PubMed.
- J. D. Humphries, P. Wang, C. Streuli, B. Geiger, M. J. Humphries and C. Ballestrem, Vinculin controls focal adhesion formation by direct interactions with talin and actin, J. Cell Biol., 2007, 179, 1043–1057 CrossRef CAS PubMed.
- A. Saez, A. Buguin, P. Silberzan and B. Ladoux, Is the Mechanical Activity of Epithelial Cells Controlled by Deformations or Forces?, Biophys. J., 2005, 89, L52–L54 CrossRef CAS PubMed.
- M. Allioux-Guérin, D. Icard-Arcizet, C. Durieux, S. Hénon, F. Gallet, J.-C. Mevel, M.-J. Masse, M. Tramier and M. Coppey-Moisan, Spatiotemporal analysis of cell response to a rigidity gradient: a quantitative study using multiple optical tweezers, Biophys. J., 2009, 96, 238–247 CrossRef PubMed.
- C.-W. Chang and S. Kumar, Vinculin tension distributions of individual stress fibers within cell-matrix adhesions, J. Cell Sci., 2013, 126, 3021–3030 CrossRef CAS PubMed.
- R. P. Johnson and S. W. Craig, F-actin binding site masked by the intramolecular association of vinculin head and tail domains, Nature, 1995, 373, 261–264 CrossRef CAS PubMed.
- J.-E. Hoffmann, Y. Fermin, R. L. Stricker, K. Ickstadt and E. Zamir, Symmetric exchange of multi-protein building blocks between stationary focal adhesions and the cytosol, eLife, 2014, 3, e02257 Search PubMed.
- A. I. Bachir, J. Zareno, K. Moissoglu, E. F. Plow, E. Gratton and A. R. Horwitz, Integrin-Associated Complexes Form Hierarchically with Variable Stoichiometry in Nascent Adhesions, Curr. Biol., 2014, 24, 1845–1853 CrossRef CAS PubMed.
- M. Yao, B. T. Goult, H. Chen, P. Cong, M. P. Sheetz and J. Yan, Mechanical activation of vinculin binding to talin locks talin in an unfolded conformation, Sci. Rep., 2014, 4, 4610 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ib00307e |
| ‡ Present address: Institut Curie, Unité Mixte de Recherche 144, Centre national de la recherche scientifique, Université P. & M. Curie, Paris 75005, France. |
| § Present address: Cell Biology and Biophysics Unit, European Molecular Biology Laboratory, Heidelberg 69117, Germany. |
| This journal is © The Royal Society of Chemistry 2016 |