
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReducing emission of carcinogenic by-products in the production of thermally reduced graphene oxide†
Ondřej
Jankovský
a,
Michal
Lojka
a,
Michal
Nováček
a,
Jan
Luxa
a,
David
Sedmidubský
a,
Martin
Pumera
b,
Jiří
Kosina
c and
Zdeněk
Sofer
*a
aDepartment of Inorganic Chemistry, University of Chemistry and Technology Prague, Technická 5, 166 28 Prague 6, Czech Republic. E-mail: zdenek.sofer@vscht.cz
bDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore
cCentral Laboratories, University of Chemistry and Technology Prague, Technická 5, 166 28 Prague 6, Czech Republic
First published on 3rd October 2016
Abstract
In the last decade, researchers have been trying to find out a simple method for large scale fabrication of high-quality graphene. A typical method for the fabrication of gram quantities of graphene materials is the oxidation of graphite to graphite oxide and consequent thermal reduction and exfoliation to reduced graphene oxide at T = ∼1000 °C. Here we show that highly reduced graphene oxide can be prepared at lower temperatures than 1000 °C while keeping the properties of graphene suitable for various electrochemical applications. The high temperature exfoliation of graphite oxide typically leads to the formation of a large amount of highly toxic volatile organic hydrocarbons such as benzene and its derivatives. The amount of volatile aromatic hydrocarbons can be reduced using low temperature exfoliation procedures that we present here. The application of a lower exfoliation temperature is highly beneficial as it also significantly reduces the etching of the graphene skeleton and the formation of toxic aromatic hydrocarbons. The effect of thermal exfoliation was investigated in detail for the temperature range of 400 °C up to 1000 °C under hydrogen as well as nitrogen atmospheres. Our findings show the route for the preparation of thermally reduced graphene oxide suitable for various electrochemical applications without the formation of toxic hydrocarbons as reaction byproducts. These findings are of high importance for the industrial scale production of thermally reduced graphene oxide.
Introduction
Due to its unique electrical, optical and mechanical properties graphene has been intensively studied since its discovery in 2004.1–3 These properties make graphene a highly promising material for a wide range of applications in modern electronics and electrochemical power sources such as supercapacitors, fuel cells or batteries which are of extreme importance for the green energy production and storage.4–8 Thanks to its very high surface area, graphene also seems to be useful for the removal of toxic ions from the environment.9–11A number of various methods for the preparation of graphene have been reported. One possibility to directly obtain graphene is the mechanical exfoliation of graphite,12 other common methods are CVD (chemical vapour deposition) techniques.13 Usually CVD-based graphene layers are of high-quality and they are important for optoelectronic or microelectronic devices. Another possibility is the exfoliation of graphite in aqueous solutions.14,15 However, for applications in the field of electrochemical power sources and composites, graphene needs to be produced in larger amounts.16–19 For this purpose, the reduction of graphene oxide seems to be the most suitable method. In this method, a graphite precursor is oxidized in an acidic environment using chlorates or permanganates to graphite oxide. Subsequently, the formed graphene oxide is reduced by different routes forming graphene. Graphene oxide can be reduced using microwaves,20 by ion bombardment21 and by chemical or thermal reduction.22,23 A number of reducing agents for the chemical reduction have been reported from which the most common are hydrazine and its derivatives,22 hydroquinone,24 complex hydrides25 or nascent hydrogen.26–28 Solvothermal or microwave assisted methods have also been used for the reduction of graphene oxide.29,30 In order to obtain reduced graphene oxide with a high C/O ratio, strong reducing reagents such as carcinogenic hydrazine must be applied.31 The concentration of the remaining oxygen functionalities has a strong impact on many properties like conductivity and electrochemical activity.32 The chemical reduction methods also usually introduce several impurities within reduced graphene oxide which can strongly influence its properties.33 Thermal reduction also exhibits an advantage over chemical reduction, since the reduced graphene oxide does not need to be separated from the chemical reduction by-products. Graphene oxide can also be reduced thermally under an inert (nitrogen) or reducing atmosphere (hydrogen). The thermal reduction of graphene oxide produces reduced graphene oxide with the highest degree of exfoliation and also the largest surface area.34,35 The in situ measurement of resistivity around the exfoliation temperature shows a significant drop of resistivity values at temperatures exceeding 200 °C, where the exfoliation of graphene oxide and the thermal decomposition of oxygen functionalities take place.36 Thermal reduction also yields reduced graphene oxide with a lower concentration of oxygen leading to higher conductivity.32 Compared to other green chemical reduction reagents (e.g. ascorbic acid), thermal reduction provides a material with a significantly higher C/O ratio. The low concentration of the remaining oxygen functionalities enhances the conductivity and improves the performance for applications in electronic and energy saving and conversion devices where it can be typically used as an electrocatalyst for hydrogen evolution or a supercapacitor electrode material. Since thermal reduction of graphene oxide is an important method for the large scale production of reduced graphene oxide, its environmental aspects must be studied in detail.
The mechanism of the thermal reduction is in general described as a process of decomposition of oxygen containing functional groups yielding carbon dioxide, carbon monoxide and water released from the structure during the heating.37 The evolved gaseous products increase the interlayer pressure bringing about the exfoliation. Recently, this view has been extended by demonstrating that the process of thermal exfoliation is much more complex and a multitude of complex compounds is released during the exfoliation apart from water and CO/CO2.38–40 At high temperatures, the formation of a large amount of various hydrocarbons was observed being dominated by highly toxic benzene as well as toluene and naphthalene. Such compounds like benzene are highly toxic and known to be carcinogenic to humans (Group 1 carcinogen). This can pose a significant issue for the production of thermally reduced graphene oxide on the industrial scale.
In this study, we examine the relationship between the exfoliation temperature, atmosphere and evolved hydrocarbons. The analysis is carried out by gas chromatography in combination with a mass spectrometer detector (GC-MS). The experiments were performed for temperatures ranging from 400 to 1000 °C using graphite oxide prepared by two most common methods, Hofmann's41 (using chlorate oxidant) and Hummers’42 (using permanganate oxidant) methods. The influence of the exfoliation atmosphere using both nitrogen and hydrogen was investigated in detail for the whole temperature range. Significant differences were observed for the exfoliation of graphite oxide at temperatures below 800 °C associated with the reduction of aromatic hydrocarbons’ concentration. The prepared thermally reduced graphene oxides were investigated in detail showing their suitability for various electrochemical applications.
Experimental
We prepared two graphite oxides from graphite microparticles (2–15 μm, 99.9995%, from Alfa Aesar). Sulfuric acid (98%), nitric acid (68%), potassium chlorate (99%), potassium permanganate (99.5%), sodium nitrate (99.5%), hydrogen peroxide (30%), hydrochloric acid (37%), silver nitrate (99.5%), barium nitrate (99.5%) and N,N-dimethylformamide (DMF) were obtained from Penta, Czech Republic. Deionized water (16.8 MΩ) was used for all syntheses.The first graphite oxide was synthesized similarly to the Hummers method and is denoted as HU-GO. Graphite (5 g) and sodium nitrate (2.5 g) were stirred with sulfuric acid (98%, 115 mL) under permanent cooling to 0 °C. While vigorously stirred, potassium permanganate (15 g) was then added over a period of two hours. During the next four hours, the reaction mixture was allowed to reach room temperature before being heated to 35 °C for 30 min. The reaction mixture was then poured into a flask containing deionized water (250 mL) and heated to 70 °C for 15 minutes. The mixture was then poured into deionized water (1 L). The unreacted potassium permanganate and manganese dioxide were removed by the addition of 3% hydrogen peroxide. The reaction mixture was then allowed to settle and decant. The obtained graphite oxide was then purified by repeated centrifugation and redispersing in deionized water until a negative reaction on sulphate ions (with Ba(NO3)2) was achieved. The graphite oxide slurry was then dried in a vacuum oven at 50 °C for 48 h before use. The composition obtained by elemental combustion analysis was 41.4 at% C, 31.8 at% O and 26.8 at% H. More details are given in the ESI (Table S1†).
The second graphite oxide prepared according to the Hofmann method is referred to as HO-GO. Sulfuric acid (98%, 87.5 mL) and nitric acid (68%, 27 mL) were added to a reaction flask (Pyrex beaker with a thermometer) containing a magnetic stir bar. The mixture was then cooled by immersion in an ice bath for 30 min. Graphite (5 g) was then added to the mixture with vigorous stirring motion to avoid agglomeration and to obtain a homogeneous dispersion. While keeping the reaction flask in the ice bath, potassium chlorate (55 g) was slowly added to the mixture (over a 30 min period) in order to avoid a sudden increase in temperature and the formation of explosive chlorine dioxide gas. Upon the complete dissolution of potassium chlorate, the reaction flask was then loosely capped to allow the escape of the gas evolved and the mixture was continuously vigorously stirred for 96 hours at room temperature. Upon completion of the reaction, the mixture was poured into 3 L of deionized water and decanted. Graphite oxide was then redispersed in HCl solution (5%, 3 L) to remove sulfate ions and repeatedly centrifuged and redispersed in deionized water until a negative reaction on chloride and sulfate ions (with AgNO3 and Ba(NO3)2 respectively) was achieved. The graphite oxide slurry was then dried in a vacuum oven at 50 °C for 48 h before use. The composition obtained by elemental combustion analysis was 47.5 at% C, 28.8 at% O and 23.7 at% H. More details are given in the ESI (Table S1†).
The thermal reduction–exfoliation of graphite oxide was performed at 400, 600, 800 and 1000 °C in a quartz glass reactor under a hydrogen and nitrogen atmosphere. 0.1 g graphite oxide was placed in a porous quartz glass capsule connected to a magnetic manipulator inside a vacuum tight tube furnace under a controlled atmosphere. The sample was flushed with nitrogen by repeated evacuation of the tube furnace to remove any traces of oxygen. Subsequently the reactor was filled with nitrogen or hydrogen, and the sample was quickly inserted by using a magnetic manipulator to the preheated furnace and held in the furnace for 12 minutes. The flow of hydrogen or nitrogen during the exfoliation procedure was set as 1000 sccm to remove the byproducts of the exfoliation procedure. The exfoliation under a nitrogen or hydrogen atmosphere and the subsequent sampling at the output of the reactor was performed using an SPME (Solid-Phase Micro Extraction) fiber for GC-MS analysis of the evolved organic species. Such a configuration minimized the retention time in the hot zone of the reactor and the immediate cooling of the carrier gas allowed the preservation of highly reactive species containing also double and triple carbon–carbon bonds.
To determine what kinds of organic species are evolved during the exfoliation of graphite oxide, gaseous products were sampled on the 85 μm Carboxen/PDMS SPME fiber (Supelco, USA) and analyzed on a gas chromatograph coupled with a mass spectrometer (GC-MS). Prior to sampling, the used SPME fiber was conditioned for 30 minutes at 240 °C under a constant flow of helium. The conditioned SPME fiber was inserted via a Teflon septum into the outline of the reactor cell and the sample was collected over 12 min. The exposed SPME fiber was thermally desorbed in the heated split/splitless injection port of the GC (Trace GC Ultra, Thermo Scientific, USA) under a constant flow of carrier gas (helium). The flow of helium was set to 1.5 mL min−1. The injection port was heated to 240 °C and worked in the split mode. The split ratio was set to the value of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. Chromatographic separations were performed on a DB-5MS UI (Agilent, USA) capillary column (60 m × 0.32 mm × 1.0 μm). The GC oven program was as follows: initial temperature 40 °C, held for 5 min, heating at 15 °C min−1 to 250 °C followed by a delay for 10 min. Masses in the range of 10 to 400 m/z in full scan mode were recorded on the ISQ (Thermo Scientific, USA) single-quadrupole MS operating at an electron impact energy of 70 eV. Acquired mass spectra were interpreted by using the electronic database NIST 14.
:10. Chromatographic separations were performed on a DB-5MS UI (Agilent, USA) capillary column (60 m × 0.32 mm × 1.0 μm). The GC oven program was as follows: initial temperature 40 °C, held for 5 min, heating at 15 °C min−1 to 250 °C followed by a delay for 10 min. Masses in the range of 10 to 400 m/z in full scan mode were recorded on the ISQ (Thermo Scientific, USA) single-quadrupole MS operating at an electron impact energy of 70 eV. Acquired mass spectra were interpreted by using the electronic database NIST 14.
To determine the total amount of the evolved organic species, gaseous products were sampled on the Anasorb® CSC sorption tube (SKC Inc., USA) connected directly into the exhaust line of the exfoliation cell. The species trapped on the active carbon were extracted with 500 μl of CS2 (>99.9%, low benzene content, Sigma-Aldrich, Czech Republic) and the CS2 extract was analyzed by gas chromatography-mass spectrometry (GC-MS). 1 μL of the CS2 extract was injected into the heated multimode injection port of the gas chromatograph 7890B (Agilent Technologies, USA). The injection port was heated up to 250 °C and operated in the split mode. The split ratio was set to a value of 1:10. The constant flow of the carrier gas (helium, purity >6.0) was set to 1.0 mL min−1. Gas chromatographic separations were performed on a HP-5MS UI capillary column (Agilent Technologies, USA; 30 m × 0.25 mm i.d. and 0.25 μm). The GC oven program was as follows: initial temperature 40 °C, held for 3 min, 15 °C min−1 to 300 °C, held for 5 min. Masses in the range of 29 to 450 Da in full scan mode were recorded on the mass spectrometer 7010 Triple-Quadrupole GC/MS (Agilent Technologies, USA) operating at an electron impact energy of 70 eV. The temperature of the ion source was set to 230 °C and the quadrupoles were heated to 150 °C.
The resistivity measurement was performed by a two-point method by placing the sample in a sapphire tube, where the sample was compressed by two copper pistons with a pressure of 5 bar. The piston was connected to a position measurement device to obtain the value of the compressed sample length. The resistivity was measured by using a 34401A digital multimeter from Keysight Technologies (USA).
The morphology was investigated using scanning electron microscopy (SEM) with a FEG electron source (Tescan Lyra dual beam microscope). The elemental composition and mapping were performed using an energy dispersive spectroscopy (EDS) analyzer (X-MaxN) with a 20 mm2 SDD detector (Oxford Instruments) and AZtecEnergy software. To conduct the measurements, the samples were placed on a carbon conductive tape. SEM and SEM-EDS measurements were carried out using a 10 kV electron beam. For the observation in transmission mode, an STEM detector integrated in an electron microscope with an applied accelerating voltage of 30 kV was used. Sample dispersion in isopropanol (1 mg mL−1) prepared by ultrasonication and drop-cast on 200 mesh TEM grids (Cu/Formvar) was used for the observation.
Combustible elemental analysis (CHNS-O) was performed using a PE 2400 series II CHNS/O analyzer (Perkin Elmer, USA). The instrument was used in CHN operating mode (the most robust and interference-free mode) to convert the sample elements to simple gases (CO2, H2O and N2). The PE 2400 analyzer automatically performed combustion, reduction, homogenization of product gases, separation and detection. An MX5 microbalance (Mettler Toledo) was used for precise weighing of the samples (1.5–2.5 mg per sample analysis). Using this procedure, the accuracy of CHN determination is better than 0.30% abs. Internal calibration was performed using N-phenyl urea.
High resolution X-ray photoelectron spectroscopy (XPS) was performed using an ESCAProbeP spectrometer (Omicron Nanotechnology Ltd, Germany) with a monochromatic aluminum X-ray radiation source (1486.7 eV). Wide-scan surveys of all elements were performed, with subsequent high-resolution scans of the C 1s and O 1s. Relative sensitivity factors were used to evaluate the carbon-to-oxygen (C/O) ratios from the survey spectra. The samples were placed in a conductive carrier made from a high purity silver bar. An electron gun was used to eliminate sample charging during the measurement (1–5 V).
An inVia Raman microscope (Renishaw, England) in backscattering geometry with a CCD detector was used for Raman spectroscopy. A DPSS laser (532 nm, 50 mW) with a 50× magnification objective was used for the measurement. Instrument calibration was achieved with a silicon reference which gives a peak position at 520 cm−1 and a resolution of less than 1 cm−1.
The thermal behaviour of the prepared thermally reduced graphene oxides was analyzed by Simultaneous Thermal Analysis (STA). The DTA and TG curves were recorded simultaneously on a Linseis STA PT1600 apparatus (heating rate 10 °C min−1) under a dynamic air atmosphere (50 mL min−1). The measurement of graphene oxide exfoliation was performed by simultaneous thermal analysis using Setsys Evolution apparatus from Setaram. The evolved gases were analyzed by using an OmniStar™ mass spectrometer from Pfeiffer Vacuum. The measurement was performed under a dynamic nitrogen atmosphere (50 mL min−1) using a heating rate of 10 °C min−1.
The surface area was measured using a sorption analyzer, Coulter SA 3100 (Beckman Coulter). The samples were outgassed for 4 hours at 95 °C under high vacuum (1 × 10−2 Pa) prior to the sorption experiments. The reason for using such a low temperature is to avoid degradation and further decomposition of oxygen functionalities. A TCD nitrogen cooled (77 K) detector was used for the evaluation of the results using BET (Brunauer, Emmett and Teller) and Kelvin equations.
X-ray powder diffraction data were collected at room temperature on a Bruker D8 Discoverer powder diffractometer with the parafocusing Bragg–Brentano geometry using CuKα radiation (λ = 0.15418 nm, U = 40 kV, I = 40 mA). Data were scanned over the angular range of 10–80° (2θ) with a step size of 0.02° (2θ). Data evaluation was performed using the software package EVA.
All glassy carbon electrodes for electrochemical measurement were cleaned by polishing them with an alumina suspension to renew the electrode surface, and then they were washed and wiped dry prior to any use. The materials were dispersed in DMF as the organic solvent to obtain a 1 mg mL−1 suspension. The suspension was then sonicated for 5 min at room temperature before every use. A cleaned GC electrode was then modified by coating it with a 2 μL aliquot of the suspension and was left to dry at ambient temperature to give a layer of randomly dispersed material on the GC surface. The modified GC electrodes, saturated Ag/AgCl reference electrode, and platinum counter electrode were then placed into an electrochemical cell containing the electrolyte solution, and the measurements were then taken. The electrolytes used were 50 mM, pH 7.2 phosphate buffer solution (PBS) as the blank buffer electrolyte and 10 mM ferro/ferri cyanide dissolved in PBS using a scan rate of 100 mV s−1. The measurement was performed using an Autolab PGSTAT 204 (Metroohm, Switzerland).
Results and discussion
Two graphite oxides (GOs) were prepared by the Hummers42 and Hofmann41 methods (see the Experimental section for details). Both GOs were further exfoliated under either a nitrogen or hydrogen atmosphere at various temperatures. Samples are termed according to their GO precursor (HU – Hummers, HO – Hofmann), exfoliation atmosphere (N2 or H2) and temperature in the silica reactor during the exfoliation (400, 600, 800 or 1000 °C). All samples were then analyzed by scanning electron microscopy (SEM), energy dispersive spectroscopy (SEM-EDS), combustion elemental analysis, high resolution X-ray photoelectron spectroscopy (XPS), Raman spectroscopy and simultaneous thermal analysis (STA). Gas chromatography-mass spectroscopy (GC-MS) was used to analyze the exfoliation products. For more details, see the Experimental section.The morphology of the obtained thermally reduced graphene oxides was investigated by SEM (Fig. S1†). The typical platelet structure was observed for all samples; however, the samples prepared at 400 °C exhibit a slightly lower degree of exfoliation. No significant differences can be observed for samples exfoliated under a hydrogen or nitrogen atmosphere. STEM images confirmed high reduction of all samples with a typical few layered graphene structure, and the images suggest a higher number of layers for the samples prepared at 400 °C, and other characterization methods shown below prove this to be the case (Fig. 1).
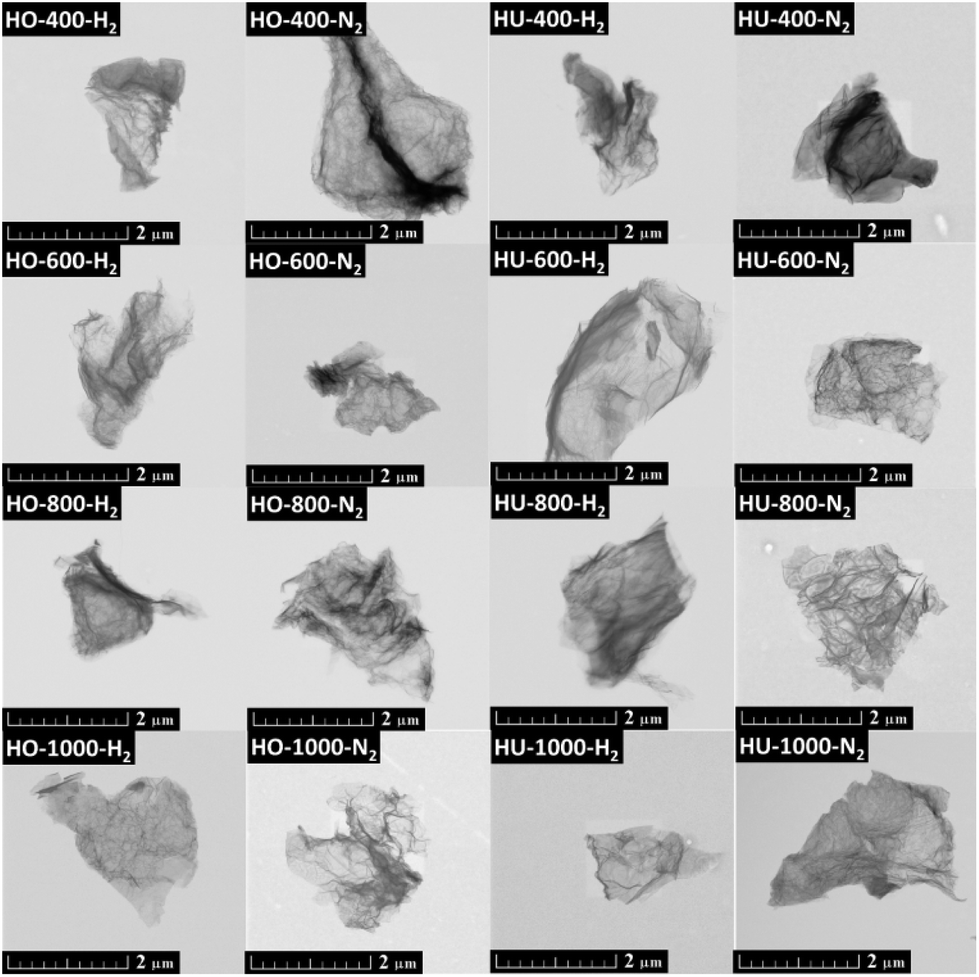 | ||
| Fig. 1 Morphology of thermally reduced graphene oxides measured using STEM. Scale bar is 2 μm. | ||
The results of combustible elemental analysis are presented in Table 1. Apart from carbon, oxygen and hydrogen, only a small amount of nitrogen was detected, especially at high temperatures. Exfoliation under a hydrogen atmosphere led to a higher content of hydrogen in thermally reduced graphene oxides. Treatment at higher temperatures also resulted in a higher level of reduction manifested by a higher C/O ratio. For 400 °C the C/O ratio (Table 2) is in the range of 5–7, while at 1000 °C it was in the range of 14–61, confirming a high level of reduction. The results are compared with other analyses. A higher C/O ratio was also obtained for samples exfoliated under a hydrogen atmosphere. Also, a higher C/O ratio was found for thermally reduced graphene oxides prepared from HU-GO compared to thermally reduced graphene oxides starting from HO-GO. This originates from different compositions of the starting graphite oxide, where HU-GO samples contain more thermally labile oxygen functionalities like ketones and carboxylic functionalities, while HO-GO samples contain predominantly more thermally stable hydroxyl groups.
| Sample | wt% C | wt% H | wt% N | wt% O |
|---|---|---|---|---|
| HO-400-H2 | 84.12 | 0.71 | 0.00 | 15.17 |
| HO-600-H2 | 89.76 | 1.32 | 0.00 | 11.56 |
| HO-800-H2 | 93.40 | 1.64 | 0.00 | 4.96 |
| HO-1000-H2 | 94.69 | 1.38 | 0.00 | 3.93 |
| HO-400-N2 | 84.12 | 0.65 | 0.00 | 15.23 |
| HO-600-N2 | 85.28 | 0.73 | 0.00 | 15.45 |
| HO-800-N2 | 85.47 | 1.18 | 0.00 | 13.35 |
| HO-1000-N2 | 90.50 | 1.05 | 0.00 | 8.45 |
| HU-400-H2 | 80.70 | 1.11 | 0.04 | 18.15 |
| HU-600-H2 | 89.47 | 1.71 | 0.00 | 8.82 |
| HU-800-H2 | 93.82 | 1.60 | 0.17 | 4.41 |
| HU-1000-H2 | 96.98 | 0.57 | 0.33 | 2.12 |
| HU-400-N2 | 80.47 | 1.05 | 0.00 | 18.48 |
| HU-600-N2 | 84.67 | 1.05 | 0.21 | 14.07 |
| HU-800-N2 | 91.71 | 1.06 | 0.27 | 6.96 |
| HU-1000-N2 | 94.15 | 0.91 | 0.13 | 4.81 |
| Sample | C/O SEM-EDS | C/O CHN-O | C/O XPS |
|---|---|---|---|
| HO-400-H2 | 18.43 | 7.39 | 7.18 |
| HO-600-H2 | 20.46 | 10.35 | 22.75 |
| HO-800-H2 | 35.45 | 25.11 | 43.25 |
| HO-1000-H2 | 36.65 | 32.13 | 44.05 |
| HO-400-N2 | 16.90 | 7.36 | 11.24 |
| HO-600-N2 | 18.14 | 7.36 | 15.31 |
| HO-800-N2 | 25.64 | 8.54 | 20.51 |
| HO-1000-N2 | 26.11 | 14.28 | 23.57 |
| HU-400-H2 | 15.67 | 5.93 | 10.31 |
| HU-600-H2 | 26.31 | 13.53 | 25.60 |
| HU-800-H2 | 31.84 | 28.37 | 52.48 |
| HU-1000-H2 | 47.86 | 60.99 | 78.60 |
| HU-400-N2 | 16.84 | 5.81 | 9.74 |
| HU-600-N2 | 21.94 | 8.02 | 15.05 |
| HU-800-N2 | 26.84 | 17.57 | 31.79 |
| HU-1000-N2 | 26.55 | 26.10 | 94.26 |
High resolution XPS was used to determine the chemical composition of the thermally reduced graphene oxide surface. The XPS survey spectra are presented in Fig. S2 and S3,† where C 1s and O 1s peaks are clearly visible at ∼284.5 eV and ∼533 eV, respectively. Also Ag 3d and Ag 3p peaks can be found in the survey spectra. These peaks originate from a silver sample holder. The survey spectra were used for the calculation of C/O ratios. The obtained results confirmed the trends observed by CHN-O analysis. The lowest C/O ratios were obtained at the lowest temperatures (∼7–11), while high C/O ratios were found at the highest temperatures (∼23–94). A higher reduction was also detected for thermally reduced graphene oxides originating from HU-GO as well as for samples reduced under a hydrogen atmosphere. The higher oxidation level of HU-GO allows for more powerful exfoliation thanks to the higher amount of gases evolved inside the structure.
In addition, high resolution XPS was also used for the detailed analysis of the C 1s to quantitatively differentiate six different carbon states present in all samples: C–C (284.4 eV); C–C/C–H (285.4 eV); C–O (286.3 eV); C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (288.0 eV); O–CO (289.0 eV) and π–π* interactions (290.5 eV). The quantitative compositions of the individual states of C 1s are listed in Table 3, while the detailed fitting of the C 1s peak is shown in Fig. S4 and S5.† The results obtained from the fitting are in good agreement with the overall chemical composition obtained by XPS.
O (288.0 eV); O–CO (289.0 eV) and π–π* interactions (290.5 eV). The quantitative compositions of the individual states of C 1s are listed in Table 3, while the detailed fitting of the C 1s peak is shown in Fig. S4 and S5.† The results obtained from the fitting are in good agreement with the overall chemical composition obtained by XPS.
| Sample | CC |
C–C/C–H | C–O | CO |
O–CO |
ππ* |
|---|---|---|---|---|---|---|
| HO-400-H2 | 63.00 | 22.06 | 9.83 | 2.36 | 1.31 | 1.14 |
| HO-600-H2 | 66.89 | 22.42 | 7.07 | 1.71 | 1.40 | 0.50 |
| HO-800-H2 | 74.19 | 17.76 | 4.71 | 1.16 | 1.06 | 1.13 |
| HO-1000-H2 | 79.13 | 14.42 | 3.57 | 0.84 | 1.33 | 0.71 |
| HO-400-N2 | 49.10 | 20.27 | 14.06 | 8.09 | 3.72 | 4.76 |
| HO-600-N2 | 55.00 | 18.48 | 12.17 | 7.18 | 4.87 | 2.30 |
| HO-800-N2 | 55.95 | 19.27 | 9.12 | 5.58 | 4.84 | 5.24 |
| HO-1000-N2 | 58.54 | 20.02 | 8.61 | 4.67 | 3.32 | 4.84 |
| HU-400-H2 | 69.38 | 18.49 | 5.97 | 2.51 | 1.72 | 1.93 |
| HU-600-H2 | 71.30 | 18.40 | 6.31 | 1.32 | 0.93 | 1.74 |
| HU-800-H2 | 78.00 | 13.57 | 4.00 | 2.08 | 1.12 | 1.24 |
| HU-1000-H2 | 81.06 | 12.10 | 3.17 | 0.68 | 1.72 | 1.26 |
| HU-400-N2 | 55.65 | 21.32 | 10.09 | 5.88 | 4.28 | 2.78 |
| HU-600-N2 | 65.65 | 17.05 | 7.10 | 4.18 | 3.01 | 3.01 |
| HU-800-N2 | 70.38 | 20.73 | 6.57 | 1.51 | 0.40 | 0.42 |
| HU-1000-N2 | 79.97 | 14.75 | 3.59 | 0.80 | 0.80 | 0.10 |
The chemical composition was also studied by SEM-EDS. Except for C and O, low concentrations of K, Mn, S, and Cl (maximally 0.2 wt%) were detected by SEM-EDS. The calculated C/O ratios based on the data from this method are summarized in Table 2. These results are in good agreement with the chemical analysis results discussed earlier: the obtained C/O ratios are similar to the results from CHN-O revealing similar trends. The higher values of C/O ratios obtained by XPS compared to SEM-EDS are likely caused by a higher surface sensitivity of XPS and also due to a relatively low sensitivity of SEM-EDS towards light elements.
Raman spectroscopy was used to obtain more information on the structure and defect density of the thermally reduced graphene oxides (Fig. S6 and S7†). Two major features corresponding to the G-band (1580 cm−1) and D-band (1350 cm−1) were found for all samples.43 The presence of the D-band indicates defects in the graphene layer, attributable to the presence of sp3 bonded carbon atoms, while the G-band resulted from sp2 bonded carbon atoms in the graphene layer.44 From the Raman spectra we calculated the D/G ratios (Table 4) indicating the density of defects. Highly reduced samples (at 1000 °C) revealed the highest D/G ratio: the D/G value of HU-1000-H2 was 1.41, while the D/G ratios for thermally reduced graphene oxides reduced at 400, 600 or 800 °C were ∼0.9. A slightly higher D/G ratio was obtained for samples originating from HU-GO, which is caused by the higher oxidation level of graphite oxide and also by the type of oxygen functionality present on the graphite oxide surface. During thermal decomposition, ketone functional groups form carbon monoxide which leads to the removal of the carbon atom from the graphene oxide skeleton and the formation of defects. A higher D/G ratio was also obtained for samples exfoliated under a hydrogen atmosphere. A higher D/G ratio for samples exfoliated in hydrogen originates from the etching of the graphene skeleton by hydrogen.40
| Sample | D/G ratio |
|---|---|
| HO-400-H2 | 0.89 |
| HO-600-H2 | 0.87 |
| HO-800-H2 | 0.91 |
| HO-1000-H2 | 1.31 |
| HO-400-N2 | 0.97 |
| HO-600-N2 | 0.93 |
| HO-800-N2 | 0.99 |
| HO-1000-N2 | 1.18 |
| HU-400-H2 | 0.92 |
| HU-600-H2 | 0.86 |
| HU-800-H2 | 0.93 |
| HU-1000-H2 | 1.41 |
| HU-400-N2 | 0.87 |
| HU-600-N2 | 0.91 |
| HU-800-N2 | 0.92 |
| HU-1000-N2 | 1.32 |
The thermal stability of the thermally reduced graphene oxides was tested using simultaneous thermal analysis (STA) under a dynamic air atmosphere (Fig. S8 and S9†). Almost all samples exhibited a very similar thermal behavior with one large exothermal effect. This effect started at ∼550 °C and was accompanied by the weight loss suggesting an onset of oxidation/combustion of the materials. This indicates only a negligible effect of the exfoliation temperature on the thermal stability of the thermally reduced graphene oxides.
Assuming the surface area 2630 m2 g−1 for a single layer graphene, the number of layers can be calculated from the surface area, which was measured using BET.28 The obtained surface areas and the calculated numbers of layers are shown in Table 5. The results show a slight increase of the exfoliation degree with the increasing exfoliation temperature. However, more significant was the influence of the graphene oxide synthesis method showing a significantly higher degree of exfoliation for samples prepared by the chlorate method in comparison with the permanganate method. No significant differences in the surface area were observed for different exfoliation atmospheres, showing its negligible effect on the degree of exfoliation. The surface areas of the starting graphite and graphene oxide were lower by two orders of magnitude (6.46 m2 g−1 graphite; 3.52 m2 g−1 HUGO; 2.97 m2 g−1 HOGO).
| Sample | Surface area (m2 g−1) | Average number of layers |
|---|---|---|
| HO-400-H2 | 657.73 | 4.0 |
| HO-600-H2 | 685.97 | 3.8 |
| HO-800-H2 | 701.83 | 3.7 |
| HO-1000-H2 | 809.97 | 3.2 |
| HO-400-N2 | 644.48 | 4.1 |
| HO-600-N2 | 682.14 | 3.9 |
| HO-800-N2 | 788.33 | 3.3 |
| HO-1000-N2 | 810.66 | 3.2 |
| HU-400-H2 | 175.15 | 15.0 |
| HU-600-H2 | 190.98 | 13.8 |
| HU-800-H2 | 212.03 | 12.4 |
| HU-1000-H2 | 357.77 | 7.4 |
| HU-400-N2 | 170.14 | 15.5 |
| HU-600-N2 | 190.88 | 13.8 |
| HU-800-N2 | 232.88 | 11.3 |
| HU-1000-N2 | 239.09 | 11.0 |
All thermally reduced graphene oxides as well as the starting graphene oxide were probed using X-ray diffraction. The diffractograms are dominated by a broad (002) reflection located around 24–26 °2θ. In the few-layer graphenes the number of layers can be calculated from the broadening of the (002) reflection using the Scherrer equation.28 In general, the lower broadening was observed for the thermally reduced graphene oxide prepared from HU-GO in comparison with HO-GO (Fig. S10†). No significant differences were observed for the different exfoliation atmospheres. The observed trends are in agreement with the measurement of the surface area by the BET method confirming a high exfoliation degree and the presence of few layer thermally reduced graphene oxides. The results are summarized in Table S2.† In addition, weak and broad reflections of the (100) and (101) planes at 42.4 °2θ and 44.5 °2θ are visible. To confirm successful reduction associated with the exfoliation, the starting graphene oxides were also measured. After the reduction, the broad peak of the (002) reflection at 12.25 °2θ for HO-GO and 10.48 °2θ for HU-GO disappeared entirely.
The conductivity of thermally reduced graphene oxide was measured by a two-point method on the powder material compressed in the measurement device by a constant force (5 bar). All samples exhibit relatively high conductivity in the range of 0.3–0.05 Ω cm. A slight decrease of specific resistivity was observed with the increase of exfoliation temperature and also a slightly lower specific resistivity was observed for the thermally reduced graphene oxide prepared from HO-GO in comparison with HU-GO. The differences between the exfoliation atmospheres were negligible. The reduction of resistivity with the increasing exfoliation temperature is closely related to the concentration of the remaining oxygen functionalities which dominantly contribute to the sample resistivity. The resistivity of the starting graphene oxide was more than one order higher due to the semiconducting properties of graphene oxide (7.92 Ω cm for HO-GO and 52.7 Ω cm for HU-GO). The resistivity of graphite used for the synthesis measured by a similar method was 0.06 Ω cm. The results of the specific resistivity measurement are summarized in ESI Table S3.†
Finally, we measured the composition of the exfoliation products for both the graphite oxides under different atmospheres and at various temperatures by GC-MS. The results of the GC-MS analysis of gases evolved during the exfoliation are shown in Fig. 2 and 3. All detected gaseous exfoliation products are shown in Table 6. Let us note that water, carbon dioxide and carbon monoxide were also formed apart from the species given in Table 6; however, they are not trapped by the SPME fiber for the subsequent GC-MS analysis.
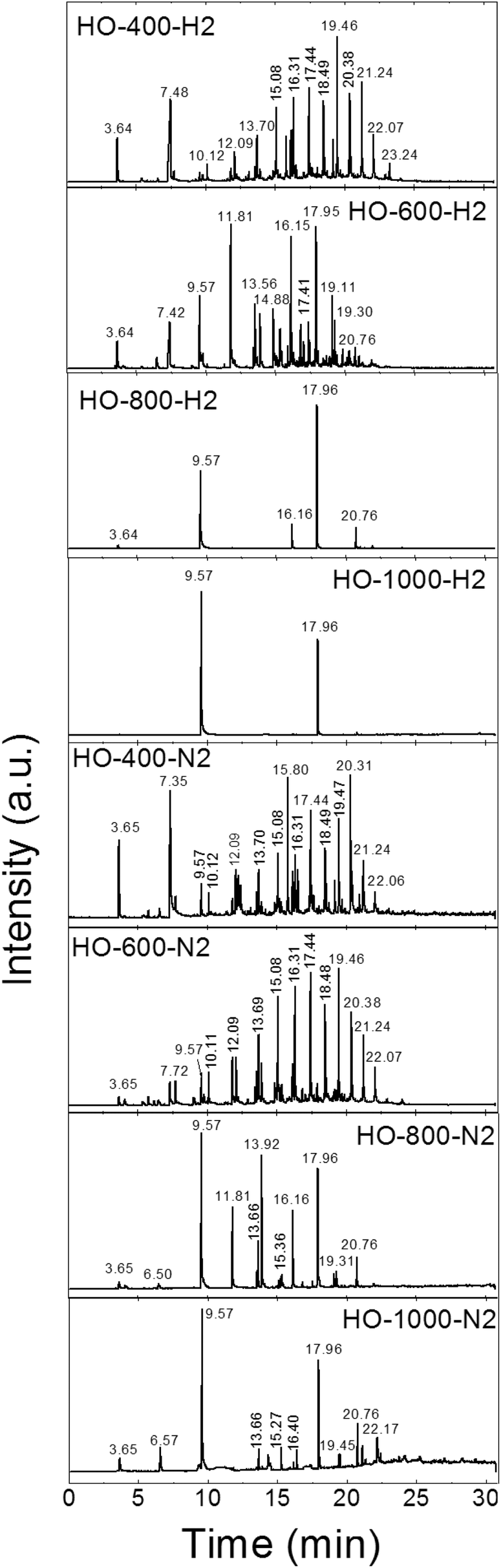 | ||
| Fig. 2 The chromatographs of the species evolved during the thermal exfoliation/reduction of HO-GO. The respective retention times are summarized in Table 6. | ||
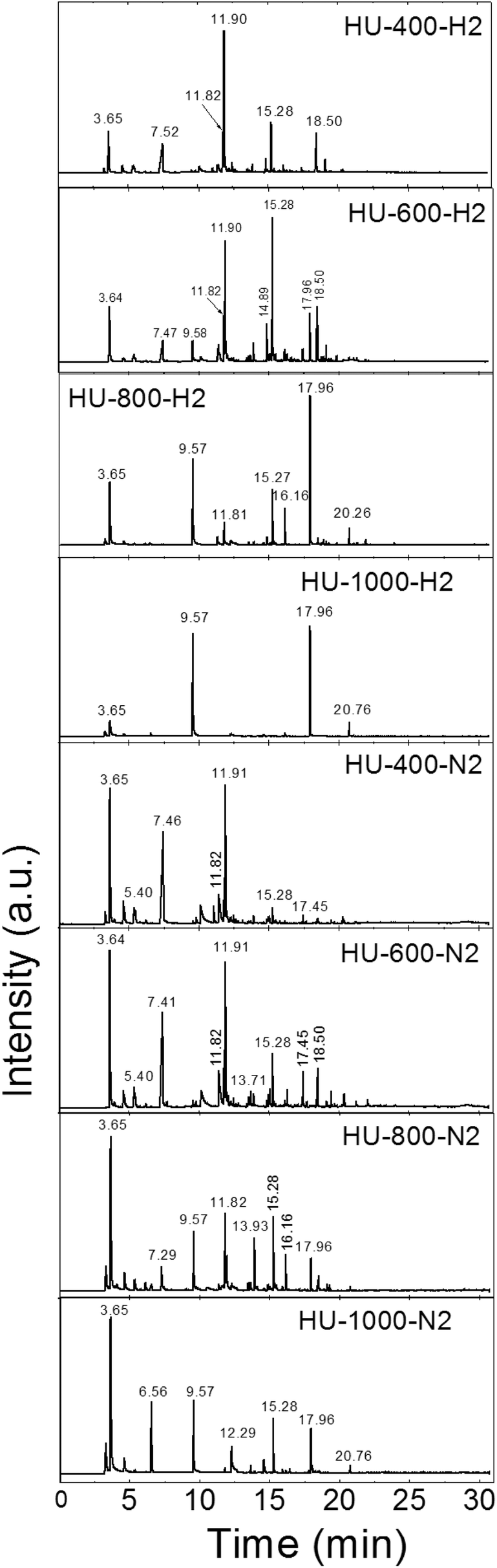 | ||
| Fig. 3 The chromatographs of the species evolved during the thermal exfoliation/reduction of HU-GO. The respective retention times are summarized in Table 6. | ||
| Retention time | Species |
|---|---|
| 3.64–3.65 | Sulfur dioxide |
| 5.40 | Acetonitrile, acetone or isopropyl alcohol |
| 6.50 | Cyclopentadiene |
| 6.56–6.57 | Carbon disulphide |
| 7.29–7.52 | Acetic acid |
| 7.72 | Hexene |
| 9.57–9.58 | Benzene |
| 10.12–10.18 | Heptene or formamide |
| 11.81–11.82 | Toluene |
| 11.90–11.91 | Dimethylformamide |
| 12.09 | Octene |
| 13.56–13.71 | Xylene or nonene |
| 13.92–13.93 | Styrene |
| 14.88–14.89 | Phenol |
| 15.08 | Decene |
| 15.27–15.28 | Benzonitrile |
| 16.16 | Cresol |
| 16.30–16.40 | Undecene |
| 17.41–17.45 | Dodecene |
| 17.96 | Naphthalene |
| 18.44–18.60 | Tridecene |
| 18.60–19.13 | Dicyanobenzene or methylnaphthalene |
| 19.30–19.47 | Tetradecene |
| 20.26–20.38 | 1-Chlordodecane or pentadecene |
| 20.76 | Acenaphthene |
| 21.24 | Hexadecene |
| 21.94 | Fluorene |
| 22.06–22.17 | Heptadecene |
| 23.24 | Octadecene |
The exfoliation mechanism of HO-GO under a hydrogen atmosphere was very similar to that of HO-400-H2 and HO-600-H2, while the mechanism for HO-800-H2 and HO-1000-H2 was entirely different. Only benzene and naphthalene were detected for HO-GO at 1000 °C. At 800 °C benzene and naphthalene were found as the major exfoliation products, but small amounts of sulfur dioxide, cresol and acenaphthene were also detected. At 600 °C all before-mentioned molecules were observed, however in significantly smaller amounts. In addition, lots of different species were detected such as acetic acid, toluene, xylene, nonane, phenol, dodecene and methylnaphthalene. Interestingly, the largest amounts were identified in the case of toluene, naphthalene, and cresol while only a minute amount of toxic benzene was detected. At 400 °C no aromatic molecules like benzene were detected in the evolved gaseous products. In this case, the major evolved products were sulfur dioxide, acetyl acid, alkanes and alkenes. The absence of benzene indicates that the minimal temperature for graphene etching by hydrogen is below 600 °C.
The mechanism of HO-GO exfoliation under a nitrogen atmosphere was relatively similar (samples HO-400-N2, HO-600-N2, HO-800-N2 and HO-1000-N2); however, broader spectra of various species were detected. Also a significantly lower amount of organic hydrocarbons is evolved in nitrogen compared to hydrogen. At 1000 °C benzene and naphthalene were also present as the main exfoliation products. In addition, peaks corresponding to sulfur dioxide, carbon disulphide, xylene, benzonitrile and acenaphthene are clearly visible from the chromatograph suggesting a slightly different mechanism. At 800 °C toluene and styrene were detected, too. At 600 °C the amount of aromatic molecules dramatically decreased and the major exfoliation products were unsaturated aliphatic hydrocarbons such as hexene, heptene, octene, nonene, decene, undecene, dodecene, tridecene, pentadecene, hexadecene and heptadecene. The formation of such large amounts of hydrocarbons indicates site etching of graphene sheets at elevated temperatures. At 400 °C the mechanism was similar to the mechanism observed at 600 °C with a low amount of aromatic and a high amount of aliphatic hydrocarbons. Lower temperatures used for exfoliation are also more suitable for environment protection in this case.
The exfoliation mechanism for the second graphite oxide, HU-GO, under a hydrogen atmosphere is again very similar to that for HU-400-H2 and HU-600-H2, while HU-800-H2 and HU-1000-H2 exhibit entirely different behavior. Benzene and naphthalene and also minor products such as sulfur dioxide and acenaphthene were detected as the major exfoliation products of HU-GO at 1000 °C. At 800 °C toluene, benzonitrile and cresol were also obtained. At 600 °C the overall amount of aromatic hydrocarbons decreased, while new species such as dimethylformamide, acetyl acid and tridecene were detected. At 400 °C dimethylformamide was the main product of exfoliation.
The mechanism of HU-GO exfoliation under a nitrogen atmosphere was relatively similar (samples HU-400-N2, HU-600-N2, HU-800-N2 and HU-1000-N2). Like in the case of HO-GO, a significantly broader spectrum of volatile organic compounds is formed, however their amounts are relatively low. At 1000 °C carbon disulphide and benzonitrile were detected in addition to benzene, naphthalene, sulfur dioxide and acenaphthene. Moreover, acetic acid, toluene and styrene were observed at 800 °C. At 600 °C, an even higher quantity of acetic acid was observed along with a large amount of dimethylformamide. Similar results were observed at 400 °C where the main products were sulfur dioxide, acetic acid and dimethylformamide suggesting that this temperature is suitable for avoiding the formation of toxic benzene and other aromatic hydrocarbons. Surprisingly, compared to HO-GO, only trace amounts of aliphatic hydrocarbons are observed. This indicates significant differences in the chemistry of the starting graphite oxides.
Compared to the nitrogen atmosphere, the hydrogen atmosphere combined with high temperatures caused simultaneous hydrogenation of graphene edges and preferential etching of carbon leading to the formation of a high amount of benzene and naphthalene. At lower temperatures, the etching of the graphene skeleton was suppressed and the concentration of aromatic hydrocarbons was significantly reduced. Except for carbon, oxygen and hydrogen, some evolved species contained also other elements such as sulfur (sulfur dioxide, carbon disulphide) and nitrogen (formamide, dimethylformamide, benzonitrile, dicyanobenzene, acetonitrile). Sulfur and nitrogen originated from the impurities in graphite oxides introduced during the oxidation of graphite and also by radical reactions under the exfoliation atmosphere (nitrogen). The presence of nitrogen in the form of nitrile groups indicates such a radical mechanism of reaction under the exfoliation atmosphere. Surprisingly, only minor quantities of oxygen containing molecules were detected, since most oxygen functionalities are decomposed to CO2 and CO. From the oxygen containing molecules, acetone, isopropanol, sulfur dioxide, acetic acid, cresol and phenol were detected. Sulfur dioxide originates from the thermal decomposition of sulfur present in graphene oxide. The sulfur is most likely present in the form of sulfuric acid esters, which decompose to sulfur dioxide during the thermal reduction of graphene oxide.33 The mechanism of oxygen containing volatile hydrocarbons’ formation is difficult to explain and can result from both the direct decomposition of oxygen functionalities on graphene and the radical reactions of exfoliation products. Tearing of the aliphatic hydrocarbons from graphene edges at low temperatures is also very unusual. This effect is dominant for HO-GO, while in the HU-GO the aromatic hydrocarbons are more frequent. This can give an idea of how the different types of oxygen functionalities can influence the etching of graphene sheet sites. The ketone groups in HU-GO form carbon oxides at elevated temperatures together with defects in the graphene skeleton from missing carbon atoms. This can cause a weakening of the graphene skeleton strength and the whole aromatic rings can be ripped off. On the other hand, more stable hydroxyl groups are more likely eliminated from the graphene skeleton by a radical reaction without inducing defects in the structure.
The quantification of hydrocarbons formed during the exfoliation procedure was performed by their sorption on activated charcoal (see the Experimental section). The detection limit of this method is slightly below 0.1 mg per kg of graphene oxide. The quantification was performed for most abundant species like benzene, toluene, ethylbenzene, xylene and naphthalene. The most interesting result is that the highest amount of hydrocarbons was observed for the thermally reduced graphene oxide prepared at 800 °C. The lower amount of hydrocarbons observed at 1000 °C can be explained by their thermal pyrolysis in the exfoliation reactor subsequently after their formation. A significantly lower amount of hydrocarbons like benzene was observed for the exfoliation at low temperatures and also under a nitrogen atmosphere in comparison with the hydrogen atmosphere. Significant differences in the amount of formed benzene and other hydrocarbons were observed for the thermal reduction of graphene oxide prepared by the chlorate method in comparison with the permanganate method. This is an important finding, since the previous results show a higher degree of exfoliation for thermally reduced graphene oxide prepared by the chlorate method. The results are summarized in Table 7.
| Sample | Benzene | Toluene | Ethylbenzene | Xylene | Naphthalene |
|---|---|---|---|---|---|
| HO-400-H2 | 4.5 | 6.2 | 3.7 | 0.0 | 0.0 |
| HO-600-H2 | 37.3 | 17.4 | 1.9 | 2.4 | 0.2 |
| HO-800-H2 | 202.5 | 1.8 | 0.0 | 0.0 | 0.8 |
| HO-1000-H2 | 144.8 | 0.4 | 0.0 | 0.0 | 0.5 |
| HO-400-N2 | 3.5 | 1.6 | 0.2 | 0.2 | 0.0 |
| HO-600-N2 | 20.3 | 6.4 | 1.7 | 0.9 | 0.0 |
| HO-800-N2 | 53.7 | 5.7 | 0.0 | 0.1 | 0.0 |
| HO-1000-N2 | 38.8 | 0.3 | 0.0 | 0.0 | 0.0 |
| HU-400-H2 | 2.6 | 1.6 | 0.3 | 0.3 | 0.0 |
| HU-600-H2 | 21.7 | 7.6 | 0.8 | 1.0 | 0.2 |
| HU-800-H2 | 186.6 | 1.2 | 0.0 | 0.0 | 0.8 |
| HU-1000-H2 | 121.9 | 0.3 | 0.0 | 0.0 | 0.4 |
| HU-400-N2 | 1.1 | 1.1 | 0.2 | 0.4 | 0.1 |
| HU-600-N2 | 10.5 | 3.5 | 1.4 | 0.8 | 0.1 |
| HU-800-N2 | 40.8 | 7.6 | 0.0 | 0.7 | 0.3 |
| HU-1000-N2 | 34.7 | 0.2 | 0.0 | 0.0 | 0.2 |
The exfoliation process was further investigated by thermal analysis using thermogravimetry (TG) combined with differential thermal analysis (DTA) coupled with mass spectrometry (MS). This method is less sensitive in comparison with GCMS, however it is suitable for the qualitative analysis of the major gaseous species formed during graphene oxide exfoliation. The MS analysis shows the majority of water, carbon dioxide and carbon monoxide evolution during the exfoliation of graphene oxide which takes place in the temperature range of 180–220 °C depending on the method of graphene oxide synthesis. The total amount of the evolved major gaseous exfoliation products can be seen from the TG curve and is several orders of magnitude higher in comparison with the amount of hydrocarbons evolved during exfoliation. The results are shown in Fig. 4.
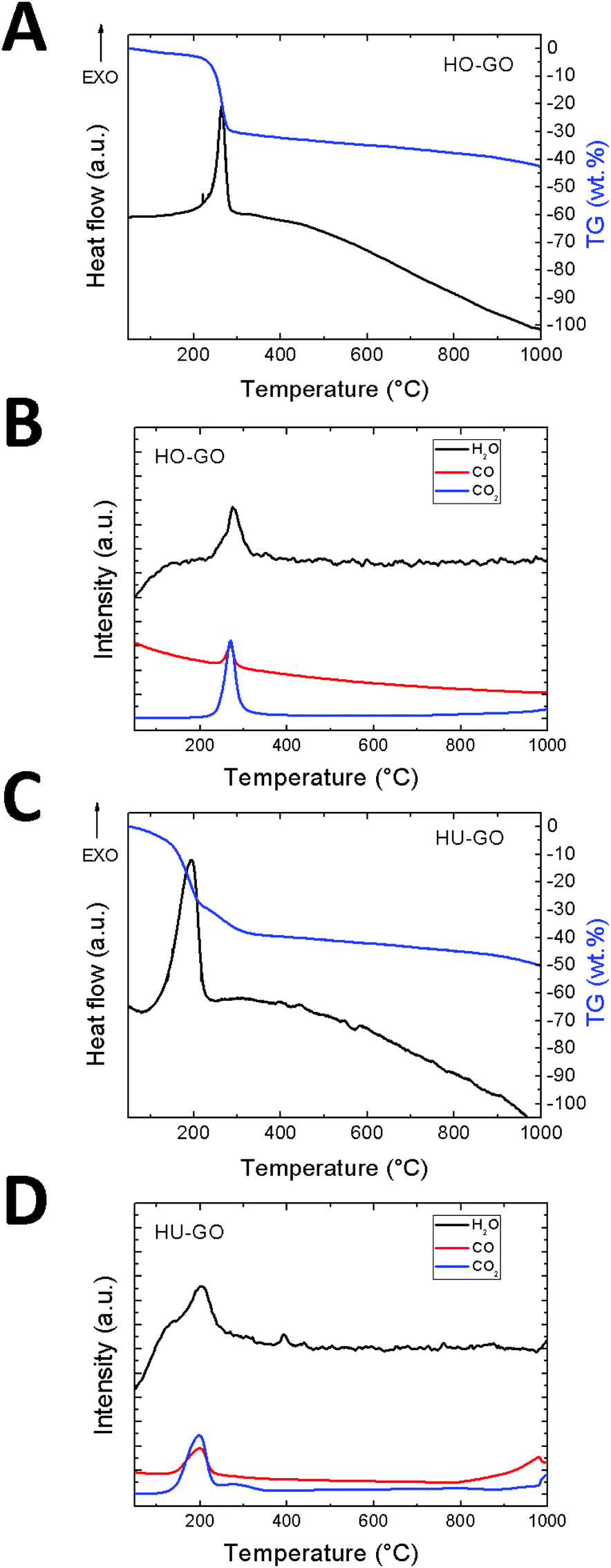 | ||
| Fig. 4 The DSC/TG analysis combined with simultaneous MS of graphene oxide exfoliated under a nitrogen atmosphere: (A) DTA and TG of HO-GO. (B) MS of HO-GO, (C) DTA and TG of HU-GO. (D) MS of HU-GO. | ||
In order to evaluate the possible application of thermally reduced graphene oxides prepared at various temperatures for electrocatalysis, we performed electrochemical characterization by cyclic voltammetry. The experiments using an [Fe(CN)6]3−/4− redox probe showed the comparable heterogeneous electron transfer (HET) rate for graphene oxide exfoliated at 600 °C as well as at higher temperatures. However the HET rate represented by the value of peak-to-peak separation was strongly influenced by the exfoliation atmosphere as well as by the method of graphite oxide synthesis. In general, the increase of HET rate is observed with the increase of the exfoliation temperature which is also associated with the well-known effect of the C/O ratio on the improvement of the HET rate. However, the most significant improvement of the HET rate is observed between the GO exfoliated at 400 °C and 600 °C. The HET rate is an important parameter for a broad range of graphene applications in energy storage and conversion as well as in electrochemical sensing applications. The values of the HET rate represented as the peak-to-peak separation values are summarized in Table 8 (the measured data are shown in Fig. 5). The comparable values revealed the possibility of lower temperature preparation of graphene which is a significantly less energy consuming process and, moreover, it brings an advantage of avoiding or at least a substantial reduction of highly toxic by-products based on aromatic hydrocarbons.
 | ||
| Fig. 5 The cyclic voltammogram for the thermally reduced graphene oxides. Scan rate 100 mV s−1, 50 mM PBS (pH = 7.2) with 10 mM K4[Fe(CN)6]. Potential is referred to as the KCl saturated Ag/AgCl electrode. | ||
| Sample | Peak-to-peak separation (mV) | Ox/red pot. (mV) |
|---|---|---|
| HO-400-H2 | 404 | 516/112 |
| HO-600-H2 | 392 | 495/103 |
| HO-800-H2 | 256 | 407/151 |
| HO-1000-H2 | 208 | 392/184 |
| HO-400-N2 | 389 | 490/101 |
| HO-600-N2 | 207 | 391/184 |
| HO-800-N2 | 206 | 390/184 |
| HO-1000-N2 | 178 | 365/187 |
| HU-400-H2 | 346 | 465/119 |
| HU-600-H2 | 248 | 393/145 |
| HU-800-H2 | 344 | 470/126 |
| HU-1000-H2 | 308 | 444/136 |
| HU-400-N2 | 514 | 557/43 |
| HU-600-N2 | 320 | 454/134 |
| HU-800-N2 | 341 | 491/150 |
| HU-1000-N2 | 320 | 457/137 |
Conclusions
We have demonstrated that the thermal exfoliation of graphite oxide leads not only to the formation of simple molecules (carbon dioxide, carbon monoxide, water), but also to many complex organic species. This process is strongly dependent on the exfoliation atmosphere, temperature as well as the method of graphite oxide synthesis. Carcinogenic aromatic hydrocarbons were in particular formed at high temperatures under a hydrogen atmosphere. This suggests a significant etching of graphene by hydrogen at an elevated temperature inducing the elimination of the whole C6 rings and their multiples like naphthalene and phenanthrene. The high amount of evolved hydrocarbons under a hydrogen atmosphere was confirmed by quantitative analysis showing over 200 mg of benzene formed per kg of graphene oxide used for exfoliation. Almost one order lower amount of hydrocarbons was formed under a nitrogen atmosphere. The highest amount of hydrocarbons was observed at 800 °C and the use of a higher temperature led to the thermal decomposition of the evolved hydrocarbons. On the other hand, thermal reduction at lower temperatures led dominantly to the formation of less toxic aliphatic compounds. Moreover lower temperatures during the large scale preparation will bring enormous cost saving. Further characterization confirmed that thermally reduced graphene oxides can be prepared even at lower temperatures with suitable properties for various electrochemical applications. The formation of toxic hydrocarbons during the synthesis of thermally reduced graphene oxide must be considered for its industrial production.Acknowledgements
The project was supported by the Czech Science Foundation (GACR No. 15-09001S and 16-05167S) and financial support was received from specific university research (MSMT No. 20-SVV/2016). M. P. acknowledges a Tier 2 grant (MOE2013-T2-1-056; ARC 35/13) from the Ministry of Education, Singapore.References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- Y. Zhang, T.-T. Tang, C. Girit, Z. Hao, M. C. Martin, A. Zettl, M. F. Crommie, Y. R. Shen and F. Wang, Nature, 2009, 459, 820–823 CrossRef CAS PubMed.
- M. Katsnelson, K. Novoselov and A. Geim, Nat. Phys., 2006, 2, 620–625 CrossRef CAS.
- X. Wang, L. Zhi and K. Müllen, Nano Lett., 2008, 8, 323–327 CrossRef CAS PubMed.
- K. S. Novoselov, V. Fal, L. Colombo, P. Gellert, M. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- F. Bonaccorso, Z. Sun, T. Hasan and A. Ferrari, Nat. Photonics, 2010, 4, 611–622 CrossRef CAS.
- N. Yang, J. Zhai, D. Wang, Y. Chen and L. Jiang, ACS Nano, 2010, 4, 887–894 CrossRef CAS PubMed.
- O. Jankovsky, P. Simek, D. Sedmidubsky, S. Huber, M. Pumera and Z. Sofer, RSC Adv., 2014, 4, 7418–7424 RSC.
- X. Deng, L. Lü, H. Li and F. Luo, J. Hazard. Mater., 2010, 183, 923–930 CrossRef CAS PubMed.
- O. Jankovský, P. Šimek, K. Klímová, D. Sedmidubský, M. Pumera and Z. Sofer, Carbon, 2015, 89, 121–129 CrossRef.
- O. Jankovský, D. Sedmidubský, P. Šimek, K. Klímová, D. Bouša, C. Boothroyd, A. Macková and Z. Sofer, Phys. Chem. Chem. Phys., 2015, 17, 25272–25277 RSC.
- A. Martinez, K. Fuse and S. Yamashita, Appl. Phys. Lett., 2011, 99, 121107 CrossRef.
- A. N. Obraztsov, Nat. Nanotechnol., 2009, 4, 212–213 CrossRef CAS PubMed.
- S. Yang, M. R. Lohe, K. Müllen and X. Feng, Adv. Mater., 2016, 28, 6213–6221 CrossRef CAS PubMed.
- A. M. Abdelkader, A. J. Cooper, R. A. W. Dryfe and I. A. Kinloch, Nanoscale, 2015, 7, 6944–6956 RSC.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 CrossRef CAS PubMed.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- R. K. Joshi, S. Alwarappan, M. Yoshimura, V. Sahajwalla and Y. Nishina, Appl. Mater. Today, 2015, 1, 1–12 CrossRef.
- C. H. A. Wong, O. Jankovský, Z. Sofer and M. Pumera, Carbon, 2014, 77, 508–517 CrossRef CAS.
- P. Šimek, Z. Sofer, O. Jankovský, D. Sedmidubský and M. Pumera, Adv. Funct. Mater., 2014, 24, 4877–4877 CrossRef.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- X. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2009, 114, 832–842 Search PubMed.
- A. B. Bourlinos, D. Gournis, D. Petridis, T. Szabó, A. Szeri and I. Dékány, Langmuir, 2003, 19, 6050–6055 CrossRef CAS.
- O. Jankovský, P. Šimek, J. Luxa, D. Sedmidubský, I. Tomandl, A. Macková, R. Mikšová, P. Malinský, M. Pumera and Z. Sofer, ChemPlusChem, 2015, 80, 1399–1407 CrossRef.
- O. Jankovský, P. Šimek, M. Nováček, J. Luxa, D. Sedmidubský, M. Pumera, A. Macková, R. Mikšová and Z. Sofer, RSC Adv., 2015, 5, 18733–18739 RSC.
- Z. Sofer, O. Jankovský, A. Libánská, P. Šimek, M. Nováček, D. Sedmidubský, A. Macková, R. Mikšová and M. Pumera, Nanoscale, 2015, 7, 10535–10543 RSC.
- V. H. Pham, H. D. Pham, T. T. Dang, S. H. Hur, E. J. Kim, B. S. Kong, S. Kim and J. S. Chung, J. Mater. Chem., 2012, 22, 10530–10536 RSC.
- W. Chen, L. Yan and P. R. Bangal, Carbon, 2010, 48, 1146–1152 CrossRef CAS.
- H. Wang, J. T. Robinson, X. Li and H. Dai, J. Am. Chem. Soc., 2009, 131, 9910–9911 CrossRef CAS PubMed.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef CAS.
- O. Jankovský, P. Marvan, M. Nováček, J. Luxa, V. Mazánek, K. Klímová, D. Sedmidubský and Z. Sofer, Appl. Mater. Today, 2016, 4, 45–53 CrossRef.
- C. K. Chua, A. Ambrosi, Z. Sofer, A. Macková, V. Havránek, I. Tomandl and M. Pumera, Chem. – Eur. J., 2014, 20, 15760–15767 CrossRef CAS PubMed.
- Z. Sofer, P. Šimek, O. Jankovský, D. Sedmidubský, P. Beran and M. Pumera, Nanoscale, 2014, 6, 13082–13089 RSC.
- B. Zhao, P. Liu, Y. Jiang, D. Pan, H. Tao, J. Song, T. Fang and W. Xu, J. Power Sources, 2012, 198, 423–427 CrossRef CAS.
- I. Jung, D. A. Dikin, R. D. Piner and R. S. Ruoff, Nano Lett., 2008, 8, 4283–4287 CrossRef CAS PubMed.
- R. Larciprete, S. Fabris, T. Sun, P. Lacovig, A. Baraldi and S. Lizzit, J. Am. Chem. Soc., 2011, 133, 17315–17321 CrossRef CAS PubMed.
- O. Jankovsky, S. Hrdlickova Kuckova, M. Pumera, P. Simek, D. Sedmidubsky and Z. Sofer, New J. Chem., 2014, 38, 5700–5705 RSC.
- Z. Sofer, P. Simek and M. Pumera, Phys. Chem. Chem. Phys., 2013, 15, 9257–9264 RSC.
- Z. Sofer, O. Jankovský, P. Simek, D. Sedmidubsky, J. Šturala, J. Kosina, R. Mikšová, A. Macková, M. Mikulics and M. Pumera, ACS Nano, 2015, 9, 5478–5485 CrossRef CAS PubMed.
- U. Hofmann and A. Frenzel, Kolloid-Z., 1934, 68, 149–151 CrossRef CAS.
- W. Hummers and R. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- D. R. Dreyer, R. S. Ruoff and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 9336–9344 CrossRef CAS PubMed.
- M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751–758 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc02491b |
| This journal is © The Royal Society of Chemistry 2016 |