DOI:
10.1039/C6GC02477G
(Critical Review)
Green Chem., 2016,
18, 5736-5750
Sustainable hybrid photocatalysts: titania immobilized on carbon materials derived from renewable and biodegradable resources
Received
4th September 2016
, Accepted 20th September 2016
First published on 22nd September 2016
Abstract
This review comprises the preparation, properties and heterogeneous photocatalytic applications of TiO2 immobilized on carbon materials derived from earth-abundant, renewable and biodegradable agricultural residues and sea food waste resources. The overview provides key scientific insights into widely used TiO2 supported on carbonaceous materials emanating from biopolymeric materials such as lignin, cellulose, cellulose acetate, bacterial cellulose, bamboo, wood, starch, chitosan and agricultural residues (biochar, charcoal, activated carbon and their magnetic forms, coal fly ash) or seafood wastes namely eggshell, clamshell and fish scales; materials that serve as a support/template for TiO2. Heightened awareness and future inspirational developments for the valorisation of various forms of carbonaceous functional materials is the main objective. This appraisal abridges various strategies available to upgrade renewable carbon-based feedstock via the generation of sustainable TiO2/carbon functional materials and provides remarks on their future prospects. Hopefully, this will stimulate the development of efficient and novel composite photocatalysts and engender the necessary knowledge base for further advancements in greener photocatalytic technologies.
 Juan Carlos Colmenares | Prof. Dr Juan Carlos Colmenares is graduated from Warsaw University of Technology in 1995 and obtained his MSc (1997) and PhD (2004) from the same University, and the scientific degree of habilitation (DSc 2015) from the Polish Academy of Sciences. His interests include materials science, nanotechnology, heterogeneous catalysis, biomass/CO2 valorisation, solar chemicals, sonochemistry, photocatalysis and water/air purification. After obtaining his PhD, he conducted postdoctoral work at the University of Cordoba, Spain (2005–2006) in Prof. Marinas's group and at the University of Southern California, Los Angeles (USA) (2006–2009) in Prof. G. A. Olah’s group. |
 Rajender S. Varma | Prof. Dr Rajender S. Varma (PhD, Delhi University 1976) was born in India. After postdoctoral research at Robert Robinson Laboratories, Liverpool, UK, he was a faculty member at Baylor College of Medicine and Sam Houston State University before joining the US Environmental Protection Agency in 1999. He has over 40 years of experience in the management of multi-disciplinary programs in areas ranging from environmentally benign alternatives for synthesis using mechanochemical, microwave- and ultrasound irradiation, to technologies for remediation of contaminated sites. Lately, he works on greener assembly of nanomaterials and the application of magnetically retrievable nanocatalysts. |
 Paweł Lisowski | Paweł Lisowski is currently a PhD candidate at the Institute of Physical Chemistry of the Polish Academy of Sciences in Poland. He raduated from Radom University of Technology and obtained his MSc (2007) in Chemical Technology with specialization in protection of the environment and post-graduate studies in Analytical Chemistry in Industry and Environmental Protection at AGH University of Science and Technology located in Krakow, Faculty of Materials Science and Ceramics. His research currently covers various aspects of rational design and synthesis of new inorganic–organic hybrid materials from renewable materials as a carbon support with photocatalytic properties using unconventional methodologies. |
1. Introduction
The use of relatively non-toxic and earth-abundant local materials in chemical conversion is a prerequisite for the development of any sustainable process. Titanium dioxide (TiO2) is one of the most commonly used photocatalysts and carbon can be accessed from a wide variety of waste resources. Carbon-based materials have been utilized in environmental remediation and now making inroads into an emerging and conceptually new area of photocatalytic materials; their unique features need to be exploited for larger scale industrial use. While tremendous progress has been made in the syntheses of porous carbon-based materials with varying structure-types, challenging opportunities remain for the functionalization of these materials in terms of the optimization of their properties for specific applications namely separation science, filtration devices, photocatalyst supports and energy storage.1–3 When compared to conventional resources, carbon materials possess outstanding properties such as hydrophobic surfaces, high surface area, large pore volume, chemical inertness, good thermal and mechanical stability, easy handling and low cost of manufacture, that will enable their enhanced use in energy-conversion and storage.1–4 The synergistic effects of TiO2 with carbon materials bearing enhanced multi-functionalities for use in heterogeneous photocatalysis, energy-storage, and solar cell applications, renders TiO2 immobilized on porous carbon as a valuable material. In the area of environmental remediation, carbon materials offer a great potential for the efficient removal of pollutants in water and air,5,6 as has been demonstrated by emerging heterogeneous photocatalysis as an alternative economical and benign technology for the purification of water and air.7,8 From a practical point of view, alternative photocatalytic materials that can match the TiO2 profile, namely versatility, inexpensiveness and abundance, and a non-toxic nature are hard to find. However, TiO2 still has some shortcomings which prevent its widespread usage. First, the large band gap energy of TiO2 (3.2 eV) limits its use exclusively to the ultraviolet region of the solar spectrum which accounts for only a very small fraction of the solar energy (about 3–5%).9–13 Second is the need to support nanostructured titania for better filtration and material recovery. Although a significant amount of research has been conducted on TiO2 photocatalysis at the laboratory scale its application on an industrial scale is limited, which needs to be addressed. To overcome the severe drawbacks of the most stable photocatalyst, TiO2, namely its low efficiency, narrow light-response range and material recycling, many strategies have been developed via modification to acquire multifunctional materials that increase its photocatalytic activity for organic pollutant treatment;14,15 comprehensive articles have documented these advancements in the field of TiO2-based photocatalysts3–9,16,17 and especially the use of carbon as a support for TiO2.18–22 Petroleum-derived polymers, when used as a support for TiO2 (e.g. high-density polyethylene (HDPE),23 polyaniline (PANI),24 resorcinol-formaldehyde (RF)25), have drawbacks such as non-renewability, higher costs, and additional contribution to pollution.26 Faced with such a dilemma, more and more attention has been paid to the development and application of natural materials as a support for TiO2. In the context of green chemistry, natural renewable materials such as lignocelluloses and raw biomass are especially attractive for their unique physicochemical properties such as good electrical conductivity, higher surface area and excellent chemical stability.27–30 We expect that such a comprehensive and critical review will provide key scientific insights into TiO2/carbon materials derived from renewable and biodegradable resources emanating from biopolymeric materials such as lignin, cellulose, cellulose acetate, bacterial cellulose, bamboo, wood, starch, chitosan and agricultural residues (biochar, charcoal, activated carbon and their magnetic forms, coal fly ash) or seafood wastes (namely eggshell, clamshell and fish scales); materials that serve as a support/template for TiO2. A keen awareness of future inspirational developments for the valorisation of various forms of carbonaceous functional materials is another objective. This review abridges different approaches or/and strategies which have been reported so far to upgrade renewable carbon-based feedstocks via the preparation of sustainable TiO2/carbon functional materials and provides comments on their future prospects that will help stimulate the development of efficient and novel composite photocatalysts; the attained knowledge-base will boost advancements in greener photocatalytic technologies.
2. Emerging role of carbonaceous materials in semiconductor photocatalysis
The semiconductor photocatalyst, TiO2, is an important material that underpins the development of critical renewable energy and environmental technologies such as photocatalytic water/air purification, hydrogen production from water splitting, and high-efficiency/low-cost solar cells.31 In photocatalysis, the electron/hole pairs are the photo-excited charge carriers on the conduction and valence bands, respectively. The free holes have strong oxidation power, while the oxygen radicals are secondary products of the electron reduction process;32 a simplified mechanism for the synergetic photocatalytic effect of TiO2/carbon support is presented in Fig. 1. TiO2 is activated directly by UV light irradiation of photon energy higher than the band gap energy of titania, forming e−/h+ pairs (Fig. 1a). More importantly, carbon plays the central role of an electron reservoir to conduct away the electrons from the e−/h+ pairs on/in TiO2, thus leading to the increased efficiency of charge separation in TiO2; the activation of TiO2, and in the meantime the activation of an electron-conductive phase (such as carbon) which activates O2 to O− ions, takes place (Fig. 1b).33
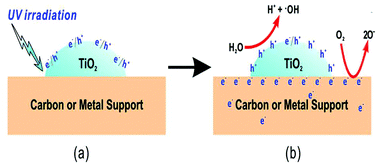 |
| | Fig. 1 Schematic illustrations of the proposed synergetic photocatalytic effect of the TiO2/carbon support. Reproduced with permission from ref. 33. | |
3. TiO2/carbon materials derived from lignocellulosic materials
Carbon materials derived from biopolymers have made considerable progress in recent years, especially in the development of molecular sieves, as photocatalyst supports and adsorbents for the storage of natural gas.26,34,35 Compared to the thermal method, photocatalytic polymerization is initiated with light and is terminated upon switching off the light. Furthermore, the molecular weight and density of the polymer can be varied through the selection of monomer and irradiation conditions. Carbonization of the carefully designed polymer provides opportunities in controlling the pore size, pore volume and bulk density of the ensuing carbon, which are important characteristics in the aforementioned applications.26,34,35 The composition of lignocellulose biomass is highly dependent on its source (Fig. 2). There is a significant variation of the lignin and (hemi)cellulose components of the lignocellulose component on whether it is derived from hardwood, softwood, or grasses, extracts, or inorganic related materials.36–38 Starch (as well as sugars), triglycerides and lignocellulose are the typical classes of feedstocks derived from biomass and are used for the production of renewable biofuels and chemicals. Lignin, a three-dimensional network macromolecule, is electronegative and has a strong affinity for the positively charged metal ions; lignin with phenolic hydroxyl-, alcoholic hydroxyl-, and carboxylic groups can react chemically with other polymers to form hybrid composites.39 On the other hand, cellulose, an organic polysaccharide comprising a linear chain of several hundreds to many thousands of β(1→4) links viaD-glucose units, is mainly used to produce paper and paperboard. It has multiple hydroxyl groups on one long chain to form hydrogen bonds with other oxygen atoms on the neighboring chain.40
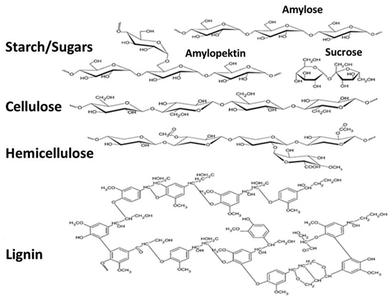 |
| | Fig. 2 Overview of the prominent fractions of the biomass feedstock. | |
3.1 TiO2/lignin
Native unmodified lignin has been used as a template for the formation of mesoporous TiO2 nanoparticles via a hydrolysis precipitation method exploiting the electronegativity and the network skeleton of lignin (see Fig. 3);41 as-synthesized composite had a BET surface area of 165.8 m2 g−1 and a pore volume of 0.312 cm3 g−1. In view of the interactions between hydroxyl groups of lignin and the surface hydroxyl groups of the TiO2 precursor, the calcined TiO2 particles have a finer crystallite size, a mesoporous structure and are well separated, to avoid grain growth (Fig. 3). However, titanium atom has a relatively low electronegativity and can react rapidly in a nucleophilic reaction medium in which lignin, a polycyclic and three-dimensional mesh organic macromolecule with several electronegative OH groups, should have strong affinity for the positively charged metal ions; lignin with phenolic hydroxyl, alcoholic hydroxyl, and carboxyl groups can chemically react with other polymers to form hybrid composites. Lignin obtainable from the black liquor waste of the paper-making process has great potential for the fabrication of TiO2 particles filled with uniformly distributed nano-pores.39,41
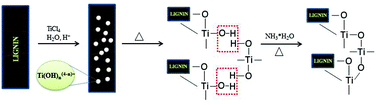 |
| | Fig. 3 Formation mechanism of mesoporous TiO2 with lignin as a template. | |
3.2 TiO2/STARBON®
Native polysaccharides are essentially non-porous and, therefore, their use is rather limited in applications where diffusion and surface interactions are required for optimum performance. Clark et al. pioneered a simple methodology to obtain highly expanded mesoporous polysaccharide materials (e.g. starch) from low surface area native starting materials.42,43 Their synthesis entails three main steps: (1) formation of a gel by heating starch in water; (2) exchange of water with a lower surface tension solvent (e.g., ethanol) at low temperature (≈5 °C); and (3) carbonization of the ensuing porous gels as-such or after drying to achieve the final mesoporous carbons (Fig. 4);42,43 additional details are available on these synthetic methodologies43–46 including TiO2 supported on STARBON-800® as shown in Fig. 5.48 Starbon® materials, derived from the controlled carbonization of different polysaccharides, exhibit chemical and surface properties that are intermediate between polysaccharides and carbons depending on the degree of carbonization;42,43 their synthesis avoids the use of templates and can be performed at a temperature of choice (e.g., 200–1000 °C). Another important feature is that these materials exhibit outstanding mesoporous textural properties, with pore volumes and sizes analogous to materials prepared via the traditional hard templating routes. The hydrophilic to hydrophobic nature of the carbonaceous product could be controlled by the degree of carbonization. Additionally, the flexible carbonization temperatures provide the possibility of tuneable surface chemistries, not accessible through hard templating (which generally needs higher temperatures, >700 °C) or soft templating processes based on polymeric organic templates (which limit the available post-processing surface functionalities). This simple and gentle process can produce a whole range of mesoporous carbon-based materials, including amorphous carbon, that can serve as excellent catalysts or catalyst supports.43–46 Interestingly, carboxylic groups can be formed on the surface after initial thermal treatment at 400 °C under an oxygen-deficient atmosphere (Fig. 5a). Then, each carboxyl group acts as an individual nucleation site for TiO2 formation which is facilitated by hydrolysis and condensation reactions promoted via sonication. Finally, the hybrid material (TiO2/STARBON®) is consolidated after thermal treatment at 400 °C in static air. These conditions preserve a pure and highly crystalline anatase phase (ca. 30 nm) leading to a reduction in the electron–hole recombination rate at the Starbon surface. TiO2 is strongly anchored to the STARBON-800® structure as no leaching is observed even after 240 min of photocatalytic degradation of phenol (Fig. 5b); STARBON-800® enhances the photoelectron conversion of TiO2 when compared to Norit and graphene oxide supports by reducing the recombination of photo-generated electron–hole pairs.48 Coating TiO2 photocatalyst on the surface of Starbon serves a dual purpose, i.e. it works as a support for nanosized TiO2 which helps bring pollutants and intermediates around the TiO2. Second, the TiO2 can effectively destroy the adsorbed pollutants thus regenerating the Starbon in situ (only 3% of the initial concentration of phenol still stayed on Starbon surface after 240 minutes of reaction in comparison with 8.7% for STARBON+P25).
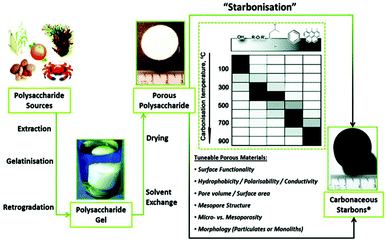 |
| | Fig. 4 A general overview of preparation of Starbons®, involving sourcing of the polysaccharide, followed by the preparation of an aqueous gel, its controlled drying and subsequent conversion to carbonaceous Starbons®. Reproduced with permission from ref. 47. | |
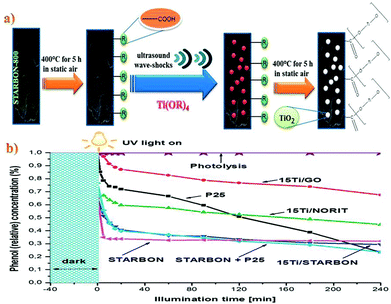 |
| | Fig. 5 (a) Schematic illustration of the plausible mechanism of the formation of TiO2/STARBON hybrid material using ultrasound-assisted wet impregnation. (b) Photocatalyst activities in aqueous phase degradation of phenol (reaction conditions: 150 mL of mother solution, 150 mg of photocatalyst, Cphenol = 50 ppm, temperature 30 °C, reaction pressure 1 bar). Reproduced with permission from ref. 48. | |
3.3 TiO2/cellulose
Novel TiO2/cellulose composites with interfacial silica and siloxane barriers have been prepared.49 Vegetable cellulosic fibers have been surface modified via the hydrolysis of tetraethoxysilane (TEOS), octyltrimethoxysilane (OTMS) or phenyltrimethoxysilane (PTMS), followed by the layer-by-layer deposition of previously synthesized TiO2 nanoparticles. Distinct properties were realised for the cellulosic nanocomposites determined by the nature of the interlayer; while hydrophilic TiO2/SiO2/cellulose composites were generated by hydrolysis of TEOS, the OTMS and PTMS treated fibres (TiO2/OTMS/cellulose and TiO2/PTMS/cellulose) showed hydrophobic characteristics. Subsequently, after sunlight irradiation, the nanocomposite containing untreated fibres acquired a yellow colour indicating the partial degradation of cellulose fibres. Conversely, all the surface modified TiO2 cellulose nanocomposites remained colourless even after several weeks of exposure to sunlight. Therefore, TiO2/cellulose nanocomposites, in view of their compatibility with polymer matrices (hydrophobic or hydrophilic) are considered as forerunners for several applications, including self-cleaning materials and air purification filters. Previous research50 has shown that the visible light activated TiO2/microcrystalline cellulose (MC) nanocatalyst was photocatalytically active in degrading nearly 90% of methylene blue (MB) in 4 h (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 Ti4+/MC synthesis conditions). As soon as electron is trapped by the molecular oxygen present on the TiO2 surface, highly active radicals are generated which are responsible for the photo-bleaching of MB molecules; this obeyed first-order reaction kinetics with reaction constants of 5.02 × 10−3 min−1. The TiO2/MC nanocomposite benefits from the synergistic effect of the physical and/or chemical interactions between the organic and inorganic components and therefore could be utilized as a photocatalyst to degrade hazardous contaminants via the formation of films that have good mechanical and UV-shielding properties and in the synthesis of transition-metal carbide materials.50 On similar lines, Zeng et al.51 prepared TiO2/cellulose composite films, which provided a cavity for the creation of TiO2 nanoparticles, and hydroxyl groups for their immobilization. The prepared materials exhibited a good photocatalytic activity for the photodegradation of high concentration of phenol (67.2 mg L−1) under weak UV light irradiation (0.56 mW cm−2). Another elegant strategy for the preparation of TiO2 nanomaterials has been described52 that applies natural cellulose whiskers, obtainable by hydrolysis of native cellulose as templates. TiO2/cellulose whisker nanoparticles exhibited quantum size effects and a lower band gap energy (2.02 eV) and displayed promising activity for the photodegradation of a methyl orange solution under weak UV light irradiation (4.3–41 mW cm−2). They exhibited good stability against photocorrosion, implying that they could be used in the photodegradation of organic pollutants. It is worth noting that the regenerated cellulose films and fibers in the wet state possess a porous structure and the hydroxyl groups assist the creation of inorganic nanoparticles in the cellulose matrix.53–56 Marques et al.57 examined sheets prepared using a mixture of synthetic TiO2/cellulose nanocomposite. The obtained composite exhibited a much higher opacity than those obtained by mechanical blending of the fibres with commercial TiO2 pigment because of the highly homogeneous distribution of TiO2 and higher specific surface area available for light scattering in sheets with the hybrid material. Such hybrids could be used as reinforcing fibers in a polymer matrix, particularly when light-barrier materials are sought. An interesting concept that involved the preparation of titanium dioxide nanocrystals on cellulose fibers (CF) by in situ hydrolysis has been reported;58 TiO2/CF composite showed enhanced adsorption capacity, good regenerability and selectivity for lead (Pb2+) removal. Moreover, the adsorption capacity of a filter bed comprising nanocomposite fibers increased 12-fold as compared to the pure cellulose fiber bed; abundant hydroxyl groups on the CF surface adsorb TiO2+ first as nucleation sites and then TiO2 nuclei are formed via hydrolysis-condensation which subsequently grow into TiO2 nanocrystals. Meanwhile, the hydroxyl groups of cellulose would preferentially adsorb onto a certain crystallographic plane of TiO2 due to anisotropy in adsorption stability. The approach described herein for material preparation and application are likely to offer an alternative versatile and greener pathway for fabricating a low cost, high efficiency engineering application as potential nanosorbents for heavy metal removal from water.
:4 Ti4+/MC synthesis conditions). As soon as electron is trapped by the molecular oxygen present on the TiO2 surface, highly active radicals are generated which are responsible for the photo-bleaching of MB molecules; this obeyed first-order reaction kinetics with reaction constants of 5.02 × 10−3 min−1. The TiO2/MC nanocomposite benefits from the synergistic effect of the physical and/or chemical interactions between the organic and inorganic components and therefore could be utilized as a photocatalyst to degrade hazardous contaminants via the formation of films that have good mechanical and UV-shielding properties and in the synthesis of transition-metal carbide materials.50 On similar lines, Zeng et al.51 prepared TiO2/cellulose composite films, which provided a cavity for the creation of TiO2 nanoparticles, and hydroxyl groups for their immobilization. The prepared materials exhibited a good photocatalytic activity for the photodegradation of high concentration of phenol (67.2 mg L−1) under weak UV light irradiation (0.56 mW cm−2). Another elegant strategy for the preparation of TiO2 nanomaterials has been described52 that applies natural cellulose whiskers, obtainable by hydrolysis of native cellulose as templates. TiO2/cellulose whisker nanoparticles exhibited quantum size effects and a lower band gap energy (2.02 eV) and displayed promising activity for the photodegradation of a methyl orange solution under weak UV light irradiation (4.3–41 mW cm−2). They exhibited good stability against photocorrosion, implying that they could be used in the photodegradation of organic pollutants. It is worth noting that the regenerated cellulose films and fibers in the wet state possess a porous structure and the hydroxyl groups assist the creation of inorganic nanoparticles in the cellulose matrix.53–56 Marques et al.57 examined sheets prepared using a mixture of synthetic TiO2/cellulose nanocomposite. The obtained composite exhibited a much higher opacity than those obtained by mechanical blending of the fibres with commercial TiO2 pigment because of the highly homogeneous distribution of TiO2 and higher specific surface area available for light scattering in sheets with the hybrid material. Such hybrids could be used as reinforcing fibers in a polymer matrix, particularly when light-barrier materials are sought. An interesting concept that involved the preparation of titanium dioxide nanocrystals on cellulose fibers (CF) by in situ hydrolysis has been reported;58 TiO2/CF composite showed enhanced adsorption capacity, good regenerability and selectivity for lead (Pb2+) removal. Moreover, the adsorption capacity of a filter bed comprising nanocomposite fibers increased 12-fold as compared to the pure cellulose fiber bed; abundant hydroxyl groups on the CF surface adsorb TiO2+ first as nucleation sites and then TiO2 nuclei are formed via hydrolysis-condensation which subsequently grow into TiO2 nanocrystals. Meanwhile, the hydroxyl groups of cellulose would preferentially adsorb onto a certain crystallographic plane of TiO2 due to anisotropy in adsorption stability. The approach described herein for material preparation and application are likely to offer an alternative versatile and greener pathway for fabricating a low cost, high efficiency engineering application as potential nanosorbents for heavy metal removal from water.
3.4 TiO2/chitosan
Chitosan (poly-β-(1→4)-2-amino-2-deoxy-D-glucose), obtained by N-deacetylation (usually over 75%) of chitin, is the next most abundant natural polysaccharide after cellulose (Fig. 6)59 and comprises the exoskeleton of insects and crustaceans (seafood processing industries’ waste products) and cell walls of fungi.60,61 Chitosan contains a large number of amino (–NH2) and hydroxyl (–OH) functional groups and possesses excellent adsorption ability for a wide variety of environmental pollutants such as heavy metals,62 dyestuffs,62 and pathogens63 and can be easily functionalized.64 Furthermore, chitosan exhibits antibacterial activity, film forming ability and a good drug delivery potential.65 The amino groups of chitosan can be easily cationized which help adsorb anionic dyes strongly via electrostatic attraction in acidic media. However, chitosan is very sensitive to pH as it can either form a gel or dissolve depending on the pH values.59–65 Additionally, chitosan has some disadvantages such as unsatisfactory mechanical properties, severe shrinkage, deformation after drying, solubility under acidic conditions, compressibility at high operating pressure and can shield and block UV radiation with degradation of textiles and leather.59,65,66 Previous research67 has showed the efficacy of a novel methyl orange imprinted core–shell TiO2/chitosan hybrid material (SICT) in selective photocatalytic degradation of methyl orange in dual-dye systems (Fig. 7). In this context, chitosan as a support for TiO2 has following advantages: (a) easy recovery and multiple reuseability; (b) selectivity for target pollutants; (c) degradation and complete mineralization of organics; and (d) synergistic effect of the photocatalysis–adsorption processes of TiO2/chitosan leading to lowering of the charge carrier recombination.68 In addition, SICT could be reused directly without further desorption and regeneration for 10 cycles while retaining 60% of its photocatalytic efficiency. The reusability of SICT would be beneficial for simplification of the operating steps and the associated cost reduction would facilitate its practical application in wastewater treatment for environmental organic pollutants. Although chitosan has shown promise as a biosorbent, the chemical stability of chitosan is unsatisfactory because the abundant free amines, with a pKa value of ∼6.5, can be protonated culminating in hydro-solubility under acidic conditions.67,68
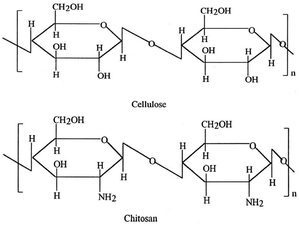 |
| | Fig. 6 Structures of cellulose and chitosan. | |
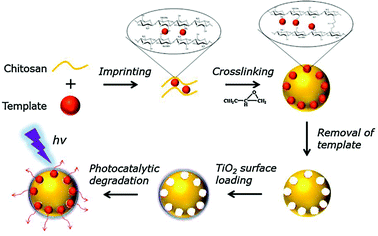 |
| | Fig. 7 Schematic illustration of the preparation process of the surface imprinted TiO2/chitosan composite. Reproduced with permission from ref. 67. | |
3.5 TiO2/cellulose acetate
Cellulose acetate (CA) is biodegradable, nontoxic and biocompatible and can be prepared by simple esterification of cellulose, which is abundant in agricultural waste such as straw and biomass residues.69–72 Cellulose acetate possess beneficial mechanical strength that facilitates its processing into films, membranes, and fibers from either melts or solutions. Further, due to paucity of hydrogen bonds and its lower crystallinity, its solubility in a wide range of organic solvents like acetone, dimethylformamide (DMF), dimethylsulfoxide (DMSO) etc. is enhanced.73 The study of a flexible mesoporous TiO2 microspheres/cellulose acetate (TCA) hybrid film (Fig. 8a),74 showed that it can be easily recycled without a decrease in its activity for the degradation of methylene blue trihydrate solution (Fig. 8b). The TCA hybrid film displays stronger adsorption and photocatalytic degradation abilities for methylene blue when compared to a cellulose acetate film under the same conditions. The TCA hybrid film with its flexible, transparent and environmentally friendly properties, can be used as a self-cleaning material thus finding suitable application in the field of wastewater treatment without leaving any photocatalyst in the reaction system. A TiO2/CA membrane prepared from DMF solution has a considerably better catalytic activity than the corresponding membrane obtained from acetone solution. By increasing the amount of TiO2 in the composite, in the photocatalytic activity of the membrane was discerned until a certain value; increasing the TiO2 content beyond that level led to lower catalytic activity.74
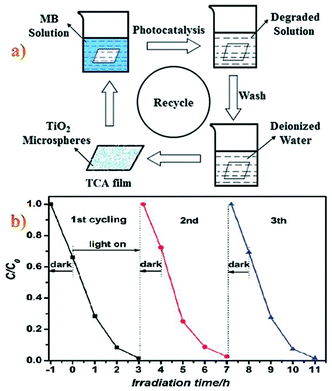 |
| | Fig. 8 (a) Schematic illustration of cellulose acetate (TCA) hybrid film recycling for use as a photocatalyst. (b) Photocatalytic activity of the sample TCA hybrid film on MB degradation with recycling (three times). Reproduced with permission from ref. 74. | |
3.6 TiO2/bacterial cellulose
Bacterial cellulose (BC), produced by acetic acid bacteria Gluconobacter xylinum, is obtainable in a pure form which requires no intensive processing to remove undesirable impurities and contaminants such as lignin, pectin and hemicellulose.75,76 BC embodies several distinguished structural features and qualities such as good mechanical properties, biocompatibility, elevated tensile strength, a higher degree of polymerization (up to 8000), high crystallinity (of 70–80%), high water content (99%) and retention capability.77,73–80 Another notable feature of BC is its high aspect ratio and abundant active functional hydroxyl groups, which make it suitable for combination with assorted nanostructures via its powerful interaction with surrounding species, such as inorganic and polymeric nanoparticles and nanowires.81 The photocatalytic mesoporous TiO2/BC hybrid nanofibers have been fabricated by surface hydrolysis with molecular precision;82 uniform and well-defined nanofibers comprise large quantities of oriented TiO2 nanoparticle arrays implanted on the BC nanofibers with diameters in the 4.3–8.5 nm range. A TiO2/BC nanocomposite has been used as a photocatalyst for methyl orange degradation under UV irradiation, and it showed higher efficiency than the commercial photo-catalyst due to its high surface area (208.17 m2 g−1). The BC template can be removed by calcination to afford mesoporous TiO2 networks composed of interconnected anatase nanowires;82,77–84 these networks exhibit considerably enhanced photocatalytic activity compared to the macroporous titania networks obtained through a similar sol–gel nanocasting procedure. The possible physical interactions between BC fibers and sol–gel solution in the TiO2/BC hybrid fibers, facilitated by sonication (750 W), are shown in Fig. 9;85 hydroxyl groups on the surface of the BC fibers provoke H-bonding interactions with the hydrophilic inorganic TiO2 sol–gel network. TiO2 nanoparticles respond to the applied voltage (−3, 0 and 3 V) of the electrostatic force microscopy (EFM) tip as the strong electrostatic dipole–dipole interactions are generated by hydrogen bonding connections between the hydroxyl groups in the cellulose and their counterparts in the inorganic oxide, thus eliciting a high electric dipole moment of TiO2 nanoparticles in the TiO2/BC hybrid fibers.
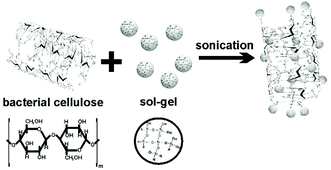 |
| | Fig. 9 Illustration of the synthesis of TiO2/BC composites. Reproduced with permission from ref. 85. | |
3.7 TiO2/wood
Wood is a natural, sustainable and abundant biopolymeric material comprised of cellulose, hemicellulose, and lignin. Wood's natural hierarchical structure is composed of well-oriented microfibers and tracheids for water, ions, and oxygen transportation during metabolism; more importantly, multiscale natural fibers from wood have unique optical properties applicable to different kinds of optoelectronics and photonic devices.86,87 However, in view of its hygroscopic and organic nature, wood is sensitive to humidity variations and fire, which can lead to dimensional and thermal instability. Wood also features numerous hydroxyl groups that confer a hydrophilic nature on the cell wall which traps the polymeric Ti–OH inside the wood samples by interaction with wood hydroxyl.86 The preparation of TiO2/wood under mild ultrasonic conditions (i.e., relatively low sonication intensity and low temperature) for a short period of time is presented in Fig. 10;87 ultrasound provides a good distribution of TiO2 throughout the wood matrix wherein these gels are deposited in the lumen by adhesion to cell walls. Consequently, the cell wall components are obstructed from access to oxygen and complete combustion is delayed, thus enhancing the thermal stability of the wood. The modified wood possesses improved the thermal and mechanical properties and dimensional stability with increasing load of inorganic TiO2 gels, as ultrasonic treatment facilitates chemical reactions between liquid and solid phases88,89 thus helping the solution to be converted to polymeric TiO2 inside the wood matrix. As each Ti is coordinated with four oxygen atoms, therefore the development of –Ti–O–Ti– chains results in three-dimensional polymeric skeletons with closed packing; wood materials with UV-resistant ability have been successfully fabricated by one-pot hydrothermal fabrication of rutile TiO2 submicrospheres on the wood surface90 as attested by ATR-FTIR spectra. On the other hand, vacuum and ultrasonic methods both enhance the dimensional stability of wood, by preventing the formation of cracks, retarding combustion of the wood matrix and diminishing biological attack.87,91 A similar hydrothermal deposition of TiO2 particles on spruce (Picea Abies) wood has been developed for the preparation of TiO2/wood composites at 75 °C in a relatively short time (up to 1 h)91 that serves the dual purpose of wood impregnation and covering the porous inner and outer surface of wood with rutile particles as discerned by ATR-FTIR and Raman subtraction spectra; the presence of titania particles on the hydrothermally treated wood surface exhibited vibrational bands of TiO2-wood interphase interactions confirming the anchoring sites of titanium ions on hemicellulose, cellulose and lignin.92
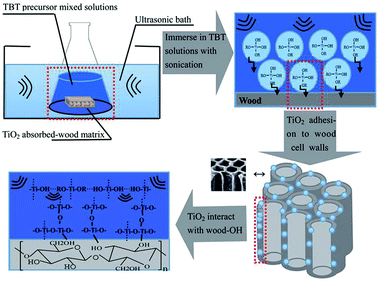 |
| | Fig. 10 Schematic of experimental set-up for preparing a TiO2/wood matrix by the ultrasonic-assisted sol–gel method. Reproduced with permission from ref. 87. | |
4. TiO2/carbon materials derived from agricultural residues
Numerous carbon sources such as biochar, activated carbon, bamboo or wood charcoal, carbon waste and even fly ash from coal-fired power plants can be utilized, as shown below.
4.1 TiO2/biochar
Biochar has been considered as a potential surrogate for activated carbon and is produced by thermal decomposition (e.g., slow pyrolysis, fast pyrolysis, hydrothermal carbonization (HTC), flash carbonization, torrefaction and gasification)93–95 of a wide range of carbon-rich biomass materials that includes woody materials, agricultural residues such as sugarcane bagasse,96 livestock manure,97 and remnants after moderate temperature (e.g., 350–700 °C) pyrolysis and carbonification of biomass in a reactor with little or no available air.98–102 Non-activated biochar, i.e., biochar that has not undergone any physical, chemical or biological modifications to improve its sorptive properties, can also be used as a precursor material for the production of activated carbon (Fig. 11). An important advantage of biochar over more conventional activated carbon is the presence of abundant surface functional groups (e.g. C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, COOH, and OH, among others). Additionally, these groups show interesting catalytic properties due to the presence of O-, N-, and S-type functional groups, which serve as a platform for the synthesis of various functionalized carbon materials;103–105 products, yields and characteristics of carbons derived from varied thermochemical conversion processes are illustrated in Fig. 11. The interaction of biochar with organic sorbates is dependent on the structural and chemical properties of the sorbates. The sorption of organic compounds by biochar, as illustrated in Fig. 12,106 occurs by: (a) pore-filling mechanisms (contributed by the majority of the surface area of biochar); (b) diffusion and partitioning mechanisms (effects of electrostatic interactions on the diffusion and equilibrium partitioning); (c) surface hydrophobic and hydrophilic interactions; (d) aromatic– and cation–π interactions (e.g. strong, non-covalent π-electron donor–acceptor (EDA) interactions); (e) electrostatic interactions (sorption of ionic and ionizable organic compounds); and (f) hydrogen bonding (sorption of polar organic compounds) or by a simultaneous occurrence of all aforementioned mechanisms.107,108 Biochar's uptake of organic solutes is a function of both surface and bulk properties of the carbonized (crystalline, graphene-like fractions) and non-carbonized fractions (non-crystalline, amorphous, organic carbon).109,110 In order to promote heterogeneous photocatalytic reactions, Kim and Kan111 developed surface-modified biochar (corn cob carbonized at 600 °C) and followed a simple sol–gel method to remove sulfamethoxazole (SMX) in water by photocatalytic oxidation using the biochar-supported TiO2 photocatalyst under UV light irradiation (15 W UV-C, 254 nm). Surprisingly, the obtained composite showed a much higher adsorption of SMX than the commercial TiO2 powder due to the hydrophobic interaction (carbon π–π interaction) between the biochar and SMX. It should be pointed out that photocatalytic oxidation of SMX using TiO2/biochar under the selected catalyst loading and irradiation time (5 g L−1 of TiO2/biochar, 6 h, pH 4) resulted in the highest removal and mineralization (91%, 81%, respectively) of SMX with negligible toxicity and accumulation of non-toxic products in water; three cycles of photocatalytic reactions using the same recycled catalyst resulted in the efficient removal of SMX (91%) confirming the high stability of the biochar-supported TiO2 photocatalyst. An ecofriendly heterogeneous photocatalysis on biochar-based materials for methylene blue (MB) photodegradation under visible light (metal halide lamp) has been reported112 wherein C-based materials induce enhancements in the photocatalytic environmental remediation of polluted water (phenol, halophenols, herbicides, dyes, azo-dyes, among others) and air (toluene, 2-propanol, among others); visible light-induced hydrogen photoproduction under solar irradiation is yet another outstanding example of photocatalytic energy storage by carbon-supported materials.
O, COOH, and OH, among others). Additionally, these groups show interesting catalytic properties due to the presence of O-, N-, and S-type functional groups, which serve as a platform for the synthesis of various functionalized carbon materials;103–105 products, yields and characteristics of carbons derived from varied thermochemical conversion processes are illustrated in Fig. 11. The interaction of biochar with organic sorbates is dependent on the structural and chemical properties of the sorbates. The sorption of organic compounds by biochar, as illustrated in Fig. 12,106 occurs by: (a) pore-filling mechanisms (contributed by the majority of the surface area of biochar); (b) diffusion and partitioning mechanisms (effects of electrostatic interactions on the diffusion and equilibrium partitioning); (c) surface hydrophobic and hydrophilic interactions; (d) aromatic– and cation–π interactions (e.g. strong, non-covalent π-electron donor–acceptor (EDA) interactions); (e) electrostatic interactions (sorption of ionic and ionizable organic compounds); and (f) hydrogen bonding (sorption of polar organic compounds) or by a simultaneous occurrence of all aforementioned mechanisms.107,108 Biochar's uptake of organic solutes is a function of both surface and bulk properties of the carbonized (crystalline, graphene-like fractions) and non-carbonized fractions (non-crystalline, amorphous, organic carbon).109,110 In order to promote heterogeneous photocatalytic reactions, Kim and Kan111 developed surface-modified biochar (corn cob carbonized at 600 °C) and followed a simple sol–gel method to remove sulfamethoxazole (SMX) in water by photocatalytic oxidation using the biochar-supported TiO2 photocatalyst under UV light irradiation (15 W UV-C, 254 nm). Surprisingly, the obtained composite showed a much higher adsorption of SMX than the commercial TiO2 powder due to the hydrophobic interaction (carbon π–π interaction) between the biochar and SMX. It should be pointed out that photocatalytic oxidation of SMX using TiO2/biochar under the selected catalyst loading and irradiation time (5 g L−1 of TiO2/biochar, 6 h, pH 4) resulted in the highest removal and mineralization (91%, 81%, respectively) of SMX with negligible toxicity and accumulation of non-toxic products in water; three cycles of photocatalytic reactions using the same recycled catalyst resulted in the efficient removal of SMX (91%) confirming the high stability of the biochar-supported TiO2 photocatalyst. An ecofriendly heterogeneous photocatalysis on biochar-based materials for methylene blue (MB) photodegradation under visible light (metal halide lamp) has been reported112 wherein C-based materials induce enhancements in the photocatalytic environmental remediation of polluted water (phenol, halophenols, herbicides, dyes, azo-dyes, among others) and air (toluene, 2-propanol, among others); visible light-induced hydrogen photoproduction under solar irradiation is yet another outstanding example of photocatalytic energy storage by carbon-supported materials.
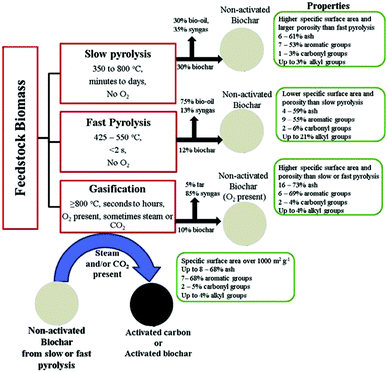 |
| | Fig. 11 Products, yields and characteristics of non-activated and physically activated biochars or carbons derived from biomass thermochemical conversion processes. Reproduced with permission from ref. 106. | |
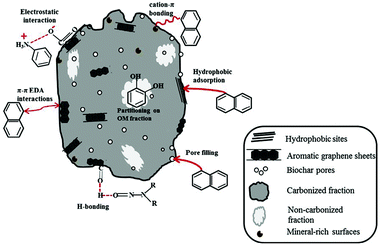 |
| | Fig. 12 Sorption mechanisms for the uptake of organic contaminants on biochar. Reproduced with permission from ref. 106. | |
4.2 TiO2/activated carbon
Activated carbon (AC), also termed active carbon, activated charcoal, or activated coal, is a porous, amorphous solid carbon material derived mainly from carbonaceous or plant-based (lignocellulosic) materials such as coconut shell,113–119 activated charcoal,120 sawdust of Tabebuia pentaphyla wood,121–124 pine sawdust,125 beech sawdust,126 peach stones,127 almond shell activated carbon,128,129 canola hull activated carbon,130 cashew nut shell activated carbon,131 water bamboo leaves,132 and bituminous coal,133 and can be used as a support for TiO2. Furthermore, activated carbon adsorbs pollutants and then releases them onto the surface of TiO2 thus delivering a higher concentration of pollutants around the TiO2 than that available in the bulk solution culminating in an enhanced degradation rate of the pollutants; intermediates produced during degradation can also be adsorbed by AC which can then undergo further oxidation.113 Nevertheless, the adsorption could generally enhance the photocatalytic rate but when the adsorption rate is much faster than the pollutant (e.g. methyl orange molecule) transfer rate, the photocatalytic kinetics are determined by the reaction rate on the TiO2 surface.134 Activated carbon has a well-developed pore structure with very large surface area and adsorption capacity and, consequently, possess a large number of active adsorption sites where organic molecules can be adsorbed before being transferred to the decomposition center of TiO2;113 oxidizing species (˙OH) generated by the photocatalyst do not have to migrate very far from the active centers of the TiO2 and degradation occurs on the catalyst surface. Using AC as a photocatalyst support conveys the pollutant molecules in close proximity to the titania active site (to come in contact with the hydroxyl radicals) for an efficient and effective photodegradation process (synergistic effect). AC can also function as a titania photosensitizer by injecting electrons into the conduction band of titania and triggering the photocatalytic formation of very reactive ˙OH radicals which are responsible for the degradation of the dyes (e.g. Basic Red 18 and Basic Red 46).130 In most cases, formate, acetate and oxalate have been detected as vital aliphatic carboxylic acid intermediates during the degradation of dyes. It is important to note that the initial formation of oxalate can increase with the illumination time, and then drop sharply. Furthermore, carboxylic acids can react directly with holes generating CO2 according to the “photo-Kolbe” reaction:
| R–COO− + TiO2(hVB+) → TiO2 + R˙ + CO2. |
In addition, photocatalytic mineralization of dyes implies the appearance of inorganic products, mainly anions, since heteroatoms are generally converted into anions at their highest oxidation state. On the other hand, TiO2 has low selectivity because intermediate products of the photooxidation of aromatic molecules are commonly photomineralized.135 In terms of the improvement of the photocatalytic properties of TiO2, Matos et al.122 investigated the changes of surface chemistry of the TiO2/activated carbon during gasification and pyrolysis. It has been found that the interaction between TiO2 and AC promotes higher concentrations of O2˙− and  radicals which induce remarkable changes in the selectivity of products. In addition, the synergistic effect could be attributed not only to a proper surface area but also to the presence of a low surface concentration of acidic oxygenated functionalities, particularly carboxylic groups. However, the surface carboxyl anions (–COO−) are stabilized by transfer of electrons to the aromatic ring of graphene layers in activated carbon. Consequently, after dissociation in the aqueous phase, carboxylic acid (e.g. acetic acid) functional groups on the AC surface coordinate with the Ti4+ metallic center of TiO2. Alternatively, this interaction could inhibit the recombination of photogenerated species in the semiconductor (e−, h+) thus forming a higher concentration of hydroxyl radical (˙OH) and superoxide anion radical (O2˙−). The influence of the O2˙− anion radical is very important because the higher the concentration of O2˙−, the higher is the production of
radicals which induce remarkable changes in the selectivity of products. In addition, the synergistic effect could be attributed not only to a proper surface area but also to the presence of a low surface concentration of acidic oxygenated functionalities, particularly carboxylic groups. However, the surface carboxyl anions (–COO−) are stabilized by transfer of electrons to the aromatic ring of graphene layers in activated carbon. Consequently, after dissociation in the aqueous phase, carboxylic acid (e.g. acetic acid) functional groups on the AC surface coordinate with the Ti4+ metallic center of TiO2. Alternatively, this interaction could inhibit the recombination of photogenerated species in the semiconductor (e−, h+) thus forming a higher concentration of hydroxyl radical (˙OH) and superoxide anion radical (O2˙−). The influence of the O2˙− anion radical is very important because the higher the concentration of O2˙−, the higher is the production of 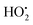 radicals that enhance the photoactivity of TiO2 in the photomineralization of 4-chlorophenol. It is worth noting that TiO2 supported on AC will degrade total organic carbon (TOC)/phenol via a photocatalytic oxidation process.128,129 Additionally, ˙OH may attack aromatic ring sites in compounds resulting in the formation of ring-opened (Fig. 13) products yielding CO2/H2O and other degradation products;136,137 breakdown of the benzene ring and its subsequent mineralization leading to CO2 and H2O could be envisaged by the decrease in TOC during the photocatalytic process.138 Moreover, AC as a support for TiO2 is effective in getting high rates of inactivate bacteria (E. coli) in the water phase.117 Interestingly, adsorbent supports provide a higher concentration of bacteria around the loaded TiO2via adsorption; the rate of E. coli inactivation in the presence of TiO2/AC catalysts is mostly determined by TiO2 particles and fits well a pseudo-first order kinetic equation. Furthermore, bacteria are inactivated on the photocatalyst surfaces and then further decomposed; the high prevailing inactivation efficiency of the composite prevents the repair of cells. The TiO2/AC composite has been used in conjunction with microwave (MW)-enhanced photodegradation of Rhodamine B (RhB) for the destruction of the conjugated structure, which was much faster than the photocatalytic degradation methods.118 A distinctive feature, that the TiO2 surface becomes more hydrophobic under MW irradiation,125 increases the probability of RhB contact with the TiO2 surface. Additional defect sites on TiO2 have been generated,139 which can increase the transition probability of e−/h+ and decrease e−/h+ recombination on the TiO2 surface. The strong interaction between TiO2 and AC can be explained by the substrate transfer to titania where it is degraded photocatalytically.140 In solution, the temperature of water and AC increases at different rates under MW irradiation due to in-core heating facilitated by MW energy which could generate locally higher temperature in some micro-surfaces of the AC particles compared to the water bulk.141
radicals that enhance the photoactivity of TiO2 in the photomineralization of 4-chlorophenol. It is worth noting that TiO2 supported on AC will degrade total organic carbon (TOC)/phenol via a photocatalytic oxidation process.128,129 Additionally, ˙OH may attack aromatic ring sites in compounds resulting in the formation of ring-opened (Fig. 13) products yielding CO2/H2O and other degradation products;136,137 breakdown of the benzene ring and its subsequent mineralization leading to CO2 and H2O could be envisaged by the decrease in TOC during the photocatalytic process.138 Moreover, AC as a support for TiO2 is effective in getting high rates of inactivate bacteria (E. coli) in the water phase.117 Interestingly, adsorbent supports provide a higher concentration of bacteria around the loaded TiO2via adsorption; the rate of E. coli inactivation in the presence of TiO2/AC catalysts is mostly determined by TiO2 particles and fits well a pseudo-first order kinetic equation. Furthermore, bacteria are inactivated on the photocatalyst surfaces and then further decomposed; the high prevailing inactivation efficiency of the composite prevents the repair of cells. The TiO2/AC composite has been used in conjunction with microwave (MW)-enhanced photodegradation of Rhodamine B (RhB) for the destruction of the conjugated structure, which was much faster than the photocatalytic degradation methods.118 A distinctive feature, that the TiO2 surface becomes more hydrophobic under MW irradiation,125 increases the probability of RhB contact with the TiO2 surface. Additional defect sites on TiO2 have been generated,139 which can increase the transition probability of e−/h+ and decrease e−/h+ recombination on the TiO2 surface. The strong interaction between TiO2 and AC can be explained by the substrate transfer to titania where it is degraded photocatalytically.140 In solution, the temperature of water and AC increases at different rates under MW irradiation due to in-core heating facilitated by MW energy which could generate locally higher temperature in some micro-surfaces of the AC particles compared to the water bulk.141
 |
| | Fig. 13 Schematic illustration of the photocatalytic degradation of phenol by TiO2/AC. | |
4.3 TiO2/activated carbon with magnetic properties
Magnetic activated carbon derived from biomass waste142,143 and soft magnetic ferrite activated carbon144 have been commonly deployed as a support for TiO2 possessing high surface areas and magnetic properties. Coconut shells or their lignocellulosic fractions, waste plant by-products that constitute a large fraction (∼35%) of the total weight of coconut fruits, are good candidate precursors for magnetic activated carbon (MAC). Recently, magnetically separable TiO2/MAC photocatalysts with high activity under solar illumination were synthesized and deployed for photocatalytic ozonation of metoprolol tartrate (MTP) in aqueous solution (50 mg L−1) with high levels of photocatalytic mineralization (70–90%);145 the MTP mineralization (i.e. conversion into CO2) rate is highly depended on the anatase content of the catalyst. This photocatalyst was easily separable due to its magnetic properties, and its reusability and stability was successfully demonstrated in a series of 10 consecutive photocatalytic ozonation runs.
4.4 TiO2/bamboo or wood charcoal
Bamboo is one of the most important non-timber forest products in the world and possesses high flexibility, high intensity, low weight, a fast growth rate and low purchasing costs. It has been extensively applied in a variety of daily applications such as indoor decoration, handicrafts, flooring construction, furniture, bamboo boats, and so forth.146,147 However, bamboo is vulnerable to attack by fungi and termites on account of its inherent hygroscopicity and therefore, prone to lose its dimensional stability.148 When exposed to an outdoor environment, unprotected bamboo shows bad decay resistance and is susceptible to attacks by fungi and insects, and degradation by moisture, air, acid rain, and sunlight thus shortening its service life and value.149,150 The term charcoal generally refers to the carbonaceous residue of wood, cellulose, bamboo, coconut shells or various industrial wastes left behind after incomplete combustion of organic matter in the absence of oxygen;151 usually it consists of carbon atoms, heteroatoms (mainly hydrogen and oxygen) and mineral matter (usually given as ash content). Wood charcoal is cheaper than AC because it doesn't need an activation process and has often been used for phenol adsorption.152 The basic feature of wood charcoal is a highly aromatic matrix formed from layers of polycyclic aromatic rings bearing a content of organic carbon (53%) with a high C/N ratio; it can selectively adsorb hydrophobic rather than polar pollutants due to its non-polar nature.153 A TiO2/bamboo charcoal composite made by a dip-dry method is a porous adsorptive material which has a high specific surface area (359.8 m2 g−1), pore volume (0.317 cm3 g−1) and average pore diameter (3.526 nm).154 Porous bamboo charcoal powder embedded in the matrix of TiO2 particles has facilitated dye absorption and the generation of electrons during exposure to light.155 Biotemplated synthesis of TiO2/wood charcoal composites developed for the synergistic removal of bisphenol A (BPA) by adsorption and photocatalytic degradation is a recent example;151 they displayed extraordinary adsorption ability for hydrophobic BPA and good photocatalytic activity (80% and 78%).
4.5 TiO2/carbon waste and residues
Recently, Dávila-Jiménez et al.156 prepared a series of TiO2/carbon composite materials from carbonized avocado kernels and sols of TiO2; their short preparation in fewer steps uses carbon with acidic groups that favors the interaction with TiO2. The most efficient composites were obtained from carbons with the largest specific surface areas and a suitable micropore–mesopore ratio, favoring the immobilization of a larger amount of TiO2 particles, regardless of the pHPZC of the carbon (pH where the carbon surface has neutral charge). Antonio-Cisneros et al.157 immobilized TiO2 on carbon emanating from residues of the plant Manihot, and this ensuing composite eliminated nearly 100% of the dye upon UV irradiation under optimal conditions; a similar investigation has been reported using carbon from Manihot dulcis waste,158 and the TiO2/carbon composite decomposed ∼50% of carminic acid under various conditions. Interestingly, the adsorption is reversible, and the dye can be partially recovered by desorption, which can be advantageous in the case of artisanal carpeting effluents.
4.6 TiO2/coal fly ash
Fly ash from coal, oil and biomass combustion are major contributors to the solid waste which is currently discarded in landfills or dumped at sea.159 Coal fly ash (CFA) is a valuable and desirable additive to cement concrete because of its spherical shape and pozzolanic properties.160,161 In other domains, coal fly ash is considered as a forerunner for several applications in adsorption,162–164 material synthesis such as zeolites,165–167 geopolymers,168–170 ceramics,171,172 and as a catalyst support.173–175 Additionally, it has been shown that the prepared TiO2/coal fly ash possesses potential advantages over the starting materials176 such as: (a) the microspherical CFA particles are easy to precipitate in water, so photocatalysts supported on CFA are easy to recycle from aqueous solution after the reaction; (b) CFA, consisting primarily of Al2O3 and SiO2 as support, can inhibit recombination of electrons and holes effectively;177,178 (c) a porous structure is formed by the 3D network with additional loading of TiO2 photocatalyst in its structure;179 (d) the cost of preparation and the sources of environment pollution can be reduced. However, it is difficult to control uniform distribution of TiO2 on CFA,180,181 which seriously restricts the activity of the photocatalyst and substrate availability. TiO2 immobilized on the surface of CFA, the TiO2/CFA composite, separates without difficulty from the treated wastewater by precipitation under quiescent conditions.179 Since CFA particles are easy to precipitate in water because of their larger size and heavier weight compared to TiO2 particles, the separation and recovery of the catalyst from water is facilitated when TiO2 is immobilized on CFA. Therefore, TiO2/CFA may be a promising material for applications in the removal of organic pollutants from water in the future.
4.7 TiO2/biomaterials for degradation of organic pollutants
A biomaterial, derived from an organism, should have the inherent advantage to identify the majority of organic dye molecules effectively due to the high biocompatibility and structural complementarity of biomaterials towards organic pollutants. Biomaterials or biomimetic materials can identify dye molecules and selectively adsorb them as exemplified by eggshell on the surface of TiO2-loaded composites.182 Various studies have shown that TiO2 loaded composites (e.g. TiO2/skeleton,182 TiO2/DENs,182 TiO2/eggshell,183 TiO2/clamshell183 and TiO2/fish scale composites184) exhibited very high photocatalytic activity due to the synergistic effect of biomaterial association and the TiO2 photocatalyst. Basically, nature-based biomaterials like fish scales are frequently used as adsorbents in biosorption processes due to their high binding capacities.185 The uptake abilities of scales from different fish species should be similar because most fish scales contain significant portions of organic protein (collagen), and the structure of collagen contains alluring functional groups, such as phosphate, carboxyl, amine and amide, that are implicated in the biosorption process.186
5. Future prospects
This critical review summarizes most of the recent efforts on the syntheses and applications of TiO2/carbon materials derived from renewable resources that, until a few years ago, were used as unique hybrid heterostructured photocatalysts for environmental remediation. TiO2/carbon functional materials, with well-designed physical and chemical properties, will not only bring many favourable features to advanced oxidation processes but will provide a model system for investigating and understanding the dependence of photocatalytic performance on materials composition and structure at various scales, including fundamental studies on reaction mechanism and kinetics. Such an attained knowledge base, in turn, will greatly stimulate the better design of highly efficient hybrid photocatalysts. There are optimistic future expectations for the TiO2/carbon materials derived from renewable resources in the field of photocatalysis and related fields of science. However, there are still a number of aspects that need to be addressed related to the rational and optimal design of materials; understanding of interphase interactions, especially under solar light conditions; novel energy-efficient processes; feedstock and production costs; issues related to carbon leaching from hybrid photocatalysts; and environmental impact concerns, to name a few. Despite much progress in photocatalysis, research on optimizing the properties of these photocatalysts is still in its infancy, as considerable challenges exist in terms of improving accurate structure–property relationships which need to be attended. Parallel to these advances, other allied applications for a greener and sustainable future are expected, namely in photodetectors, hybrid organic–inorganic solar cells, self-cleaning materials, air purification filters, photocorrosion-resistant materials, energy storage, CO2 reduction, selective organic transformations and water splitting for hydrogen generation. Developments in these associated fields may unveil their enhanced understanding thus harnessing the power of these fascinating nanostructured materials.
Disclaimer
The views expressed in this article are those of the authors and do not necessarily represent the views or policies of the US Environmental Protection Agency. Any mention of trade names or commercial products does not constitute an endorsement or recommendation for use.
Acknowledgements
Prof. Dr Juan C. Colmenares gratefully acknowledges support from COST Action FP1306 for networking and possibilities for meetings and future students exchange. Paweł Lisowski would like also to thank the National Science Centre (NCN) in Poland for the research project 2015/17/N/ST5/03330.
References
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS PubMed.
- Y. H. Ng, S. Ikeda, M. Matsumura and A. Amal, Energy Environ. Sci., 2012, 5, 9307–9318 CAS.
- B. H. Nguyen, V. H. Nguyen and D. L. Vu, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2015, 6, 1–13 Search PubMed.
- K. R. Reddy, M. Hassan and V. G. Gomes, Appl. Catal., A, 2015, 489, 1–16 CrossRef CAS.
- G. Liu, L. Wang, H. G. Yang, H.-M. Cheng and G. Q. Lu, J. Mater. Chem., 2010, 20, 831–843 RSC.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. O'Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS.
- M. D. Hernandez-Alonso, F. Fresno, S. Suarez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 CAS.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- J. Virkutyte and R. S. Varma, RSC Adv., 2012, 2, 2399–2407 RSC.
- J. Virkutyte and R. S. Varma, RSC Adv., 2012, 2, 1533–1539 RSC.
- J. Virkutyte, B. Baruwati and R. S. Varma, Nanoscale, 2010, 2, 1109–1111 RSC.
- J. Virkutyte and R. S. Varma, New J. Chem., 2010, 34, 1094–1096 RSC.
- M. Pelaez, B. Baruwati, R. S. Varma, R. Luque and D. D. Dionysiou, Chem. Commun., 2013, 49, 10118–10120 RSC.
- G. Varshney, S. R. Kanel, D. Kempisty, V. Varshney, A. Agrawal, E. Sahle-Demessie, R. S. Varma and M. N. Nadagouda, Coord. Chem. Rev., 2016, 306, 43–64 CrossRef CAS.
- M. N. Chong, B. Jin, C. W. K. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- S. Malato, P. Fernández-Ibáñez, M. I. Maldonado, J. Blanco and W. Gernjak, Catal. Today, 2009, 147, 1–59 CrossRef CAS.
- R. Leary and A. Westwood, Carbon, 2011, 49, 741–772 CrossRef CAS.
-
C. O. Ania, L. F. Velasco and T. Valdes-Solis, Novel Carbon Adsorbents, Elsevier, 2012, pp. 521–547 Search PubMed.
-
J. L. Faria and W. Wang, Carbon Materials for Catalysis, John Wiley & Sons, Inc., 2008, pp. 481–506 Search PubMed.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- R. J. White, V. Budarin, R. Luque, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 3401–3418 RSC.
- Q. F. Xu, Y. Liu, F.-J. Lin, B. Mondal and A. M. Lyons, ACS Appl. Mater. Interfaces, 2013, 5, 8915–8924 CAS.
- L. Su and Y. X. Gan, Composites, Part B, 2012, 43, 170–182 CrossRef CAS.
- X. Wang, X. Xue, X. Liu, X. Xing, Q. Li and J. Yang, Mater. Chem. Phys., 2015, 153, 117–126 CrossRef CAS.
- S. Singh, H. Mahalingam and P. K. Singh, Appl. Catal., A, 2013, 462–463, 178–195 CrossRef CAS.
- Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893–1909 RSC.
- S. Dutta, A. Bhaumik and K. C.-W. Wu, Energy Environ. Sci., 2014, 7, 3574–3592 CAS.
- M.-M. Titirici, R. J. White, C. Falco and M. Sevilla, Energy Environ. Sci., 2012, 5, 6796–6822 Search PubMed.
- I. Delidovich, K. Leonhard and R. Palkovits, Energy Environ. Sci., 2014, 7, 2803–2830 CAS.
- S. Lacombe and T. Pigot, Catal. Sci. Technol., 2016, 6, 1571–1592 CAS.
- H. Park, H.-I. Kim, G.-h. Moon and W. Choi, Energy Environ. Sci., 2016, 9, 411–433 CAS.
- J. Shi, Chem. Rev., 2013, 113, 2139–2181 CrossRef CAS PubMed.
- A. Brandt, J. Gräsvik, J. P. Halletta and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- H. Kobayashi and A. Fukuoka, Green Chem., 2013, 15, 1740–1763 RSC.
- R. F. Perez and M. A. Fraga, Green Chem., 2014, 16, 3942–3950 RSC.
- X. Chen, D.-H. Kuo, D. Lu, Y. Hou and Y.-R. Kuo, Microporous Mesoporous Mater., 2016, 223, 145–151 CrossRef CAS.
- V. Budarin, J. H. Clark, J. J. E. Hardy, R. Luque, K. Milkowski, S. J. Tavener and A. J. Wilson, Angew. Chem., Int. Ed., 2006, 45, 3782–3786 CrossRef CAS PubMed.
- V. Budarin, J. H. Clark, J. J. E. Hardy, R. Luque, K. Milkowski, S. J. Tavener and A. J. Wilson, Angew. Chem., 2006, 118, 3866–3870 CrossRef.
- V. L. Budarin, J. H. Clark, R. Luque and D. J. Macquarrie, Chem. Commun., 2007, 634–636 RSC.
- J. H. Clark, V. Budarin, T. Dugmore, R. Luque, D. J. Macquarrie and V. Strelko, Catal. Commun., 2008, 9, 1709–1714 CrossRef CAS.
- R. Luque, C. S. K. Lin, C. Du, D. J. Macquarrie, A. Koutinas, R. Wang, C. Webb and J. H. Clark, Green Chem., 2009, 11, 193–200 RSC.
- M.-M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250–290 RSC.
- J. C. Colmenares, P. Lisowski and D. Łomot, RSC Adv., 2013, 3, 20186–20192 RSC.
- G. Goncalves, P. A. A. P. Marques, R. J. B. Pinto, T. Trindade and C. P. Neto, Compos. Sci. Technol., 2009, 69, 1051–1056 CrossRef CAS.
- J. Virkutyte, V. Jegatheesan and R. S. Varma, Bioresour. Technol., 2012, 113, 288–293 CrossRef CAS PubMed.
- J. Zeng, S. Liu, J. Cai and L. Zhang, J. Phys. Chem. C, 2010, 114, 7806–7811 CAS.
- S. Liu, D. Tao, H. Bai and X. Liu, J. Appl. Polym. Sci., 2012, 126, 1–9 CrossRef.
- J. Zeng, R. Li, S. Liu and L. Zhang, ACS Appl. Mater. Interfaces, 2011, 3, 2074–2079 CAS.
- H. Rollins, F. Lin, J. Johnson, J. Ma, J. Liu, M. Tu, D. DesMarteau and Y. Sun, Langmuir, 2000, 16, 8031–8036 CrossRef CAS.
- S. Liu, L. Zhang, J. Zhou and R. Wu, J. Phys. Chem. C, 2008, 112, 4538–4544 CAS.
- D. Ke, S. Liu, K. Dai, J. Zhou, L. Zhang and T. Peng, J. Phys. Chem. C, 2009, 113, 16021–16026 CAS.
- P. A. A. P. Marques, T. Trindade and C. P. Neto, Compos. Sci. Technol., 2006, 66, 1038–1044 CrossRef CAS.
- Y. Li, L. Cao, L. Li and C. Yang, J. Hazard. Mater., 2015, 289, 140–148 CrossRef CAS PubMed.
- M. N. V. R. Kumar, React. Funct. Polym., 2000, 46, 1–27 CrossRef CAS.
- H. Orelma, I. Filpponen, L. S. Johansson, J. Laine and O. J. Rojas, Biomacromolecules, 2011, 12, 4311–4318 CrossRef CAS PubMed.
- C. D. Tran, S. Duri, A. Delneri and M. Franko, J. Hazard. Mater., 2013, 252–253, 355–366 CrossRef CAS PubMed.
- D. Reddy and S. M. Lee, Adv. Colloid Interface Sci., 2013, 201–202, 68–93 CrossRef CAS PubMed.
- S. Mallick, S. Sharma, M. Banerjee, S. S. Ghosh, A. Chattopadhyay and A. Paul, ACS Appl. Mater. Interfaces, 2012, 4, 1313–1323 CAS.
- V. K. Mourya and N. N. Inamdar, React. Funct. Polym., 2008, 68, 1013–1051 CrossRef CAS.
- L. Cordero-Arias, S. Cabanas-Polo, H. Gao, J. Gilabert, E. Sanchez, J. A. Roether, D. W. Schubert, S. Virtanene and A. R. Boccaccini, RSC Adv., 2013, 3, 11247–11254 RSC.
- W. A. Lee, N. Pernodet, B. Li, C. H. Lin, E. Hatchwell and M. H. Rafailovich, Chem. Commun., 2007, 4815–4817 RSC.
- G. Xiao, H. Su and T. Tan, J. Hazard. Mater., 2015, 283, 888–896 CrossRef CAS PubMed.
- M. A. Nawi, A. H. Jawad, S. Sabar and W. S. W. Ngah, Desalination, 2011, 280, 288–296 CrossRef CAS.
- J. Simon, H. P. Müllera, R. Kocha and V. Müller, Polym. Degrad. Stab., 1998, 59, 107–115 CrossRef CAS.
- N. Y. Abou-Zeid, A. I. Waly, N. G. Kandile, A. A. Rushdy, M. A. El-Sheikh and H. M. Ibrahim, Carbohydr. Polym., 2011, 84, 223–230 CrossRef CAS.
- D. Wang, G. Sun and L. W. Yu, Carbohydr. Polym., 2011, 83, 1095–1100 CrossRef CAS.
- B. E. Dale, J. Chem. Technol. Biotechnol., 2003, 78, 1093–1103 CrossRef CAS.
- A. Wittmar, D. Vorata and M. Ulbricht, RSC Adv., 2015, 5, 88070–88078 RSC.
- X. Jin, J. Xu, X. Wang, Z. Xie, Z. Liu, B. Liang, D. Chen and G. Shen, RSC Adv., 2014, 4, 12640–12648 RSC.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- P. Ross, R. Mayer and M. Benziman, Microbiol. Rev., 1991, 55, 35–58 CAS.
- D. Klemm, D. Schumann, U. Udhardt and S. Marsch, Prog. Polym. Sci., 2001, 26, 1561–1603 CrossRef CAS.
- A. Svensson, E. Nicklasson, T. Harrah, B. Panilaitis, D. L. Kaplan, M. Brittberg and P. Gatenholm, Biomaterials, 2005, 26, 419–431 CrossRef CAS PubMed.
- W. Hu, S. Chen, J. Yang, Z. Li and H. Wang, Carbohydr. Polym., 2014, 101, 1043–1060 CrossRef CAS PubMed.
- H. S. Barud, T. Regiani, R. F. C. Marques, W. R. Lustri, Y. Messaddeq and S. J. L. Ribeiro, J. Nanomater., 2011, 721631 Search PubMed.
- J. Huang and Y. Gu, Curr. Opin. Colloid Interface Sci., 2011, 16, 470–481 CrossRef.
- D. P. Sun, J. Z. Yang and X. Wang, Nanoscale, 2010, 2, 287–292 RSC.
- D. Y. Zhang and L. M. Qi, Chem. Commun., 2005, 2735–2737 RSC.
- G. Yang, J. J. Xie, Y. X. Deng, Y. G. Bian and F. Hong, Carbohydr. Polym., 2012, 87, 2482–2487 CrossRef CAS.
- J. Gutierrez, A. Tercjak, I. Algar, A. Retegi and I. Mondragon, J. Colloid Interface Sci., 2012, 377, 88–93 CrossRef CAS PubMed.
- B. Unger, M. Bucker, S. Reinsch and T. Hubert, Wood Sci. Technol., 2013, 47, 83–104 CrossRef CAS.
- B. Wang, M. Feng and H. Zhan, RSC Adv., 2014, 4, 56355–56360 RSC.
- B. T. T. Chu, G. Tobias, C. G. Salzmann, B. Ballesteros, N. Grobert, R. I. Todd and M. L. Green, J. Mater. Chem., 2008, 18, 5344–5349 RSC.
- S. Mallakpour, M. Dinari and V. Behranvand, RSC Adv., 2013, 3, 23303–23308 RSC.
- Q. Sun, Y. Lu, H. Zhang, H. Zhao, H. Yu, J. Xu, Y. Fu, D. Yang and Y. Liu, Mater. Chem. Phys., 2012, 133, 253–258 CrossRef CAS.
- P. Pori, A. Vilcnik, M. Petric, A. S. Skapin, M. Mihelcic, A. S. Vuk, U. Novak and B. Orel, Appl. Surf. Sci., 2016, 372, 125–138 CrossRef CAS.
- J. Li, H. Yu, Q. Sun, Y. Liu, Y. Cui and Y. Lu, Appl. Surf. Sci., 2010, 256, 5046–5050 CrossRef CAS.
- S.-H. Kong, S.-K. Loh, R. T. Bachmann, S. A. Rahim and J. Salimon, Renewable Sustainable Energy Rev., 2014, 39, 729–739 CrossRef CAS.
- S. Ren, H. Lei, L. Wang, Q. Bu, S. Chen and J. Wu, RSC Adv., 2014, 4, 10731–10737 RSC.
- J. Han, X. Wang, J. Yue, S. Gao and G. Xu, Fuel Process. Technol., 2014, 122, 98–106 CrossRef CAS.
- Y. Yao, B. Gao, M. Inyang, A. R. Zimmerman, X. Cao, P. Pullammanappallil and L. Yang, Bioresour. Technol., 2011, 102, 6273–6278 CrossRef CAS PubMed.
- C. J. Atkinson, J. D. Fitzgerald and N. A. Hipps, Plant Soil, 2010, 337, 1–18 CrossRef CAS.
- F. Lian, F. Huang, W. Chen, B. Xing and L. Zhu, Environ. Pollut., 2011, 159, 850–857 CrossRef CAS PubMed.
- Y. Yu and H. Wu, Energy Fuels, 2010, 24, 5660–5668 CrossRef CAS.
- T. R. Brown, M. M. Wright and R. C. Brown, Biofuels, Bioprod. Biorefin., 2011, 5, 54–68 CrossRef CAS.
- D. A. Laird, R. C. Brown, J. E. Amonette and J. Lehmann, Biofuels, Bioprod. Biorefin., 2009, 3, 547–562 CrossRef CAS.
- O. Mašek, P. Brownsort, A. Cross and S. Sohi, Fuel, 2013, 103, 151–155 CrossRef.
- I. Velo-Gala, J. J. Lopez-Penalver, M. Sanchez-Polo and J. Rivera-Utrilla, Appl. Catal., B, 2013, 142–143, 694–704 CrossRef CAS.
- J. Matos, Top. Catal., 2015, 1–9 Search PubMed.
- R. A. Shawabkeh, M. Al-Harthi and S. M. Al-Ghamdi, Energy Sources, Part A, 2014, 36, 93–103 CrossRef CAS.
- M. Inyang and E. Dickenson, Chemosphere, 2015, 134, 232–240 CrossRef CAS PubMed.
- P. Zhang, H. Sun, L. Yu and T. Sun, J. Hazard. Mater., 2013, 244–245, 217–224 CrossRef CAS PubMed.
- C. Chen, W. Zhou and D. Lin, Bioresour. Technol., 2015, 179, 359–366 CrossRef CAS PubMed.
- B. Chen, D. Zhou and L. Zhu, Environ. Sci. Technol., 2008, 42, 5137–5143 CrossRef CAS PubMed.
- M. Keiluweit, P. S. Nico, M. G. Johnson and M. Kleber, Environ. Sci. Technol., 2010, 44, 1247–1253 CrossRef CAS PubMed.
- J. R. Kim and E. Kan, J. Environ. Manage., 2016, 180, 94–101 CrossRef CAS PubMed.
- J. Matos, Top. Catal., 2016, 59, 394–402 CrossRef CAS.
- X. Wang, Y. Liu, Z. Hu, Y. Chen, W. Liu and G. Zhao, J. Hazard. Mater., 2009, 169, 1061–1067 CrossRef CAS PubMed.
- M. Khraisheh, J. Kim, L. Campos, A. H. Al-Muhtaseb, G. M. Walker and M. AlGhouti, Environ. Eng. Sci., 2013, 30, 515–526 CrossRef CAS PubMed.
- M. Toyoda, Y. Nanbu, T. Kito, M. Himno and M. Inagaki, Desalination, 2003, 159, 273–282 CrossRef CAS.
- Y. Li, S. Zhang, Q. Yu and W. Yin, Appl. Surf. Sci., 2007, 253, 9254–9258 CrossRef CAS.
- L. Youji, M. A. Mingyuan, W. Xiaohu and W. Xiaohua, J. Environ. Sci., 2008, 20, 1527–1533 CrossRef.
- H. Zhong, Y. Shaogui, J. Yongming and S. Cheng, J. Environ. Sci., 2009, 21, 268–272 CrossRef.
- C. Andriantsiferana, E. F. Mohamed and H. Delmas, Environ. Technol., 2014, 35, 355–363 CrossRef CAS PubMed.
- B. Ayoubi-Feiz, S. Aber, A. Khataee and E. Alipour, Environ. Sci. Pollut. Res., 2014, 21, 8555–8564 CrossRef CAS PubMed.
- J. Matos, J. M. Chovelon, T. Cordero and C. Ferronato, Open Envrion. Eng. J., 2009, 2, 21–29 CrossRef CAS.
- J. Matos, A. Garcia and P. S. Poon, J. Mater. Sci., 2010, 45, 4934–4944 CrossRef CAS.
- J. Matos, A. Garcia, T. Cordero, J.-M. Chovelon and C. Ferronato, Catal. Lett., 2009, 130, 568–574 CrossRef CAS.
- J. Matos, E. Garcia-Lopez, L. Palmisano, A. Garcia and G. Marci, Appl. Catal., B, 2010, 99, 170–180 CrossRef CAS.
- M. Asiltürk and S. Şener, Chem. Eng. J., 2012, 180, 354–363 CrossRef.
- A. E. Eliyas, L. Ljutzkanov, I. D. Stambolova, V. N. Blaskov, S. V. Vassilev, E. N. Razkazova-Velkova and D. R. Mehandjiev, Cent. Eur. J. Chem., 2013, 11, 464–470 CAS.
- T. S. Jamil, M. Y. Ghaly, N. A. Fathy, T. A. Abd el-halim and L. Österlund, Sep. Purif. Technol., 2012, 98, 270–279 CrossRef CAS.
- A. Omri, S. D. Lambert, J. Geens, F. Bennour and M. Benzina, J. Mater. Sci. Technol., 2014, 30, 894–902 CAS.
- A. Omri and M. Benzina, Environ. Monit. Assess., 2014, 186, 3875–3890 CrossRef CAS PubMed.
- N. M. Mahmoodi, M. Arami and J. Zhang, J. Alloys Compd., 2011, 509, 4754–4764 CrossRef CAS.
- S. Ragupathy, K. Raghu and P. Prabu, Spectrochim. Acta, Part A, 2015, 138, 314–320 CrossRef CAS PubMed.
- D. Huang, Y. Miyamoto, T. Matsumoto, T. Tojo, T. Fan, J. Ding, Q. Guo and D. Zhang, Sep. Purif. Technol., 2011, 78, 9–15 CrossRef CAS.
- L. F. Velasco, J. B. Parra and C. O. Ania, Appl. Surf. Sci., 2010, 256, 5254–5258 CrossRef CAS.
- Y. J. Li, Z. J. Song, Z. P. Li, Y. Z. Ouyang and W. B. Yan, Chem. J. Chin. Univ., 2007, 28, 1710–1715 CAS.
- J. M. Herrmann, C. Duchamp, M. Karkmaz, B. T. Hoai, H. Lachheb, E. Puzenat and C. Guillar, J. Hazard. Mater., 2007, 146, 624–629 CrossRef CAS PubMed.
- G. Xue, H. Liu, Q. Chen, C. Hills, M. Tyrer and F. Innocent, J. Hazard. Mater., 2011, 186, 765–772 CrossRef CAS PubMed.
- W. T. Tsai, M. K. Lee, T. Y. Su and Y. M. Chang, J. Hazard. Mater., 2009, 168, 269–275 CrossRef CAS PubMed.
- L. Lagunas-Allué, M. T. Martinez-Soria, J. S. Asensio, A. Salvador, C. Ferronato and J. M. Chovelon, Appl. Catal., B, 2010, 98, 122–131 CrossRef.
- S. Horikoshi, H. Hidaka and N. Serpone, Environ. Sci. Technol., 2002, 36, 1357–1366 CrossRef CAS PubMed.
- J. Matos, J. Laine and J. M. Herrmann, Appl. Catal., B, 1998, 18, 281–291 CrossRef CAS.
- L. L. Bo, X. Quan, S. Chen, H. M. Zhao and Y. Z. Zhao, Water Res., 2006, 40, 3061–3068 CrossRef CAS PubMed.
- A. L. Cazetta, O. Pezoti, K. C. Bedin, T. L. Silva, A. P. Junior, T. Asefa and V. C. Almeida, ACS Sustainable Chem. Eng., 2016, 4, 1058–1068 CrossRef CAS.
- A. M. Balu, B. Baruwati, E. Serrano, J. Cot, J. Garcia-Martinez, R. S. Varma and R. Luque, Green Chem., 2011, 13, 2750–2758 RSC.
- S. Wang and S. Zhou, Appl. Surf. Sci., 2010, 256, 6191–6198 CrossRef CAS.
- D. H. Quinonesa, A. Reya, P. M. Álvareza, F. J. Beltrána and P. K. Plucinski, Appl. Catal., B, 2014, 144, 96–106 CrossRef.
- S. H. Lin, L. Y. Hsu, C. S. Chou, J. W. Jhang and P. Wu, J. Anal. Appl. Pyrolysis, 2014, 107, 9–16 CrossRef CAS.
- Y. Xiao, Q. Zhou and B. Shan, J. Bridge. Eng., 2010, 15, 533–541 CrossRef.
- Y. Yu, Z. Jiang, G. Wang, G. Tian, H. Wang and Y. Song, Wood Sci. Technol., 2012, 46, 781–790 CrossRef CAS.
- K. Lu, J. Wood Sci., 2006, 52, 173–178 CrossRef CAS.
- S. Chang, T. Yeh and J. Wu, Polym. Degrad. Stab., 2001, 74, 551–557 CrossRef CAS.
- L. Luo, Y. Yang, M. Xiao, L. Bian, B. Yuan, Y. Liu, F. Jiang and X. Pan, Chem. Eng. J., 2015, 262, 1275–1283 CrossRef CAS.
- S. Mukherjee, S. Kumar, A. K. Misra and M. Fan, Chem. Eng. J., 2007, 129, 133–142 CrossRef CAS.
- M. B. Fernandes and P. Brooks, Chemosphere, 2003, 53, 447–458 CrossRef CAS PubMed.
- C. Da-li, J. Shen-xue and Z. Qi-sheng, For. Stud. China, 2012, 14, 240–245 CrossRef.
- C.-S. Chou, C.-Y. Chen, S.-H. Lin, W.-H. Lu and P. Wue, Adv. Powder Technol., 2015, 26, 711–717 CrossRef CAS.
- M. M. Dávila-Jiménez, M. P. Elizalde-González, E. García-Díaz, V. Marín-Cevada and J. Zequineli-Pérez, Appl. Catal., B, 2015, 166–167, 241–250 CrossRef.
- C. M. Antonio-Cisneros, M. M. Dávila-Jiménez, M. P. Elizalde-González and E. García-Díaz, Food Chem., 2015, 173, 725–732 CrossRef CAS PubMed.
- C. M. Antonio-Cisneros, M. M. Dávila-Jiménez, M. P. Elizalde-González and E. García-Díaz, Int. J. Mol. Sci., 2015, 16, 1590–1612 CrossRef CAS PubMed.
- S. Muhammad, E. Saputra, H. Sun, J. de C. Izidoro, D. A. Fungaro, H. M. Ang, M. O. Tade and S. Wang, RSC Adv., 2012, 2, 5645–5650 RSC.
- H. Cho, D. Oh and K. Kim, J. Hazard. Mater., 2005, 127, 187–195 CrossRef CAS PubMed.
- V. Heaquet, P. Ricou, I. Lecuyer and P. Le Cloirec, Fuel, 2001, 80, 851–856 CrossRef.
- S. B. Wang, Y. Boyjoo and A. Choueib, Chemosphere, 2005, 60, 1401–1407 CrossRef CAS PubMed.
- S. B. Wang, Y. Boyjoo, A. Choueib and Z. H. Zhu, Water Res., 2005, 39, 129–138 CrossRef CAS PubMed.
- S. Wang and H. Wu, J. Hazard. Mater., 2006, 136, 482–501 CrossRef CAS PubMed.
- K. S. Hui and C. Y. H. Chao, Microporous Mesoporous Mater., 2006, 88, 145–151 CrossRef CAS.
- H. Tanaka, H. Eguchi, S. Fujimoto and R. Hino, Fuel, 2006, 85, 1329–1334 CrossRef CAS.
- M. Gross-Lorgouilloux, P. Caullet, M. Soulard, J. Patarin, E. Moleiro and I. Saude, Microporous Mesoporous Mater., 2010, 131, 407–417 CrossRef CAS.
- Sindhunata, J. S. J. van Deventer, G. C. Lukey and H. Xu, Ind. Eng. Chem. Res., 2006, 45, 3559–3568 CrossRef CAS.
- J. G. S. van Jaarsveld, J. S. J. van Deventer and G. C. Lukey, Mater. Lett., 2003, 57, 1272–1280 CrossRef CAS.
- S. Wang, L. Li and Z. H. Zhu, J. Hazard. Mater., 2007, 139, 254–259 CrossRef CAS PubMed.
- E. Furlani, S. Bruckner, D. Minichelli and S. Maschio, Ceram. Int., 2008, 34, 2137–2142 CrossRef CAS.
- G. Qian, Y. Song, C. Zhang, Y. Xia, H. Zhang and P. Chui, Waste Manage., 2006, 26, 1462–1467 CrossRef CAS PubMed.
- S. B. Wang, G. Q. Lu and H. Y. Zhu, Chem. Lett., 1999, 28, 385–386 CrossRef.
- S. Wang and G. Q. Lu, Stud. Surf. Sci. Catal., 2007, 167, 275–280 CrossRef CAS.
- S. B. Wang, Environ. Sci. Technol., 2008, 42, 7055–7063 CrossRef CAS PubMed.
- X. Cui, J. Shi, Z. Ye, Z. Zhang, B. Xu and S. Chen, Int. J. Photoenergy, 2014, 823078 Search PubMed.
- D. Zhao, C. Chen, Y. Wang, W. Ma, J. Zhao, T. Rajh and L. Zang, Environ. Sci. Technol., 2008, 42, 308–314 CrossRef CAS PubMed.
- W. Kim, T. Tachikawa, T. Majima and W. Choi, J. Phys. Chem. C, 2009, 113, 10603–10609 CAS.
- J.-W. Shi, S.-H. Chen, S.-M. Wang, P. Wu and G.-H. Xu, J. Mol. Catal. A: Chem., 2009, 303, 141–147 CrossRef CAS.
- B. Gao, P. S. Yap, T. M. Lim and T.-T. Lim, Chem. Eng. J., 2011, 171, 1098–1107 CrossRef CAS.
- Y.-T. Yu, Powder Technol., 2004, 146, 154–159 CrossRef CAS.
- J. Wang, C. Li, X. Luan, J. Li, B. Wang, L. Zhang, R. Xu and X. Zhang, J. Mol. Catal. A: Chem., 2010, 320, 62–67 CrossRef CAS.
- X. Chen, C. Li, J. Wang, J. Li, X. Luan, Y. Li, R. Xu and B. Wang, Mater. Lett., 2010, 64, 1437–1440 CrossRef CAS.
- L.-N. Ho, S.-A. Ong and Y. L. See, Water, Air, Soil Pollut., 2012, 223, 4437–4442 CrossRef CAS.
- W. T. Liu, Y. Zhang, G. Y. Li, Y. Q. Miao and X. H. Wu, J. Fish Biol., 2008, 72, 1055–1067 CrossRef.
- S. Mustafiz, M. S. Rahaman, D. Kelly, M. Tango and M. R. Islam, Energy Sources, 2003, 25, 905–916 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b and
Paweł
Lisowski
*a
b and
Paweł
Lisowski
*a















 radicals which induce remarkable changes in the selectivity of products. In addition, the synergistic effect could be attributed not only to a proper surface area but also to the presence of a low surface concentration of acidic oxygenated functionalities, particularly carboxylic groups. However, the surface carboxyl anions (–COO−) are stabilized by transfer of electrons to the aromatic ring of graphene layers in activated carbon. Consequently, after dissociation in the aqueous phase, carboxylic acid (e.g. acetic acid) functional groups on the AC surface coordinate with the Ti4+ metallic center of TiO2. Alternatively, this interaction could inhibit the recombination of photogenerated species in the semiconductor (e−, h+) thus forming a higher concentration of hydroxyl radical (˙OH) and superoxide anion radical (O2˙−). The influence of the O2˙− anion radical is very important because the higher the concentration of O2˙−, the higher is the production of
radicals which induce remarkable changes in the selectivity of products. In addition, the synergistic effect could be attributed not only to a proper surface area but also to the presence of a low surface concentration of acidic oxygenated functionalities, particularly carboxylic groups. However, the surface carboxyl anions (–COO−) are stabilized by transfer of electrons to the aromatic ring of graphene layers in activated carbon. Consequently, after dissociation in the aqueous phase, carboxylic acid (e.g. acetic acid) functional groups on the AC surface coordinate with the Ti4+ metallic center of TiO2. Alternatively, this interaction could inhibit the recombination of photogenerated species in the semiconductor (e−, h+) thus forming a higher concentration of hydroxyl radical (˙OH) and superoxide anion radical (O2˙−). The influence of the O2˙− anion radical is very important because the higher the concentration of O2˙−, the higher is the production of  radicals that enhance the photoactivity of TiO2 in the photomineralization of 4-chlorophenol. It is worth noting that TiO2 supported on AC will degrade total organic carbon (TOC)/phenol via a photocatalytic oxidation process.128,129 Additionally, ˙OH may attack aromatic ring sites in compounds resulting in the formation of ring-opened (Fig. 13) products yielding CO2/H2O and other degradation products;136,137 breakdown of the benzene ring and its subsequent mineralization leading to CO2 and H2O could be envisaged by the decrease in TOC during the photocatalytic process.138 Moreover, AC as a support for TiO2 is effective in getting high rates of inactivate bacteria (E. coli) in the water phase.117 Interestingly, adsorbent supports provide a higher concentration of bacteria around the loaded TiO2via adsorption; the rate of E. coli inactivation in the presence of TiO2/AC catalysts is mostly determined by TiO2 particles and fits well a pseudo-first order kinetic equation. Furthermore, bacteria are inactivated on the photocatalyst surfaces and then further decomposed; the high prevailing inactivation efficiency of the composite prevents the repair of cells. The TiO2/AC composite has been used in conjunction with microwave (MW)-enhanced photodegradation of Rhodamine B (RhB) for the destruction of the conjugated structure, which was much faster than the photocatalytic degradation methods.118 A distinctive feature, that the TiO2 surface becomes more hydrophobic under MW irradiation,125 increases the probability of RhB contact with the TiO2 surface. Additional defect sites on TiO2 have been generated,139 which can increase the transition probability of e−/h+ and decrease e−/h+ recombination on the TiO2 surface. The strong interaction between TiO2 and AC can be explained by the substrate transfer to titania where it is degraded photocatalytically.140 In solution, the temperature of water and AC increases at different rates under MW irradiation due to in-core heating facilitated by MW energy which could generate locally higher temperature in some micro-surfaces of the AC particles compared to the water bulk.141
radicals that enhance the photoactivity of TiO2 in the photomineralization of 4-chlorophenol. It is worth noting that TiO2 supported on AC will degrade total organic carbon (TOC)/phenol via a photocatalytic oxidation process.128,129 Additionally, ˙OH may attack aromatic ring sites in compounds resulting in the formation of ring-opened (Fig. 13) products yielding CO2/H2O and other degradation products;136,137 breakdown of the benzene ring and its subsequent mineralization leading to CO2 and H2O could be envisaged by the decrease in TOC during the photocatalytic process.138 Moreover, AC as a support for TiO2 is effective in getting high rates of inactivate bacteria (E. coli) in the water phase.117 Interestingly, adsorbent supports provide a higher concentration of bacteria around the loaded TiO2via adsorption; the rate of E. coli inactivation in the presence of TiO2/AC catalysts is mostly determined by TiO2 particles and fits well a pseudo-first order kinetic equation. Furthermore, bacteria are inactivated on the photocatalyst surfaces and then further decomposed; the high prevailing inactivation efficiency of the composite prevents the repair of cells. The TiO2/AC composite has been used in conjunction with microwave (MW)-enhanced photodegradation of Rhodamine B (RhB) for the destruction of the conjugated structure, which was much faster than the photocatalytic degradation methods.118 A distinctive feature, that the TiO2 surface becomes more hydrophobic under MW irradiation,125 increases the probability of RhB contact with the TiO2 surface. Additional defect sites on TiO2 have been generated,139 which can increase the transition probability of e−/h+ and decrease e−/h+ recombination on the TiO2 surface. The strong interaction between TiO2 and AC can be explained by the substrate transfer to titania where it is degraded photocatalytically.140 In solution, the temperature of water and AC increases at different rates under MW irradiation due to in-core heating facilitated by MW energy which could generate locally higher temperature in some micro-surfaces of the AC particles compared to the water bulk.141