
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homogeneous vinyl ester-based synthesis of different cellulose derivatives in 1-ethyl-3-methyl-imidazolium acetate†
L. P.
Hinner
,
J. L.
Wissner
,
A.
Beurer
,
B. A.
Nebel
and
B.
Hauer
*
Institut für Technische Biochemie, Allmandring 31, Universtität Stuttgart, D-70569 Stuttgart, Germany. E-mail: bernhard.hauer@itb.uni-stuttgart.de
First published on 15th August 2016
Abstract
A homogeneous acylation of cellulose with different vinyl esters in the biodegradable and less toxic ionic liquid 1-ethyl-3-methyl-imidazolium acetate ([EMIM]OAc) is described for the first time. The reaction proceeds in the absence of any additional catalyst and glucose- and cellulose-esters with chain lengths of C8 to C16 are accessible by using equimolar amounts of acyl donor. Cellulose esters with a degree of substitution (DS) in the range of 0.9–3.0 were synthesised successfully. Different reaction parameters like reaction time, temperature and amount of substrate were systematically changed and analysed by NMR, IR and HPLC-GPC. The highest DS was achieved at 80 °C and a reaction time of 2 hours. Taking into consideration the literature, the DS and degree of polymerisation (DP) of fatty acid chloride and vinyl ester-based synthesis routes were compared. Similar DS-values were obtained, but the DP was significantly reduced during the synthesis using fatty acid chlorides in [BMIM]Cl. As an undesirable side reaction, acetates from [EMIM]OAc are bound to the cellulose backbone. The quantity of bound acetate groups during vinyl ester-based synthesis rose with decreasing polarity of the substrates but overall proved to be much lower compared to the literature described anhydride or fatty acid chloride based synthesis routes in [EMIM]OAc. This novel process was extended by using further acyl donors like vinyl benzoate, pivalate and 2-ethylhexanoate to demonstrate the applicability of the vinyl ester-based cellulose modification in [EMIM]OAc. [EMIM]OAc was recycled with an efficiency of ∼90% and reused for subsequent syntheses.
Introduction
Cellulose as a part of plant cell walls is the most abundant biopolymer on Earth. With an estimated annual biosphere production of 9 × 1010 tons, cellulose is a remarkable natural resource, which is nowadays mainly used for the production of paper and pulp or is simply burned in order to produce heat.1 In contrast, other applications based on the polymeric character of cellulose are also applied like for example the production of cellulose acetate. Thermoplastic and other kinds of polymeric materials based on cellulose have a great potential for reducing the dependency of our modern society on fossil fuels.Cellulose consists of several anhydroglucose units (AGU), which are β-(1→4)-glycosidically linked, thereby forming long polymer chains and a very strong hydrogen bond network with both intra- and inter-chain hydrogen bonds. This strong hydrogen bond network makes it very difficult to process cellulose because cellulose is not soluble in common organic solvents.2
The modification of cellulose, among others, can be achieved by esterification of the free cellulose hydroxy groups. However, the present technologies for cellulose modifications are mainly limited to heterogeneous reaction processes, where cellulose is not solubilised and an excess of acyl donor is required.3 Due to the strong steric hindrance of non-solubilised cellulose, such strategies are limited to the use of short acyl chains (C2–C4). In contrast, a homogeneous modification procedure of cellulose, where cellulose is solubilised, leads to benefits like control of the amount and distribution of substituents along the polysaccharide backbone, where even longer, sterically challenging acyl chains can be introduced.4 A homogeneous modification of cellulose can be realised in ionic liquids capable of solubilising cellulose.5–7
Parameters like degree of substitution (DS) and degree of polymerization (DP) are important to characterize the modified cellulose. The DS defines how many alcohol groups of cellulose are modified (n = 0–3). For thermoplastic applications of cellulose acetate, a DS of 2.5 is often applied because this value allows thermoplastic processability. However, the desired DS is dependent on the particular applications of produced materials and thus a good synthesis should permit a huge variety of modifications. The, e.g., heterogeneous process for cellulose acetate production is a multi-step process where cellulose is first fully acetylated (DS = 3) followed by a partial deacetylation step. A homogeneous synthesis protocol would allow an active DS adjustment by using different amounts of acyl donors.
The esterification of cellulose is usually carried out with highly reactive and corrosive carboxylic acid chlorides or anhydrides.4 The usage of anhydrides leads to high conversion rates but large amounts of acyl donor react to yield undesirable carboxylic acid side products. In addition, especially long-chain fatty acid anhydrides are highly nonpolar, which results in a minor solubility and a reduced mass transfer in polar cellulose solvent systems. In the case of carboxylic acid chlorides, acid scavengers are needed to avoid accumulation of highly reactive hydrogen chloride.8 Despite this, the usage of acetyl chloride in combination with some halogen containing ionic liquids leads to significant degradation of the cellulose backbone and the ionic liquid.9 In this context, the ionic liquid [EMIM]OAc displays a halogen free alternative with a high cellulose dissolution capability. In addition, [EMIM]OAc is, in comparison with halogen containing ionic liquids, less toxic and even biodegradable.10 [EMIM]OAc shows further benefits like non-volatility and a low melting point, but is, in this combination, not a complete inert solvent because it leads to a slight acetylation of 1.5 mol% of all cellulose hydroxy groups.11
Additionally, it was shown that the usage of anhydrides or chlorinated substrates as an acetylation agent led to the activation of the ionic liquid acetates and thereby to an increased acetylation of cellulose.10,12 This effect hampers the control of the substitution pattern of cellulose in the environmentally friendly ionic liquid [EMIM]OAc and limits its industrial application.
To reduce the acetylation effect induced by acyl donors and catalysed by [EMIM]OAc, the usage of less reactive acyl donors is recommended. A promising alternative to anhydride or acyl chloride based syntheses is the transesterification of cellulose originating from vinyl esters. A homogeneous vinyl ester-based synthesis of cellulose ester was already applied in a DMSO/tetrabutylammonium fluoride (TBAF) system, yielding cellulose esters with a high DS.13 As a significant drawback, the reaction suffers from long reaction times, up to days, and high substrate excess, necessary to accomplish high DS-values. In addition, the difficult recycling process of the DMSO/TBAF system and its flammability hamper industrial scale applications.12
Recently, a vinyl ester-based cellulose modification was carried out in a heterogeneous reaction system in DMSO with NaOH or KOH being used as catalysts. This cellulose ester process requires high substrate concentrations and is limited to short acyl chains of C2 to C4. However, due to low steric hindrance, synthesis was achieved in only five minutes.14 In contrast to this heterogeneous process, the vinyl ester-based synthesis of starch acetate in alkaline ionic liquids like [BMIM]OAc was described to be homogeneous.15 The catalytic property of some low molecular salts like acetates for transesterification forming starch acetates is a well-known principle.16 The catalytic principle in the context of ionic liquids was first described by Shogren and Biswas and recently used for the synthesis of cellulose acetate in [EMIM]OAc based on isopropenyl acetate as the acyl donor.15 This reaction was shown to proceed in minutes, but still high acyl donor concentrations are required.
In the present study, the first vinyl ester-based synthesis of cellulose derivatives in [EMIM]OAc is demonstrated. A variety of cellulose esters using long chain fatty acids, aromatic, branched and sterically challenging substrates, like pivalate, as acyl donors are accessible. This process leads to a high DS requiring a low amount of acyl donor, it ensures a constant high degree of polymerisation and allows significantly lower acetate activation in comparison with the anhydride based synthesis in [EMIM]OAc. Apart from these facts the vinyl ester-based synthesis shows benefits like high conversion rates, volatile side products, no need for acid scavengers, reusability of [EMIM]OAc, mild reaction conditions, low toxicity and biodegradable solvents.
Results and discussion
Vinyl ester-based synthesis in [EMIM]OAc
As a model reaction for initial parameter optimisation, glucose laurates were synthesised based on vinyl esters in [EMIM]OAc and product identification was carried out by LC-MS/ESI analysis. The highest yields of glucose dilaurate, glucose tri-laurate and glucose tetralaurate using 3 mol vinyl laurate per mol glucose were achieved after 4 hours at 60 °C (S1†). Longer reaction times and higher reaction temperatures resulted in a decrease in glucose laurate yield probably owing to unknown side reactions (S2†). In addition to glucose as a model substrate, the esterification of cellulose with different vinyl esters, especially vinyl laurate, in [EMIM]OAc was also investigated (Fig. 1). 3% w/w of cellulose, solubilised in [EMIM]OAc, was esterified with different amounts of vinyl laurate (2–5 mol per AGU) at 80 °C for 4 hours under continuous stirring. The cellulose laurates were analysed by FT-IR and showed specific cellulose laurate signals. In the range of 3200–3500 cm−1 O–H stretching, at 2852–2920 cm−1 CH2 and CH3 signals of the alkane chains, at 1740 cm−1 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of ester groups and at 1049 cm−1 a further significant cellulose signal representing C–C, C–OH, C–H ring and side group vibrations of the cellulose backbone appear (S6†).17 In addition, 1H-NMR spectra confirm the formation of cellulose laurate with signals of the AGU at 3.5–5.5 ppm. Furthermore, shifts at 2.2–2.4 ppm induced by the protons of the α-carbon and shifts at 1.4–1.7 ppm caused by the β-carbon of the esterified laurate were observed (Fig. 2). Alkane protons of cellulose laurate shifted at 1.1–1.35 ppm and the terminal methyl group at 0.85–0.9 ppm (Fig. 2).
O stretching of ester groups and at 1049 cm−1 a further significant cellulose signal representing C–C, C–OH, C–H ring and side group vibrations of the cellulose backbone appear (S6†).17 In addition, 1H-NMR spectra confirm the formation of cellulose laurate with signals of the AGU at 3.5–5.5 ppm. Furthermore, shifts at 2.2–2.4 ppm induced by the protons of the α-carbon and shifts at 1.4–1.7 ppm caused by the β-carbon of the esterified laurate were observed (Fig. 2). Alkane protons of cellulose laurate shifted at 1.1–1.35 ppm and the terminal methyl group at 0.85–0.9 ppm (Fig. 2).
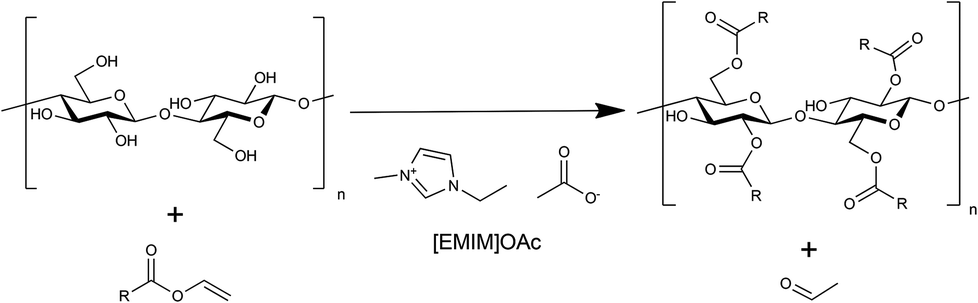 | ||
| Fig. 1 Reaction scheme of vinyl ester-based cellulose esterification. | ||
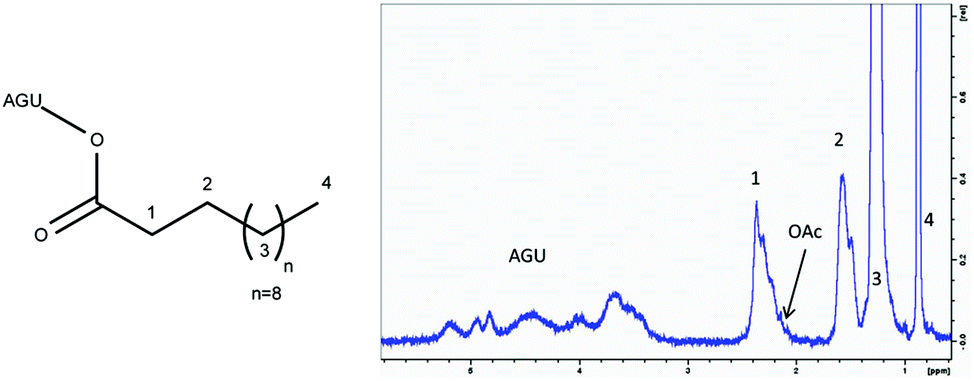 | ||
| Fig. 2 Structure and corresponding 1H-NMR of cellulose laurate. Cellulose laurate was synthesised in [EMIM]OAc using 3 mol per AGU vinyl laurate at 80 °C for 4 hours. Every peak is labeled to allow comparison with the structure drawn on the left side of the figure. The “OAc”-marked peak belongs to the α-carbon of additional bound acetate esters. | ||
Equimolar concentration of acyl donor (3 mol per AGU) resulted in a DS of 2.4, representing a conversion of 80% of cellulose hydroxy groups. Increasing the amount of acyl donor to 4 mol per AGU led to a higher DS of 2.9, while a further increase of substrate to 5 mol per AGU did not result in a higher DS (Table 1). All produced cellulose laurates were soluble in [EMIM]OAc and product separation was achieved by the addition of water. In comparison with the DMSO/TBAF reaction system, based on 10 mol per AGU vinyl laurate to obtain a DS of 2.6, the present process with significantly lower acyl donor concentrations resulted in even higher DS-values.13 Furthermore, the vinyl ester-based synthesis in [EMIM]OAc is as efficient with regard to the amount of acyl donor as the synthesis using fatty acid chloride in [BMIM]Cl. For instance, it has been reported that 5 mol steroyl chloride per AGU led to DS values of 2.6.8 In this case it has to be mentioned that a potentially higher steric hindrance in the case of a longer chain length (C18) may cause a lower synthesis efficiency. For a better understanding we synthesised cellulose laurate in [BMIM]Cl at 80 °C using lauroyl chloride and achieved a DS-value of 2.3, showing comparable potency of vinyl ester-based synthesis in [EMIM]OAc (DS 2.4) (Table 2). We further investigated the influence of the reaction temperature and time under equimolar reaction conditions. At reaction temperatures of 40–60 °C and 4 h reaction time the vinyl ester-based synthesis in [EMIM]OAc created DS-values lower than 2.1. The highest DS in the range of 2.3 to 2.4 was achieved at 70 °C or 80 °C, respectively, while a further increase to 90 °C and 110 °C resulted in lower DS values in the range of 1.9 to 2.2 (Fig. 3). The temperature optimum between 70 °C and 80 °C differed from the previously investigated temperature optimum in the synthesis of glucose laurate synthesis at 60 °C (S1†). Those differences can be explained by a higher viscosity during the reaction, which can result in lower mass transfer rates of solutions of cellulose in [EMIM]OAc compared to glucose in [EMIM]OAc. The optimised reaction temperature of 80 °C has already been applied for the synthesis of cellulose esters using fatty acid chlorides.8,9 The vinyl ester-based synthesis in [EMIM]OAc could be carried out at lower temperatures between 40 °C and 50 °C, enabling even more mild reaction conditions. However, this requires a longer reaction time and/or a higher acyl donor concentration to achieve a comparable degree of esterification (Fig. 3). The influence of the reaction time was investigated at an optimum temperature of 80 °C. During the first 30 minutes, the reaction proceeded fast and led to a DS-value of 2.1. After 60 minutes, the reaction was finished and DS-values between 2.1 and 2.4 were measured. No further increase in the DS of cellulose laurate could be detected up to 4 hours (Fig. 4). The reaction was more than 10-fold faster compared to vinyl ester-based synthesis in DMSO/TBAF where a reaction time of 70 hours was needed.13 We conclude that the reaction temperature and time have a great influence on the reaction speed and degree of substitutions of the produced cellulose ester.
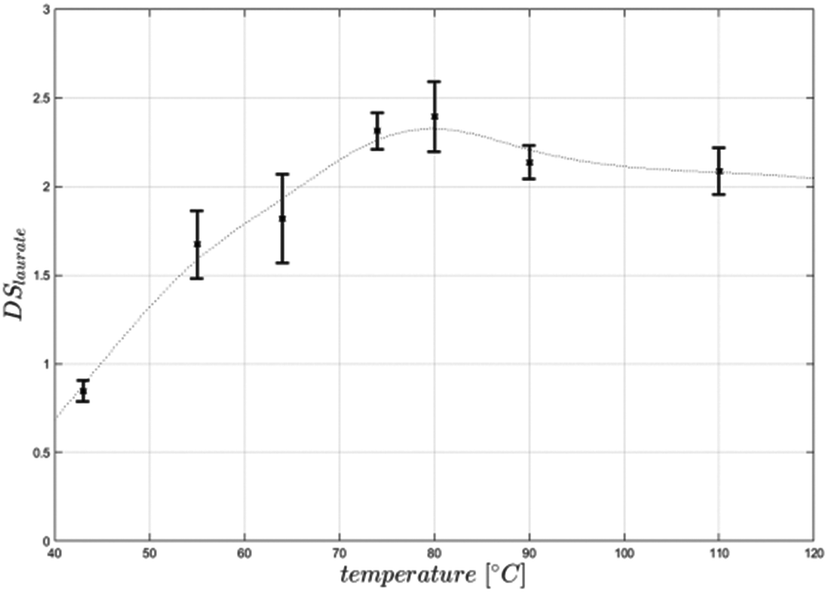 | ||
| Fig. 3 Temperature dependence of cellulose laurate synthesis in [EMIM]OAc. Cellulose laurate was synthesised in [EMIM]OAc with a reaction time of 4 hours, using 3 mol per AGU vinyl laurate. The reaction was carried out in triplicate. | ||
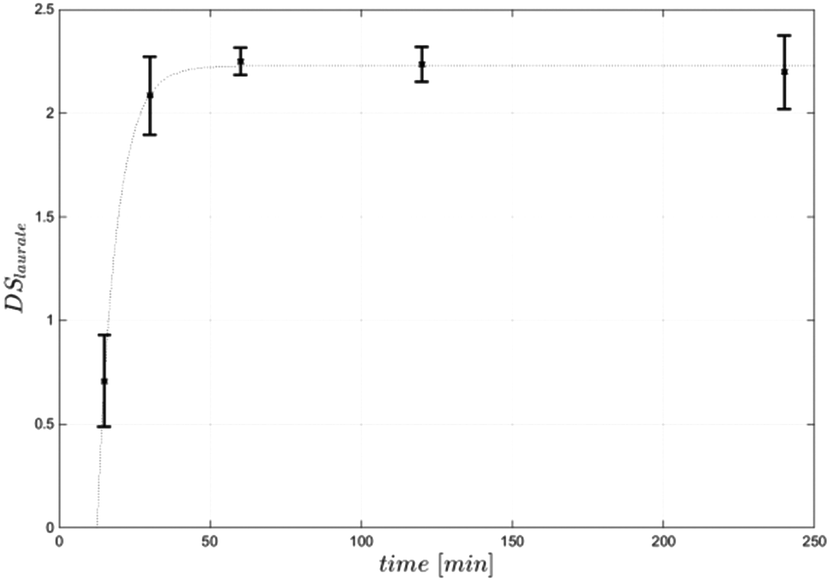 | ||
| Fig. 4 Reaction kinetics of cellulose laurate synthesis at 80 °C. Cellulose laurate was synthesised in [EMIM]OAc using 3 mol per AGU vinyl laurate at 80 °C. The reaction lasted for 4 hours and at different time points samples were collected. The reaction was carried out in triplicate. | ||
| Product | Vinyl laurate [mol per AGU] | DS |
|---|---|---|
| Cellulose laurate | 2 | 1.6 |
| 3 | 2.4 | |
| 4 | 2.9 | |
| 5 | 2.8 |
| Compound | Ionic liquid | Vinyl laurate [mol per AGU] | Temp. [°C] | Time [h] | DS | M w [g mol−1] | M n [g mol−1] |
|---|---|---|---|---|---|---|---|
| Vinyl laurate | [EMIM]OAc | 3 | 80 | 4 | 2.4 | 280![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 368 368 |
89936 |
| Lauroyl chloride | [BMIM]Cl | 3 | 80 | 2 | 2.3 | 8743 | 5630 |
Comparison with fatty acid chloride based synthesis routes
To demonstrate the differences of the present process and product quantification and qualification in comparison with the existing synthesis strategies, several experiments based on lauroyl chloride in [BMIM]Cl were carried out and compared to the vinyl ester-based synthesis in [EMIM]OAc. For both synthesis strategies similar DS values were observed using equimolar acyl donor concentration (Table 2). We obtained a slightly higher DS value of lauroyl chloride modified cellulose than that described in the literature by Barthel and co-workers.9 These data are more in accordance with results published by Huang et al., where cellulose stearates with DS values of 2.6 in [BMIM]Cl were synthesised.8 Those differences could be explained by different stirring conditions, causing different mass transfer behaviour. An important observation, besides comparable DS values discussed above, is the significant differences in molecular weight distributions between different synthesised cellulose derivatives. Relative to polystyrene standards, cellulose laurates synthesised using lauroyl chloride displayed a reduced molecular weight distribution. The number average molar mass (Mn) is about 15 times lower and the mass average molar mass (Mw) is actually 32 times lower compared to cellulose laurates synthesised from vinyl laurate (Table 2). It has to be mentioned that these values were calculated relative to polystyrene standards being, in comparison with cellulose laurate, non-dendritic polymers. This can lead to an inaccuracy in calculated values. Nevertheless, differences in molar mass distributions of various synthesised cellulose laurate derivatives with similar DS values in a magnitude of 15 to 32 are surprising and indicate that cellulose laurate synthesis in [BMIM]Cl with lauroyl chloride distinctly decreases the DP (Table 2). The degradation of the cellulose backbone with carboxylic acid chlorides has already been discussed in the literature.8,18 In addition, the reduction of the degree of polymerisation has already been shown using acetyl chloride in the context with other halogen containing ionic liquids, such as 1-ethyl-3-methylimidazolium chloride ([EMIM]Cl).9 This reduction may be caused by the formation of highly reactive hydrogen chloride or by reactive lauroyl chloride itself. We showed that the vinyl ester-based synthesis in [EMIM]OAc resulted in a much higher DP. We assume that the non-acidic and volatile acetaldehyde is produced instead of highly reactive by-products (Fig. 1). Furthermore, the vinyl ester itself is less reactive in comparison with the carboxylic acid chloride causing gentler reaction conditions and thus a reduced depolymerisation of the cellulose backbone.Acetylation of cellulose as an undesirable side reaction
Previous work demonstrated that the cellulose–[EMIM]OAc system undergoes different side reactions.4 For instance, the acetylation of cellulose at low concentrations in [EMIM]OAc was reported in the absence of derivatising reagents. Even without any further basic catalyst, the imidazolium acetate containing ionic liquid can catalyse a deprotonation, which can lead to the formation of carbene species followed by a modification of reducing end-groups of cellulose. However, in the absence of an additional base the side reaction mentioned above is very slow and expires in days.4 Another side reaction, which shifted into our focus, is the formation of mixed anhydrides after introducing chlorinated substrates or anhydrides into the synthetic approach, which results in increased acetylation of cellulose.4,12 Furthermore it was reported that introducing propionic anhydride as an acyl donor, [EMIM]OAc catalyses the corresponding cellulose ester, but with a majority of acetylated hydroxy groups and only a small amount of propion esters.12 The acetylation of cellulose in [EMIM]OAc was analysed by 1H-NMR. The α-carbon of the cellulose laurate carbonyl group, synthesised with fatty acid chlorides in [BMIM]Cl, showed a chemical shift between 2.10 ppm and 2.50 ppm (Fig. 5). In contrast to cellulose esters with no additional acetylation, increased acetylation of the remaining and unmodified hydroxyl groups was obtained in [EMIM]OAc. This could be demonstrated by a second peak at 2.15 ppm, which refers to cellulose acetate methyl groups (Fig. 5). It is worth mentioning that the determination of the acetate degree of substitution (DSacetate) by 1H-NMR is not feasible for highly substituted long chain cellulose esters (C8–C12), because signals can hardly be separated. However, in the case of cellulose esters with a lower DS, peaks are separated in the corresponding 1H-NMR spectra and thus the DSacetate can be calculated.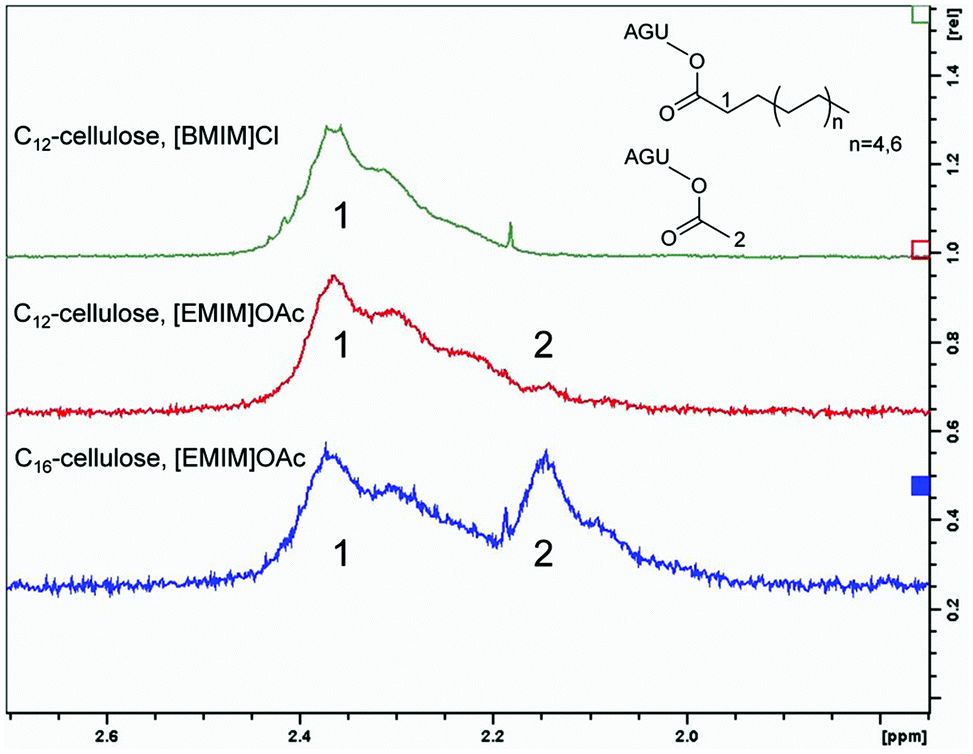 | ||
| Fig. 5 Signals of the α-carbon of cellulose fatty acid ester and cellulose acetate in 1H-NMR-spectra from different syntheses. The synthesis of cellulose laurate (C12) was carried out in [BMIM]Cl using 3 mol per AGU lauroyl chloride and in [EMIM]OAc using 3 mol per AGU vinyl laurate. For the synthesis of cellulose palmitate in [EMIM]OAc 3 mol per AGU vinyl palmitate was used. The signal of the fatty acid ester α-carbon is indicated with 1, while the signal of the α-carbon of cellulose acetate is indicated with 2. | ||
We compared the DSacetate after syntheses of cellulose laurates using lauroyl chloride, lauric anhydride and vinyl laurate in [EMIM]OAc. In accordance with the literature, we detected a majority of DSacetate towards DSlaurate using lauroyl chloride and lauric anhydride, respectively.10 The highly reactive lauroyl chlorides created 88.2% acetylation while lauric anhydride gave 83.3% acetylation of all substituted hydroxyl groups (DStotal) (Table 3). Vinyl laurate induced an acetylation of only 11.1%, which implies that the majority of 88.9% of all OH-groups is modified by the introduced acyl donor. These results show that the usage of vinyl esters allows a better control of the substitution pattern of cellulose in [EMIM]OAc in comparison with fatty acid chloride or anhydride based syntheses. In addition, the usage of fatty acid chlorides and anhydrides with equimolar substrate concentrations caused a significantly reduced DSacyl donor of 0.3 compared to the vinyl laurate synthesis, where the DSacyl donor is higher than 2.1. Furthermore, the amount of additional acetate groups varied not only between fatty acid chlorides, anhydrides and vinyl esters, but also different side chains resulted in differences in %-DSacetate. Esterification with sterically challenging and highly nonpolar vinyl palmitate gave a higher additional acetylation of 20.0%, while the application of shorter and less nonpolar vinyl laurate or vinyl octanoate resulted in significantly lower acetylation, of 11.1% and 6.3%, respectively (Fig. 5 and Table 3). In addition, a %-DSacetate of 4.2% to 4.5% of cellulose was achieved using the shorter and less nonpolar vinyl pivalate and vinyl benzoate (Table 3). However, the moderate nonpolar but branched and sterically demanding vinyl 2-ethylhexyloate leads to a significantly smaller DSacyl donor of 0.9 and a combined elevated %-DSacetate of 18.1% (Table 3). Such results suggest that polarity and steric properties have a great influence on additional acetylation.
| Acyl donor | Acyl donor [mol per AGU] | DSacyl donor | DSacetate | DStotal | %-DSacetate [%] |
|---|---|---|---|---|---|
| Vinyl octanoate (C8) | 2 | 1.5 | 0.1 | 1.6 | 6.3 |
| Vinyl laurate (C12) | 2 | 1.6 | 0.2 | 1.8 | 11.1 |
| Vinyl palmitate (C16) | 2 | 1.6 | 0.4 | 2.0 | 20.0 |
| Vinyl benzoate | 3 | 2.3 | 0.1 | 2.4 | 4.2 |
| Vinyl pivalate | 3 | 2.1 | 0.1 | 2.2 | 4.5 |
| Vinyl 2-ethylhexyloate | 3 | 0.9 | 0.2 | 1.1 | 18.1 |
| Lauroyl chloride | 3 | 0.04 | 0.3 | 0.34 | 88.2 |
| Lauric anhydride | 3 | 0.06 | 0.3 | 0.36 | 83.3 |
Regarding the reaction time, it can be speculated that the activation of acetates in [EMIM]OAc using vinyl esters is much slower compared to anhydrides showing a reduced acetylation of cellulose. The acylation of cellulose with vinyl esters is faster compared to the undesirable activation of acetate. Decreasing the polarity of introduced vinyl esters resulted a low solubility as well as reduced mass transfer in polar [EMIM]OAc and therefore a slower reaction. Assuming that the vinyl ester-based acetate activation proceeds at the phase interface and is faster than the mass transfer of nonpolar acyl donor, an increased acetylation of the cellulose backbone, as observed, is consequential. For sterically demanding substrates like vinyl 2-ethylhexyloate, the acylation is hindered which supports acetylation. Apart from these assumptions, it can be pointed out that the vinyl ester-based synthesis allows a gentle modification of cellulose with a comparably low acetylation. Especially if unbranched, relatively polar vinyl esters are used, acetylation is limited to low DSacetate values.
Additional modes of functionalisation
Besides fatty acids, other functional groups were also tested to modify cellulose and to show the universal applicability of the developed vinyl ester-based cellulose modification. We synthesised cellulose benzoates with a DSbenzoate of 2.3 and a DSacetate of 0.1. The corresponding IR-spectrum showed characteristic cellulose benzoate peaks at 1711 cm−1 (CO in OC–O–R), 1264 cm−1 (C–O in OC–O–R), 3067 cm−1 (benzene ring C–H stretching), 1601 cm−1 and 1451 cm−1 (aromatic CC) and 707 cm−1 for out of plane C–H bending of monosubstituted benzene (S5†). 1H-NMR also confirmed the formation of cellulose benzoate at δ = 6.80–8.20 (phenyl protons, 5H) and δ = 2.80–5.50 ppm (proton of cellulose backbone, 7H). In addition, a small signal at δ = 1.55–2.10 ppm appeared, which was considered to be the proton of the corresponding methyl group of acetate. In addition to cellulose benzoate, the sterically challenging cellulose pivalate with a DSpivalate of 2.1 and a DSacetate of 0.1 was synthesised using the corresponding vinyl esters (Table 3). The seven protons of the cellulose pivalate backbone gave 1H-NMR signals at δ = 2.80–5.50 ppm, while the protons of methyl groups showed signals at δ = 1.30–0.90 ppm (9H) (Fig. 6A).19 IR-spectrometry also confirmed the formation of cellulose pivalate and showed characteristic bands at 2972 cm−1 and 2875 cm−1 (C–H in CH3 groups, stretching vibrations), at 1732 cm−1 (CO in OC–O–R), at 1461 cm−1 and 1481 cm−1 (C–H in CH3, asymmetric deformation vibration), and at 1397 cm−1 and 1367 cm−1 (t-butyl groups) (S4†).20
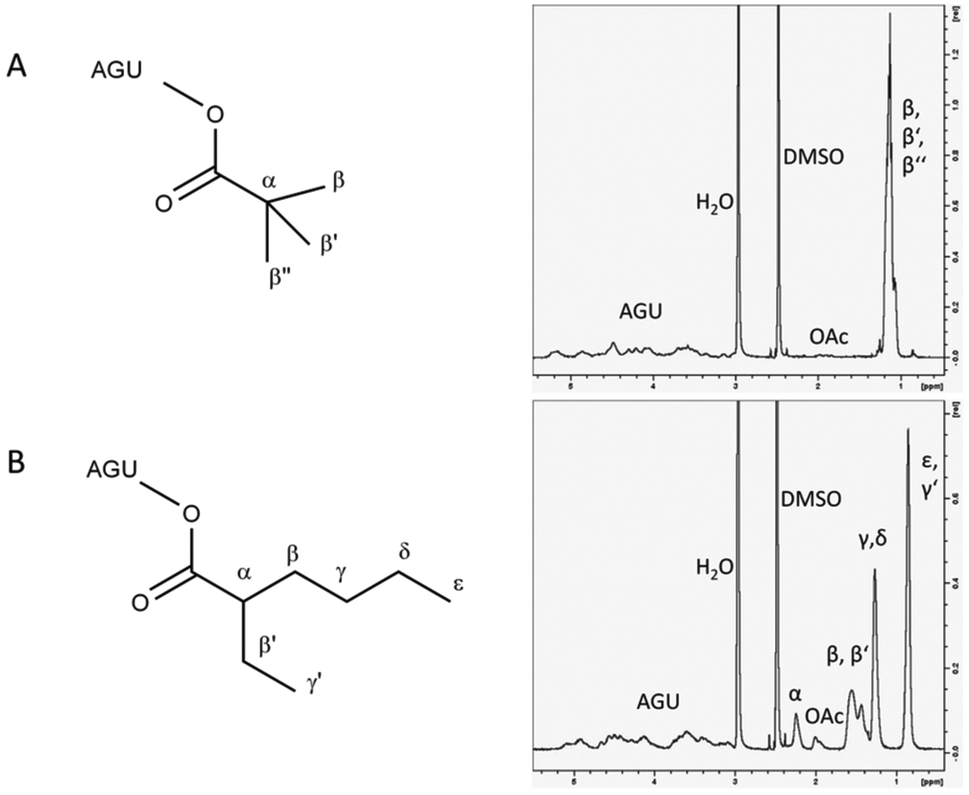 | ||
| Fig. 6 Structure and 1H-NMR of cellulose pivalate (A) and cellulose 2-ethylhexyloate (B). Cellulose pivalate and cellulose 2-ethylhexyloate were synthesised in [EMIM]OAc using 3 mol per AGU of the corresponding vinyl ester. The “OAc” peak belongs to the α-carbon of additional bound acetate esters. | ||
The cellulose 2-ethylhexyloate with a DS2-ethylhexyloate of 0.9 and a DSacetate of 0.1 was also prepared. 1H-NMR confirmed the formation of cellulose 2-ethylhexyloate. The two terminal methyl groups (ε,γ’) displayed signals at δ = 0.95–0.70 ppm (6H), the protons of the γ- and δ-carbon of hexyl groups gave δ = 1.35–1.10 ppm (4H) and the protons of the β-carbon of the hexyl group and the β’-carbon of the ethyl side chain led to a peak at δ = 1.65–1.35 ppm (4H) (Fig. 6B). At δ = 2.10–1.85 ppm a signal from the α-carbon of the additional bound acetate appeared in the 1H-NMR spectrum (Fig. 6B). The IR spectrum of cellulose 2-ethylhexyloate showed distinctive signals at 3500 cm−1 (O–H), signals between 2872 cm−1 and 2958 cm−1 (C–H) and a band at 1732 cm−1 (CO in OC–O–R) (S3†). The vinyl ester-based synthesis in [EMIM]OAc enabled the gentle synthesis of a huge variety of cellulose esters with high and moderate degrees of substitution. The products were soluble in the ionic liquid and product separation was achieved by the addition of water. Not only fatty acid esters with different chain lengths, even aromatic, bulky and branched cellulose modifications were successfully synthesised.
Upscaling, product separation and IL reusability
To investigate the product separation and reusability of [EMIM]OAc, the novel synthesis was performed in a larger scale using 200 g of [EMIM]OAc, 4 mol vinyl laurate per AGU (34 g) and 3% w/w cellulose (6.2 g) at 80 °C (1. synthesis) (Table 4). The reaction was performed for 4 hours and quenched afterwards by the addition of 200 ml of water leading to immediate product precipitation followed by decantation to separate product and the ionic liquid/water mixture. The solid product was washed with methanol, and dried at 60 °C giving 18.8 g cellulose laurate with a DSlaurate of 2.3 (Table 4). The degree of substitution is lower compared to syntheses, which were performed on a smaller scale (Table 1). Those differences might be explained as due to poor mixing in the used up-scaling equipment. To recover the ionic liquid out of the aqueous mixture, the system was heated up to 100 °C for 4 hours to evaporate the water. It was possible to recover 186 g of the ionic liquid, which represents a recycling efficiency of 93% (Table 5). After a repeated synthesis with a recycled ionic liquid (2. synthesis), using 5.8 g cellulose and 32 g vinyl laurate (Tables 4 and 5) a DS of 2.4, a product yield of 21 g and a recycling efficiency of 88% were obtained. Barthel and Heinze published a significantly smaller yield using lauroyl chloride where 0.35 g cellulose laurate (DS = 1.54) was obtained using 0.5 g cellulose and 2.14 ml lauroyl chloride.9 The higher yield from the present synthesis might be explained by less depolymerisation of cellulose ester and a higher degree of substitution. In detail, a decreased degree of polymerization, induced by fatty acid chlorides, leads to a complex mixture of cellulose laurate oligomers and polymers, which complicate the downstream process (product recovery) and can negatively influence the mass balance of the whole process. Small oligomers of cellulose esters might be partial soluble in purification solvents like water and methanol and thus get washed out. In the following synthesis–recycling cycle (3. synthesis) the DS of produced cellulose laurate decreased to 1.8 and, after a further synthesis–recycling cycle (4. synthesis), to 1.6. Simultaneously, the amount of cellulose laurate obtained decreased to 12 g and 10 g, respectively (Table 4). A similar effect has already been observed for the phosphorylation of starch in 1-butyl-3-methylimidazolium chloride ([BMIM]Cl) where the obtained DS decreased from 0.55 to 0.45 within 5 recycling cycles.21 The reduced DS can be caused by the loss of catalytic performance of the ionic liquid or can be explained by the formation of side products. Investigations to understand and prevent the mechanism of reduced synthesis efficiency after each recycling step have to be performed in further studies.| Reactor run | [EMIM]OAc input [g] | [EMIM]OAc recycled [g] | Recycling efficiency [%] |
|---|---|---|---|
| 1. Synthesis | 200 | 186 | 93 |
| 2. Synthesis (1. recycl.) | 186 | 163 | 88 |
| 3. Synthesis (2. recycl.) | 163 | 156 | 96 |
| 4. Synthesis (3. recycl.) | 156 | 137 | 88 |
In general, the recycling of the ionic liquid is a key parameter for an ecologically friendly and economically feasible process. In that context, King et al. discussed the possibility of distilling [EMIM]OAc under special conditions, but they discussed this option very sceptically regarding the purity and corrosiveness of the recycled ionic liquid.22 Therefore, evaporation strategies for ionic liquids seem inappropriate for [EMIM]OAc.
In our case we have chosen the often applied anti-solvent strategy where the addition of a solvent leads to product precipitation and enables separation.9,23,24 Afterwards the evaporation of the volatile anti-solvent recovers the ionic liquid. The performed recycling experiments using water as an anti-solvent demonstrate a recycling efficiency of about 90%. In contrast to the synthesis efficiency, the recycling efficiency is not hampered by each recycling step. In addition, the ability of the ionic liquid to solubilize cellulose is not negatively affected. The usage of other anti-solvents like ethanol or acetone might be cost saving alternatives due to lower boiling points24 but these solvents enhance the hazardousness of the process due to their flammability. The loss of about 10% w/w ionic liquid can be explained by the residual ionic liquid, which remained in the crude product after the water induced precipitation and was washed out during the methanol purification step.
Experimental
Materials
The utilized α-cellulose, vinyl laurate, vinyl pivalate, vinyl benzoate, trifluoroacetic acid-d (d-TFA), lauroyl chloride and chloroform-d (CDCl3) were purchased by Sigma-Aldrich, Taufkirchen, Germany. Vinyl octanoate, vinyl 2-ethylhexyloate, vinyl palmitate and lauric anhydride were obtained from TCI-Deutschland GmbH, Eschborn, Germany.[BMIM]Cl and [EMIM]OAc were received from IoLiTec-Ionic Liquids Technologies GmbH, Heilbronn, Germany. ReadyCal-Kit Poly(styrene) Mp 474–2520000 Da and gel permeation column SDV, linear M 10 μM, 8 × 300 mm were purchased from Polymer Standard Service GmbH, Mainz, Germany. DMSO-d6 was purchased from Euriso-Top GmbH, Saarbrücken, Germany.
Measurements
The DS of cellulose ester was analysed by 1H-NMR on a 500 MHz NMR-spectrometer (Bruker, Avance 500) in CDCl3, or a mixture of CDCl3 and dimethyl sulfoxide-d6 depending on the solubility of the particular modified cellulose. Those polymers, which were soluble in pure DMSO-d6, were analysed at 100 °C on a 700 MHz NMR-spectrometer (Bruker, Ascend). 10 μg μl−1 cellulose ester was solubilised in the corresponding NMR-solvent. 1.67% of trifluoroacetic acid (d-TFA) was added to shift hydroxyl protons of cellulose or residual water signals into a region of non-interest or to deuterate hydroxy groups as reported previously.25,26 The DSfatty acid of cellulose octanoate, -laurate and -palmitate was calculated from the 1H-NMR spectrum from the integral of the terminal methyl groups (Imethyl) at 0.85–0.90 ppm and the area of the AGU signals (IAGU) at 3.50–5.50 ppm according to: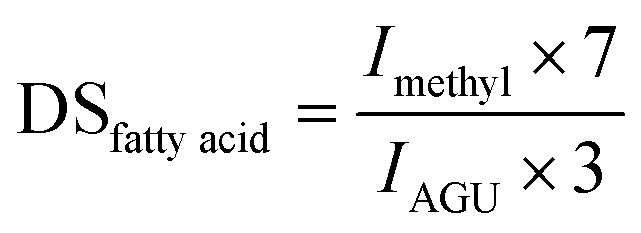
In the case of cellulose 2-ethylhexyloate the integral of the two terminal methyl groups (I2methyl) at 0.78–0.92 ppm, for cellulose pivalate the integral of the three terminal methyl groups (I3methyl) at 0.90–1.35 ppm and for cellulose benzoate the area of the phenyl protons (Iphenyl) at 6.90–8.10 ppm were used to determine the corresponding DS-values consistent with the following equations:

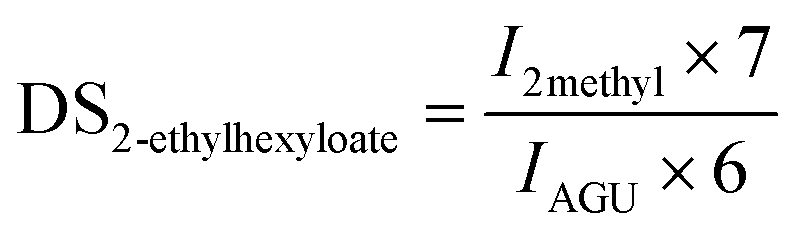
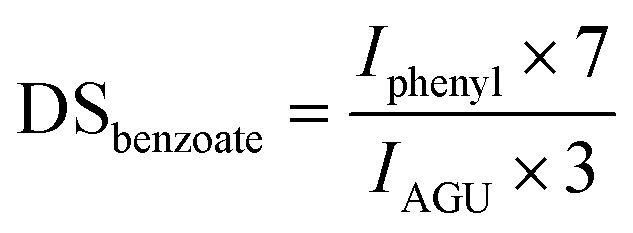
For cellulose pivalate, ethylhexyloate, benzoate and for low to medium substituted cellulose fatty acid esters also the DSacetate was calculated by using the area of the acetate methyl groups (Iacetate) at 1.85–2.07 ppm in relation to the integral of AGU (IAGU) at 3.50–5.50 ppm according to the equation:
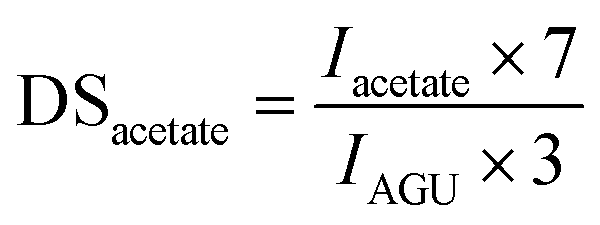
The FT-IR spectra were recorded with a FT-IR spectrometer (Bruker, Vector 22, equipped with a MKII golden gate single reflection diamond ATR-system).
Gel permeation chromatography was performed using an Agilent Technology 1260 Infinity ELSD-HPLC system (Agilent, Santa Clara, USA) using a SDV, linear M 10 μM, 8 × 300 mm, column. The flow rate and temperature were 1.5 mL min−1 and 40 °C. THF was used as the eluent and the solvent for cellulose ester dissolution. The method stayed isocratic for 10 minutes. For molecular weight calibration, ReadyCal-Kit Poly(styrene) Mp 474–2520000 Da from PSS (Mainz, Germany) was used. The calculation of molecular weight distribution (Mw, Mn) was carried out using Agilent Cirrus GPC multi detector software.
Glucose laurates (mono-, di-, tri-, tetra-) were separated using an Agilent Eclipse XDP (CN-column, 150 × 4.6 mm, 5 μm). The flow rate and temperature were 0.8 mL min−1 and 35 °C. The method is isocratic at 5% ACN and 95% water (0.1% v/v formic acid) for the first 3 minutes. Afterwards an ACN–water (0.1% v/v formic acid) gradient increased from 3% ACN to 98% ACN in 15 minutes and stayed isocratic for 5 minutes. Glucose laurates were analysed by HPLC-ELSD (Agilent Technology 1260 Infinity HPLC-ELSD system) and identified by HPLC-MS (Agilent Technologies 6130 Quadrupole MS). The LC-MS was operated in API-ES mode under the following conditions: drying gas flow 10 L min−1, nebuliser 40 psig, drying gas temperature 300 °C, and +/− Vcap 3000. The system was used in positive and negative scan modes with a fragmentor voltage of 70 eV to cover values between m/z 100 and 1500. The ELSD was operated at a nebuliser temperature of 90 °C, an evaporator temperature of 40 °C and a gas flow of 1 SLM. Quantification of glucose laurates was performed by using the peak area of the detector response because no standard of glucose laurates was available.
Acetylation of glucose in [EMIM]OAc
Glucose was solubilised in [EMIM]OAc to receive 350 mg of a 0.83 mmol mg−1 solution. For investigations regarding the temperature dependency of the reaction, 2 mol vinyl laurate per mol glucose was added to the solution and shaken in an Eppendorf thermomixer comfort at 600 rpm at different temperatures between 40 and 90 °C for 22 hours. In addition, the time kinetics of the reaction was analysed using 3 mol vinyl laurate per mol glucose at 60 °C. Afterwards the samples were diluted by a factor of 4 with acetonitrile and analysed by HPLC.Acylation of cellulose in [EMIM]OAc
Dissolving of 3% w/w cellulose was achieved in [EMIM]OAc at 80 °C and mixed with a KPG-stirrer up to 12 h to accomplish complete dissolution. The reaction was started by the addition of 3 mol vinyl ester, lauric anhydride, or lauroyl chloride per AGU and mixed for 4 hours at 80 °C. 1 mL samples were taken at different time points and cooled to 4 °C. The polymer was separated from the ionic liquid by the addition of 50% w/w water followed by centrifugation. The supernatant was discarded and the cellulose pellet was washed with an excess amount of methanol and subsequently dried at 60 °C. The amount of introduced vinyl esters (2–5 mol per AGU) and the reaction temperature (40–110 °C) were varied systematically to investigate their influence. In the case of cellulose 2-ethylhexyloate and cellulose pivalate, the product was dissolved in methanol and precipitated with an excess amount of water and then dried by lyophilisation for 14 hours.Upscaling and reusability
The upscaling was performed similarly to a described standard protocol, but using higher amounts of educts, solvents and a 1 litre reaction flask (Table 4). After the addition of water the product was separated from the ionic liquid/water mixture by filtration. The ionic liquid was recycled by evaporating the water from the ionic liquid/aqueous mixture at 100 °C for 4 hours. The recycled ionic liquid was weighed and reused for an additional synthesis.Acylation of cellulose in [BMIM]Cl
The acylation of cellulose in [BMIM]Cl was carried out according to Barthel and Heinze,9 but using a KPG-stirrer to assure sufficient stirring of the highly viscous cellulose solution.Conclusions and outlook
In this study we developed a process that allows for the first time the synthesis of a variety of cellulose esters using the minor toxic and biodegradable ionic liquid [EMIM]OAc. The described synthetic strategy represents a new approach for homogeneous and gentle production of cellulose esters with a sensitive control over substitution pattern with even sterically demanding nonpolar side chains. In contrast to the fatty acid chloride based syntheses in halogen containing ionic liquids, the novel vinyl ester-based synthesis in [EMIM]OAc leads to a much higher DP. The acetylating character of [EMIM]OAc towards cellulose is significantly lower using vinyl esters in contrast to anhydrides or chlorinated acyl donors and allows a better regulation of the substitution pattern of cellulose. A variety, like branched and bulky side chains, fatty acids of C8–C16 chain length and aromatic groups, can be introduced into the cellulose backbone. The ionic liquid [EMIM]OAc was recycled with an efficiency of about 90% and reused for three additional synthesis–recycling cycles. Investigations towards further upscaling and additional recycling studies of this reaction to examine the commercial applicability of the process will be carried out and may enable the implementation of a new homogeneous environmentally friendly modification of cellulose. These efforts will possibly attenuate the dependence of our society on fossil fuels for the synthesis of different kinds of polymeric materials.Acknowledgements
We would like to thank the Bundesministerium für Bildung und Forschung for the funding within the framework of Cluster Bioindustrie 2021.References
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS PubMed.
- T. Liebert, Cellulose Solvents – Remarkable History, Bright Future, ACS Symposium Series, Washington, DC, 2010, vol. 1033 Search PubMed.
- J. Zhang, J. Wu, Y. Cao, S. Sang, J. Zhang and J. He, Cellulose, 2008, 16, 299–308 CrossRef.
- M. Gericke, P. Fardim and T. Heinze, Molecules, 2012, 17, 7458–7502 CrossRef PubMed.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- R. P. Swatloski, R. D. Rogers and J. D. Holbrey, Univ. Alabama, US 6824599, 2004, 2 Search PubMed.
- T. Liebert and T. Heinze, BioResources, 2008, 3, 576–601 Search PubMed.
- K. Huang, J. Xia, M. Li, J. Lian, X. Yang and G. Lin, Carbohydr. Polym., 2011, 83, 1631–1635 CrossRef CAS.
- S. Barthel and T. Heinze, Green Chem., 2006, 8, 301–306 RSC.
- S. Köhler, T. Liebert, M. Schöbitz, J. Schaller, F. Meister, W. Günther and T. Heinze, Macromol. Rapid Commun., 2007, 28, 2311–2317 CrossRef.
- S. K. Karatzos, L. A. Edye and R. M. Wellard, Cellulose, 2011, 19, 307–312 CrossRef.
- S. Dorn, Jena, Univ., Diss., 2009 Search PubMed.
- T. Heinze, R. Dicke, A. Koschella, A. H. Kull, E.-A. Klohr and W. Koch, Macromol. Chem. Phys., 2000, 201, 627–631 CrossRef CAS.
- X. Cao, S. Sun, X. Peng and L. Zhong, J. Agric. Food Chem., 2013, 61, 2489–2495 CrossRef CAS PubMed.
- R. L. Shogren and A. Biswas, Carbohydr. Polym., 2010, 81, 149–151 CrossRef CAS.
- R. Dicke, Cellulose, 2004, 11, 255–263 CrossRef CAS.
- M. Fan, D. Dai and B. Huang, Fourier transform infrared spectroscopy for natural fibres, InTech, 2012 Search PubMed.
- M. C. V. Nagel and T. Heinze, Polym. Bull., 2010, 65, 873–881 CrossRef CAS.
- D. Xu, B. Li, C. Tate and K. J. Edgar, Cellulose, 2010, 18, 405–419 CrossRef.
- M. Hesse, H. Meier and B. Zeeh, Spektroskopische Methoden in der organischen Chemie, 2005, vol. 2 Search PubMed.
- W. Xie and L. Shao, Starch/Staerke, 2009, 61, 702–708 CrossRef CAS.
- A. W. T. King, J. Asikkala, I. Mutikainen, P. Järvi and I. Kilpeläinen, Angew. Chem., Int. Ed., 2011, 50, 6301–6305 CrossRef CAS PubMed.
- K. Shill, S. Padmanabhan, Q. Xin, J. M. Prausnitz, D. S. Clark and H. W. Blanch, Biotechnol. Bioeng., 2011, 108, 511–520 CrossRef CAS PubMed.
- J. Shi, K. Balamurugan, R. Parthasarathi, N. Sathitsuksanoh, S. Zhang, V. Stavila, V. Subramanian, B. A. Simmons and S. Singh, Green Chem., 2014, 16, 3830 RSC.
- C. Buchanan, K. Edgar, J. Hyatt and A. Wilson, Macromolecules, 1991, 24, 3050–3059 CrossRef CAS.
- G. Samaranayake and W. G. Glasser, Carbohydr. Polym., 1993, 22, 79–86 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Influence of temperature and time on the production of glucose laurates in [EMIM]OAc. IR-spectra of cellulose 2-ethylhexyloate, cellulose pivalate, and cellulose benzoate. See DOI: 10.1039/c6gc02005d |
| This journal is © The Royal Society of Chemistry 2016 |