
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneous photocatalytic organic synthesis: state-of-the-art and future perspectives
Donia
Friedmann†
a,
Amer
Hakki†
b,
Hyejin
Kim
c,
Wonyong
Choi
c and
Detlef
Bahnemann
de
aParticles and Catalysis Research Group, School of Chemical Engineering, Tyree Energy Building, The University of New South Wales, Sydney NSW 2052, Australia. E-mail: donia.friedmann@unsw.edu.au
bDepartment of Chemistry, King's College, University of Aberdeen, Aberdeen AB24 3UE, UK. E-mail: a.hakki@abdn.ac.uk
cSchool of Environmental Science and Engineering/Department of Chemical Engineering, Pohang University of Science and Technology (POSTECH), Pohang, Korea. E-mail: wchoi@postech.edu
dLaboratory “Photoactive Nanocomposite Materials”, Saint-Petersburg State University, Ulyanovskaya str. 1 Peterhof, Saint-Petersburg, 198504 Russia. E-mail: detlef.bahnemann@spbu.ru
eLaboratory “Photocatalysis and Nanotechnology”, Institut fuer Technische Chemie, Gottfried Wilhelm Leibniz Universitaet Hannover, Callinstrasse 3, D-30167 Hannover, Germany. E-mail: bahnemann@iftc.uni-hannover.de
First published on 6th September 2016
Abstract
Heterogeneous photocatalytic systems have the potential to provide a green organic synthesis route for a number of industrially important chemicals. Issues remain with lack of selectivity. In this paper, a review is presented on achievements in this field. Parallels are drawn between systems optimised for heterogeneous photocatalytic organic degradation and heterogeneous catalytic organic synthesis. There is much fundamental knowledge that is still missing in this field of research. Parameters that can be manipulated are reaction solvent, pH, photon energy, chosen photocatalyst and its specific properties, and perhaps the use of more than one photocatalyst. Screening of photocatalysts for specific reactions and adapting the reaction conditions may achieve the best selectivity. Unlike the popular case of photocatalysts for organic degradation, the photocatalysts for organic synthesis should be highly customised on a case-by-case basis. Attention should be given to photocatalysts with the potential to be activated by the visible light spectrum, in order to achieve cost effectiveness of the heterogeneous photocatalytic organic synthesis.
 Donia Friedmann | Dr Donia Friedmann received her bachelor and PhD of Chemical Engineering from the University of New South Wales in Sydney, Australia in 1996 and 2000 respectively. Since then she has worked both in academia and industry as a researcher, predominately in the areas of particle science and technology, with a focus on heterogeneous photocatalysis for environmental applications. She has 38 publications and a h-index of 25. A career highlight is being awarded the Alexander von Humboldt Postdoctoral Research Fellowship in 2005 to carry out research work at the University of Leibniz in Hannover, Germany with Prof. Detlef Bahnemann's group. |
 Amer Hakki | Dr rer. nat. Amer Hakki is currently a Research Fellow in the Department of Chemistry at the University of Aberdeen (United Kingdom). He received his PhD degree in Technical Chemistry in 2013 from the Gottfried Wilhelm Leibniz University Hannover, Germany, after working under the guidance of Prof. Detlef W. Bahnemann. He then completed postdoctoral training at the same institute with Prof. Bahnemann. Amer Hakki research interests are centered on photocatalysts and their environmental applications as well as solar energy conversion reactions. |
 Hyejin Kim | Hyejin Kim received a BS in Chemistry at Chonnam National University (Gwangju, Korea) in 2015 and currently pursuing a MS in Environmental Engineering under the supervision of Prof. Wonyong Choi at POSTECH (Pohang, Korea). Her scientific interests include organic photocatalyst synthesis and environmental applications. |
 Wonyong Choi | Wonyong Choi received B.S. from Seoul National University (Korea) in 1988 and Ph.D. from CALTECH (USA) in 1996. He joined POSTECH in 1998. His research interests are mainly focused on semiconductor photocatalysis and photochemistry for solar energy conversion and environmental applications. Dr Choi has published more than 230 articles with over 24 |
 Detlef Bahnemann | Prof. Dr rer. nat. habil. Detlef Bahnemann (PhD in Chemistry TU Berlin 1981, Habilitation in Technical Chemistry Leibniz University Hannover 2012) is currently Head of the Research Unit “Photocatalysis and Nanotechnology” at the Institute of Technical Chemistry of the Leibniz University Hannover in Germany and also Director of the Research Institute Nanocomposite Materials for Photonic Applications at Saint Petersburg State University. His main research topics include photocatalysis, photoelectrochemistry, solar chemistry and photochemistry focussed on synthesis and physical–chemical properties of semiconductor and metal nanoparticles. His more than 300 publications have been cited more than 27 |
Introduction
Organic chemicals are essential for the manufacture of a great number of products and other chemicals including pharmaceuticals, pesticides, and food additives which are an integral part of everyday life. Chemical industries have their established organic synthesis routes and manufacturing processes in place that have not changed greatly over the years. However, with increasing knowledge with regards to the environmental impact of some reagents, catalysts, generated by-products and waste solvents, environmental policies have become more stringent. At the same time, there is an ever increasing need to consider energy costs to remain competitive and important safety issues associated with the handling of toxic and highly reactive reagents. Subsequently, chemical manufacturers have had to consider improvements at each step of their processes including alternative ‘green’ synthesis routes and ‘green’ processing methods.Conventional industrial routes for many important organic chemicals typically require harsh operating conditions, such as high temperature and pressure. Thus, the development of photocatalytic synthesis routes which rely on light as an energy source to drive chemical reactions under much milder reaction conditions is highly desirable. Moreover, photocatalytic systems match the needs of green engineering in which fewer processing steps are achieved by employing multiple-catalysts or so called ‘one-pot’ reactions. These photocatalytic systems may be homogeneous or heterogeneous systems. Heterogeneous systems utilise a solid phase photoactive semiconductor as photocatalyst whereas homogeneous systems may employ photosensitive molecules which are soluble in the reaction media such as photoactive dyes.
Semiconductor heterogeneous photocatalysis generates both oxidising and reducing species on a single particle at the same time. It is therefore suited for the synthesis of organics either through oxidative or reductive pathways or through the combination of both pathways. In the literature there are many examples of heterogeneous photocatalytic organic synthesis via oxidative pathways1–6 and via reductive pathways.7–11 The potential to utilise visible light for photocatalyst activation could mean even greater economical and environmental advantages. This has been demonstrated for the sunlight-induced functionalisation of heteroaromatic bases with aldehydes.12 However, the photostability of these kind of photocatalysts has to be taken into account. Both homogeneous and heterogeneous photocatalytic organic synthesis methods remain predominantly at the research stage, with few commercial applications.13 Further development of this route as a viable ‘greener’ alternative for high temperature and pressure methods is highly desirable.
There has been a number of reviews on photocatalytic organic synthesis. Publications by Shiraishi and Hirai14,15 are of particular significance to this area of research as are those by Palmisano et al.2,13,16 Recently, two general reviews have been published by Cherevatskaya and Koenig,17 Vorontsov and Arsentyev.18 Other relevant published reviews have been more specific, for example the review by Lu and Yao19 focussed on oxidation reactions, in particular alcohols oxidation, aromatic hydroxylation and alkene oxidation. The review by Valenzuela et al.20 was specific to reduction reactions. Ohtani et al.21 reviewed photocatalytic synthesis of cyclic amino acids. Molinari et al.22,23 focused on the reduction and partial oxidation of organic compounds in membrane reactors. Munir et al.24 reviewed the development of photocatalysts for selective and efficient organic transformations. Hakki et al.25 reviewed the synthesis of nitrogen containing compounds. More recently the use of 1-D nanostructures for improved selectivity has gained more attention and has been reviewed by Weng et al.26 and Han et al.27 A mini-review has been published on core–shell nanostructure photocatalysts for improved selectivity.28 Visible light photocatalysis for organic transformations has been reviewed by Chen et al.29 and Lang et al.30 Photocatalysis for the selective transformations of biomass derived compounds has been reviewed by Colmenares and Luque.31 Li et al.32 reviewed and reported on recent advances in the selective heterogeneous photocatalytic valorisation of lignin-based compounds into value-added chemicals. González-Béjar et al.33 contributed a book chapter on light driven catalysis published in Green Chemistry which covered a number of relevant reactions and mechanisms. There have also been some recently published books and book chapters on general photocatalysis such as those by Colmenares and Xu,34 Yuan et al.35 and Imamura et al.36
Another important and relevant area of research to heterogeneous photocatalytic organic synthesis is the vast number of work on the photocatalytic reduction of CO2. This topic is both important from a CO2 capturing perspective and for its potential to produce an array of useful chemicals. Significant advances have been made in this field and such processes and reactions are much better understood. Li et al. reviewed the state of the art of CO2 photocatalytic reduction applications utilising hierarchical nano/micro photocatalysts which usually provide the advantages of large surface areas, high CO2 adsorption capacities and fast mass transport.37 They discussed the development of various hierarchical photocatalysts including 3D hierarchical microspheres, hetero-structured nanocomposites, yolk/shell structures and hollow structures. Liu et al. have reviewed the activities of combined TiO2 semiconductor nanocatalysts under solar light for the reduction of CO2.38 Both reviews by Li et al. and Liu et al. have concluded that more studies are still needed for such systems.37,38 Liu et al. also identified several materials and systems based on TiO2 semiconductors for the electrophotocatalytic CO2 reduction to methane.
Given the recent surge of activity in the field of photocatalytic organic synthesis, an updated, encompassing review is needed. In this review here, we highlight achievements in the field of heterogeneous photocatalytic organic synthesis and discuss the kinetics, mechanisms, selectivity and yields of a number of studied reactions, with discussions on the related semiconductor photochemistry. In doing so, we also highlight knowledge gaps that exist in these areas of science and their interdependence, and discuss strategies to improve photocatalysts selectivity and the process in general.
Interestingly, Ravelli et al.39 carried out a Life Cycle Analysis (LCA) and an Environmental Assessment Tool for Organic Synthesis (EATOS) to assess the environmental burden associated with some functionalisation reactions of nitrogen-heteroaromatics using TiO2 (solar) photocatalysis and compared these with the same reactions under thermal conditions. In that study, the thermal processes were evaluated to give a better environmental performance than their photocatalytic counterparts, while the simplicity of photocatalysis was highlighted. Such studies are essential for better decision making. A better understanding and improvements of photocatalytic organic synthesis efficiencies, a reduced solvent usage and the reliance on solar energy, will help pave the way for the further development of this route as a viable ‘greener’ alternative for high temperature and pressure chemical manufacturing methods.
Background
Heterogeneous semiconductor photocatalysis
Semiconductor photocatalysis involves the activation of a semiconductor by light, as shown schematically in Fig. 1. This light needs to have energy greater than the semiconducting photocatalyst's bandgap, that is, the energy between the valence band (VB, where electrons are bound to individual atoms) and the conduction band (CB, where the electrons are free to move in the atomic lattice of the material). The thermodynamics of the photocatalytic oxidation and reduction half reactions that can take place are thus determined by the potential of CB electrons and that of VB holes, that is, the positions of the band edges of a given photocatalyst. It is therefore essential to take into account the reduction potentials E of the substrate, and of several other intermediates that are formed during the photocatalytic reaction.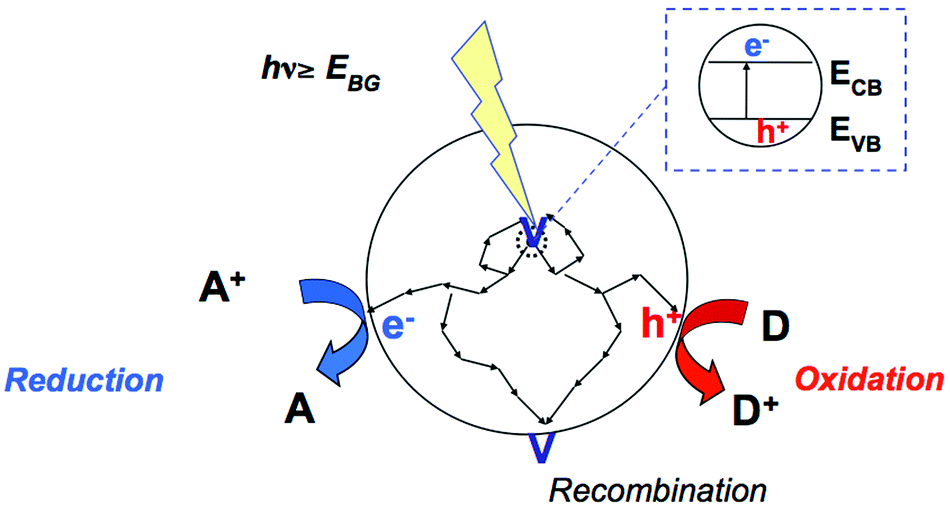 | ||
| Fig. 1 Schematic of photoactivation of semiconductor and generation of electron hole pair. h+: hole, e−: electron, a: electron acceptor, d: electron donor. cb: conduction band, vb: valence band, ebg: band gap energy, hν: energy of impinging photon. | ||
The reduction potential E of the couples M/M˙− refers to reactions described by reactions (R1) and (R2). These reactions refer usually to one-electron reductions vs. the standard hydrogen electrode.
| oxidant + e− → reductant (for example Cl˙/Cl−) | (R1) |
| oxidant + e− + nH+ → reductant (for example ˙OH, H+/H2O) | (R2) |
The VB holes of most metal oxide semiconductors are highly oxidising and can directly oxidise surface adsorbed species. When the holes oxidise adsorbed water or surface hydroxyl groups, they form highly oxidising ˙OH radicals. These ˙OH radicals can then take part in various oxidation reactions, on or near the surface of the photocatalyst. On the other hand the photogenerated CB electrons typically reduce dioxygen, if present in the system, and generate radicals which also take part in oxidation reactions. However, the photogenerated electrons can also reduce other ions or species in the system if this is thermodynamically possible.
Semiconductor heterogeneous photocatalysis has been studied for a number of years, predominately for the treatment of wastewaters40 or contaminated air,41 and hydrogen production by water splitting.42 For the treatment of water and air, the oxidising radicals can degrade the organic pollutants by successive oxidation reactions and can typically achieve the complete mineralisation of organic contaminants.43
From the vast number of studies in this field it has been concluded that heterogeneous semiconductor photocatalysis for environmental remediation is mostly suited for the degradation of waste streams containing low concentrations of pollutants that are highly hazardous and typically difficult to treat with conventional technologies.43,44 For water splitting, the small driving force for H2 generation is an issue with TiO2 photocatalysts.45
On the fundamental level, heterogeneous photocatalytic processes are on the way to be well understood. The photocatalytic activity of a semiconductor is the result of an interplay between phase composition, electronic structure, particle size, exposed surface area, degree of aggregation, mobility of charge carriers, presence of impurities, amount and kind of defects, adsorption of molecules from gas or liquid phases, lateral interactions between adsorbed species, and the nature of solvent used.43 Various modifications to the TiO2 photocatalyst, being one of the most researched photocatalysts, have been made to achieve better efficiencies, for example by doping with noble metals and metal ions.46 Such modifications have been useful in obtaining incremental increases in efficiencies, but not enough to allow commercialisation and acceptance of TiO2 photocatalysis as a competitive technology for environmental applications or water splitting.
Application of heterogeneous photocatalysis to synthesise organic compounds is equally not very common.13 This research field is still developing and fundamental research in this area is continuing. It is clear thus far that an array of useful organic compounds can be synthesised using this route, both through oxidative and reductive pathways, this review highlights key results and findings. We also include a separate section on heterogeneous photocatalytic organic synthesis of polymers.
Overview of heterogeneous photocatalytic organic transformations
In photocatalytic reactions the semiconductor particle behaves practically as a microelectrode kept always under open circuit potential with the anodic and cathodic current being equal in magnitude. Thus two reactions, an oxidation and a reduction, must proceed simultaneously on the same particle surface (otherwise the particle would be charged, eventually leading to the overall reaction being stopped). Hoffman47 described the photon as a traceless agent, and the transfer of the photogenerated electron and hydrogen as basic steps in the context of a photocatalytically driven process, occurring either simultaneously or in two steps.In the photocatalytic organic synthesis process, the presence of electron or hole scavengers is always necessary when the desired product is formed via reaction(s) with VB holes and CB electrons, respectively. However, in many cases, the photocatalytically produced intermediates at both VB and CB edges are substrates for further catalytic reactions at the surface of the employed semiconductor which may result in a desired final product, at a given selectivity. In the following sections photocatalytic organic transformations involving oxidative reactions are described as are those involving reductive reactions.
Photocatalytic organic transformations involving oxidative reactions
Much effort has been devoted to the application of TiO2 for the selective photocatalytic oxidation of a broad range of organic compounds including hydrocarbons, aromatic compounds, and alcohols. Fox and co-workers were one of the earliest researchers who have placed great emphasis on the photocatalytic organic transformation especially on the photocatalytically induced oxygenation of various organic compounds.48–50 These authors have found that the products obtained employing TiO2 as the photocatalyst are different from those obtained electrochemically on metal electrodes as well as from those generated when a homogeneous photocatalyst was used.Almquist and Biswas51 studied the photocatalytic oxidation of cyclohexane on TiO2 in various solvents to determine the effect of the solvent media (refer to the reaction sequence in Fig. 2). Selectivity to cyclohexanol and cyclohexanone and reaction rates were found to be dependent on adsorption, the type of solvent and the partially oxidised solvent species on the photocatalyst surface. In non-polar solvents, selectivity was low with cyclohexanol preferentially adsorbing onto TiO2 and being completely mineralised to CO2. In polar solvents, selectivity was higher since cyclohexanol adsorbed on TiO2 to a lesser extent due to competition with the solvent.
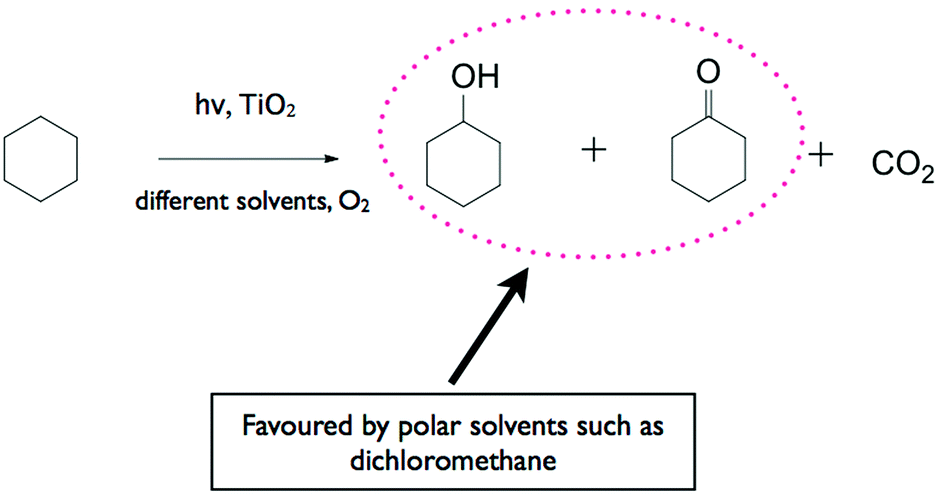 | ||
| Fig. 2 Reaction sequence of the photocatalytic oxidation of cyclohexane on TiO2 in various solvents to determine the effect of the solvent media. Polar solvents favour the desired products cyclohexanone and cyclohexanol, complete oxidation to CO2 occurs in non-polar solvents. | ||
Mu et al.52 have also studied the oxidation of liquid cyclohexane using acetonitrile as a solvent. The effects of catalyst loading, temperature, radiant flux, and reactant concentration were examined. An 83% selectivity of cyclohexanone was achieved with cyclohexanol (5%) and CO2 (12%) being the other products.
Park and Choi54 have investigated the effects of various parameters (electron acceptor, photocatalyst surface modification, and the combination of photocatalysts) on the direct synthesis of phenol from benzene using photocatalytic oxidation processes. They have found that the addition of Fe3+, H2O2, Fe3+ + H2O2, or polyoxometalate highly enhanced the phenol production yield and selectivity in TiO2 suspensions. Moreover, modification of the TiO2 surface either by platinisation or by fluorination also increased the yield of the photocatalytically produced phenol.
Palmisano et al.2 demonstrated the effect of the substituent group of benzene derivatives on selectivity to hydroxylated compounds. The substituents studied were either electron withdrawing groups (EWG) (nitrobenzene, cyanobenzene, benzoic acid, 1-phenylethanone), electron donor groups (EDG) (phenol, phenylamine, N-phenylacetamide) or a combination of EWG and EDG (4-cholorophenol). The substituent group was found to determine selectivity to ortho and para mono-hydroxy derivatives. The competing mineralisation of the parent compound and intermediates was found to be important for compounds containing an EWG due to the strong interaction of these molecules with the TiO2 surface.
Yoshida et al.4 reported direct hydroxylation of benzenes to phenols using Pt/TiO2 suspended in water containing a high substrate concentration. For example they studied a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v benzene/water reaction solutions in which selectivity for hydroxylation of benzenes was greatly improved when the reaction was conducted in the absence of molecular O2. In these reactions on Pt/TiO2 photocatalysts, protons (H+) were used as electron acceptors instead of molecular O2, accompanied by production of H2 on Pt co-catalysts.
:1 v/v benzene/water reaction solutions in which selectivity for hydroxylation of benzenes was greatly improved when the reaction was conducted in the absence of molecular O2. In these reactions on Pt/TiO2 photocatalysts, protons (H+) were used as electron acceptors instead of molecular O2, accompanied by production of H2 on Pt co-catalysts.
Soana et al.55 studied the photocatalytic oxidation of naphthalene in aqueous solution. The products were (E,Z)-2-formylcinnamaldehydes and 1,4-naphthoquinone and traces of naphthols. They also studied the 1-substituted naphthalenes (both with an electron-withdrawing and electron-donating group). The results showed a similar product distribution indicating that there is no direction effect of the substituent on the aromatic ring. They postulated a mechanism involving the transfer of a hydroxy group to naphthalene followed by coupling with superoxide radical. Ohno et al.56 also studied the photocatalytic oxidation of naphthalene, however in a mixed solution of acetonitrile and water using various kinds of TiO2 powders as the photocatalysts and molecular oxygen as the electron acceptor. Their main product was 2-formylcinnamaldehyde. According to Lu and Yao,19 difficulties encountered during the hydroxylation of aromatics using heterogeneous photocatalysis, such as unclear mechanisms, and a complex interplay of various factors on achieving desired selectivity, continue to hinder industrial applications of such a process.
The photocatalytic oxidation of primary, secondary, and tertiary as well as aromatic alcohols by TiO2 particles suspended in their aqueous solutions has been studied extensively.64–69 Wang et al.68,69 have reported that the photocatalytic oxidation pathway of methanol from its aqueous solution, depends on the molecular species adsorbed at the TiO2 surface (reactions (R3)–(R8)). According to their measurements, the authors concluded that at a critical molar ratio between water and methanol of approximately 300, water is the dominant surface species and the oxidation pathway is via the photocatalytically generated ˙OH radicals (refer to reactions (R5)–(R6)). If the water content is lower than this critical ratio, the direct oxidation of methanol by the photogenerated holes will be the predominant process at the TiO2 surface (refer to reactions (R7), (R8)).
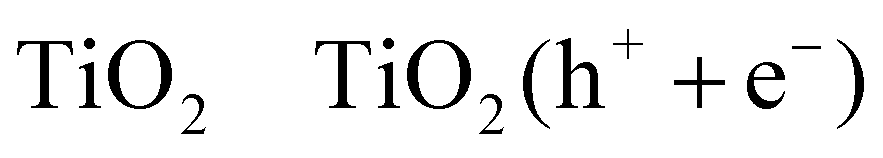 | (R3) |
| TiO2 (h+) + OHs− → TiO2 + ˙OHs | (R4) |
| TiO2 (h+) + H2Os → TiO2 + ˙OHs + Haq+ | (R5) |
| ˙OHs + RCH2OH → R˙CHOH + H2O | (R6) |
| TiO2 (h+) + RCH2OH → TiO2 + R˙CHOH + Haq+ | (R7) |
| R˙CHOH + O2 → RCHO + HO2˙ | (R8) |
Molinari et al. demonstrated the TiO2 photocatalytic conversion of geraniol, citronellol, trans-2-penten-1-ol and 1-pentanol and achieved >70% selectivity.70 This study provided insights into the mechanistic processes of the alcohol partial oxidation. It highlighted the necessity of alcohol adsorption and the inhibiting competitive effect of water on alcohol adsorption and subsequently its conversion. The effect of the alcohol chain on reactivity was also studied, with the longer chains being more susceptible to the inhibiting effect of water content. From studies on the photocatalytic degradation of organic contaminants, it has been shown that small changes to the molecule structure strongly influence photocatalyst performance.71
Bellardita et al.1 demonstrated the photocatalytic synthesis of piperonal from piperonyl alcohol (refer to Fig. 3). The best selectivity was around 35%. Other products detected were CO2 and trace amounts of 1,3-bis(3,4-(methylenedioxy)benzyl) ether. This latter was due to the coupling of alcohol molecules at higher piperonal concentrations. Photocatalytic oxidation of benzene to phenol with TiO2 in aqueous media has been performed by many researchers, but selectivities were significantly lower (20%) compared to those achieved by Bellardita et al.1 and references within.
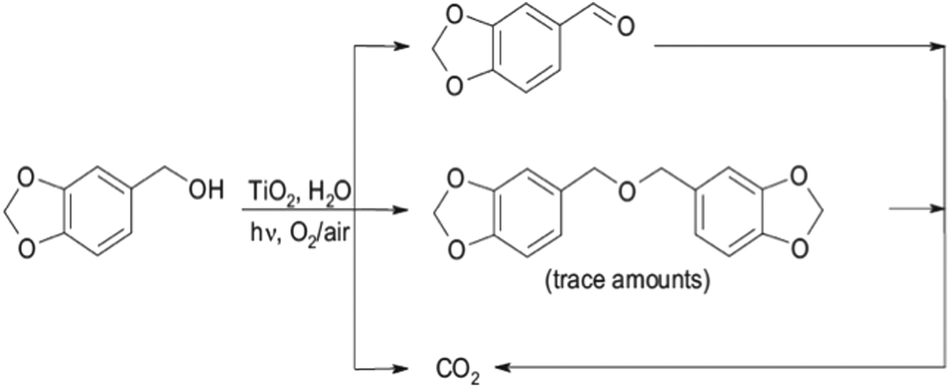 | ||
| Fig. 3 Reaction sequence for the photocatalytic synthesis of piperonal from piperonyl alcohol.1 | ||
Augugliaro et al.72 studied the photocatalytic production of vanillin at room temperature in aqueous medium starting from different educts as trans-ferulic acid, isoeugenol, eugenol or vanillyl alcohol employing commercial or home prepared TiO2 samples as photocatalysts. The selectivity to vanillin ranged from 1.4 to 21 mol% with respect to the starting substrate. Moreover, the same research group was able to enhance the yield of the photocatalytically produced vanillin by combining the photocatalytic system with a pervaporation separation process.73 The utilisation of a highly selective membrane allowed the continuous recovery of vanillin by pervaporation from the reacting solution so that its oxidative degradation was largely avoided and the yield was substantially enhanced.
Zhang et al. studied the aerobic oxidation of glycerol in water using visible light activated photocatalysts.74 Selectivity was achieved by the use of sol–gel encapsulated photocatalytic species in silica based matrices. The combination of water as solvent, the use of visible light as the driving energy source and ambient conditions address the required criteria for achieving green chemical process. Colmenares et al.75 reported on an interesting magnetically separable TiO2/maghemite-silica nanocomposite photocatalyst for the selective oxidation of benzyl alcohol. They achieved an unprecedented selectivity towards benzaldehyde of 90% in acetonitrile at a benzyl alcohol conversion of ca. 50%. This was superior in terms of activity to any other supported transition metal catalysts reported to date.75
Photocatalytic organic synthesis of nitrogen containing compounds
Ohtani et al.76 demonstrated the photocatalytic N-alkylation of ammonia (in alcohol) to amines using Pt–TiO2 as photocatalyst. The same authors77 also reported the formation of secondary amines from primary amines in aqueous solutions employing platinised TiO2 photocatalysts, with some oxygenated products including alcohols and aldehydes ((R9)–(R12) in Fig. 4). Selectivity was found to be dependent on the Pt loading on TiO2. Starting from diamines, cyclic secondary amines can also be photocatalytically synthesised.78–80 Fox and co-workers48 reported the photocatalytic oxidation of several primary aliphatic amines in anhydrous acetonitrile to form symmetrical N-alkylidene amines.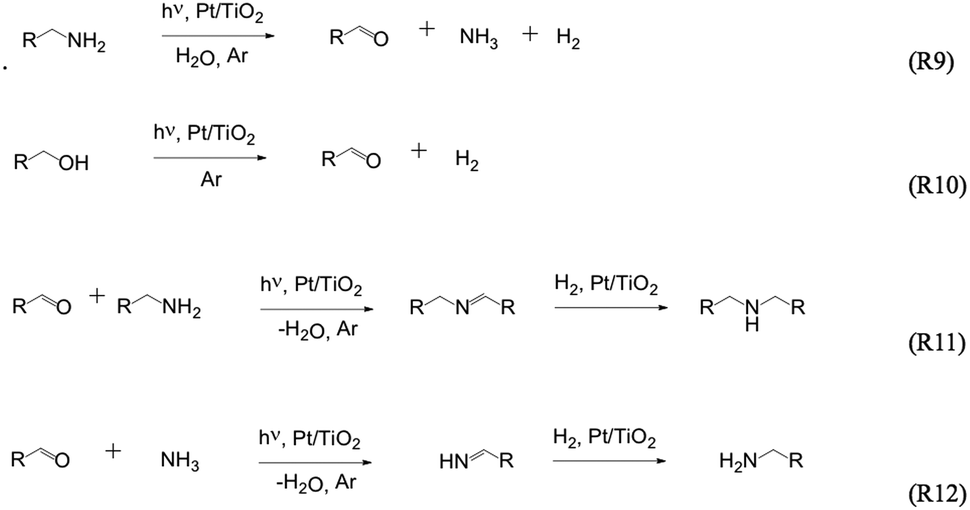 | ||
| Fig. 4 The formation of secondary amines from primary amines in aqueous solutions employing platinised TiO2 photocatalysts.77 | ||
Amino acids including glycine, alanine, serine, aspartic acid, and glutamic acid were also photocatalytically obtained when methane–ammonia–water mixtures were irradiated in the presence of Pt–TiO2 as photocatalyst.81,82 Onoe and Kawai83 also synthesised amino acids and amines starting from ammonia using Pt-CdS photocatalysts.
The photocatalytic reduction of nitroaromatic compounds has been studied by several groups.84–86 Mechanistically, the light-induced six-electron reduction of the nitro compound occurs via a sequence of electron transfer, protonation, and dehydration reactions (refer to Fig. 5). Alcohol solvents, which also act as sacrificial agents, take part in such reactions.87 The oxidation of alcohol solvents leads to the formation of hydroxyalkyl radicals which are known to be powerful reducing agents (E° more negative than −1.00 V vs. the NHE).88
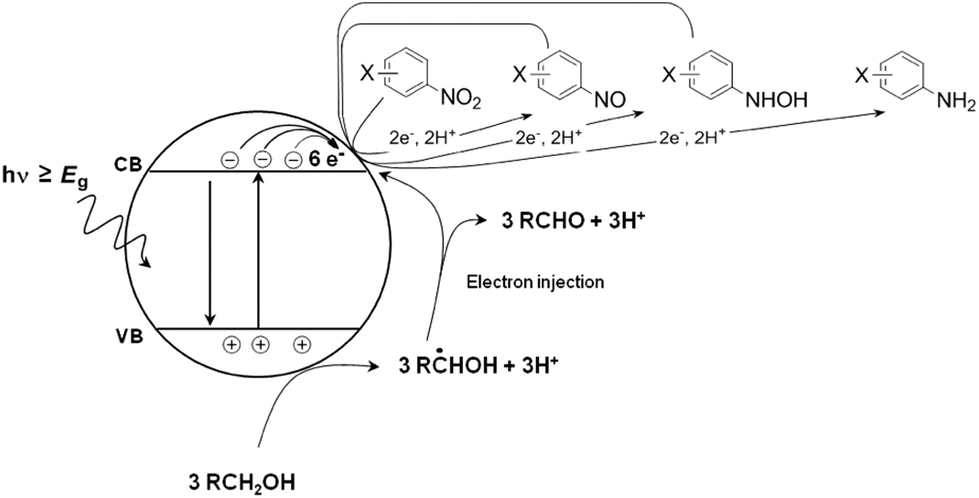 | ||
| Fig. 5 Light-induced six-electron reduction of the nitro compound occurs via a sequence of electron transfer, protonation, and dehydration reactions. | ||
Brezova et al.85 studied the influence of the solvent viscosity and polarity on the rate of the photocatalytic reduction of 4-nitrophenol to 4-aminophenol in different alcohols. The reduction rate decreased by increasing the viscosity of the employed alcohol whereas it increased with increasing the polarity of the alcohol. Recently, the successful chemoselective photocatalytic reduction of various nitroaromatic compounds (carrying reducible groups other than NO2) to the corresponding aminobenzenes in the presence of TiO2 suspended in acetonitrile with oxalic acid acting as a sacrificial reagent was reported.86 Tada et al.89,90 studied the effect of metal doping of the photocatalyst on the reduction of nitroaromatic compounds. The selectivity of the Ag/TiO2-photocatalysed reduction was rationalised on the basis of the selective adsorption of the nitroaromatic compounds on the modified catalyst surfaces and the restriction of the product, that is, aniline from readsorbing.
Imines also have been formed under illumination of alcoholic solutions of nitrobenzene in the presence of TiO2. The reaction showed high selectivity when C1–C3 alcohols were used. Higher alcohols led to the formation of a mixture of aniline and imines.91 Hakki et al.87 showed that the selectivity of this reaction is also dependent on the surface properties of the employed TiO2 photocatalyst where the surface Lewis acidity plays an important role. Anatase has a much higher Lewis acidity than rutile, which promoted imine selectivity. Rutile showed a higher selectivity towards the formation of aromatic amino compound. Lang et al.6 discussed the selective formation of imines in a two-step process, by aerobic photocatalytic oxidation of amines, a selective oxygenation step to generate aldehyde intermediates and a subsequent condensation step to form the imine products (refer to Fig. 6).
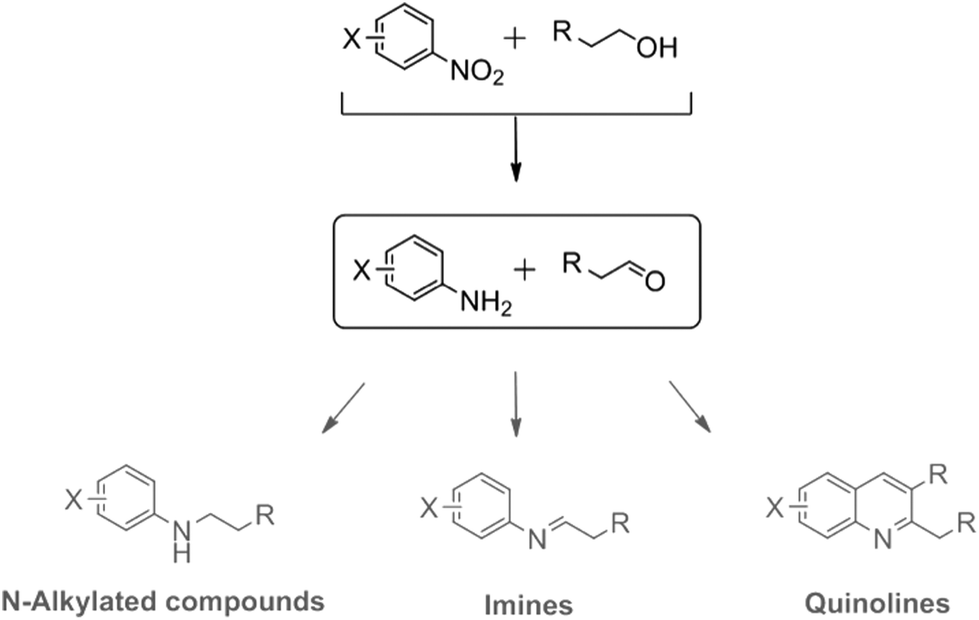 | ||
| Fig. 6 The selective formation of imines in a two-step process: (i) aerobic photocatalytic oxidation of amines to generate aldehyde intermediates and (ii) condensation step to form the imine products.6 | ||
The addition of olefins to trisubstituted imines yields homoallyl amines. This has been demonstrated by Kisch et al. using CdS-based photocatalysts.9,92–95 For example, Pehlivanugullari et al.9 studied the synthesis of unsaturated alpha-cyano-homoallylamines from imines and olefins photocatalysed by silica and cellulose supported cadmium sulphide.
Examples of photocatalytic cyclisation reactions include the study by Shiraishi et al.96 who synthesised benzimidazoles from 1,2-diaminobenzene using Pt–TiO2 photocatalysts in alcohol solution. Hakki et al.11,87 studied the photocatalytic formation of quinolines starting with nitroaromatic compounds in alcohols in the presence of TiO2. They found that the surface Brønsted acid sites strongly affected the selectivity of the products, enhancing the yield of the quinolines.11 The photocatalytic formation of quinolines has also been successfully achieved using a hybrid organic–inorganic materials in which the organic acid was fixed into the pores of mesoporous silica-titania composites.97
Heterogeneous photocatalytic synthesis of organic polymers
Photochemistry enables polymer synthesis by initiating a chain process which carries free radicals as active species in the polymerisation process. In the general process of photoinduced radical polymerisation, the role of light is to activate photoinitiators via electron transfer reactions to generate reactive species that subsequently react with monomers to yield polymer products.98 Over the past decades, heterogeneous semiconductor photocatalysis has been employed as a viable method to initiate the polymerisation process.99–112 In such processes, a semiconductor is excited by light absorption and the photogenerated electrons and holes activate the initiators via reductive and oxidative steps, respectively, as illustrated in Fig. 7. | ||
| Fig. 7 Photoinitiated free radical polymerisation and atom transfer radical polymerisation (atrp) using heterogeneous photocatalysis (m: monomer, r-x: alkyl halide). | ||
Free radical polymerisation initiated by heterogeneous photocatalysis
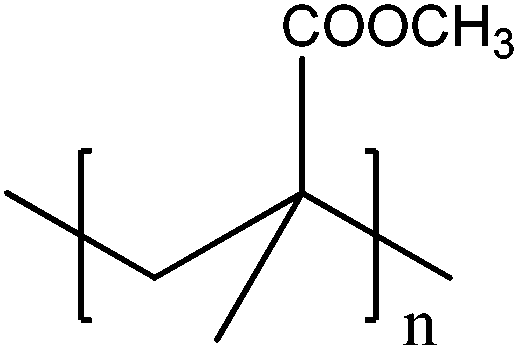
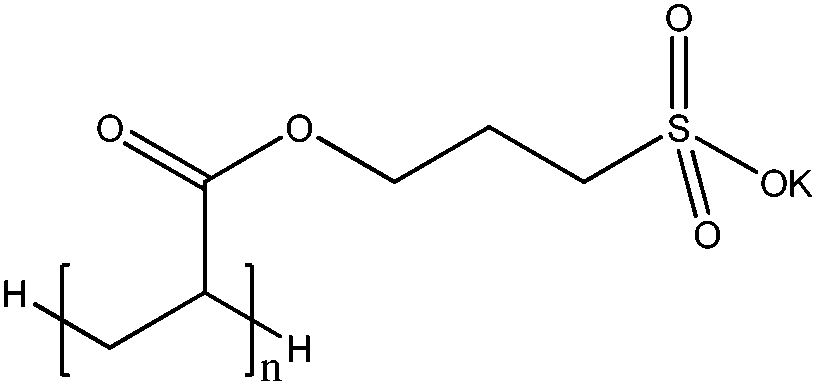
Table 1 summarises the free radical polymerisation using a heterogeneous photocatalyst that activates co-initiators such as triethylamine (TEA) and diphenyliodonium (Ph2I+) salt. Ni et al.99 demonstrated the photo-polymerisation of methyl methacrylate (MMA) in aqueous suspensions of commercial TiO2. Photogenerated holes can directly react with the monomer or H2O molecules to generate the activated monomer (radical species) and OH radicals which lead to the synthesis of polymethylmethacrylate (PMMA) through a free radical chain mechanism. As a co-initiator that is oxidatively activated, TEA is oxidised by the photoinitiated hole at the nitrogen nonbonding electron pair. The resulting TEA radical cation (Et3N˙+) abstracts a hydrogen atom from another TEA molecule, forming α-amine radical (Et2NEt˙) that induces the free radical polymerisation. Kiskan et al.101 showed free radical polymerisation of MMA using mesoporous carbon nitride (mpg-C3N4) as a visible light photocatalyst in the presence of amine co-initiators. Similarly, Wang et al.102 studied the use of porous-conjugated polymer B-(Boc-CB)2-BO as a heterogeneous photocatalyst under visible light to initiate the polymerisation of MMA.| Catalyst | Irradiation | Initiation step | Polymerisation step | Ref. |
|---|---|---|---|---|
| TiO2 | Mercury lamp (365 nm) | h+ + MMA → ˙MMA | ˙MMA + nMMA → PMMA | 99 |
| h+ + H2O → ˙OH | ˙OH + MMA → OH-MMA˙ | |||
| OH-MMA˙ + nMMA → PMMA | ||||
|
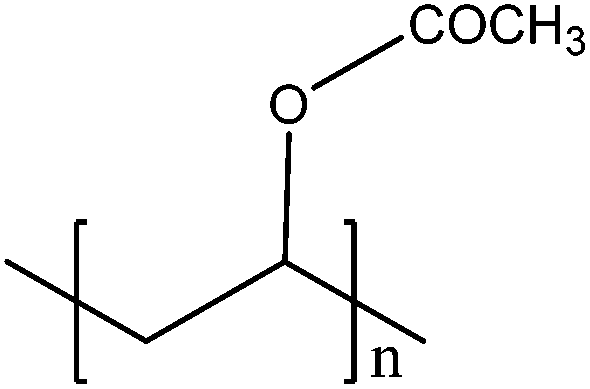
100 | |||
| Mercury lamp (365 nm) | RCOOH + h+ → R˙ + CO2 + H+ | R˙ + nVAc → PVAc (polyvinylacetate) | ||
|
101 | |||
| mpg-C3N4 | 300 W Xe lamp (>420 nm) | h+ + Et3N → Et3N˙+ | Et2NEt˙ + nMMA → PMMA | |
| Et3N˙+ + Et3N → Et3NH+ + Et2NEt˙ | ||||
| polyHIPE | 23 W household energy saving lamp | h+ + Et3N → Et3N˙+ | Et2NEt˙ + nMMA → PMMA | 102 |
| Et3N˙+ + Et3N → Et3NH+ + Et2NEt˙ | ||||
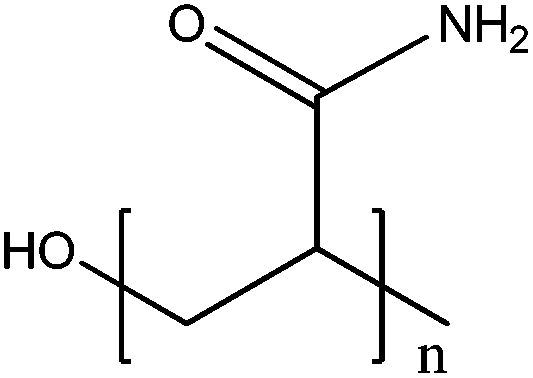
| ZnO, Fe/ZnO | >350 nm | h+ + H2O → ˙OH | ˙OH + AA → OH-AA˙ | 103 |
| OH-AA˙ + nAA → PAA (polyacrylamide) | ||||
|
||||
| e− + Ph2I+ → Ph2I˙ | Ph˙ + nMMA → PMMA | |||
| Ph2I˙ → PhI + Ph˙ | Et2NEt˙ + nMMA → PMMA | |||
| h+ + Et3N → Et3N˙+ | ||||
| Et3N˙+ + Et3N → Et3NH+ + Et2NEt˙ | ||||
| Fe3O4 | >350 nm | ROOH + h+ → R˙ + CO2 + H+ | R˙ + nMMA → PMMA | 104 |
| h+ + Et3N → Et3N˙+ | Et2NEt˙ + nMMA → PMMA | |||
| Et3N˙+ + Et3N → Et3NH+ + Et2NEt |
A diphenyliodonium salt has been frequently employed as a co-initiator that is reductively activated. The iodonium ion can be reduced by a photoinduced electron to a diphenyliodonium radical that is subsequently decomposed to generate a phenyl radical. Dadashi-Silab et al.103 have shown that ZnO and Fe-doped ZnO can initiate free radical photo-polymerisation of MMA. This group also tested a possibility of cationic polymerisation and found that this system generates radicals only, not cationic species from the iodonium salt. Another photo-initiation process employs alkyl radicals that are generated from the hole-induced decarboxylation of carboxylic acids. Weng et al.100 reported vinylacetate polymerisation initiated by alkyl radicals generated from butyric acid. A similar photo-polymerisation process was also developed using iron oxide nanoparticles (Fe3O4) capped with lauric acid.104
Atom transfer radical polymerisation initiated by heterogeneous photocatalysis
Atom transfer radical polymerisation (ATRP) is initiated by a reversible redox reaction using transition metal complex and alkyl halide.113 In this reaction, a reduced transition metal complex activates an alkyl halide by transferring a halide atom to the transition metal complex thereby generating an alkyl radical, which subsequently reacts with monomers to initiate radical polymerisation (see Fig. 7).In recent years, semiconductor photocatalysts have been employed to initiate ATRP reaction by reducing transition metal complexes such as CuII and FeIII (see Table 2). Yan et al.105 reported that TiO2 nanoparticles can be used to reduce a CuII–ligand complex by photoinduced electrons for initiating the ATRP process under UV light while using methanol as a hole scavenger. The light intensity and quantity of photocatalyst needed to be optimised for a maximal yield. ZnO and Fe-doped ZnO were also applied to the ATRP process for the synthesis of PMMA.108 Hybrid TiO2 nanocomposites were employed to utilise visible light. Dye-sensitised TiO2 successfully synthesised polysulfopropylmethacrylate by adjusting the ratio of [CuII]/[CuI].106 TiO2/rGO nanocomposites were utilised to initiate the ATRP reaction under visible light.107 The presence of reduced graphene oxide facilitated scavenging of conduction band electrons while retarding electron hole recombination. As alternative initiators of the ATRP process, organic semiconductors and metal organic framework (MOF) were also tested. Mesoporous carbon nitride can induce vinyl monomer polymerisation by reducing CuII to CuI in the ATRP process.109 MOF can be employed for ATRP by using photoactive organic chromophore ligands for visible light absorption.110 An iron complex was employed as an alternative to the Cu-complex due to its abundance and environmently-friendly nature. Fe2O3 and TiO2/C3N4 as photocatalysts can reduce FeIII to FeII which then reacts with the alkyl halide to generate alkyl radicals, which initiate ATRP.111,112
| Catalyst | Irradiation | Initiation step | Polymerisation step | Ref. |
|---|---|---|---|---|
| TiO2 | 330 nm | e− + CuII–L → CuI–L | R˙ + nSPMA → PSPMA (poly sulfopropyl methacrylate) | 105 |
|
||||
| dye-sensitised TiO2 | Xenon Lamp (220 nm–1200 nm) | CuI–L + R–X → X–CuII– | R˙ + nSPMA → PSPMA | 106 |
| L + R˙(L: 2,2′-bipyridyl) | ||||
| TiO2/RGO | LED (visible light) | e− + CuII–L → CuI–L | R˙ + nMMA → PMMA | 107 |
| CuI–L + R–X → X–CuII–L + R˙(L: 2,2′-bipyridyl) | ||||
| ZnO, Fe/ZnO | 8 W BLB (350 nm) | e− + CuII–L → CuI–L | R˙ + nMMA → PMMA | 108 |
| CuI–L + R–X → X–CuII–L + R˙(L: phthalocyanine) | ||||
| mpg-C3N4 | Sunlight, 8 W BLB (350 nm) | e− + CuII–L → CuI–L | R˙ + nMMA → PMMA | 109 |
| CuI–L + R–X → X–CuII–L + R˙(L: PMDETA) | ||||
| MOF (NNU-35) | Xenon lamp (520 nm) | e− + CuII–L → CuI–L | R˙ + nMMA → PMMA | 110 |
| CuI–L + R–X → X–CuII–L + R˙(L: PMDETA) | ||||
| e− + CuII–L → CuI–LCuI–L + R–X → X–CuII–L + R˙(L: PMDETA) | ||||
| Fe2O3 | 500 W Hg lamp (300 nm–450 nm) | e− + FeIII–L → FeII–L | R˙ + nMMA → PMMA | 111 |
| FeII–L + R–X → X–FeIII–L + R˙(L:Triphenylphosphine | ||||
| TiO2/C3N4 | Sunlight, 500 W Hg lamp | e− + FeIII–L → FeII–LFeII–L + R–X → X–FeIII–L + R˙(L: PMDETA) | R˙ + nMMA → PMMA | 112 |
The heterogeneous photocatalyst can be easily recovered and reused in the polymer synthesis process and the photoactivation method enables the polymer synthesis to occur under mild reaction conditions. However, this approach suffers from low conversion yields and is easily hindered by the presence of dioxygen that scavenges free radicals. To prevent inhibition by O2, many strategies have been developed.114 Examples of such strategies include (i) carrying out the reaction in the absence of oxygen, with limited practical applications; (ii) aiming to avoid the formation of peroxyl radicals or to avoid reinitiating the polymerisation from them; and (iii) using alternative photocuring monomers, photocationic systems, and hybrid technology.
Improving the selectivity of photocatalytic organic synthesis
Modifying VB and CB potentials
A major issue with heterogeneous photocatalytic process for organic synthesis by oxidation is the highly oxidising environment that is produced and generally its non-selectivity, with products and reactants being lost due to complete mineralisation. The oxidation of an organic molecule may occur either directly by the photogenerated holes or via an indirect paths, in which, hydroxyl radicals (˙OH) are formed via the oxidation of the adsorbed water or OH group, which then in turn oxidise the organic substrate. Thus, minimising the mineralisation of products and reactants in photocatalytic oxidation reactions to achieve good selectivity can be controlled by controlling the reduction potential of band gap edges of a given photocatalyst.It is well known that different semiconductors may have significantly different CB and VB potentials. These will determine the thermodynamic feasibility of which reactions can proceed. Fig. 8 shows the CB and VB potentials of various semiconductors. On the other hand, the standard reduction potentials for different organics is dependent on the reaction medium, and given the reactivity of intermediate species, this data is not always available.57
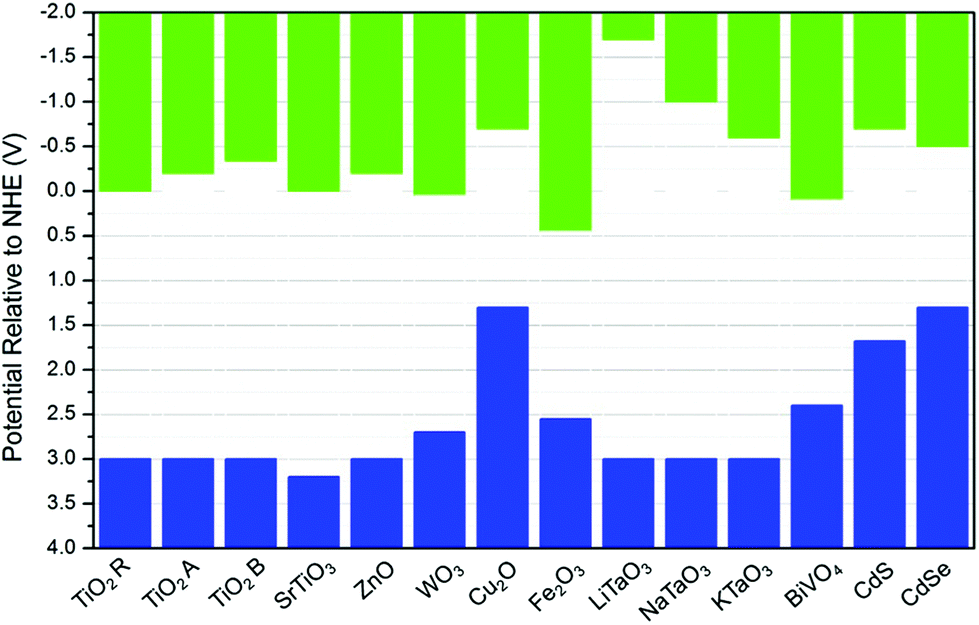 | ||
| Fig. 8 Bandgap energies of different semiconductors and relative edge energies, i.e., vb (blue columns) and cb (green columns) energies, relative to the normal hydrogen electrode (NHE) at pH = 0. The values for TiO2 were obtained from ref. 119. r, a, and b refer to rutile, anatase, and brookite, respectively. The values of MTaO3 were obtained from ref. 120, the remaining values were obtained from ref. 121–123. | ||
Additionally, shifts in semiconductor band edge potentials are expected to occur with changes in pH and solvents. This is expected to lead to altered surface phenomena and charge transfer events.115 Changes at the interface are expected to be photocatalyst as well as solvent dependent. For TiO2, the most studied photocatalyst, it in known that a Nernstian pH dependence exists for band edge potentials, while for CdS for example, the slope of the shift of band edges with pH has been found to be lower than the Nernstian dependence.115 Matsumura and co-workers found that the flat band potential of CdS was more cathodic with a decrease in pH, a fact that would favour proton reduction.116 It is important to note that the bulk structure of non-porous semiconductor photocatalysts is not expected to change significantly with pH variations. When considering the effect of the solvent on band edge potentials, for a polycrystalline TiO2 electrode it has been discussed that the main difference with respect to Vfb when immersed in an aqueous and a non-aqueous solution is the potential absence of a proton adsorption–desorption equilibrium. Generally, different behaviour of semiconductor flatband potentials is expected for protic and aprotic solvents.117,118 For TiO2, Vfb was found to be significantly more positive for water and non-aqueous protic solvents (MeOH and EtOH) than for non-aqueous aprotic solvents (MeCN, DMF and THF).115
The band levels of a given semiconductor material may be adjusted if its crystalline size is within a certain dimension where quantum effects are evident. Typically, when a semiconductor particle falls below a critical radius of approximately 10 nm, according to the material, the charge carriers begin to exhibit quantum mechanical behaviour.124–127 Under these conditions, the bandgap of the quantised semiconductor is larger compared to the bulk material, and the VB potential and CB potential shift accordingly. Hence, quantised semiconductor materials have larger redox potentials. This may result in increased photoefficiencies for systems in which the charge transfer is rate-limiting or thermodynamically not feasible. In the case of CdSe, for example, the reduction potential for nanocrystals with an average diameter of 3.0, 5.4, and 7.0 nm is −1.57, −0.91, and −0.80 V, respectively (versus SHE).128 This shift will significantly enhance the reduction power of the material with smaller particles in comparison with the bigger one. Holmes et al.,129 suggested that the activity of CdSe for protons reduction is a direct function of the energetics of the particles. In their work they have shown a constant decay of H2 evolution rate with increasing the particle size of CdSe which correlates very well with the expectation from Gerischer theory for electron transfer at illuminated semiconductor–electrolyte interfaces.129 The same consideration can also be applied for the hole-transfer kinetics involving the valence band and the electron donor. However, because of the small electron effective mass (me = 0.13mo) versus the significantly larger hole mass (mh = 1.14mo), most of the band gap increase is seen as a shift in the conduction band to more negative potentials (vs. NHE) rather than a shift in the valence band to more positive potentials.130
The VB and CB potentials may also be different depending on the crystalline nature of the semiconductor materials.87 For example, for TiO2, brookite nanorods have a flat band potential that is 140 mV cathodically shifted compared to anatase nanoparticles.119 The flat band potential of rutile is 200 mV anodically shifted compared to anatase. Since the VB edge of TiO2 has been reported to be almost constant at 3.0 V vs. NHE at pH 0 regardless of the crystalline phase120 this means that the photogenerated electrons in the CB of brookite, rutile and anatase have different potentials, and are most reducing for brookite, and least reducing for rutile. Thus, taking this into consideration, while evaluating the thermodynamic driving forces for the reactions of interest, improved selectivity may be achieved by appropriate selection of the crystalline phase. The same logic can be applied when considering the type of semiconductor that is chosen for a given reaction.
Tripathy et al.127 have shown that a strong change in the main reaction product of the photocatalytic oxidation of toluene, that is, benzoic acid versus benzaldehyde, can be achieved depending on the electronic properties of TiO2 (anatase, rutile, Ru doped). The main reaction products they obtained are shown in Fig. 9. The anatase based nanotubes lead to a distribution of products with benzoic acid as the main product (ca. 71%) and benzaldehyde and benzyl alcohol being the minor products. When rutile nanotubes were used as photocatalysts the main reaction product was benzaldehyde (ca. 76%) with benzoic acid and benzyl alcohol as byproducts. Moreover, more drastic changes were observed for Ru-doped rutile based photocatalysts on which the main reaction product was benzaldehyde (89.07%), and the formation of benzoic acid was completely suppressed.
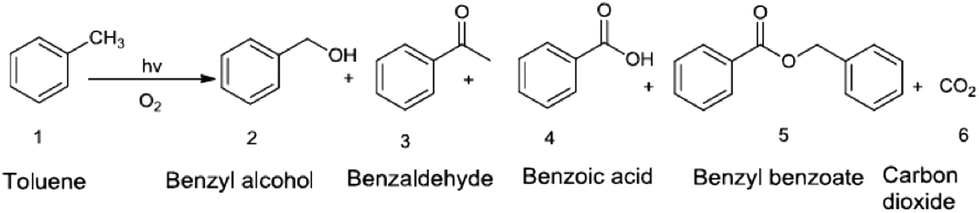 | ||
| Fig. 9 Main reaction products formed upon the photocatalytic oxidation of toluene according to ref. 127. | ||
Tripathy et al. attributed these differences in the observed selectivity to the position of the CB of the three photocatalysts which affected their ability to reduce molecular oxygen to O2˙−. Using the luminol test,131 they confirmed that the generation of O2˙− species was highest for the anatase nanotubes followed by rutile, whereas for the Ru-doped material O2˙− species was not detectable. The presence of Ru was said to suppress O2˙− formation at the CB of TiO2 (Fig. 10a) since Ru3+/4+ states in TiO2 are situated 0.4 eV below the CB of anatase.127 A proposed mechanism is shown in Fig. 10b.
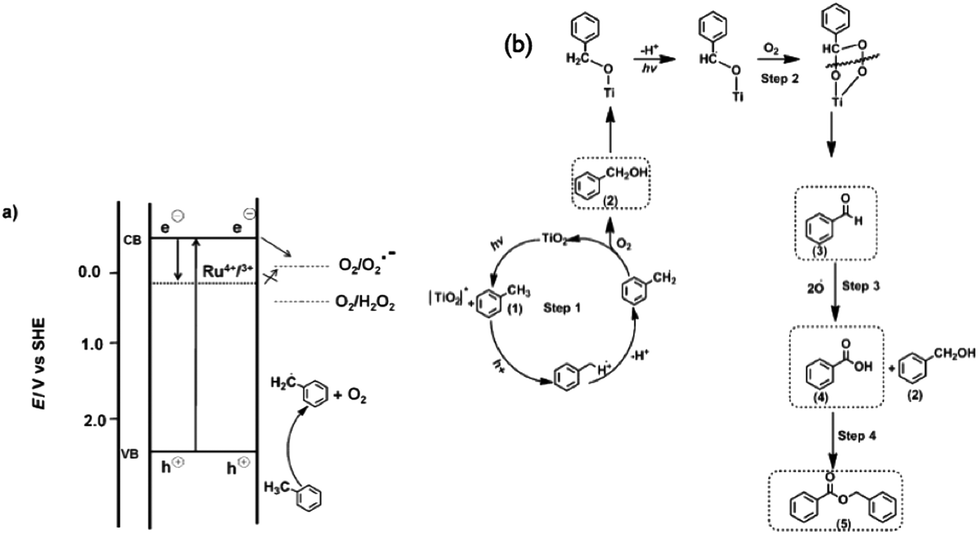 | ||
| Fig. 10 (a) Illustration of the TiO2 band positions during the photocatalytic oxidation of toluene (anatase and rutile) and ruthenium-doped TiO2 nanotubes under UV irradiation. cb = conduction band, vb = valence band. (b) Proposed reaction pathways for the photocatalytic toluene oxidation.127 | ||
Another example is the selective photocatalytic hydroxylation of benzene to phenol in water containing molecular O2. Tomita et al.132 reported that platinised tungsten oxide (Pt/WO3) photocatalytically produced phenol from benzene with high selectivity (for example, 74% at 69% of benzene conversion) that is much higher than that on TiO2 photocatalysts which generate CO2 as a main product. This difference in the selectivity was assigned to the difference in the potential of the CB electrons between WO3 and TiO2. Their results confirmed that photoexcited electrons on the Pt/WO3 photocatalysts mainly generate H2O2 from molecular O2 through a two-electron reduction. The formed H2O2 did not significantly contribute to the undesirable peroxidation of the phenol produced. In contrast, the photogenerated CB electrons on TiO2 are efficient to produce oxygen radical species, such as O2˙− or HO2˙, which contributed to the successive oxidation of phenol and other intermediates to CO2 reducing the selectivity for phenol. Moreover, the authors confirmed, depending on the results of reactions using 18O-labeled O2 and H2O, that the holes generated on Pt/WO3 reacted primarily with H2O molecules, even in the presence of benzene in aqueous solution, selectively generating ˙OH radicals that subsequently reacted with benzene to produce phenol. In contrast, benzene was directly oxidised by the photogenerated holes on TiO2 which consequently lowered the selectivity for phenol by TiO2. Thus, the two unique features of Pt/WO3, the absence of reactive oxygen radical species from O2 and the ability to selectively oxidise water to form ˙OH, were said to be the most likely reasons for the highly selective phenol production. The proposed mechanisms are illustrated in Fig. 11.
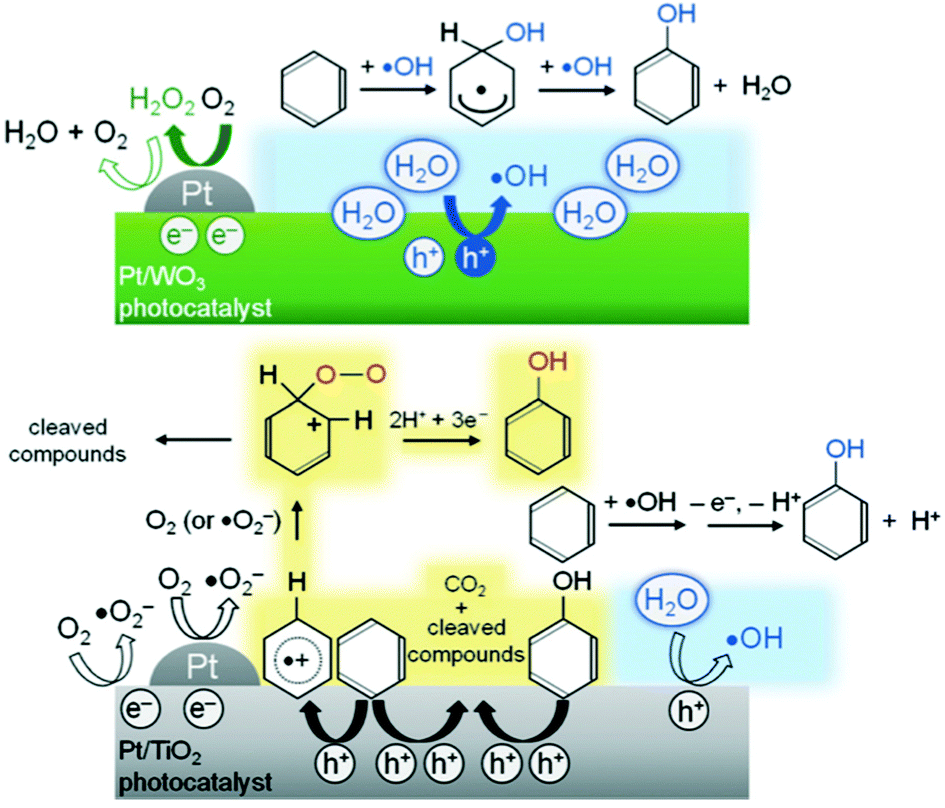 | ||
| Fig. 11 Proposed reaction mechanisms for phenol production over Pt/WO3 and Pt/TiO2 photocatalysts.132 | ||
The mechanism of aromatic ring hydroxylation over illuminated TiO2 was investigated in detail by Yuzawa et al.133 They proposed two kinds of electrophilic active species depending on the reaction condition: (i) a surface oxygen radical in neutral or acidic conditions and (ii) a hydroxyl radical in basic condition, both of which were produced by a photogenerated hole on the surface of TiO2. In both cases, these active species can attack the aromatic ring to form an intermediate, followed by the formation of a hydroxylated product.
Excitation wavelength effect on reaction selectivity
The excitation wavelength may be another means of affecting the potentials of the photogenerated charge carriers, more so the CB electrons. Depending on the energy of the impinging light onto the photocatalyst and the electronics of the energy levels of a given semiconductor, different excited states may be created depending on the resulting transitions. Again, it may be possible to selectively match the irradiation wavelength to achieve thermodynamics that are favourable for the organic transformations of interest. Yoshida and co-workers4,133,134 have shown that aromatic ring hydroxylation of benzene derivatives, by using water as an oxidant over platinum-loaded titanium oxide photocatalyst, proceeded with high selectivity under the light of selected wavelength. Lower selectivity of phenols, produced via photocatalytic toluene or p-xylene hydroxylation over 0.1 wt% Pt/TiO2, was obtained under illumination with 365 ± 20 nm light but improvements of both phenols yield and selectivity were obtained when the incident light was limited to 405 ± 20 nm in wavelength. The reasoning was that the photoexcitation at around 405 nm occurred at the surface levels of the semiconductor. These photogenerated charge carriers were said to contribute more to the desired reaction compared to the photogenerated charge carriers in the deeper layers formed by the interband excitation at 365 nm. The lower selectivity at 365 nm was assumed to be a result of electron transfer from toluene to TiO2, which was assigned as an absorption band at around 360 nm. This excitation was said to generate benzyl radicals to form dibenzyl.Ke et al.135 showed that there was a correlation between the reduction ability of Au–CeO2 and the illumination wavelength (and size of the Au deposits on CeO2). The reduction ability of the Au–CeO2 particles was said to be due to surface plasmon resonance of the gold nanoparticles. When irradiated, the Au-NPs absorbed the energy and then abstracted hydrogen from the solvent isopropanol forming Au–H species on the Au-NP surface. The shorter the wavelength of illumination, the stronger the reduction ability of the resulting Au–H species.
Visible light irradiation (λ > 450 nm) of Pt nanoparticles supported on Degussa P25 TiO2 (Pt/P25 catalyst) promoted efficient and selective aerobic oxidation of aniline to nitrosobenzene.136 Under UV irradiation azobenzene was reported to be the major product and nitrosobenzene was scarcely detected.137 A difference in mechanism may be described schematically in Fig. 12. Under visible light irradiation, intra-band electronic excitation of Pt atoms occurs, these electrons are then transferred to the anatase CB on which O2 is reduced to O2˙−, promoting photocatalytic cycles. The O2˙− attracts the H atom, which is removed from aniline upon its deprotonation on the Lewis base site on the Pt nanoparticles, and this produces a hydroperoxide species. The subsequent reaction between the anilino anion and hydroperoxide species gives rise to nitrosobenzene and water and completes the photocatalytic cycle.
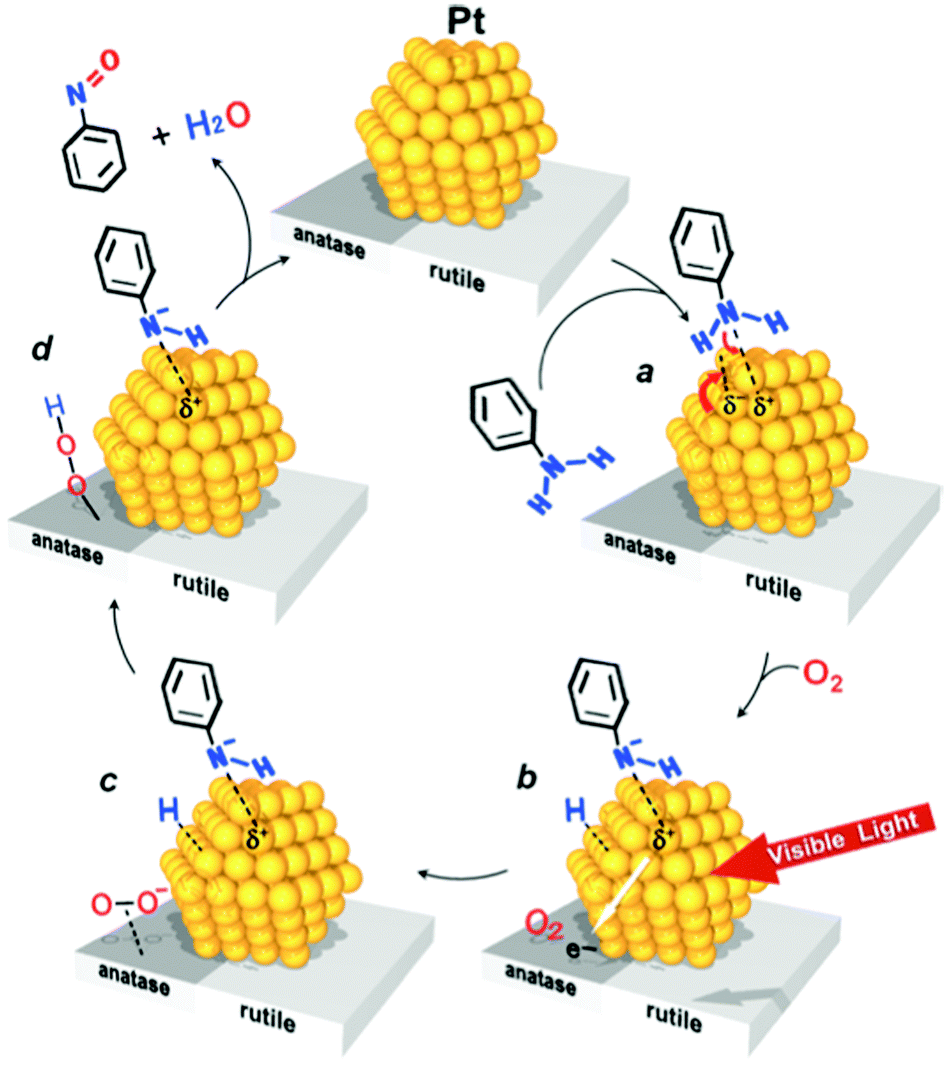 | ||
| Fig. 12 Aerobic oxidation of aniline to nitrosobenzene under visible light irradiation (>450 nm). | ||
Modifying photocatalyst surface properties
As mentioned earlier, once activated, TiO2 photocatalysts provide a highly oxidising environment and may lead to the mineralisation of the products and their precursors. The right reaction conditions need to be investigated to limit this mineralisation. Typically, highly adsorbed species are well mineralised. Achieving control over adsorption is hence another approach to affect selectivity as demonstrated by Shiraishi et al.15 when using microporous TiO2 structures. Adsorption is also strongly linked to surface properties such as zeta potential, which can be controlled by solution pH. Other surface properties that are inherent to the material such as functional groups lead to preferential adsorption. Within a sub-group of photocatalysts, heterogeneous systems with well-defined textural characteristics may also represent a suitable means to tailor the selectivity of photocatalytic processes.5 The use of one-dimensional nanostructure based materials continues to receive more attention as a method to achieve improved selectivity of organic photocatalytic synthesis.26,27 The use of NaTaO3 photocatalysts showed better efficiencies for the dehydrogenation coupling of isopropanol and hydrogenation coupling of acetone. The intermolecular hydrogen transfer from isopropanol to acetone was believed to be promoted due to an important role of the sodium ion in facilitating the proton transfer between the oxidation and reduction sites.138In order to ensure highly efficient and selective photocatalytic reactions, there are two important aspects which need to be controlled: (i) the adsorption of the reactants on the photocatalyst surface (ii) the desorption of products from the photocatalyst surface. This means that an abundant and selective supply of educts to the active sites is to be enhanced with a simultaneous restriction of the product(s) readsorption.
Ohtani et al. highlighted the importance of surface adsorption on selectivity from their studies on the synthesis of cyclic secondary amines starting from diamines.78–80 Ohtani et al.80 found that the optical purity of the product pyridine 3-carboxylic acid (PCA) was dependent on whether the photocatalyst was TiO2 or CdS. TiO2 resulted in the formation of an excess of the L-isomer while CdS based photocatalysts results in mostly racemic PCA.80 The difference in selectivity for the optical purity was attributed to the difference in the position of the amino group in L-Lys undergoing oxidative attack by the hole (h+). Metal doping improved the selectivity of the reduction stage but not the oxidation stage, and on its own was not sufficient to improve the overall selectivity.79 Hence, by modifying the physicochemical properties of the TiO2 surface, the mode of adsorption of L-Lys on TiO2, can be affected, resulting in better control over selectivity of optically pure PCA.79
Metal deposition can increase the adsorption of organic substrates onto the photocatalyst surface.89,90 This is also useful for reactions other than dehalogenation. Tada et al.89,90 reported an increase in the amount of adsorbed nitrobenzene when using Ag and Pt-Ag/TiO2 compared to bare TiO2, while restricting the product aniline from re-adsorbing. This selective adsorption of nitrobenzene resulted in a considerable increase in the activity and the selectivity of its photocatalytic reduction to aniline.
Canlas and co-workers139 have shown that indiscriminately reactive catalyst surfaces can be made reactant shape-selective through the use of partial overcoating with an inert oxide. For example, TiO2 coated with a porous very thin Al2O3 layer is a selective photocatalyst for both reduction and oxidation reactions (schematically shown in Fig. 13). Using this photocatalyst, nitrobenzene and benzyl alcohol were photocatalytically reduced and oxidised, respectively, while the ortho methylated derivatives did not react. The selection mechanism arises from size-sieving based on the ability of the reactants to adsorb on the active catalyst surface.139
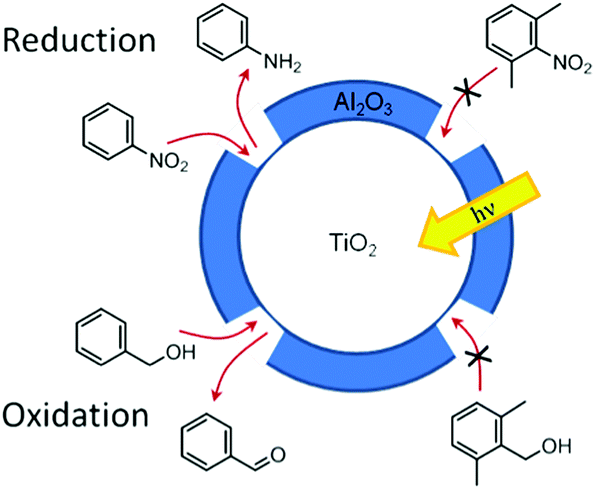 | ||
| Fig. 13 Selective photocatalytic oxidations and reductions of mixtures carried out over TiO2 covered with templated nanocavities.139 | ||
Surface defects and acidity
For the photocatalytic formation of quinolines through cyclisation reactions, Hakki et al.11 found that the surface Brønsted acid sites affect the selectivity of the products, enhancing the yield of the quinolines. Hakki et al. supported their observation by direct addition of a Brønsted acid (p-toluene sulfonic acid (p-TsOH) (5 mol%)) into the reaction dispersions in which they found a significant increase of quinoline yield from approximately 6% up to approximately 47%.The formation of quinolines through cyclisation has also been successfully achieved using mesoporous silica–titania composites with high selectivity of up to 53% when only small amounts of arenesulfonic acid were imbedded inside the pores of the silica. This can be compared to 6% in the case of pure TiO2 and 18% in the case of silica modified TiO2.97 The yield of substituted benzaldehydes and benzoic acid formed by the photocatalytic oxidation of substituted toluenes in acetonitrile, with a TiO2 photocatalyst, was dramatically improved by the addition of small amounts of sulfuric acid.140 Hakki et al.87 also showed that for imine selectivity, Lewis acidity played an important role. This was again dependent on the type of photocatalyst. Anatase had a much higher Lewis acidity than rutile, which promoted imine selectivity. Rutile on the other hand showed a higher selectivity towards the formation of aromatic amino compounds.
Solvent effects
The photocatalyst's surface properties are not the only factors that affect the adsorption of substrates. An employed solvent will also strongly influence the interaction between the surface of the photocatalyst and the substrate. Almquist and Biswas51 studied the photocatalytic oxidation of cyclohexane using TiO2 in various solvents to determine the effect of the solvent media. The solvents studied were cyclohexanol, acetone, isopropanol, dichloromethane, chloroform, carbon tetrachloride, benzene, and n-hexane. Selectivity to cyclohexanol and cyclohexanone and reaction rates were found to be dependent on adsorption, the type of solvent and the partially oxidised solvent species on the photocatalyst surface. In non-polar solvents, selectivity was low with cyclohexanol preferentially adsorbing onto TiO2 and being completely mineralised to CO2. In polar solvents, selectivity was higher since cyclohexanol adsorbed on TiO2 to a lesser extent due to competition with the solvent. Another example on the important role of solvent was provided by Colmenares et al. who studied a TiO2/maghemite–silica nanocomposite photocatalyst and reported better conversion and selectivity in acetonitrile for the photocatalytic oxidation of benzyl alcohol to benzylaldehyde compared to aqueous conditions.75When water is used as the solvent in heterogeneous photocatalytic reactions, it is easily adsorbed on the surface of the photocatalysts and is oxidised by the photogenerated holes forming highly oxidising hydroxyl radicals. These highly oxidising conditions restrict the ability to control the reaction selectivity of photocatalytic oxidation reactions. Since many organic compounds have limited solubility in water, an organic solvent is most often used during photocatalytic organic synthesis, for example, acetonitrile and various alcohols. Acetonitrile cannot be oxidised by the photogenerated holes of common semiconductors, and does not therefore take part in photocatalytic synthesis mechanisms. Moreover, its weak basicity was suggested to play a role in suppressing a proton transfer during the selective epoxidation of olefins with O2 and minimised undesirable products such as cyclohexenol.15 Augugliaro et al. have recently investigated the use of dimethyl carbonate as a green organic solvent.141
For photocatalytic reduction reactions, it could be highly advantageous to have a sacrificial reagent which scavenges the photogenerated holes.87,142–144 For photocatalytic reductive reactions, the adsorption of both the electron acceptor and hole scavenger are essential, therefore competitive adsorption between these two species could be important. Tan et al. reported an optimum ratio of formic acid (hole scavenger) and Se(VI) ions in aqueous solution, for the reduction reaction due to competitive adsorption between the selenate and formate ions.143
Additionally, in the case of employing alcohols as holes scavengers, the photogenerated CB electrons are not the only reducing agents present in the reaction media. Alcohol solvents are often themselves oxidised during the photocatalytic reactions producing reducing radicals.144,145 Brezova et al.85 studied the photocatalytic reduction rates of nitroaromatic compounds, and found that the reductive conversion was the fastest in methanol, followed by ethanol and then n-propanol. The same trend was observed for the photocatalytic reduction of Cd2+ (ref. 146) and Se(VI) reduction.145 Asmus et al.147 showed that the yield of the α-hydroxyalkyl radicals was greater for methanol than ethanol which in turn was greater than that from n-propanol. Thus the findings by Chenthamarakshan et al.146 and Tan et al.145 of increased reaction rates correlate with an increase in the formation of reducing radicals.147 Hence the rate at which α-hydroxyalkyl radicals are produced may also affect selectivity of photocatalytic organic synthesis.
The oxidation of the alcohols involves the abstraction of a hydrogen atom from the α-position carbon to produce α-hydroxyalkyl radicals148 as shown in reaction (R13).
| R–CH2–OH + OH˙ → R–˙CH–OH + H2O | (R13) |
The reduction potential of the produced radicals depends on the parent alcohol. The redox potentials of the radicals formed from the solvents methanol, ethanol and 2-propanol are shown in Fig. 14. The difference in reduction potentials of the radicals generated from these solvents can affect observed photocatalytic reaction rates.
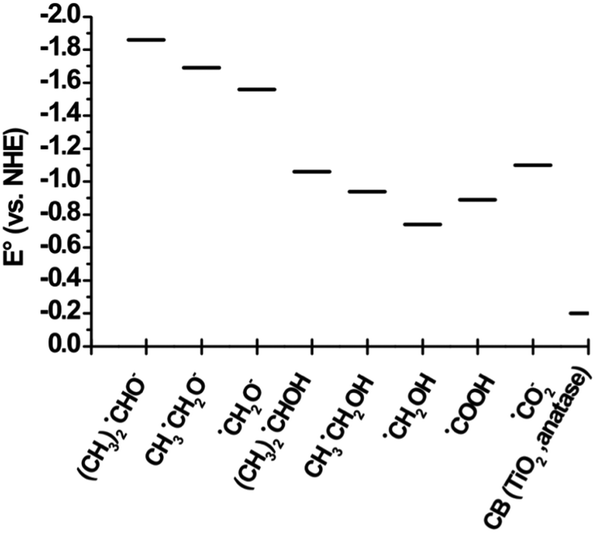 | ||
| Fig. 14 Reduction potentials of α-hydroxyl radicals149,150 compared with the cb potential of TiO2 (anatase) determined at pH 0. | ||
The fate of the generated α-hydroxyalkyl radicals is also dependent on the relative position compared to the CB potential of the employed photocatalyst. Fig. 14 shows the reduction potential of α-hydroxyl radicals in acidic and basic forms149 compared with the CB potential of TiO2 (anatase) determined at pH 0. As can be seen, these radicals are more powerful reducing agents with very negative reduction potentials vs. the NHE compared to the photogenerated TiO2 CB electrons.
In the absence of O2 and the presence of a nitroaromatic compound, the α-hydroxyalkyl radical may take part in a number of reactions (refer to reactions (R14)–(R26)). The α-hydroxyalkyl radical may (i) inject an electron into the CB of the photocatalyst forming the corresponding carbonyl compound and proton (R16). This process is referred to as “current-doubling” in photoelectrochemistry and has been observed in many related systems including the photoanodic oxidation of alcohols on TiO2,151 (ii) it may be reoxidised by the trapped photogenerated holes forming, again, the corresponding carbonyl compound and proton (R17), or (iii) they may directly react with the nitro group to form carbonyl compound, proton, and nitroaromatic radical anion152(R21); the latter may trap an electron from the CB forming the corresponding nitroso compound (R22).
Ferry and Glaze153 suggested that the CB electrons were the principal species driving the photocatalytic reduction of nitroaromatic compounds to aminoaromatic compounds. They showed that the reduction rates of the nitroaromatic compounds in illuminated TiO2 slurries containing MeOH or i-PrOH were almost equal, while in the absence of TiO2, these reduction rates measured, using pulse radiolysis techniques employing isopropoxyl radicals were more than 16 times higher than those obtained for methoxyl radicals. Nevertheless, the possibility remains that the reduction process involves a combination of agents, that is, the photocatalyst surface and reducing radicals derived from the primary or secondary alcohols as electron donors.
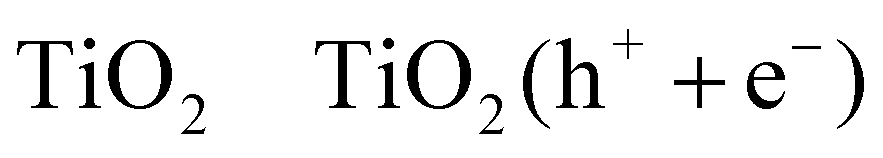 | (R14) |
| 3TiO2 (h+) + 3RCH2OH → 3TiO2 + 3R˙CHOH + 3H+ | (R15) |
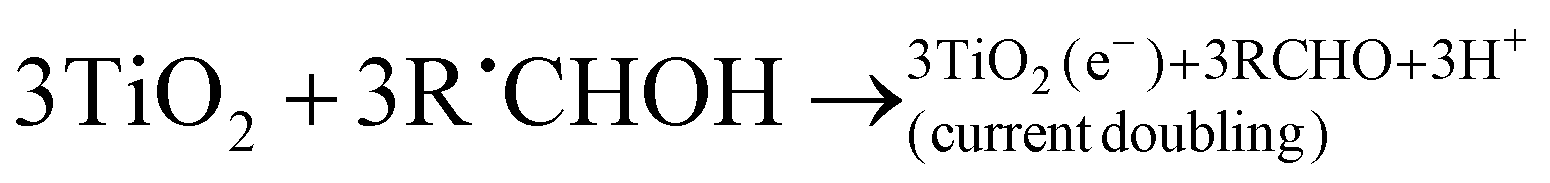 | (R16) |
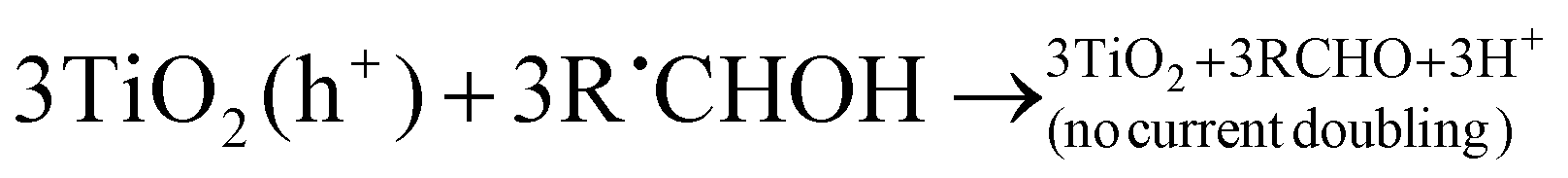 | (R17) |
| Ar–NO2 + 2TiO2(e−) + 2H+ → Ar–NO + H2O | (R18) |
| Ar–NO + 2TiO2(e−) + 2H+ → Ar–NHOH | (R19) |
| Ar–NHOH + 2TiO2(e−) + 2H+ → Ar–NH2 + H2O | (R20) |
| Ar–NO2 + R˙CHOH → Ar–NO2−˙ + RCHO + H+ | (R21) |
| Ar–NO2−˙ + TiO2(e−) + 2H+ → Ar–NO + H2O | (R22) |
| 6TiO2 (h+) + 3RCH2OH → 6TiO2 + 3RCHO + 6H+ | (R23) |
| Ar–NO2 + 6TiO2(e−) + 6H+ → Ar–NH2 + 2H2O + 6TiO2 | (R24) |
| 3TiO2 (h+) + 3RCH2OH → 3TiO2 + 3RCHO + 6H+ | (R25) |
| Ar–NO2 + 3TiO2(e−) + 3e− + 6H+ → Ar–NH2 + 2H2O + 3TiO2 | (R26) |
Regardless of which reduction pathway is more likely, the reduction of one molecule of nitroaromatic compound to the corresponding aminoaromatic compound is accompanied with the simultaneous formation of three carbonyl compound molecules that are produced by the oxidation of three alcohol molecules by the photogenerated holes. However, the number of required photons is different according to the reduction pathway.
If the nitroaromatic compound is reduced only by the photogenereted CB electrons (no current doubling and no direct reduction with the α-hydroxyalkyl radicals, see reactions (R18)–(R20)), six photons are required to photogenerate the required six electrons (see reactions (R23) and (R24)). On the other hand, only three photons are required in case of the contribution of the α-hydroxyalkyl radicals via either direct reduction of the nitroaromatic compound or injection of the electron in the CB of TiO2 (current doubling) (see reactions (R25) and (R26)). In (R26) 3e− refers to the electrons coming from R˙CHOH either via direct reaction with the nitroaromatic compound or via injection of its electron into the CB, that is, a current doubling.
It is worth mentioning here that β-hydroxyalkyl radicals may be formed upon the photocatalytic oxidation of alcohols that do not have an α-hydrogen atom such as tert-butanol and tert-pentanole. However, these radicals have a very low reduction power (more positive reduction potential) to be able to compete with the CB electron for the reduction of the organic substrate such nitroaromatic compounds. Additionally, unfavourable steric effects154 may explain the reported moderate reactivity of tert-butyl alcohol radicals.57
Solvents have differing properties such as viscosity, polarity, polarisability, and hence they have a different ability to stabilise the charged intermediate species. Hecht and Fawcett described how the solvent properties can affect the electron transfer kinetics and hence the observed photocatalytic reaction rates.155 Solvent choice can therefore provide another means of controlling selectivity. Soana et al. reported the effect of an organic solvent to slow down the reaction and improve selectivity.55 Solvent purity is also an important issue. When studying the transfer hydrogenation of Schiff bases, (N-benzylidenebenzylamine (BdBA) and N-benzylideneaniline), Ohtani et al.80 showed that when water impurities were present in 2-propanol solvent, hydrolysis of N-benzylidenebenzylamine (BdBA) occurred, with the subsequent formation of an undesirable product (N-benzylpropyl-2-amine). Addition of molecular sieve 3A to the reaction mixture improved the selectivity to DBA (dibenzylamine) from BdBA, by absorbing contaminant water and thereby inhibiting the hydrolysis of BdBA.
Multi-catalyst approaches for photocatalytic organic synthesis
For catalytic systems, multi-catalyst (dual catalyst) systems where the catalysts are present as separate entities, working sequentially or in tandem have been used to achieve selective organic synthesis. There are many examples of successful applications in the literature156 and references within. There are also patented dual catalyst systems, highlighting the potential usefulness and the possibility of commercialisation of such an approach.157,158 While reported work on heterogeneous multi-photocatalysts is rare, given the promising results with catalytic systems, the exploration of this approach to improve photocatalytic process selectivity and reactivity is warranted.Issues with a multi-catalyst approach are selecting the appropriate catalysts, compatibility with other catalysts and reagents, solvents and intermediates generated during the course of the reaction. In nature biological processes, enzyme architecture facilitates multiple reaction scenarios156 Some of these issues can be avoided by adding the catalyst sequentially to the reaction media. In cooperative catalysis, both the catalysts are present at the onset of the reaction, and share the same catalytic cycle, activating two different functional groups cooperatively to achieve the bond formation steps. In relay or sequential catalysis, the two catalysts do not act during the same catalytic cycle, both catalysts are present at the onset of the reaction and are compatible.133
For photocatalytic degradation of organic contaminants, systems containing both rutile and anatase crystalline phases have been shown to offer better efficiencies due to favourable electronic interactions between the two phases of the same semiconductor, as a result of appropriate CB and VB positioning.159 For the photocatalytic oxidation of naphthalene, Ohno et al.56 reported an increase in activity of pure rutile particles when these were physically mixing with a small amount of small-sized anatase particles, (which were inactive for the photocatalytic oxidation of naphthalene). Ohno et al.56 explained the results as being due to the synergism between rutile and anatase particles where the holes are transferred from anatase particles to rutile particles, and the naphthalene is mainly oxidised on rutile particles while oxygen is mainly reduced on anatase particles. Mechanistically, this is feasible since Soana et al. postulated the photocatalytic oxidation of naphthalene to involve a mechanism involving the transfer of a hydroxy group to naphthalene followed by coupling with superoxide radical to produce 2-formylcinnamaldehyde.55
In the oxygenated systems, photogenerated holes and hydroxyl radicals (HO˙), which are formed via the reaction of adsorbed water molecules or hydroxyl groups with the photogenerated holes on TiO2, are not the only oxidising species in the system. The superoxide radical (O2˙−), that is, generated by the reaction of molecular oxygen with the photogenerated electrons, is also an active oxidising species and is responsible for the decrease of the selectivity of the photocatalytic oxidation of organic substrates. Thus inhibiting the formation of this species may result in increased selectivity. To overcome the production of the superoxide radical Marotta et al. have used Cu2+ cations as electron scavengers.160 Cupric ions can be reduced to Cu0 by CB electrons since the standard redox potential of Cu2+/Cu0 couple is 0.337 V (vs. NHE) which is more positive than that of the CB edge. The advantage of employing this redox couple is the possibility of regeneration of the cupric ions via the reoxidation of the metallic copper in a dark run as can be explained in Fig. 15.
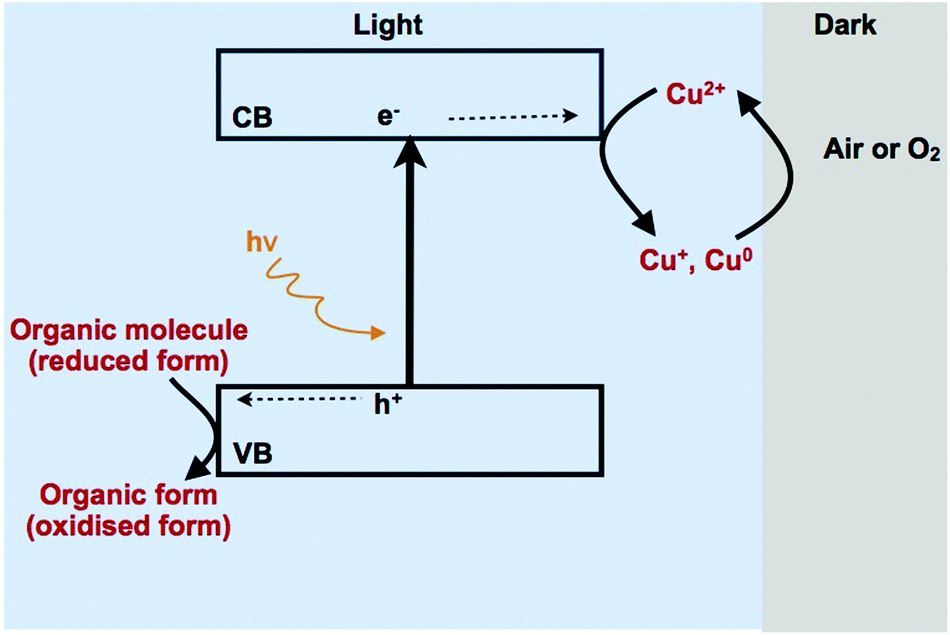 | ||
| Fig. 15 Mechanism depicting the simultaneous photocatalytic oxidation of organic species and the reduction of Cu2+.160 | ||
The authors reported an approximately 50% selectivity for the photocatalytic oxidation of benzyl alcohol to benzaldehyde. Spasiano et al. have developed TiO2/Cu2+/solar radiation system for the selective oxidation of benzyl alcohol to benzaldehyde in water in a solar pilot plant with compound parabolic collectors reactor (CPC).161 They have found that the oxidation rates are strongly influenced by the initial cupric ions concentration, incident solar irradiance and temperatures. They reported 53.3% yield for benzaldehyde with respect to the initial benzyl alcohol concentration (63.4% of selectivity). This is one of the very rare examples of employing the photocatalytic method for organic synthesis in a pilot scale.
Other examples of dual semiconductors in the literature are those involving heterojunctions between two different semiconductors such as in capped and coupled semiconductor systems.126,162,163 These dual-photocatalyst systems require the direct contact between the two semiconductors, which allows the transfer of charges between them. This can be taken advantage of to separate charge carriers and reduce electron hole recombination. For organic synthesis applications, careful knowledge of the oxidation potentials required for specific reactions, may allow for designing a system containing two specific semiconductors with favourable CB and VB levels, to drive certain reactions or inhibit others. Liu et al.28 presented a mini-review on the use of core–shell nanostructures for selective organic transformations.
Tsukamoto et al.64 studied the dual semiconductor WO3/TiO2 photocatalysts for the photocatalytic oxidation of alcohols in water. These dual photocatalyst systems were shown to promote the selective oxidation of alcohols to aldehydes and showed higher catalytic activity than pure TiO2. The high aldehyde selectivity was explained by the change in adsorption properties of the WO3/TiO2 photocatalyst compared to TiO2 rather than electronic effects. The suppression of the aldehyde decomposition was said to be due to its reduced adsorption on TiO2. A schematic is shown in Fig. 16.
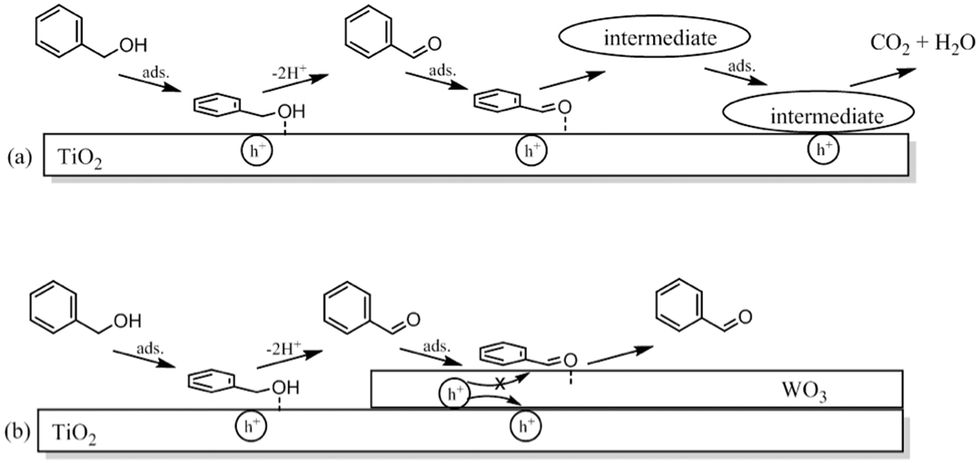 | ||
| Fig. 16 Schematic representation of the photocatalytic oxidation reactions of benzaldehyde on (a) TiO2 and (b) WO3/TiO2 photocatalysts.64 | ||
A selectivity enhancement in alcohol photooxidation using TiO2 covered with Nb2O5 has also been demonstrated. The coverage of TiO2 surface with Nb2O5 enhanced the selective partial oxidation of various alcohols, including primary and secondary alcohols.63 Unlike the case of WO3 modified TiO2, the authors attributed the enhancement in the selectivity to the inhibition in the generation of O3− by modifying the surface of TiO2 with Nb2O3 as their ESR studies have shown. In fact O3− is known to be active even at room temperature thus it has higher activity toward the complete oxidation of organic molecules in comparison with O2− which is stable up to ca. 423 K.
Modification of TiO2 with Au nanoparticles dramatically decreases the amount of OH-groups on its surface.164,165 Surface OH groups play an important role on both the adsorption and charge carriers trapping steps in the photocatalytic systems.166 Ide et al.165 reported that the presence of Au nanoparticles on the surface of Aeroxide P25 suppresses the total photocatalytic oxidation of phenol in aqueous media to CO2 with hydroquinone being the main product. This difference in the selectivity is neither attributed to the improved charge separation efficiency, due to the sink of the photogenerated electrons to the metal nanoparticles, nor to the plasmonic effect of the Au nanoparticles. The difference in selectivity is attributed to a drop of the affinity of the produced hydroquinone to adsorb on Au-modified TiO2. This reduced affinity results from the decrease in the OH groups on the surface of TiO2 upon modifying with Au. Similarly, Ide et al. have reported an enhancement in the selectivity of the photocatalytic oxidation of cyclohexane towards the formation of cyclohexanone and cyclohexanol employing Au-modified Fe/Ni/TiO2 as photocatalyst.167
A high level of efficient and selective sunlight-induced cyclohexane oxidation has also been obtained on TiO2 (P25) modified with iron oxide.168 The presence of iron oxide efficiently prevented the interactions between bulky molecules, that is, cyclohexane and the partially oxidised products, with the VB holes on the P25 surface. Interestingly, the photocatalytic oxidation of cyclohexane over FeO/TiO2 was substantially improved to give a higher yield without any loss in the selectivity when the reaction was conducted under a CO2 atmosphere. Similar improvements by adding CO2 to the system have been reported for the oxidation of aqueous benzene to phenol over TiO2-supported gold nanoparticles.169
Magdziarz et al. used a new green and energy efficient sonophotodeposition synthesis method for the preparation of selective iron doped TiO2/zeolite based photocatalytic materials.170 Sonophotodeposition does not involve the use of strong chemical reducing agents and it can be carried out under mild reaction conditions within a short time. It involves the use of a sonication probe and a Xenon lamp as a sun imitating light source. Magdziarz et al. were the first to report on the successful use of sonophotodeposition for the deposition of a non-noble metal (iron) on the surface of TiO2/zeolite. The photocatalysts were tested for the oxidation of benzyl alcohol into benzaldehyde and showed better results, in terms of alcohol conversion and yield of benzaldehyde, in comparison with the photocatalysts prepared by an ordinary wet-impregnation method.170
Ruberu et al. demonstrated the photocatalytic alcohol dehydrogenation and hydrogenolysis driven by the visible region of sunlight using M (Pt, Pd) nanoparticles deposited on CdS1−xSex (0 < x < 1) nanorods.171 Employing these photocatalysts, benzyl alcohol was converted under sunlight illumination to benzaldehyde, with toluene and H2 as byproducts. Relative benzaldehyde, H2, and toluene amounts were strongly affected by the structure and the composition of the photocatalyst. Under selected conditions, CdS-Pt favoured dehydrogenation (H2) over hydrogenolysis (toluene) 8:1, whereas CdS0.4Se0.6-Pd favoured hydrogenolysis over dehydrogenation 3:1. As shown in Fig. 17, the photocatalytic conversion of benzyl alcohol can undergo two photocatalytic pathways. The first one favours alcohol dehydrogenation and produces benzaldehyde and molecular hydrogen; while the other pathway favours alcohol hydrogenolysis and produces toluene and molecular oxygen O2. However, both pathways are thermodynamically uphill. The authors explained the different selectivities between semiconductor-Pt (which favoured H2) and semiconductor-Pd (which favoured toluene) in terms of the known reactivity of Pt and Pd surfaces. Pd is known to strongly adsorb hydrogen atoms “protons” and promote reduction reactions. Thus, H2 gas produced during dehydrogenation quickly adsorbs onto the Pd surface, forming Pd–H reduction sites for the conversion of benzaldehyde into toluene.
 | ||
| Fig. 17 Schematic formation of benzaldehyde and toluene upon dehydrogenation or hydrogenolysis of benzyl alcohol. | ||
Finally, Han et al.'s novel studies on ternary multidimensional systems for the selective photohydrogenation of nitro aromatic compounds provide a unique and clever approach for taking advantage of the properties of the individual components to produce highly tailored functional photocatalytic materials.172 In their work, Han et al. produced a ternary hierarchical nanostructure, CdS-1D ZnO-2D GR, made up of CdS-sensitised 1D ZnO nanorod arrays on a 2D graphene (GR) sheet, which served as an efficient visible-light-driven photocatalyst. This nanostructure aimed at improving solar energy capture and conversion. The ternary structure combined the fast electron transport of 1D ZnO nanorods, the excellent electron conductivity of 2D GR and the intense visible-light absorption of CdS. The matched energy levels of CdS, ZnO and GR efficiently increased photogenerated charge carriers separation and transfer. The unique combination also provided high chemical stability and prevented ZnO and CdS from photocorrosion.172 Such novel materials and the fact that they were synthesised at low temperature open up new opportunities for designing highly effective and sought after solar driven photocatalysts.
Conclusions and outlook
The application of heterogeneous semiconductor photocatalysis to organic synthesis presents more difficult challenging issues compared to applications such as the degradation of organic contaminants. All kinds of photocatalytic applications are based on the photoinduced charge transfers occurring on semiconductor interface with electrons and holes utilised as reductants and oxidants, respectively. The key issue in utilising photocatalysis for selective organic synthesis is how to control the ways of interfacial charge transfer so that only the specific functional groups in substrate organic molecules can be selectively transformed while the rest of the molecular structure remains intact. Given the strong oxidation power of VB holes photogenerated in oxide semiconductors that are popular as a stable photocatalyst (e.g., TiO2, ZnO, WO3), VB holes tend to oxidise and degrade the whole molecules non-selectively.At present, there are many knowledge gaps and technological difficulties in the research field of organic synthesis using heterogeneous photocatalytic process. It is envisaged that, similar to organic degradation processes,173 individual photocatalysts will provide improved selectivity for selective reactions. It is expected that each photocatalyst needs to be optimised for specific organic synthesis reaction case by case since the selectivity control should depend on the molecular structure and property of the specific organic substrate as well as the photocatalyst properties. Studies are needed to understand the link between the properties of catalyst surface and substrate molecules and the desired selectivity, in order to control the typical over-oxidation that occurs with photocatalytic organic synthesis. Perhaps considering other photocatalysts may be needed to circumvent the highly oxidising nature of photoactivated TiO2. CB and VB potentials may need to be matched with oxidation potentials of organics of interest (perhaps in various solvents). Examining the use of one or more photocatalysts in one reactive system to achieve improved selectively and reaction kinetics may be another novel approach. This has seen success in catalytic synthesis reactions. While there is still much experimentation to be done, this is justified by the potential gains of developing a green, potentially solar driven organic synthesis process.
Acknowledgements
This work was supported by Global Research Laboratory program (2014K1A1A2041044) funded by the Korea government (MSIP) through NRF. Parts of the present study were performed within the Project “Establishment of the Laboratory “Photoactive Nanocomposite Materials”” No. 14.Z50.31.0016 supported by a Mega-grant of the Government of the Russian Federation.References
- M. Bellardita, V. Loddo, G. Palmisano, I. Pibiri, L. Palmisano and V. Augugliaro, Appl. Catal., B, 2014, 144, 607 CrossRef CAS
.
- G. Palmisano, M. Addamo, V. Augugliaro, T. Caronna, A. Di Paola, E. López, V. Loddo and M. Schiavello, Catal. Today, 2007, 122(1–2), 118 CrossRef CAS
- S. Higashida, A. Harada, R. Kawakatsu, N. Fujiwara and M. Matsumura, Chem. Commun., 2006, 2804 RSC
- H. Yoshida, H. Yuzawa, M. Aoki, K. Otake, H. Itoh and T. Hattori, Chem. Commun., 2008, 4634 RSC
- A. Maldotti and A. Molinari, Top. Curr. Chem., 2011, 303, 185 CrossRef CAS PubMed
- X. Lang, H. Ji, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 3934 CrossRef CAS PubMed
- C. Pruden, J. Pross and Y. Li, J. Org. Chem., 1992, 57, 5087 CrossRef
- K. Park, H. Joo, K. Ahn and K. Jun, Tetrahedron Lett., 1995, 36, 5943 CrossRef CAS
- H. Pehlivanugullari, E. Sumer and H. Kisch, Res. Chem. Intermed., 2007, 33, 297 CrossRef CAS
- L. Cermenati, C. Richter and A. Albini, Chem. Commun., 1998, 805 RSC
- A. Hakki, R. Dillert and D. Bahnemann, Catal. Today, 2009, 144, 154 CrossRef CAS
- T. Caronna, C. Gambarotti, L. Palmisano, C. Punta, M. Pierini and F. Recupero, J. Photochem. Photobiol., A, 2007, 189, 322 CrossRef CAS
- G. Palmisano, V. Augugliaro, M. Pagliaro and L. Palmisano, Chem. Commun., 2007, 33, 3425 RSC
- Y. Shiraishi and T. Hirai, J. Photochem. Photobiol., C, 2008, 9(4), 157 CrossRef CAS
- Y. Shiraishi and T. Hirai, J. Jpn. Pet. Inst., 2012, 55(5), 287 CrossRef CAS
- G. Palmisano, E. García-López, G. Marcì, V. Loddo, S. Yurdakal, V. Augugliaro and L. Palmisano, Chem. Commun., 2010, 46(38), 7074 RSC
- M. Cherevatskaya and B. Koenig, Russ. Chem. Rev., 2014, 83(3), 183 CrossRef
- A. Vorontsov and A. Arsentyev, Curr. Org. Chem., 2013, 7(21), 2459 CrossRef
- H. Lu and J. Yao, Curr. Org. Chem., 2014, 18(10), 1365 CrossRef CAS
- M. Valenzuela, E. Albiter, O. Ríos-Bernÿ, I. Córdova and S. Flores, J. Adv. Oxid. Technol., 2010, 3(3), 321 Search PubMed
- B. Ohtani, B. Pal and S. Ikeda, Catal. Surv. Asia, 2003, 7(2–3), 165 CrossRef CAS
- R. Molinari, P. Argurio and C. Lavorato, Curr. Org. Chem., 2013, 17(21), 2516 CrossRef CAS
-
R. Molinari, P. Argurio and C. Lavorato, in Membrane Reactors for Energy Applications and Basic Chemical Production, ed. A. Basile, L. Pi Paola, F. Hai and V. Piemonte, Woodhead Publishing in Energy, Cambridge, UK, 2015, ch. 20, pp. 605–639 Search PubMed
- S. Munir, D. Dionysiou, S. Khan, S. Shah, B. Adhikari and A. Shah, J. Photochem. Photobiol., B, 2015, 148, 209 CrossRef CAS PubMed
- A. Hakki, R. Dillert and D. Bahnemann, Curr. Org. Chem., 2013, 17(21), 2482–2502 CrossRef CAS
- B. Weng, S. Liu, Z.-R. Tang and Y.-J. Xu, RSC Adv., 2014, 4(25), 12685 RSC
- C. Han, S. Liu, Z.-R. Tang and Y.-J. Xu, Curr. Org. Chem., 2015, 19(6), 484 CrossRef CAS
- S. Liu, N. Zhang and Y.-J. Xu, Part. Part. Syst. Charact., 2014, 31(5), 540 CrossRef CAS
- J. Chen, J. Cen, X. Xu and X. Li, Catal. Sci. Technol., 2016, 6, 349 CAS
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473 RSC
- J. Colmenares and R. Luque, Chem. Soc. Rev., 2014, 43, 765 RSC
- S.-H. Li, S. Liu, J. Colmenares and Y.-J. Xu, Green Chem., 2016, 18, 594 RSC
- M. González-Béjar, J. Pérez-Prieto and J. Scaianoa, Green Chem., 2012, 71–106 Search PubMed
-
J. Colmenares and Y.-J. Xu, Heterogeneous Photocatalysis: From Fundamentals to Green Applications, Springer-Verlag, Berlin, Heidelberg, 2016 Search PubMed
-
L. Yuan, N. Zhang, Y.-J. Xu and J. Colmenares, in Heterogeneous Photocatalysis: From Fundamentals to Green Applications, ed. J. Colmenares and Y.-J. Xu, Springer-Verlag, Berlin Heidelberg, 2016, ch. 8, pp. 249–282 Search PubMed
-
K. Imamura and H. Kominami, in Heterogeneous Photocatalysis, ed. J. Colmenares and Y.-J. Xu, Springer, Berlin, Heidelberg, 2015, pp. 283–320 Search PubMed
- X. Li, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 2603 RSC
- H. Liu, A. Q. Dao and C. Fu, J. Nanosci. Nanotechnol., 2016, 16, 3437 CrossRef CAS PubMed
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Appl. Catal., B, 2010, 99(3–4), 442 CrossRef CAS
- D. Bahnemann, Sol. Energy, 2004, 7(4), 445 CrossRef
- Y. Paz, Adv. Chem. Eng., 2009, 36, 289 CAS
- M. Ni, M. Leun, D. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11(3), 401 CrossRef CAS
- D. Friedmann, C. Mendive and D. Bahnemann, Appl. Catal., B, 2010, 99(3–4), 398 CrossRef CAS
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. Bahnemann, Chem. Rev., 2014, 114(19), 9919 CrossRef CAS PubMed
- K. Maeda, A. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49(24), 4096 CrossRef CAS PubMed
- H. Park, H.-I. Kim, G.-H. Moon and W. Choi, Energy Environ. Sci., 2016, 9, 411 CAS
- N. Hoffman, J. Phys. Org. Chem., 2015, 28(2), 121 CrossRef
- M. Fox and J. Younathan, Tetrahedron, 1986, 42, 6285 CrossRef CAS
-
M. Fox and M. Chanon, Photoinduced Electron Transfer, Elsevier Science Publishers, Amsterdam, 1988 Search PubMed
- M. Fox and A. Abdel-Wahab, Tetrahedron Lett., 1990, 31(32), 4533 CrossRef CAS
- C. Almquist and P. Biswas, Appl. Catal., A, 2001, 214, 259 CrossRef CAS
- W. Mu, J.-M. Herrmann and P. Pichat, Catal. Lett., 1989, 3(1), 73 CrossRef CAS
- M. Fujihira, Y. Satoh and T. Osa, Nature, 1981, 293, 206 CrossRef CAS
- H. Park and W. Choi, Catal. Today, 2005, 101(3–4), 291 CrossRef CAS
- F. Soana, M. Sturini, L. Cermenati and A. Albini, J. Chem. Soc., Faraday Trans. 2, 2000, 4699 Search PubMed
- T. Ohno, K. Tokieda, S. Higashida and M. Matsumura, Appl. Catal., A, 2003, 244, 383 CrossRef CAS
- P. Wardman, J. Phys. Chem. Ref. Data, 1989, 8(4), 1637 CrossRef
- T. Hirakawa, C. Koga, N. Negishi, K. Takeuchi and S. Matsuzawa, Appl. Catal., B, 2009, 87, 46 CrossRef CAS
- T. Kawai and T. Sakata, Nature, 1980, 286, 474 CrossRef CAS
- F. Hussein, G. Pattenden, R. Rudham and J. Russell, Tetrahedron Lett., 1984, 25, 3363 CrossRef CAS
- F. Hussein and R. Rudham, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 2817 RSC
- C.-Y. Wang, R. Pagel, D. Bahnemann and J. Dohrmann, J. Phys. Chem. B, 2004, 108, 14082 CrossRef CAS
- S. Furukawa, T. Shishido, K. Teramura and T. Tanaka, ACS Catal., 2012, 2(1), 175 CrossRef CAS
- D. Tsukamoto, M. Ikeda, Y. Shiraishi, T. Hara, N. Ichikuni, S. Tanaka and T. Hirai, Chem. – Eur. J., 2011, 17, 9816 CrossRef CAS PubMed
- V. Augugliaro, T. Caronna, V. Loddo, G. Marci, G. Palmisano, L. Palmisano and S. Yurdaka, Chem. – Eur. J., 2008, 14, 4640 CrossRef CAS PubMed
- T. Kandiel, R. Dillert and D. Bahnemann, Photochem. Photobiol. Sci., 2009, 8, 683 CAS
- L. Palmisano, V. Augugliaro, M. Bellardita, A. Di Paola, E. Lopez, V. Loddo, G. Marci, G. Palmisano and S. Yurdakal, ChemSusChem, 2011, 4, 1431 CrossRef CAS PubMed
-
C.-Y. Wang, H. Gorenzin and M. Shultz, Abstracts of Papers of the American Chemical Society, 2004, vol. 227, p. U811 Search PubMed
- C.-Y. Wang, H. Groenzin and M. Shultz, J. Am. Chem. Soc., 2004, 126, 8094 CrossRef CAS PubMed
- A. Molinari, M. Montoncello, H. Rezala and A. Maldotti, Photochem. Photobiol. Sci., 2009, 8(5), 613 CAS
- W. Irawaty, D. Friedmann, J. Scott, P. Pichat and R. Amal, Catal. Today, 2011, 178, 51 CrossRef CAS
- V. Augugliaro, G. Camera-Roda, V. Loddo, G. Palmisano, L. Palmisano, F. Parrino and M. Puma, Appl. Catal., B, 2012, 111–112, 555 CrossRef CAS
- G. Camera-Roda, V. Augugliaro, A. Cardillo, V. Loddo, G. Palmisano and L. Palmisano, Chem. Eng. J., 2013, 224, 136 CrossRef CAS
- Y. Zhang, R. Ciriminna, G. Palmisano, Y.-J. Xu and M. Pagliaro, RSC Adv., 2014, 4(35), 18341 RSC
- J. Colmenares, W. Ouyang, M. Ojeda, E. Kuna, O. Chernyayeva, D. Lisovytskiy, S. De, R. Luque and A. Balu, Appl. Catal., B, 2016, 183, 107 CrossRef CAS
- B. Ohtani, H. Osaki, S. Nishimoto and T. Kagiya, Tetrahedron Lett., 1986, 27, 2019 CrossRef CAS
- B. Ohtani, H. Osaki, S. Nishimoto and T. Kagiya, Chem. Lett., 1985, 1075 CrossRef CAS
- S. Nishimoto, B. Ohtani, T. Yoshikawa and T. Kagiya, J. Am. Chem. Soc., 1983, 105, 7180 CrossRef CAS
- B. Pal, S. Ikeda, H. Kominami, Y. Kera and B. Ohtani, J. Catal., 2003, 217, 152 CAS
- B. Ohtani, S. Tsuru, S. Nishimoto, T. Kagiya and K. Izawa, J. Org. Chem., 1990, 55, 5551 CrossRef CAS
- H. Reiche and A. Bard, J. Am. Chem. Soc., 1979, 101, 3127 CrossRef CAS
- W. Dunn, Y. Aikawa and A. Bard, J. Am. Chem. Soc., 1981, 103, 6893 CrossRef CAS
- J. Onoe and T. Kawai, J. Chem. Soc., Chem. Commun., 1987, 1480 RSC
- F. Mahdavi, T. Bruton and Y. Li, J. Org. Chem., 1993, 58, 744 CrossRef CAS
- V. Brezova, A. Blazkova, I. Surina and B. Havlinova, J. Photochem. Photobiol., A, 1997, 107, 233 CrossRef CAS
- K. Imamura, K. Hashimoto and H. Kominami, Chem. Commun., 2012, 48, 4356 RSC
- A. Hakki, R. Dillert and D. Bahnemann, Phys. Chem. Chem. Phys., 2013, 5, 2992 RSC
- H. Schwarz and R. Dodson, J. Phys. Chem., 1989, 93, 409 CrossRef CAS
- H. Tada, T. Ishida, A. Takao, S. Ito, S. Mukhopadhyay, T. Akita, K. Tanaka and H. Kobayashi, ChemPhysChem, 2005, 6, 1537 CrossRef CAS PubMed
- H. Tada, A. Takao, T. Akita and K. Tanaka, ChemPhysChem, 2006, 7, 1687 CrossRef CAS PubMed
- O. Rios-Berny, S. Flores, I. Cordova and M. Valenzuela, Tetrahedron Lett., 2010, 51, 2730 CrossRef CAS
-
H. Kisch and M. Hopfner, in Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH Verlag GmbH, Germany, 2008, pp. 232–275 Search PubMed
- H. Kisch, Angew. Chem., Int. Ed., 2013, 52, 812 CrossRef CAS PubMed
- M. Hopfner, H. Weiss, D. Meissner, F. Heinemann and H. Kisch, Photochem. Photobiol. Sci., 2002, 1, 696 CAS
- H. Keck, W. Schindler, F. Knoch and H. Kisch, Chem. – Eur. J., 1997, 3, 1638 CrossRef CAS
- Y. Shiraishi, M. Ikeda, D. Tsukamoto, S. Tanaka and T. Hirai, Chem. Commun., 2011, 47, 4811 RSC
- A. Hakki, R. Dillert and D. Bahnemann, ACS Catal., 2013, 3, 565 CrossRef CAS
- Y. Yagci, S. Jockusch and N. Turro, Macromolecules, 2010, 43, 6245 CrossRef CAS
- X. Ni, J. Ye and C. Dong, J. Photochem. Photobiol., A, 2006, 181, 19 CrossRef CAS
- Z. Weng, X. Ni, D. Yang, J. Wang and W. Chen, J. Photochem. Photobiol., A, 2009, 201, 151 CrossRef CAS
- B. Kiskan, J. Zhang, X. Wang, M. Antonietti and Y. Yagci, ACS Macro Lett., 2012, 1, 546 CrossRef CAS
- Z. Wang, K. Landfester and K. Zhang, Polym. Chem., 2014, 5, 3559 RSC
- S. Dadashi-Silab, A. Asiri, S. Khan, K. Alamry and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1500 CrossRef CAS
- S. Dadashi-Silab, Y. Yar, H. Acar and Y. Yagci, Polym. Chem., 2015, 6, 1918 RSC
- J. Yan, B. Li, F. Zhou and W. Liu, ACS Macro Lett., 2013, 2, 592 CrossRef CAS
- B. Li, B. Yu and F. Zhou, Macromol. Rapid Commun., 2014, 35, 1287 CrossRef CAS PubMed
- A. Bansal, A. Kumar, P. Kumar, S. Bojja, A. Chatterjee, S. Ray and S. Jain, RSC Adv., 2015, 5, 21189 RSC
- S. Dadashi-Silab, M. Tasdelen, A. Asiri, S. Khan and Y. Yagci, Macromol. Rapid Commun., 2014, 35, 454 CrossRef CAS PubMed
- S. Dadashi-Silab, M. Tasdelen, B. Kiskan, X. Wang, M. Antonietti and Y. Yagci, Macromol. Chem. Phys., 2014, 215, 675 CrossRef CAS
- Y. Liu, D. Chen, X. Li, Z. Yu, Q. Xia, D. Liang and H. Xing, Green Chem., 2016, 18, 1475 RSC
- L.-C. Liu, M. Lu, Z.-H. Hou, G.-X. Wang, C.-A. Yang, E.-X. Liang, H. Wu, X.-L. Li and Y.-X. Xu, J. Appl. Polym. Sci., 2015, 132, 42389 Search PubMed
- E. Liang, M. Lu, Z. Hou, Y. Huang, B. He, G. Wang, L. Liu, H. Wu and M. Zhong, J. Appl. Polym. Sci., 2016, 133, 42891 CrossRef
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS PubMed
- S. Ligon, B. Husár, H. Wutzel, R. Holman and R. Liska, Chem. Rev., 2014, 114, 557 CrossRef CAS PubMed
- A. Puga, Coord. Chem. Rev., 2016, 315, 1 CrossRef CAS
- M. Matsumura, M. Hiramoto, T. Lehara and H. Tsubomura, J. Phys. Chem., 1984, 88(2), 248 CrossRef CAS
-
K. Rajeshwar, in Semiconductor Electrodes and Photoelectrochemistry, ed. S. Licht, Wiley-VCH, Weinheim, Germany, 2003, pp. 1–57 Search PubMed
- G. Redmond and D. Fitzmaurice, J. Phys. Chem., 1993, 97, 1426 CrossRef CAS
- T. Kandiel, A. Feldhoff, L. Robben, R. Dillert and D. Bahnemann, Chem. Mater., 2010, 22(6), 2050 CrossRef CAS
- H. Kato and A. Kudo, J. Phys. Chem. B, 2001, 105(19), 4285 CrossRef CAS
- M. Graetzel, Nature, 2001, 414, 338 CrossRef PubMed
-
R. Memming, Semiconductor Electrochemistry, Wiley-VCH, Weinheim, 2001 Search PubMed
- S. Chen and L. W. Wang, Chem. Mater., 2012, 24, 3659 CrossRef CAS
- M. Hoffmann, S. Martin, W. Choi and D. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS
- A. Hoffman, H. Yee, G. Mills and M. Hoffmann, J. Phys. Chem., 1992, 96, 5540 CrossRef CAS
- D. Beydoun, R. Amal, G. Low and S. McEvoy, J. Nanopart. Res., 1999, 1(4), 439 CrossRef CAS
- J. Tripathy, K. Lee and P. Schmuki, Angew. Chem., Int. Ed., 2014, 53(46), 12605 CAS
- C. Wang, M. Shim and P. Guyot-Sionnest, Science, 2001, 291, 2390 CrossRef CAS PubMed
- M. Holmes, T. Townsend and F. Osterloh, Chem. Commun., 2011, 48(3), 371 RSC
- I. Robel, M. Kuno and P. V. Kamat, J. Am. Chem. Soc., 2007, 129, 4136 CrossRef CAS PubMed
- NSCU, Department of Chemistry, http://ncsu.edu/project/chemistrydemos/Light/Luminol.pdf accessed 01/06/2016.
- O. Tomita, B. Ohtani and R. Abe, Catal.: Sci. Technol., 2014, 4, 3850 CAS
- H. Yuzawa, M. Aoki, K. Otake, T. Hattori, H. Itoh and H. Yoshida, J. Phys. Chem. C, 2012, 116(48), 25376 CAS
- H. Yuzawa and H. Yoshida, Chem. Commun., 2010, 46, 8854 RSC
- X. Ke, X. Zhang, J. Zhao, S. Sarina, J. Barry and H. Zhu, Green Chem., 2013, 15(1), 236 RSC
- Y. Shiraishi, H. Sakamoto, K. Fujiwara, S. Ichikawa and T. Hirai, Am. Chem. J., 2014, 4(8), 2418 CAS
- C. Karunakaran, S. Senthilvelan and S. Karuthapandian, J. Photochem. Photobiol., A, 2005, 172(2), 207 CrossRef CAS
- B. Cao, J. Zhang, J. Zhao, Z. Wang, P. Yang, H. Zhang, L. Li and Z. Zhu, ChemCatChem, 2014, 6(6), 1673 CrossRef CAS
- C. Canlas, J. Lu, N. Ray, N. Grosso-Giordano, S. Lee, J. Elam, R. Winans, R. Van Duyne, P. Stair and J. Notestein, Nat. Chem., 2012, 4(12), 1030 CrossRef CAS PubMed
- D. Worsley, A. Mills, K. Smith and M. Hutchings, Chem. Commun., 1995, 1119–1120 RSC
- V. Augugliaro, G. Camera-Roda, V. Loddo, G. Palmisano, L. Palmisano, J. Soria and S. Yurdakal, J. Phys. Chem. Lett., 2015, 6(10), 1968 CrossRef CAS PubMed
- T. Tan, C. Yip, D. Beydoun and R. Amal, Chem. Eng. J., 2003, 95(1), 179 CrossRef CAS
- T. Tan, D. Beydoun and R. Amal, J. Mol. Catal. A: Chem., 2003, 202(1–2), 73–85 CrossRef CAS
- V. Nguyen, R. Amal and D. Beydoun, Chem. Eng. Sci., 2003, 58(19), 4429 CrossRef CAS
- T. Tan, D. Beydoun and R. Amal, J. Photochem. Photobiol., A, 2003, 159(3), 273 CrossRef CAS
- C. Chenthamarakshan, H. Yang, Y. Ming and K. Rajeshwar, J. Electroanal. Chem., 2000, 494, 79 CrossRef CAS
- K. Asmus, H. Mockel and A. Henglein, J. Phys. Chem., 1973, 77(10), 1218 CrossRef CAS
- M. Kaise, H. Nagai, K. Tokuhashi, S. Kondo, S. Nimura and O. Kikuchi, Langmuir, 1994, 10(5), 1345 CrossRef CAS
- J. Lilie, G. Beck and A. Henglein, Berichte der Bunsengesellschaft für physikalische Chemie, 1971, 75(5), 465 CrossRef
- J. Butler and A. Henglein, Radiation Physical Chemistry, 1980, 5, 603 Search PubMed
- S. Yamagata, S. Nakabayashi, K. Sancier and A. Fujishima, Bull. Chem. Soc. Jpn., 1988, 61, 3429 CrossRef CAS
- V. Jagannadham and S. Steenken, J. Am. Chem. Soc., 1984, 106, 6542 CrossRef CAS
- J. Ferry and W. Glaze, Langmuir, 1998, 14(13), 3551 CrossRef CAS
- H. Fischer, Chem. Rev., 2001, 101(12), 3581 CrossRef CAS PubMed
- M. Hecht and W. Fawcett, J. Phys. Chem., 1995, 99, 1311 CrossRef CAS
- H. Pellissier, Tetrahedron, 2013, 69, 7171 CrossRef CAS
-
A. Jones, Direct Epoxidation Process Using a Mixed Catalyst System, US6307073B1, 2002 Search PubMed
-
R. Brady, S.-C. Kao, F. Karol and T. Nemzek, Mixed Catalyst System, US6069213A, 2000 Search PubMed
- G. Li, C. Richter, R. Milot, L. Cai, C. Schmuttenmaer, R. Crabtree, G. Brudvig and V. Batista, Dalton Trans., 2009, 45, 10078 RSC
- R. Marotta, I. Di Somma, D. Spasiano, R. Andreozzi and V. Caprio, Chem. Eng. J., 2011, 172(1), 243 CrossRef CAS
- D. Spasiano, L. del Pilar Prieto Rodriguez, J. Olleros, S. Malato, R. Marotta and R. Andreozzi, Appl. Catal., B, 2013, 136–137, 56 CrossRef CAS
- I. Bedja and P. Kamat, J. Phys. Chem., 1995, 99, 9182 CrossRef CAS
- D. Beydoun, R. Amal, G. Low and S. McEvoy, J. Phys. Chem. B, 2000, 104(18), 4387 CrossRef CAS
- J. Carneiro, C.-C. Yang, J. Moma, J. Moulijn and G. Mul, Catal. Lett., 2009, 129(1–2), 12 CrossRef CAS
- Y. Ide, R. Ogino, M. Sadakane and T. Sano, ChemCatChem, 2013, 5(3), 766 CrossRef CAS
- D. Bahnemann, M. Hilgendorff and R. Memming, J. Phys. Chem. B, 1997, 101(21), 4265 CrossRef CAS
- Y. Ide, N. Kawamoto, Y. Bando, H. Hattori, M. Sadakane and T. Sano, Chem. Commun., 2013, 49(35), 3652 RSC
- Y. Ide, H. Hattori, S. Ogo, M. Sadakane and T. Sano, Green Chem., 2012, 14(5), 1264 RSC
- Y. Ide, N. Nakamura, H. Hattori, R. Ogino, M. Ogawa, M. Sadakane and T. Sano, Chem. Commun., 2011, 47(41), 11531 RSC
- A. Magdziarz, J. Colmenares, O. Chernyayeva, K. Kurzydłowski and J. Grzonka, ChemCatChem, 2016, 8, 536 CrossRef CAS
- T. Ruberu, N. Nelson, I. Slowing and J. Vela, J. Phys. Chem. Lett., 2012, 3(19), 2798 CrossRef CAS
- C. Han, Z. Chen, N. Zhang, J. Colmenares and Y.-J. Xu, Adv. Funct. Mater., 2015, 25(2), 221 CrossRef CAS
- J. Ryu and W. Choi, Environ. Sci. Technol., 2008, 42, 294 CrossRef CAS PubMed
Footnote |
| † These authors contributed equally to manuscript. |
| This journal is © The Royal Society of Chemistry 2016 |