
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-free, visible-light-mediated, decarboxylative alkylation of biomass-derived compounds†
Johanna
Schwarz
and
Burkhard
König
 *
*
Institute of Organic Chemistry, University of Regensburg, Universitätsstraße 31, 93053 Regensburg, Germany. E-mail: Burkhard.Koenig@chemie.uni-regensburg.de; Tel: (+49)-941-943-4575
First published on 26th May 2016
Abstract
This work describes a mild, environmentally friendly method to activate natural carboxylic acids for decarboxylative alkylation. After esterification of biomass-derived acids to N-(acyloxy)phthalimides, the active esters are cleaved reductively by photocatalysis to give alkyl radicals, which undergo C–C bond formation with electron-deficient alkenes. This reaction is catalyzed by the organic dye eosin Y and green light (535 nm) and the scope of acids includes abundant amino acids, α-oxy acids and fatty acids which are available from renewable resources.
Introduction
Carboxylic acids are among the most abundant, renewable feedstocks on our planet. They are non-toxic, stable and inexpensive and therefore valuable starting materials for “green chemistry”.1 Due to the shortage of fossil fuels and rising energy demand, alternative sources of raw materials are gaining importance.2 In order to generate platform chemicals and high-value chemicals like pharmaceuticals from biomass-derived compounds, the carboxy group can be targeted as a chemo- and regioselective leaving group for C–C bond formation reactions.3In principle, decarboxylative reactions are known for a long time, but old protocols (e.g. according to Hunsdiecker,4 Barton5 and Kolbe6) are not suitable for cross-coupling reactions between acids and other substrates under benign conditions. Over the last two decades, many transition metal-catalysed, decarboxylative C–C and C–X coupling methods have been reported by Gooßen,7 Myers8 and many others.9 Even though these methods are versatile in application and often compatible with multiple step reactions, they require high temperatures as well as palladium or copper reagents. Moreover, they are usually limited to decarboxylation of Csp2–COOH or Csp–COOH bonds.
Recently, first photoredox-mediated processes were developed for the activation of the carboxy group. The usage of photocatalysts and visible light enables reactions under mild conditions and low energy consumption. During the last few years, photocatalytic, decarboxylative reactions like arylations,10 vinylations,11 allylations,12 alkynylations,13 fluorinations14 and hydrodecarboxylations15 have been reported by MacMillan and other groups.16 Also different methods for decarboxylative alkylations have already been investigated.17 Here, alkyl radicals which are generated from carboxylic acids upon extrusion of CO2 react with activated alkenes, typically Michael acceptors. The alkyl radicals can either be formed by esterification of carboxylic acids and subsequent reductive cleavage of the ester bond (Scheme 1a)17c,18 or by oxidative cleavage of the acid itself (Scheme 1b).17a These methods have already been reported by different groups, but expensive and toxic transition metal catalysts are needed in all cases and the scope of carboxylic acids is limited.
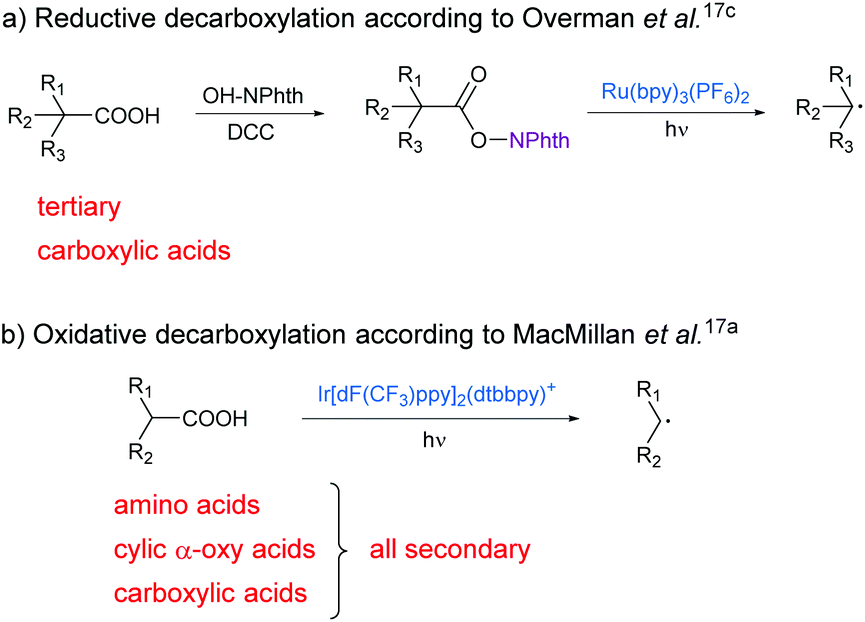 | ||
| Scheme 1 Photocatalytic generation of an alkyl radical from carboxylic acids for subsequent coupling with electron-deficient alkenes. | ||
Herein, we report for the first time a metal-free, photo-catalytic, decarboxylative alkylation which is applicable for a broad variety of natural carboxylic acids involving amino acids, α-oxy acids and fatty acids (Scheme 2). Therefore, N-(acyloxy)-phthalimides were synthesized from the corresponding acids according to a simple method developed by Okada,18b,19 analogously to Overman's approach. These active esters undergo C–C bond formation with electron-deficient alkenes under irradiation with green light in the presence of DIPEA and the organic, non-toxic and cheap dye eosin Y. Thus, valuable chemicals can be obtained from renewable biomass under eco-friendly and mild conditions.
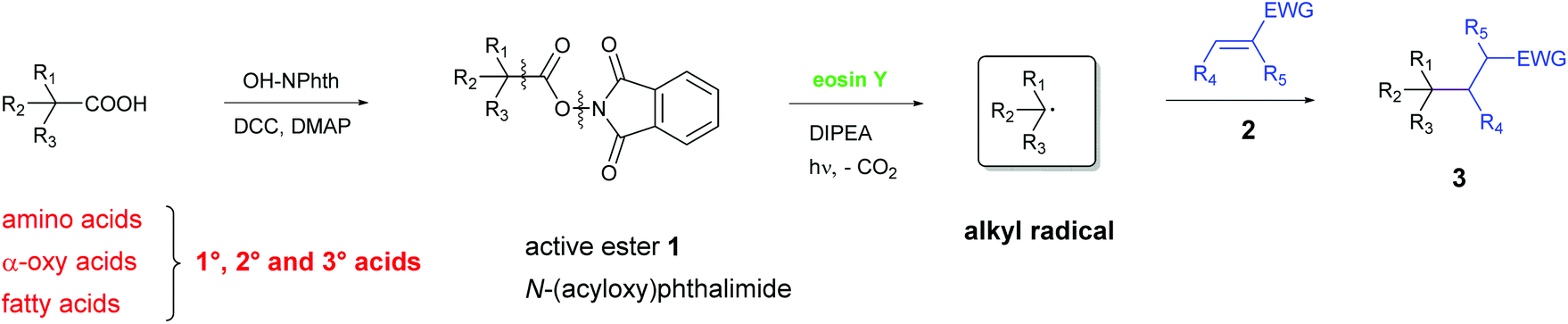 | ||
| Scheme 2 Our work: a general approach for the decarboxylative alkylation of natural compounds by reductive cleavage of N-(acyloxy)phthalimide 1. | ||
Results and discussion
Synthesis and scope
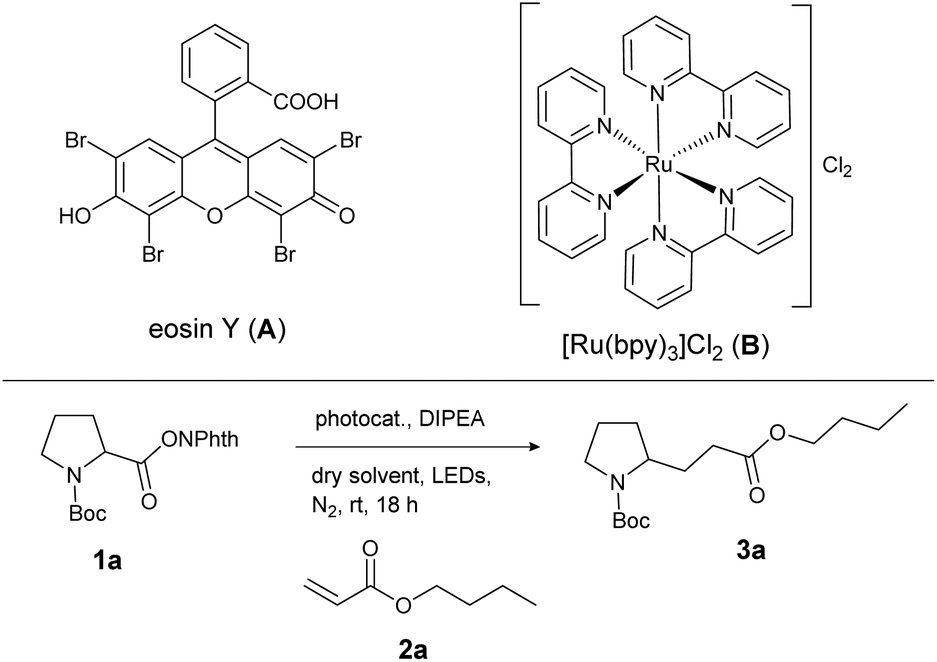
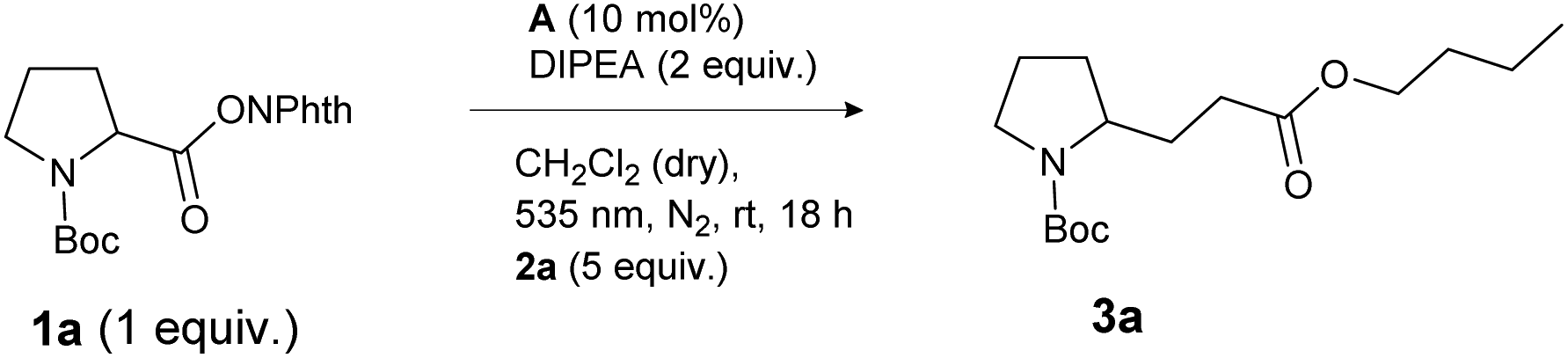
For investigation of the reaction conditions of the decarboxylative alkylation, the N-(acyloxy)phthalimide of N-Boc-protected proline (1a) and n-butyl acrylate (2a) served as test substrates. A mixture of both the compounds, the base DIPEA and a homogeneous photocatalyst was irradiated with LEDs under a nitrogen atmosphere. For this reaction, no heterogeneous catalysts have been investigated, although an application would be conceivable.20 First, the catalytic activity of the photocatalysts eosin Y (A) and [Ru(bpy)3]Cl2 (B) in different solvents was screened (Table 1). The inexpensive organic dye A enabled higher yields than the metal catalyst B in almost every solvent (except DMF: Table 1, entries 2 and 7). As [Ru(bpy)3]Cl2 is known to be a stronger reductant than eosin Y,21 the reason for the better performance of the metal-free catalyst A is probably its better solubility and stability in these solvents. Decomposition of B was indicated by darkening of the reaction mixtures after several minutes. The best product yield of 96% was obtained with photocatalyst A and CH2Cl2 as the solvent (Table 1, entry 9). For further optimization, different reaction conditions and control reactions were investigated (Table 2). Reducing the amount of catalyst to 5 mol% (Table 2, entry 1) or the amount of alkene from 5 to 2 eq. (Table 2, entry 4) resulted in lower yields of about 85%. Also a shorter reaction time (Table 2, entry 2) or the use of a smaller amount of base (Table 2, entry 3) had a clearly negative impact. However, running the reaction in the presence of air (Table 2, entry 5) gave the same yield as after degassing the reaction mixture (Table 1, entry 9). Measuring the oxygen concentration during the non-degassed reaction showed that the oxygen was consumed within two hours (see the ESI†). Control experiments without light, DIPEA or catalyst led to no product formation at all, regardless of whether oxygen was present or not (Table 2, entry 6 to 11).|
|
|||
|---|---|---|---|
| Entry | Photocatalytic system | Solvent | Yieldb [%] |
| a Reactions were performed using 1 equiv. 1a, 5 equiv. 2a and 2 equiv. DIPEA. b Determined by GC analysis using naphthalene as an internal standard. | |||
| 1 | [Ru(bpy)3]Cl2 (2 mol%, 455 nm) | DMSO | 30 |
| 2 | [Ru(bpy)3]Cl2 (2 mol%, 455 nm) | DMF | 47 |
| 3 | [Ru(bpy)3]Cl2 (2 mol%, 455 nm) | CH3CN | 60 |
| 4 | [Ru(bpy)3]Cl2 (2 mol%, 455 nm) | CH2Cl2 | 18 |
| 5 | [Ru(bpy)3]Cl2 (2 mol%, 455 nm) | THF | 7 |
| 6 | Eosin Y (10 mol%, 535 nm) | DMSO | 30 |
| 7 | Eosin Y (10 mol%, 535 nm) | DMF | 37 |
| 8 | Eosin Y (10 mol%, 535 nm) | CH3CN | 65 |
| 9 | Eosin Y (10 mol%, 535 nm) | CH2Cl2 | 96 |
| 10 | Eosin Y (10 mol%, 535 nm) | THF | 53 |
|
|
||
|---|---|---|
| Entry | Modification | Yielda [%] |
| a Determined by GC analysis using naphthalene as an internal standard. | ||
| 1 | 5 mol% A | 85 |
| 2 | 9 h irradiation time | 64 |
| 3 | 1 equiv. DIPEA | 26 |
| 4 | 2 equiv. 2a | 83 |
| 5 | Air atmosphere | 96 |
| 6 | No base | 0 |
| 7 | No light | 0 |
| 8 | No photocatalyst | 0 |
| 9 | Air atmosphere, no base | 0 |
| 10 | Air atmosphere, no light | 0 |
| 11 | Air atmosphere, no photocatalyst | 0 |
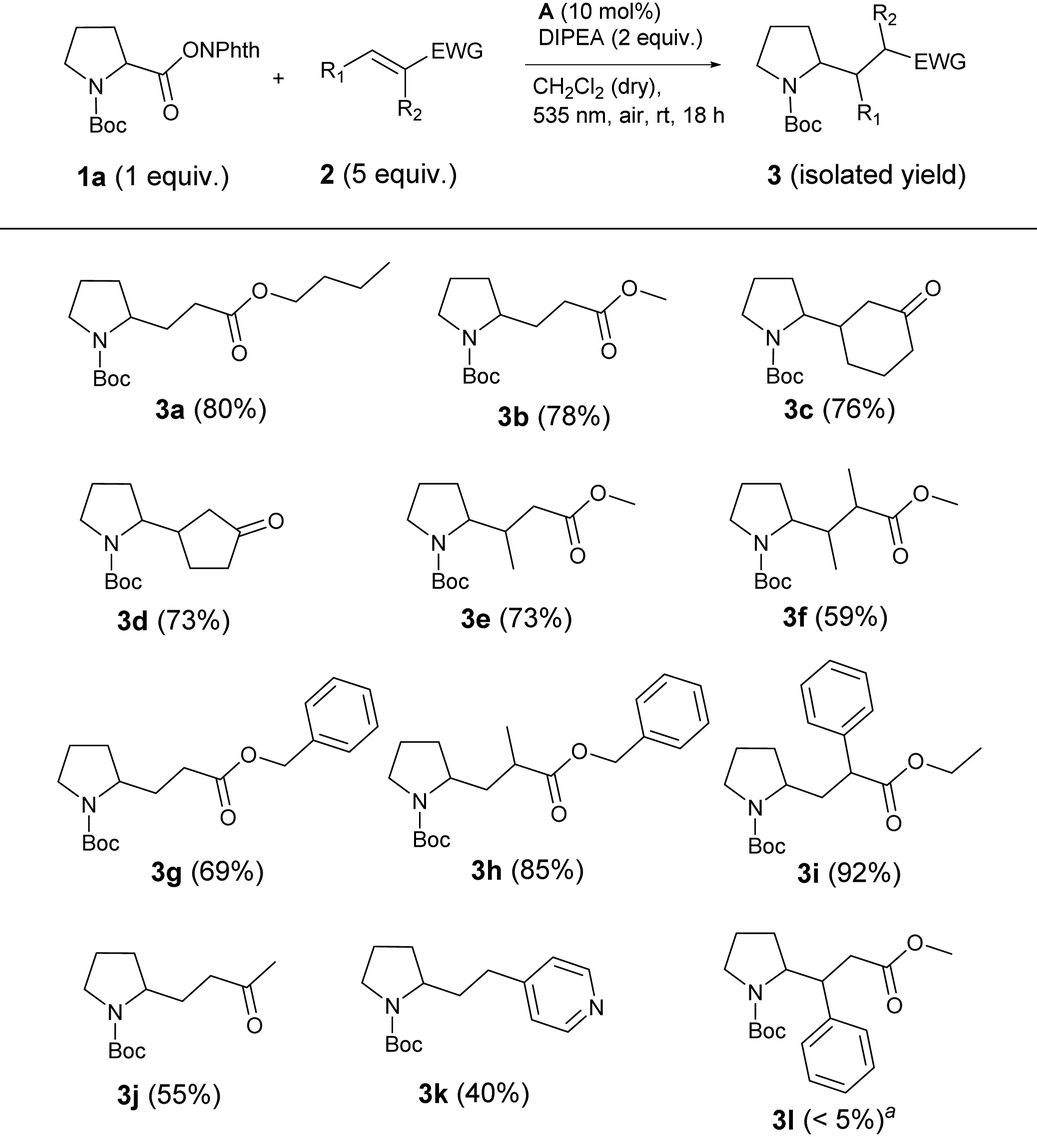
With these optimized reaction conditions, the scope and limitations of the decarboxylative alkylation were explored. Therefore, the reaction of different electron-deficient alkenes with active ester 1a was investigated (Table 3). The expected cross coupling products were observed in moderate to excellent yields when α,β-unsaturated esters and ketones were used as coupling partners. Unsubstituted or β-methylated aliphatic Michael acceptors gave the desired products 3a, 3b and 3e in good yields of 73 to 80%. Benzylic, α,β-unsaturated esters 2g and 2h were suitable reaction partners in the same way. Also reactions with cyclic ketones gave products 3d and 3e in about 75% yield. However, introduction of a phenyl ring at the attacked carbon inhibited the formation of product 3l almost completely, presumably due to the steric hindrance by the bigger substituent at the β-position. In contrast, if the phenyl ring was positioned on the α-carbon of the Michael acceptor, the best yield of 92% (3i) was observed. This result indicates that after the attack of the N-Boc proline fragment at the double bond, a radical is formed at the α-position, which is stabilized by an adjacent phenyl ring in the case of 3i. Next to Michael acceptors, the electrophilic, heteroaromatic styrene derivative vinylpyridine (2k) also yielded the corresponding product 3k in 40% yield.
| a Product not isolated. Yield estimated by 1H-NMR. |
|---|
|
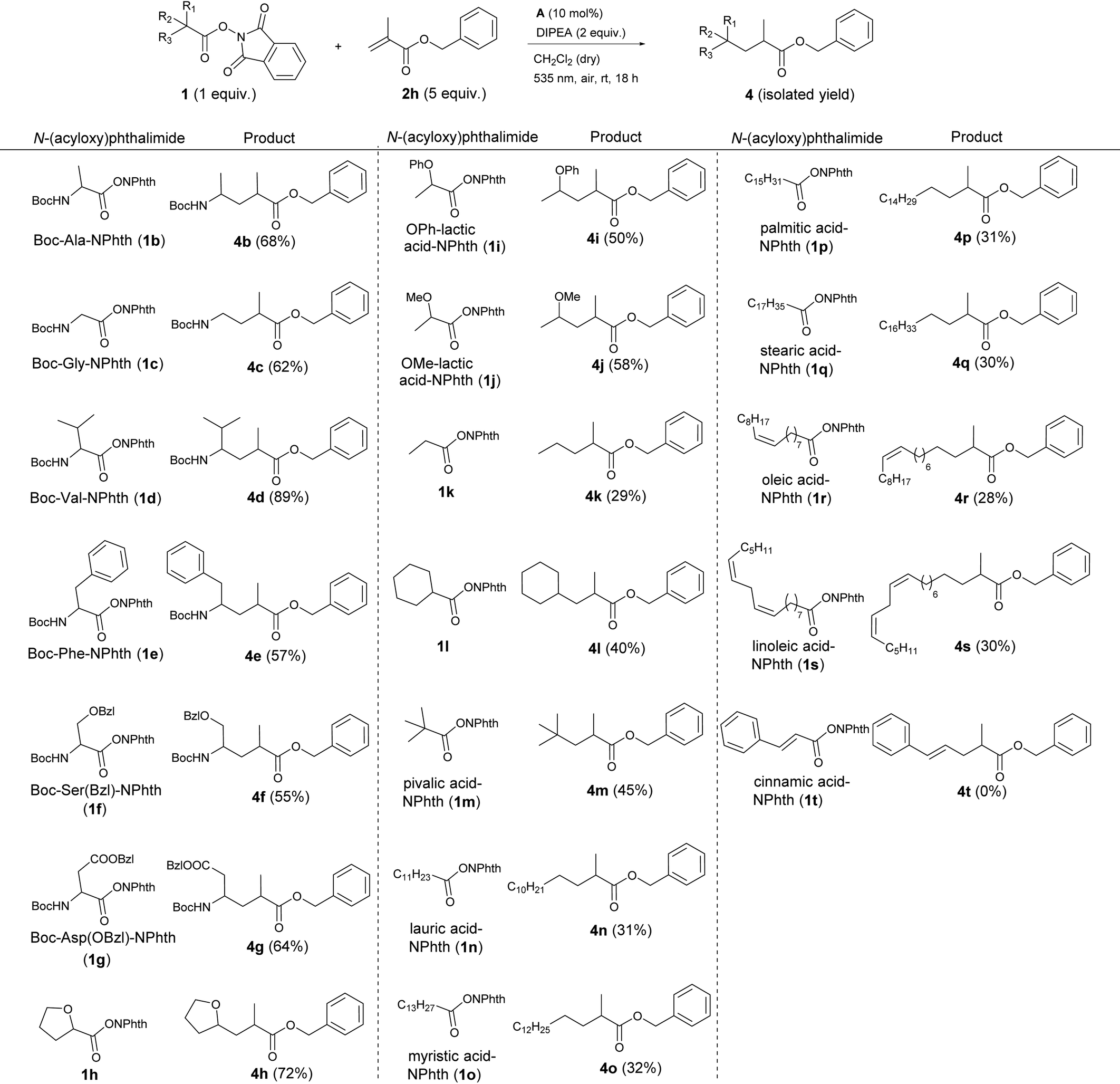
After establishing the scope of activated alkenes, N-(acyloxy)phthalimide 1 was varied (Table 4). Here, benzyl methacrylate 2h served as a model alkene and again, the optimized conditions of Table 2, entry 5 were applied. A broad variety of esterified acids, including readily available, biomass-derived compounds could be employed. This photocatalytic cross-coupling reaction works on the one hand for primary (1c, 1k, 1n–s), secondary (1b, 1c–j, 1l) as well as for tertiary carboxylic acids (1l) and on the other hand for different substrate classes like amino acids (1b–g), α-oxy acids (1h–j) and even fatty acids (1n–s).
|
The active esters of simple, Boc-protected, secondary amino acids proline (1a, Table 3), alanine (1b) or valine (1d) gave the highest yields (68–89%). The N-(acyloxy)phthalimide of the primary amino acid glycine showed slightly less product formation (4c, 62%). Also amino acid derivatives with aromatic side chains like phenylalanine (1e) or benzyl-protected serine (1f) and aspartic acid (1g) yielded the corresponding products 4e–g in about 60% yield. As a typical natural α-hydroxy acid, lactic acid with different protecting groups was investigated. Here, the phenoxy- (1i) and methoxy-protected derivatives (1j) gave 50 and 58% yield, respectively, and fewer coupling products than the cyclic α-oxy starting material 1h (72%).
Applying active esters of simple carboxylic acids without any heteroatom at the α-position to the carboxy moiety also resulted in product formation, although the yields decreased to 30–45% (4k–s). This decline in efficiency indicates that the stability of the alkyl radical generated after decarboxylation is the decisive point for successful product formation. In the case of amino and α-oxy compounds, the radical is stabilized by electron-donation from the lone pair of the heteroatom to the singly occupied p orbital of the neighboring radical centered carbon, which explains the higher yields.22 Considering the decarboxylative alkylation of N-(acyloxy)phthalimides of simple carboxylic acids, secondary (1l, 40%), and even more, tertiary carboxylic acids (1m, 45%) are more prone to decarboxylation than primary, linear carboxylic acids (1k, 1n–s; all about 30%). Again, tertiary alkyl radicals are more stable than secondary and especially primary radicals, which is in good accordance with the observed yields.22b Nonetheless, several fatty acids, which are widespread in nature could be applied for this photocatalytic reaction. Saturated (1n–q), mono-unsaturated (1r) and di-unsaturated (1s) fatty acid derivatives with different chain lengths (12–18 C) all gave products 4n–s in about 30% yield. The reaction with the active ester of cinnamic acid (1t) yielded no product, because the vinylic radical intermediate, which is formed upon decarboxylation is apparently not stable enough to undergo further reactions. In summary, we could show that cheap and abundant compounds from renewable biomass are suitable starting materials for this cross-coupling reaction.
Mechanistic investigations
Based on the report by Overman et al.,17c the known photochemistry of eosin Y23 and our results, we propose the following mechanism for the decarboxylative alkylation of N-(acyloxy)phthalimides with electron-deficient alkenes (Scheme 3): Irradiation of a photocatalyst (PC) with visible light generates the excited catalyst PC*, which is reductively quenched by the sacrificial electron donor DIPEA to give DIPEA+˙. Regeneration of the PC is presumably achieved by reduction of N-(acyloxy)phthalimide 1a, which gives the corresponding radical anion. Splitting of the N–O bond and subsequent elimination of CO2 generates alkyl radical 1a˙. This radical can attack the double bond of Michael acceptor 2a to form the intermediate 3a˙. It is assumed that the hydrogen atom abstraction from DIPEA+˙ or the solvent finally enables the formation of product 3a.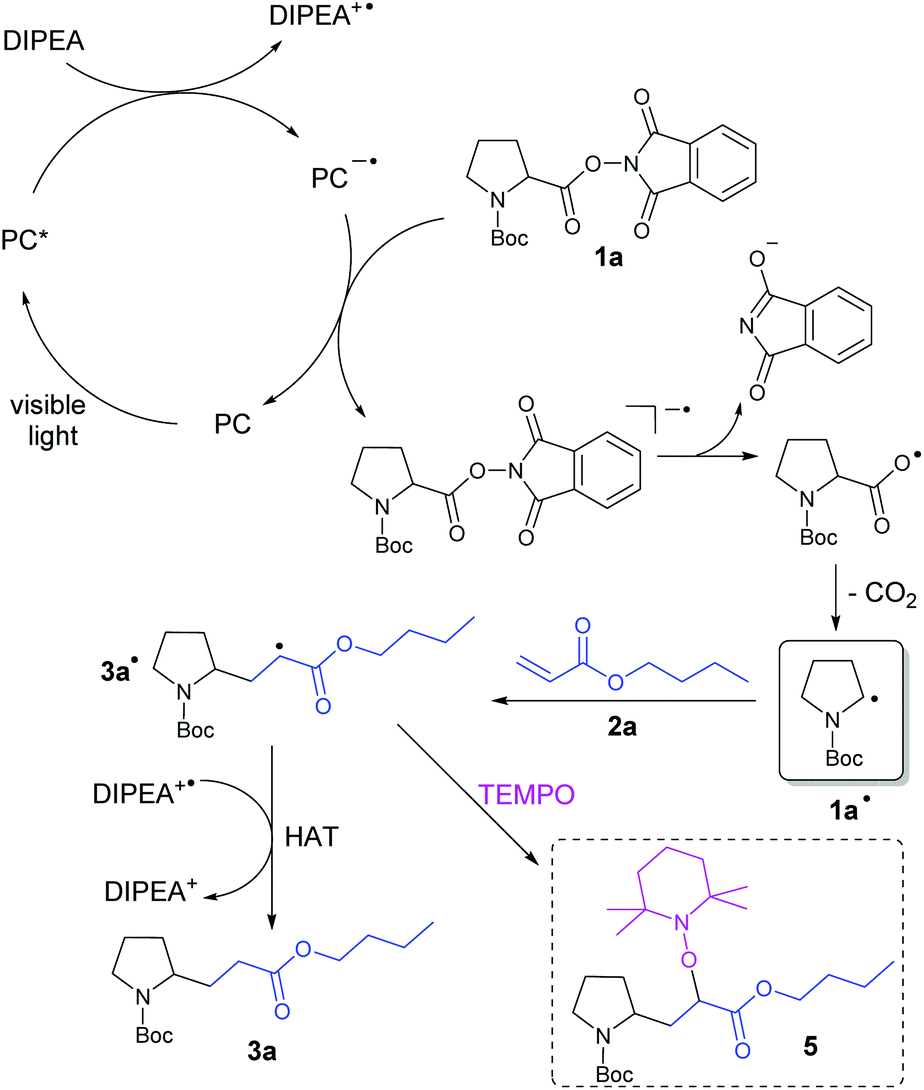 | ||
| Scheme 3 Proposed reaction mechanism for the photocatalytic, decarboxylative alkylation of N-(acyloxy)phthalimide 1a with n-butylacrylate (2a). | ||
This mechanistic proposal was confirmed by several spectroscopic studies. Single electron transfer (SET) from DIPEA to the excited PC should be exergonic according to the redox potentials of DIPEA (+0.72 V vs. SCE in CH3CN)24 and the excited PC A (eosin Y*/eosin Y˙−: +0.83 V vs. SCE in CH3CN–H2O, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)21b or B (Ru2+*/Ru+: +0.77 V vs. SCE in CH3CN)25 and is a well-documented process in the literature.21,26 Furthermore, this step was confirmed by luminescence quenching experiments with both photocatalysts. In the case of eosin Y, fluorescence quenching could hardly be observed with DIPEA, N-(acyloxy)-phthalimide 1a or alkene 2a (see the ESI† for graphs and further details). This indicates that excited PC A does rather react from the triplet than from the singlet state which was already shown by our group for another reaction.27 For verification of this triplet reactivity, transient absorption measurements were performed, due to the short triplet lifetime of eosin Y (τT = 320 ± 10 ns).27a Performing fluorescence experiments with [Ru(bpy)3]Cl2 (B) revealed that the emission of B* is quenched by the electron donor DIPEA, which proves SET between both species (Fig. 1). Upon titration of B* with 1a or 2a, no quenching was observed (see the ESI†).
:1)21b or B (Ru2+*/Ru+: +0.77 V vs. SCE in CH3CN)25 and is a well-documented process in the literature.21,26 Furthermore, this step was confirmed by luminescence quenching experiments with both photocatalysts. In the case of eosin Y, fluorescence quenching could hardly be observed with DIPEA, N-(acyloxy)-phthalimide 1a or alkene 2a (see the ESI† for graphs and further details). This indicates that excited PC A does rather react from the triplet than from the singlet state which was already shown by our group for another reaction.27 For verification of this triplet reactivity, transient absorption measurements were performed, due to the short triplet lifetime of eosin Y (τT = 320 ± 10 ns).27a Performing fluorescence experiments with [Ru(bpy)3]Cl2 (B) revealed that the emission of B* is quenched by the electron donor DIPEA, which proves SET between both species (Fig. 1). Upon titration of B* with 1a or 2a, no quenching was observed (see the ESI†).
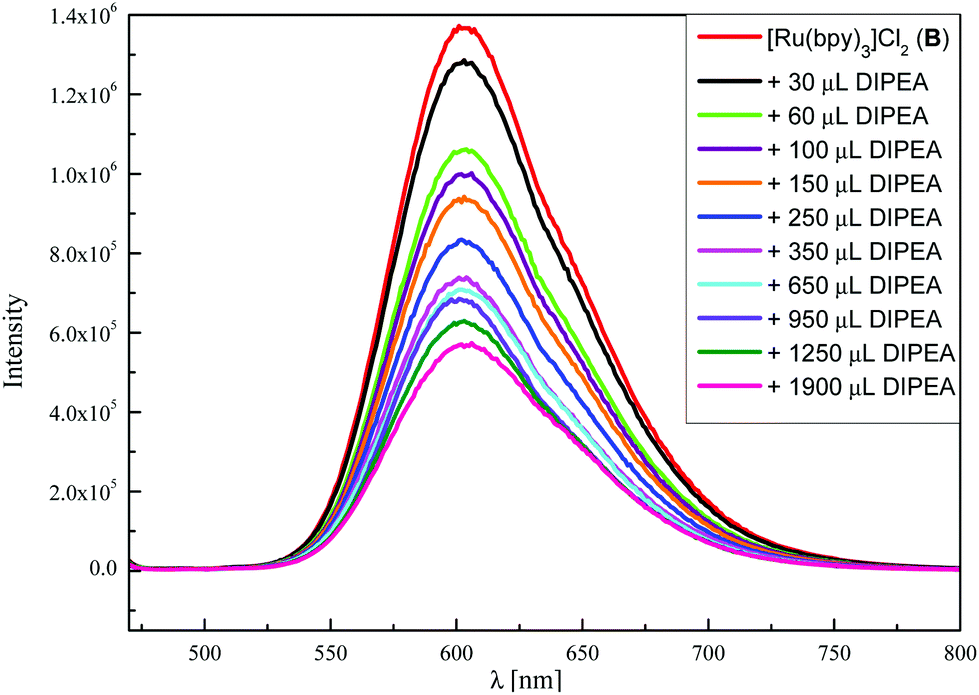 | ||
| Fig. 1 Fluorescence quenching of [Ru(bpy)3]Cl2 (B, 15.0 μM in CH3CN) upon titration with DIPEA (100 mM in CH3CN). | ||
The next step of the catalytic cycle was assumed to be the regeneration of the PC by SET from PC˙− to 1a. Cyclic voltammetry measurements of the active ester 1a (−1.20 V vs. SCE in CH3CN, see the ESI†) showed that reduction by the strongly reducing PC B (Ru+/Ru2+: −1.33 V vs. SCE in CH3CN)21a is thermodynamically feasible. Although redox potentials are not directly comparable with each other in the case of PC A (eosin Y˙−/eosin Y: −1.06 V vs. SCE in CH3CN–H2O, 1:1),21b this catalyst is also known to be a powerful reductant. Eosin Y shows an absorption maximum at 527 nm which corresponds to an energy of 2.35 eV. After intersystem crossing to its reactive triplet state, 1.89 eV are still available for photocatalytic reactions.21b To confirm the formation of radical 3a˙ during the reaction, this intermediate was captured by the persistent radical TEMPO giving product 5 (Scheme 3), which was determined by LC-MS analysis (see the ESI†). Furthermore, the quantum yield of the photocatalytic model reaction was determined (see the ESI†). The low value of Φ = 2.6 ± 0.5% corresponds to the relatively long reaction times of 18 h and indicates that efficient radical chain processes are very unlikely in this mechanism.28 For further insight, the stability of the photocatalyst eosin Y has been investigated by measuring UV-vis absorption spectra during the reaction (see the ESI†). Over the reaction time of 18 h, the catalyst slowly degrades and a precipitate occurs, which slows down the product formation after some hours (see the ESI† for the time course of the reaction). However, the remaining photocatalytic active species is sufficient to reach almost complete conversion within the reaction time.
Experimental
General procedure for the synthesis of N-(acyloxy)phthalimides 1
Starting materials 1 were synthesized by using a slightly modified procedure based on the reports by Reiser et al.18a and Overman et al.17c The respective carboxylic acid (8.00 mmol, 1.0 equiv.), N-hydroxy-phthalimide (1.43 g, 8.80 mmol, 1.1 equiv.), N,N′-dicyclohexyl-carbodiimide (1.98 g, 9.60 mmol, 1.2 equiv.) and 4-dimethyl-aminopyridine (0.98 g, 0.80 mmol, 0.1 equiv.) were mixed in a flask with a magnetic stirring bar. Dry THF (40 mL) was added and the orange reaction mixture was stirred for 15 h at rt. The white precipitate was filtered off and the solution was concentrated by evaporation of the solvent. Purification by column chromatography on flash silica gel (CH2Cl2 or CH2Cl2/CH3OH = 9:1) gave the corresponding product.
General procedure for the photocatalytic decarboxylative alkylation
In a 5 mL crimp cap vial with a stirring bar, eosin Y (A, 19.4 mg, 0.03 mmol, 0.1 equiv.) and N-(acyloxy)phthalimide 1 (0.30 mmol, 1.0 equiv.) were added. After addition of DIPEA (102 μL, 0.60 mmol, 2.0 equiv.), the corresponding olefin 2 (1.50 mmol, 5.0 equiv.) and dry CH2Cl2 (4 mL), the vial was capped to prevent evaporation. The reaction mixture was stirred and irradiated through the vials’ plane bottom side using green LEDs (535 nm) for 18 h at rt. The reaction mixture of two vials with the same content was combined and diluted with a saturated aqueous solution of NaHCO3 (20 mL). It was extracted with EA (3 × 20 mL) and the combined organic phases were washed with brine (20 mL), dried over Na2SO4 and concentrated under vacuum. Purification of the crude product was performed by automated flash column chromatography (PE/EA = 19:1 to 1:1) yielding the corresponding product 3 or 4 as colorless oil.
Conclusions
In conclusion, we have developed a metal-free, photocatalytic method for the decarboxylative alkylation of biomass-derived compounds. The advantage of this procedure over other methods reported in the literature is the broad substrate scope including cheap and abundant α-amino acids, α-oxy acids and fatty acids as well as primary, secondary and tertiary substrates. In addition, the reactions can be carried out under mild, environmentally friendly conditions with the metal-free, organic dye eosin Y as the photocatalyst. The carboxylic acids are activated by esterification to N-(acyloxy)phthalimides and then reductively cleaved upon irradiation with green light. This generates alkyl radicals, which undergo cross-coupling with electron-deficient alkenes. Thereby, largely new compounds for the synthesis of pharmaceuticals or fine chemicals are generated. The method contributes to the ongoing efforts replacing fossil fuels by renewable feedstocks in the synthesis of chemical intermediates.Acknowledgements
We thank the German Science Foundation (DFG, GRK 1626, Chemical Photocatalysis) for financial support for this work.References
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- (a) A. Pfennig, Chem. Ing. Tech., 2007, 79, 2009–2018 CrossRef CAS; (b) R. Watson, in Ressourcenverknappung, 50 Schlüsselideen der Zukunft, Springer, Berlin Heidelberg, 2014, pp. 24–27 Search PubMed.
- E. Scott, F. Peter and J. Sanders, Appl. Microbiol. Biotechnol., 2007, 75, 751–762 CrossRef CAS PubMed.
- H. Hunsdiecker and C. Hunsdiecker, Chem. Ber., 1942, 75, 291–297 CrossRef.
- (a) D. H. R. Barton, D. Bridon, I. Fernandaz-Picot and S. Z. Zard, Tetrahedron, 1987, 43, 2733–2740 CrossRef CAS; (b) D. H. R. Barton, D. Crich and W. B. Motherwell, J. Chem. Soc., Chem. Commun., 1983, 939–941 RSC.
- (a) H. Kolbe, Liebigs Ann. Chem., 1849, 69, 257–294 CrossRef; (b) H. Kolbe, Liebigs Ann. Chem., 1848, 64, 339–341 CrossRef.
- (a) L. J. Gooßen, G. Deng and L. M. Levy, Science, 2006, 313, 662–664 CrossRef PubMed; (b) N. Rodriguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC.
- (a) A. G. Myers, D. Tanaka and M. R. Mannion, J. Am. Chem. Soc., 2002, 124, 11250–11251 CrossRef CAS PubMed; (b) D. Tanaka, S. P. Romeril and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 10323–10333 CrossRef CAS PubMed.
- R. Shang and L. Liu, Sci. China: Chem., 2011, 54, 1670–1687 CrossRef CAS.
- (a) Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257–5260 CrossRef CAS PubMed; (b) Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832–1835 CrossRef CAS PubMed; (c) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed; (d) Y. Jin, M. Jiang, H. Wang and H. Fu, Sci. Rep., 2016, 6, 20068 CrossRef PubMed.
- (a) A. Noble and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602–11605 CrossRef CAS PubMed; (b) A. Noble, S. J. McCarver and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 624–627 CrossRef CAS PubMed.
- (a) C. Hu and Y. Chen, Org. Chem. Front., 2015, 2, 1352–1355 RSC; (b) S. B. Lang, K. M. O'Nele, J. T. Douglas and J. A. Tunge, Chem. – Eur. J., 2015, 21, 18589–18593 CrossRef CAS PubMed; (c) S. B. Lang, K. M. O'Nele and J. A. Tunge, J. Am. Chem. Soc., 2014, 136, 13606–13609 CrossRef CAS PubMed.
- (a) J. Yang, J. Zhang, L. Qi, C. Hu and Y. Chen, Chem. Commun., 2015, 51, 5275–5278 RSC; (b) F. Le Vaillant, T. Courant and J. Waser, Angew. Chem., Int. Ed., 2015, 54, 11200–11204 CrossRef CAS PubMed; (c) H. Huang, G. Zhang and Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 7872–7876 CrossRef CAS PubMed; (d) Q.-Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 11196–11199 CrossRef CAS PubMed.
- (a) S. Ventre, F. R. Petronijevic and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 5654–5657 CrossRef CAS PubMed; (b) X. Wu, C. Meng, X. Yuan, X. Jia, X. Qian and J. Ye, Chem. Commun., 2015, 51, 11864–11867 RSC.
- (a) C. Cassani, G. Bergonzini and C.-J. Wallentin, Org. Lett., 2014, 16, 4228–4231 CrossRef CAS PubMed; (b) J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340–11348 CrossRef CAS PubMed.
- J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS PubMed.
- (a) L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS PubMed; (b) D. W. Manley, R. T. McBurney, P. Miller, R. F. Howe, S. Rhydderch and J. C. Walton, J. Am. Chem. Soc., 2012, 134, 13580–13583 CrossRef CAS PubMed; (c) G. Pratsch, G. L. Lackner and L. E. Overman, J. Org. Chem., 2015, 80, 6025–6036 CrossRef CAS PubMed; (d) Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338–3341 CrossRef CAS PubMed; (e) Y. Miyake, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2013, 49, 7854–7856 RSC.
- (a) G. Kachkovskyi, C. Faderl and O. Reiser, Adv. Synth. Catal., 2013, 355, 2240–2248 CrossRef CAS; (b) K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, J. Am. Chem. Soc., 1991, 113, 9401–9402 CrossRef CAS.
- K. Okada, K. Okubo, N. Morita and M. Oda, Tetrahedron Lett., 1992, 33, 7377–7380 CrossRef CAS.
- (a) M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341–357 CrossRef CAS; (b) U. I. Gaya, Heterogeneous Photocatalysis Using Inorganic Semiconductor Solids, Springer, Netherlands, 2014 Search PubMed; (c) B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 5985–5992 CrossRef CAS; (d) M. Schiavello, Electrochim. Acta, 1993, 38, 11–14 CrossRef CAS; (e) X. Ning, S. Meng, X. Fu, X. Ye and S. Chen, Green Chem., 2016 10.1039/c6gc00572a; (f) Q. Wang, J. Li, Y. Bai, J. Lian, H. Huang, Z. Li, Z. Lei and W. Shangguan, Green Chem., 2014, 16, 2728–2735 RSC; (g) J. Yuan, J. Wen, Y. Zhong, X. Li, Y. Fang, S. Zhang and W. Liu, J. Mater. Chem., 2015, 3, 18244–18255 RSC; (h) J.-J. Zhou, R. Wang, X.-L. Liu, F.-M. Peng, C.-H. Li, F. Teng and Y.-P. Yuan, Appl. Surf. Sci., 2015, 346, 278–283 CrossRef CAS.
- (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC.
- (a) J. Hioe and H. Zipse, Org. Biomol. Chem., 2010, 8, 3609–3617 RSC; (b) M. L. Coote, C. Y. Lin and H. Zipse, The Stability of Carbon-Centered Radicals, John Wiley & Sons, 2009 Search PubMed; (c) J. S. Wright, H. Shadnia and L. L. Chepelev, J. Comput. Chem., 2009, 30, 1016–1026 CrossRef CAS PubMed.
- M. Majek, F. Filace and A. J. V. Wangelin, Beilstein J. Org. Chem., 2014, 10, 981–989 CrossRef CAS PubMed.
- (a) V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS; (b) G. J. Barbante, N. Kebede, C. M. Hindson, E. H. Doeven, E. M. Zammit, G. R. Hanson, C. F. Hogan and P. S. Francis, Chem. – Eur. J., 2014, 20, 14026–14031 CrossRef CAS PubMed.
- C. Creutz and N. Sutin, Inorg. Chem., 1976, 15, 496–499 CrossRef CAS.
- (a) D. M. Schultz and T. P. Yoon, Science, 2014, 343 Search PubMed; (b) K. Zeitler, Angew. Chem., Int. Ed., 2009, 121, 9969–9974 CrossRef; (c) J. Hu, J. Wang, T. H. Nguyen and N. Zheng, Beilstein J. Org. Chem., 2013, 9, 1977–2001 CrossRef PubMed.
- (a) A. U. Meyer, T. Slanina, C.-J. Yao and B. König, ACS Catal., 2015, 369–375 Search PubMed; (b) A. Penzkofer, A. Beidoun and M. Daiber, J. Lumin., 1992, 51, 297–314 CrossRef CAS.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of starting materials, spectroscopic investigations, analytical data and 1H- and 13C-NMR spectra. See DOI: 10.1039/c6gc01101b |
| This journal is © The Royal Society of Chemistry 2016 |