
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A dual-walled cage MOF as an efficient heterogeneous catalyst for the conversion of CO2 under mild and co-catalyst free conditions†
Yun-Hu
Han
ab,
Zhong-Yuan
Zhou
ac,
Chong-Bin
Tian
a and
Shao-Wu
Du
*a
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, P. R. China. E-mail: swdu@fjirsm.ac.cn; Fax: +86-591-83709470
bGraduate University of Chinese Academy of Sciences, Beijing 100039, P. R. China
cCollege of Chemistry, Fuzhou University, Fuzhou 350001, P. R. China
First published on 26th April 2016
Abstract
A novel 3D → 3D interpenetrated Zn-polyhedral MOF [Zn6(TATAB)4(DABCO)3(H2O)3]·12DMF·9H2O (1) (H3TATAB = 4,4′,4′′-s-triazine-1,3,5-triyl-tri-p-aminobenzoic acid, DABCO = 1,4-diazabicyclo[2.2.2]octane) based on a dual-walled icosahedral cage has been assembled by incorporating zinc paddlewheel units with nitrogen-rich tritopic carboxylate ligands. Owing to the high density of Lewis acid active sites and affinity to CO2 of the cage, this material exhibits an excellent catalytic efficiency in the cycloaddition of propylene oxide with CO2 into propylene carbonate under mild conditions (100 °C and ambient CO2 pressure) with a high yield of 99% over 16 h. Moreover, the catalytic reaction is environmentally friendly without any need for co-catalysts and solvents.
Introduction
The development of efficient, environmentally friendly catalysts for CO2 transformation into economically competitive products has been a longstanding goal for chemists, because CO2 is inexpensive and an abundant renewable C1 feedstock.1,4f However, CO2 is also a significant risk to the global climate. There are several possible approaches to reduce the levels of CO2 in the atmosphere which include the removal of CO2 from air or post-combustion of industrial point sources (i.e. flue-gas capture) and transformation of these into desirable products.2 A promising strategy for CO2 fixation is coupling with epoxides to form five-membered cyclic carbonates, which are currently used as valuable chemicals in numerous applications such as electrolyte components in lithium batteries, and intermediates for the production of plastics, pharmaceuticals, polar aprotic solvents, and fine chemicals.3 Although some homogeneous catalysts have been used for the formation of cyclic carbonates in industry under mild conditions, catalyst separation and disposal present both environmental and economic drawbacks.4 Meanwhile, heterogeneous catalysts e.g. metal oxides, zeolites, titanosilicates, and ion-exchanged resins have also been explored to catalyse the cycloaddition of CO2 with epoxides.5 However, those catalytic reactions are usually performed at high temperatures (>100 °C) and/or under CO2 pressures (>30 atm of CO2), which increase the cost of reaction processes and energy loss.5 Therefore, to explore high efficiency heterocatalysis for the coupling reactions of epoxides with CO2 under mild conditions is a priority to minimize cost and reduce environmental pollution.Metal–organic frameworks (MOFs), in particular those composed of high-connectivity metal–organic polyhedral cages have recently shown great potential for CO2 chemical conversion. Polyhedral MOFs have unique advantages including the available control of pore shape and dimensionality, tailored chemical environment and extra-large surface area.6 Most importantly, these materials contain confined nanospace that allows the facile access of substrates to the catalytically active sites located within the cage.7 Although several polyhedral MOFs have been evaluated as heterogeneous Lewis acid catalysts for chemical conversion of CO2, the catalytic processes usually require either high temperature and/or pressure to achieve good reaction yields,8 or the presence of co-catalysts such as nBu4NBr (TBAB), which is considered to be an environmentally unfriendly reagent.9 The underlying cause of this low activity could be the low density of Lewis acid active sites in these MOF materials. One way to overcome this is to increase the density of Lewis acid active sites by decorating the vertexes, and/or edges, and/or faces of polyhedral cages in MOFs with catalytically active centers.10 For example, Ma et al. have succeeded in increasing the density of catalytically active sites by the substitution of the facial ligand in the cage of MOF-505 with a metalloporphyrin ligand.10a The modified cuboctahedral cage shows high catalytic activity for the cycloaddition of CO2 and epoxide under ambient conditions, with twice the efficiency of its prototypal MOF-505.10a However, a co-catalyst TBAB was still needed in this case. Herein, we report a novel 3D → 3D interpenetrated Zn-polyhedral MOF, [Zn6(TATAB)4(DABCO)3(H2O)3]·12DMF·9H2O (1) (H3TATAB = 4,4′,4′′-s-triazine-1,3,5-triyl-tri-p-aminobenzoic acid, DABCO = 1,4-diazabicyclo[2.2.2]octane) based on a dual-walled Zn48 cage. The high density of Lewis acid active sites in the dual-walled cage and the enriched CO2-philic groups in the ligand enable 1 to function as an efficient catalyst for the cycloaddition of propylene oxide with CO2 into propylene carbonate under mild and co-catalyst free conditions.
Experimental section
General information
All reagents were commercially purchased and used without any further purification. The purity of all gases is 99.999%. TGA was performed on a TGA/NETZSCH STA449C instrument heated from 40 to 800 °C under a nitrogen atmosphere at a heating rate of 10 °C min−1. Powder X-ray diffraction was recorded on a PANalytical X'pert PRO X-ray diffraction system using Cu Kα radiation in the 2θ range of 5–50°. The Fourier transform infrared spectra using KBr pellets were collected on a Spectrum-One FT-IR spectrophotometer in the range of 4000–400 cm−1. Elemental analyses (C, H, N) were performed with an Elementar Vario EL III Analyser. Gas adsorption measurement was performed in the ASAP (Accelerated Surface Area and Porosimetry) 2020 System. Gas chromatography (GC) measurements were analysed using a GC-Smart (SHIMADZU) spectrometer and a flame ionization detector (FID). Products were analysed by Gas Chromatography-Mass Spectrometry (4000GC-MS, Varian-Agilent).Preparation of [Zn6(TATAB)4(DABCO)3(H2O)3]·12DMF·9H2O (1)
A mixture of Zn(NO3)2·6H2O (0.60 mmol, 178.20 mg), H3TATAB (0.45 mmol, 218.25 mg) and DABCO (0.3 mmol, 66.06 mg) was sealed in a 20 mL Teflon-lined stainless steel vessel with 6 mL of N,N′-dimethylformamide (DMF). The mixture was heated to 115 °C for 4 hours and maintained at this temperature for 3 days. Then the reaction system was cooled slowly to room temperature for another 3 days. The colorless cubic crystals of 1 were collected, washed with DMF and CH2Cl2 and then dried in air (yield 85% based on Zn(NO3)2·6H2O). Elemental analysis calcd (%) for 1 C150H204N42O48Zn6 (3755.79): C 47.97, H 5.47, N 15.66; found: C 47.89, H 5.22, N 15.45. FT-IR (KBr, cm−1) (see ESI Fig. S1†): 3289s, 1602s, 1490s, 1377vs, 1305s, 1244s, 1176s, 1053w, 1014w, 918w, 898w, 860s, 802s, 786s, 734w, 705w, 619w, 595w, 568w, 507w, 470w, 424w.Single-crystal X-ray diffraction study
Single-crystal X-ray diffraction data were collected on a Rigaku Diffractometer with a Mercury CCD area detector (Mo Kα: λ = 0.71073 Å) at room temperature. Crystal Clear software was used for data reduction and empirical absorption correction. The structure was solved by direct methods using SHELXTL and refined by full-matrix least-squares on F2 using SHELX-97 program.11a Metal atoms in the compound were located from the E-maps, and other non-hydrogen atoms were located in successive difference Fourier syntheses. All non-hydrogen atoms were refined anisotropically. The organic hydrogen atoms were positioned geometrically. The cavities were filled with highly disordered lattice solvent molecules that could not be completely mapped by single-crystal X-ray diffraction, which are often observed in high-symmetry structures.12 Thus the SQUEEZE routine of PLATON was applied to remove scattering contributions from solvent molecules. In addition, because some atoms (C1, C2) of DABCO lie in the special position, the atoms are in statistical distribution at a special position. The reported refinements are of the guest-free structure by the SQUEEZE routine.11b The final formula of 1 was derived from the squeeze result combined with the elemental analysis and TGA data. The elemental analysis suggests that there are 48 DMF and 36 H2O molecules located in the cavity per unit cell (2280 electrons) which is well consistent with the squeeze result (2303 electrons calculated using Platon Software). Also, the formula can be further supported by the TGA results (vide infra). Crystallographic data and other pertinent information for 1 are summarized in Table S2.† CCDC number for 1 is 1450479.General procedure for the cycloaddition of CO2 to cyclic carbonate catalysed by 1
In a 50 mL stainless-steel autoclave with a magnetic stirring bar in the absence of a solvent and a co-catalyst under CO2 pressure, with a catalyst of 0.21 or 0.42 mol% per paddlewheel unit (desolvation and dehydration under vacuum at 120 °C) and epoxides (20 mmol) were added. The reaction was carried out under 1 atm CO2 and at 100 °C for an appropriate time. The reuse experiments were carried out for the cycloaddition of propylene oxide with CO2 under similar conditions. The catalyst was retrieved by filtration, washed and soaked with DMF and CH2Cl2 (ca. 5 × 10 mL), and reactivated as mentioned above prior to being used for the next catalytic cycle. The products were monitored by using a GC-Smart spectrometer (DM-1 column, L × I. D. 30 m × 0.25 μm; injector temperature 220 °C) and identified by the comparison of GC retention times and mass spectra with those of the authentic samples. All the yields were based on epoxide.Results and discussion
Colorless cubic crystals of 1 were prepared under solvothermal conditions via the reaction of Zn(NO3)3·6H2O salt with the organic linkers H3TATAB and DABCO at 115 °C in DMF. A single crystal X-ray diffraction study reveals that 1 crystallizes in the highly symmetric cubic space group Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) . The asymmetry unit of 1 consists of two quarters of a Zn2+ ion, one-third of a TATAB3− ligand and one-fourth of a DABCO and a coordinated water molecule. It also contains 3/4 of DMF and 3/4 of water solvent molecules, as determined by CHN elemental analysis and the TGA results. Both the crystallographic independent Zn2+ ions adopt a square pyramidal coordination geometry with the basal plane being occupied by four carboxylate oxygen atoms and the apical position being taken by a nitrogen atom from DABCO in Zn1 or a coordinated water molecule in Zn2 (see ESI Fig. S2†). The Zn1 and Zn2 atoms are bridged by four bridging carboxylate groups from four different TATAB3− ligands, forming a {Zn2(O2C)4} paddlewheel secondary building unit (SBU) with a short Zn–Zn distance of 2.94 Å.
. The asymmetry unit of 1 consists of two quarters of a Zn2+ ion, one-third of a TATAB3− ligand and one-fourth of a DABCO and a coordinated water molecule. It also contains 3/4 of DMF and 3/4 of water solvent molecules, as determined by CHN elemental analysis and the TGA results. Both the crystallographic independent Zn2+ ions adopt a square pyramidal coordination geometry with the basal plane being occupied by four carboxylate oxygen atoms and the apical position being taken by a nitrogen atom from DABCO in Zn1 or a coordinated water molecule in Zn2 (see ESI Fig. S2†). The Zn1 and Zn2 atoms are bridged by four bridging carboxylate groups from four different TATAB3− ligands, forming a {Zn2(O2C)4} paddlewheel secondary building unit (SBU) with a short Zn–Zn distance of 2.94 Å.
The most fascinating topological feature of 1 is the dual-walled cage motif, in which one cage is encapsulated by another cage (Fig. 1c), a result from the interpenetration of two independent but identical 3D cage nets. Compound 1 is built from an icosahedral Zn24 cage (denoted as Zn24-A) where the paddlewheel {Zn2(O2C)4} dimer residue at its twelve vertexes and TATAB3− ligands occupy eight of the twenty trigonal planes (Fig. 1a). The cubic packing of non-space-filling icosahedral cages through face- and edge-sharing creates another Zn24 cage (denoted as Zn24-B) in the gap of each of the eight neighbouring Zn24-A cages. Compared to Zn24-A, the Zn24-B cage is also defined by twelve {Zn2(O2C)4} dimers and eight TATAB3− ligands, except for the six pairs of adjacent {Zn2(O2C)4} vertices, each of which is further connected by an additional DABCO ligand, resulting in a highly distorted icosahedral cage (Fig. 1b).
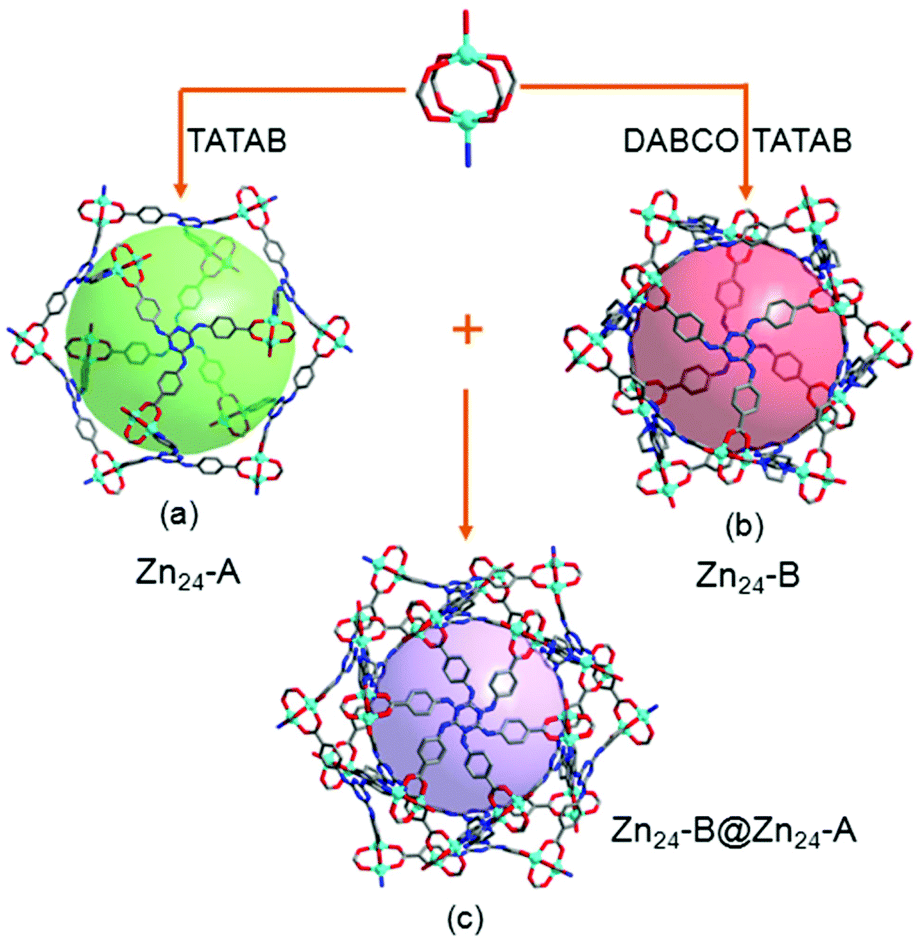 | ||
| Fig. 1 (a) View of the Zn24-A cage (the distance between opposite vertices is ca. 34.4 Å). (b) View of the Zn24-cage (the distance between opposite vertices is ca. 30.2 Å). (c) View of the dual-walled cage motif of 1. | ||
Further analysing the structure of 1 reveals that these two types of cages are alternatively arranged to form a 3D network in which each cage of one type is encircled by eight cages of the other type and vice versa (Fig. 2a and b). Two identical 3D cage networks are interpenetrated in such a complicated way that every Zn24-B cage in one net is completely encapsulated by a Zn24-A cage of the other net and vice versa, resulting in a 3D framework with double-walled cages (Fig. 2c). Remarkably, each Zn24-A cage in one net is interlaced with eight Zn24-A cages belonging to the other net through all its ligand-decorated trigonal planes, that is, each ligand defined plane of one Zn24-A cage is interlocked by another Zn24-A cage with face-to-face π–π interactions between the central cores of TATAB3− ligands stacked in an eclipsed fashion (centroid-to-centroid distance: 3.412). The propagation of octuplet polycatenation leads to a 3D extended polyhedral architecture of 1, which is one of the rare examples of 3D polycatenanes by mechanically linking coordination cages,13 and is the second example that also features a cage-within-cage motif.13f
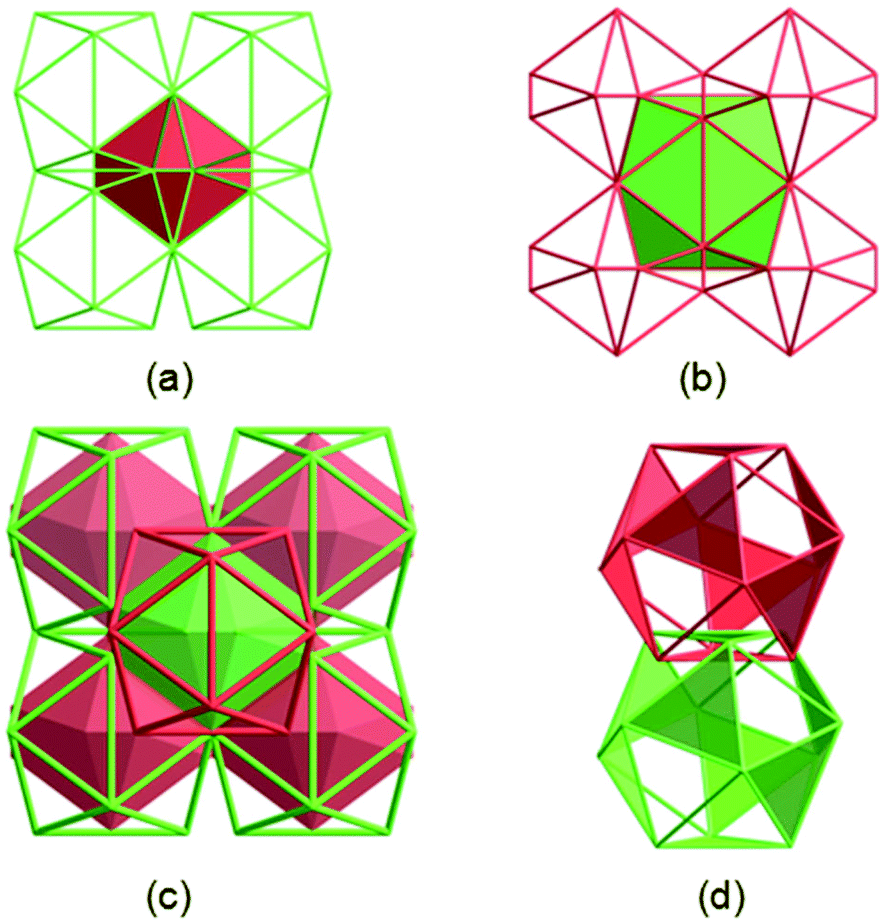 | ||
| Fig. 2 (a) View of the Zn24-B (pink polyhedron) generated by eight Zn24-A cages. The other four Zn24-A cages behind are not shown. (b) View of the Zn24-A cage generated by eight Zn24-B cages. The other four Zn24-B cages behind are not shown. (c) The interpenetration of two identical nets of 1 showing a cage-within-cage motif. (d) One of the octuplet catenations showing the interlocking between two Zn24-A coming from different nets. | ||
In 1, the dual-walled cage is ca. 3.2 nm in diameter and each contains 48 Zn2+ Lewis acid catalytic sites and 48 –NH– Lewis base sites that are able to interact with CO2. The combination of high density of catalytic sites and CO2-philic groups in 1 may guarantee an outstanding CO2 chemical fixation performance. Removal of the guest molecules reveals that the effective free volume of 1, calculated by PLATON analysis, is 64.5% of the crystal volume (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 175.9 Å3 of the 23534.6 Å3 unit cell volume). The phase purity of 1 was checked by powder XRD and recorded at room temperature (see ESI Fig. S3†). The peak positions of the simulated pattern closely match those of the experimental ones, indicating phase purity of the as-synthesized sample. The TGA and variable temperature XRD measurements were performed to study the thermal stability of 1. The TGA curve of 1 shows a continuous weight loss without an obvious plateau from 40 to 500 °C (see ESI Fig. S4†). After removing the lattice solvents by treating 1 under vacuum at 150 °C for 24 hours, the TGA curve of the desolvated sample shows almost no weight loss from 0 to ca. 300 °C. Thus, the weight loss of 27.1% from 0 to 300 °C corresponds to the loss of lattice DMF and water molecules (calcd 27.6%). The variable temperature XRD experiment also indicates that 1 is stable up to ca. 200 °C, above this temperature, the framework starts to decompose (see ESI Fig. S3†).
175.9 Å3 of the 23534.6 Å3 unit cell volume). The phase purity of 1 was checked by powder XRD and recorded at room temperature (see ESI Fig. S3†). The peak positions of the simulated pattern closely match those of the experimental ones, indicating phase purity of the as-synthesized sample. The TGA and variable temperature XRD measurements were performed to study the thermal stability of 1. The TGA curve of 1 shows a continuous weight loss without an obvious plateau from 40 to 500 °C (see ESI Fig. S4†). After removing the lattice solvents by treating 1 under vacuum at 150 °C for 24 hours, the TGA curve of the desolvated sample shows almost no weight loss from 0 to ca. 300 °C. Thus, the weight loss of 27.1% from 0 to 300 °C corresponds to the loss of lattice DMF and water molecules (calcd 27.6%). The variable temperature XRD experiment also indicates that 1 is stable up to ca. 200 °C, above this temperature, the framework starts to decompose (see ESI Fig. S3†).
The permanent porosity of 1 was evaluated by measuring N2 adsorption isotherms at 77 K using activated materials. The resulting N2 isotherms exhibited typical type I behaviour, indicative of microporosity (see ESI Fig. S5†). From the isotherm, the BET/Langmuir surface area for 1 was estimated to be 61.4/87.4 m2 g−1. Single-component gas adsorption isotherms (CO2 and N2) were measured at 273 K for 1. Compound 1 displays a high CO2 capacity (49.1 cm3 g−1) at 769 mmHg and 273 K (see ESI Fig. S6†). This is in contrast to the N2 uptake capacity observed for 1 (5.3 cm3 g−1) at the same temperature and under the same pressure. Furthermore, the initial uptake in the low-pressure region of the CO2 isotherm at 273 K is much steeper than that observed for N2. These results indicate the high affinity of the MOF framework to CO2, which demonstrates the potential of the MOF for CO2 physisorption and CO2/N2 separation. The high CO2 uptake of 1 is due to the strong hydrogen bonding between the NH group of the TATAB3− ligand and the CO2 molecule.14
Given that 1 possesses high density of Lewis acid sites and excellent affinity to CO2, we decided to investigate the catalytic capability of 1 as a Lewis acid catalyst for the cycloaddition of CO2 with epoxides to form cyclic carbonates. As shown in Table 1, compound 1 demonstrates highly efficient catalytic activity for cycloaddition of propylene oxide with CO2 into propylene carbonate at 100 °C under 1 atm CO2 pressure with a yield of 99% over 16 h without any co-catalyst (Table 1, entry 1). At 10 atm CO2 pressure, the reaction completes in only one hour but finishes in six hours if half the amount of catalyst is used (Table 1, entries 2 and 3). In view of the fact that 1 has a high affinity for CO2 and a low affinity for N2, we also carried out the catalytic reaction under simulated dry flue-gas conditions. When the pressure ratio of N2 and CO2 is 8/2 and the total pressure is 1 atm, the yield of propylene carbonate is 28% (Table 1, entry 4). But when the pressure ratio of N2 and CO2 is 2/8, the yield of propylene carbonate is 67% (Table 1, entry 5). These results indicate again that 1 can selectively absorb CO2 over N2 and the presence of N2 has little interference in the chemical fixation of CO2. In the absence of compound 1, no propylene carbonate was found (Table 1, entry 6). Significantly, the catalytic reaction is environmentally friendly as no co-catalyst and solvent were needed and the reaction was carried out under milder conditions compared to other MOF systems (Table S1†).
| Entry | Catalyst (amt (mol%)) | T (°C) | P (atm) | t (h) | Conv. (%) | Yieldb (%, based on epoxide) |
|---|---|---|---|---|---|---|
| a Reaction conditions: epoxide (20.0 mmol) with a catalyst per paddlewheel unit. b The yields were determined by GC with n-butanol as an internal standard. | ||||||
| 1 | 1 (0.42) | 100 | 1 | 16 | 100 | 99 |
| 2 | 1 (0.42) | 100 | 10 | 1 | 100 | 99 |
| 3 | 1 (0.21) | 100 | 10 | 6 | 99 | 98 |
| 4 | 1 (0.42) | 100 | 1 (PN2/PCO2 = 8/2) | 16 | 30 | 28 |
| 5 | 1 (0.42) | 100 | 1 (PN2/PCO2 = 2/8) | 16 | 70 | 67 |
| 6 | Blank | 100 | 1 | 16 | — | — |
The excellent catalytic activity of 1 for the chemical fixation of CO2 encouraged us to further explore the generality of this catalytic system. Chemical fixation of CO2 with epoxides substituted with different functional groups under similar conditions was investigated (Table 2). A high catalytic activity was observed for the cycloaddition of epichlorohydrin and butylene oxide with CO2 into butylene carbonates at 100 °C and 1 atm CO2 pressure with yields of 95% and 91%, respectively over 16 h (Table 2, entries 2 and 3). Even for the large styrene oxide, the yield of the cycloaddition is still up to 89% (Table 2, entry 4). However, further increasing the size of epoxide substrates resulted in a dramatic decrease in the yield of cyclic carbonates. The yields of cycloaddition dropped down to 50% (Table 2, entry 5) and 8% (Table 2, entry 6) for benzyl phenyl glycidyl ether and trans-2,3-diphenylethylene oxide. It is speculated that the limited diffusion of the large-sized epoxide substrate into the void space within the dual-walled cage of 1 is responsible for these low yields.13e,15,16
| Entry | Epoxides | Products | Conv. (%) | Yieldb (%, based on epoxides) | TONc |
|---|---|---|---|---|---|
| a Reaction conditions: epoxides (20.0 mmol), 1 (0.42 mol% per paddlewheel unit), CO2 (1 atm), 100 °C and 16 hours. b The yields were determined by GC with n-butanol as an internal standard. c TON = moles of aimed product/moles of active metal sites. | |||||
| 1 |
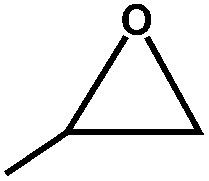
|

|
100 | 99 | 246 |
| 2 |
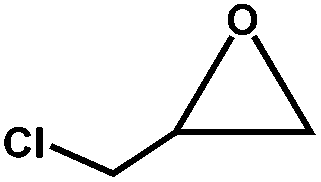
|
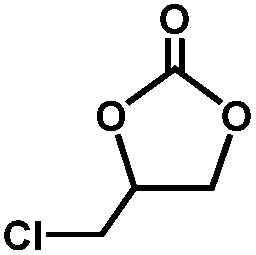
|
98 | 95 | 158 |
| 3 |
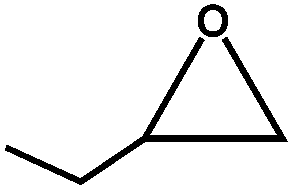
|
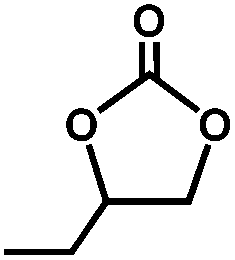
|
92 | 91 | 152 |
| 4 |

|
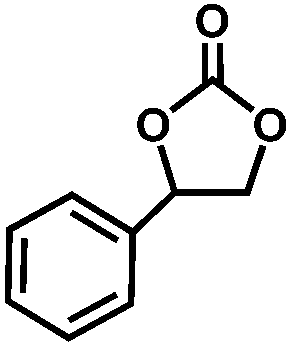
|
90 | 89 | 148 |
| 5 |

|
|
50 | 50 | 83 |
| 6 |
|
|
8 | 8 | 13 |
In addition to high catalytic activity, reusability and stability are also very important for heterogeneous catalytic systems. The recycling experiments were conducted using 1 as a catalyst in the cycloaddition of propylene oxide with CO2 under similar conditions. The catalyst after one cycle can be easily recovered by centrifugation and subsequently used in the successive runs for six cycles without an appreciable loss of its catalytic performance (Fig. 3). A 96% yield of cyclic carbonate was obtained in the sixth run. The powder XRD of the recovered catalyst was identical to that of the freshly prepared sample, indicating the stability of 1 during the catalytic reaction (see ESI Fig. S7†). The reusability of 1 was also tested through recycle reaction kinetics for the first and second cycle (see ESI Fig. S8†). In both reaction cycles, similar activities are clearly exhibited. To confirm the heterogeneous nature of 1, the catalytic reaction was stopped after 3 hours and the reaction mixture was filtered while still hot to remove the catalyst. The reaction was then continued for another 13 hours, during which the yield of propylene carbonate remained unchanged (see ESI Fig. S8†).
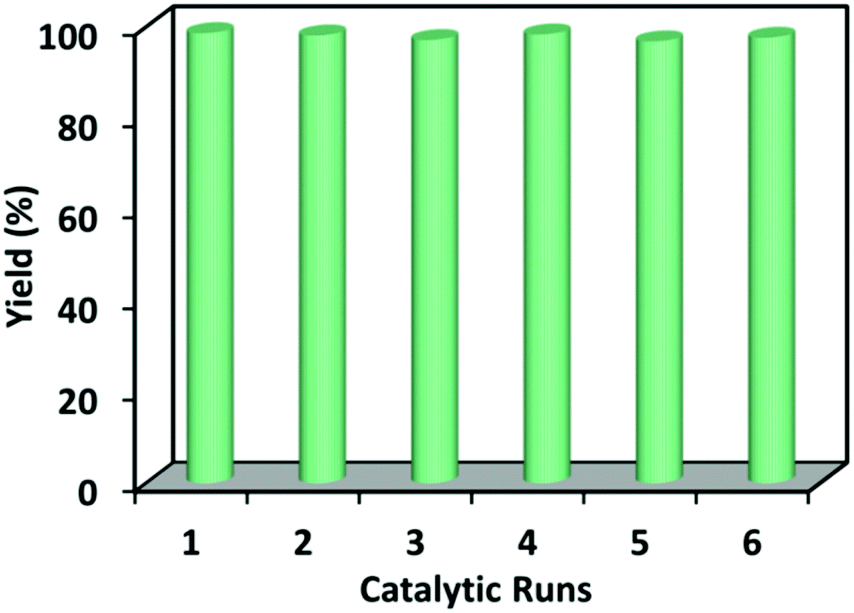 | ||
| Fig. 3 The catalytic cycles of compound 1 for the cycloaddition of CO2 and propylene epoxide reaction. | ||
Besides the high density of Lewis acid active sites, the good catalytic activity of 1 may also be attributed to the existence of amine-functionalized ligands. Previous research has shown that the introduction of Lewis basic functional groups such as amines into MOFs can not only increase the CO2 affinity but also improve the carbonate yields due to the synergistic effect of the Lewis acid–base pair.17 Accordingly, a tentative mechanism can be proposed for the cycloaddition of epoxide and CO2 into cyclic carbonate catalysed by 1 bearing an acid–base pair. First, CO2 is activated at the basic amine sites forming a carbamate species, which then reacts with epoxides that are adsorbed on the adjacent Zn2+ Lewis acidic sites to form ring opening intermediates. Second, ring-closure through an intramolecular nucleophilic attack by the oxyanion at the carbon center of CO2 occurs to generate a cyclocarbonate.
Conclusions
In summary, we have synthesized a 3D → 3D interpenetrated polyhedral MOF featuring a fascinating cage-within-cage motif and a rarely observed octuplet polycatenation. Due to the high density of Lewis acid sites and the excellent affinity of the cage to CO2, compound 1 exhibits good performance for the cycloaddition of propylene oxide with CO2 into propylene carbonate at mild temperature and under 1 atm CO2 pressure with a yield of 99% over 16 h. Furthermore, it is the first MOF that is able to catalyse cycloaddition of CO2 to epoxides under mild and solvent-free conditions without the help of any co-catalyst.Acknowledgements
We thank the National Basic Research Program of China (973 Program, 2012CB821702), the National Natural Science Foundation of China (21233009 and 21571175) and the State Key Laboratory of Structural Chemistry, the Fujian Institute of Research on the Structure of Matter, and the Chinese Academy of Sciences for financial support.Notes and references
- (a) M. Ed. Aresta, Carbon Dioxide as Chemical Feedstock, WileyVCH, Weinheim, Germany, 2010 Search PubMed; (b) I. Omae, Catal. Today, 2006, 115, 33–52 CrossRef CAS.
- (a) A. Goeppert, M. Czaun, G. K. S. Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833–7853 RSC; (b) A. Kumar, D. G. Madden, M. Lusi, K. J. Chen, E. A. Daniels, T. Curtin, J. J. Perry IV and M. J. Zaworotko, Angew. Chem., Int. Ed., 2015, 54, 14372–14377 CrossRef CAS PubMed; (c) X. C. Kang, Q. G. Zhu, X. F. Sun, J. Y. Hu, J. L. Zhang, Z. M. Liu and B. X. Han, Chem. Sci., 2016, 7, 266–273 RSC; (d) Z. H. He, Q. L. Qian, J. Ma, Q. L. Meng, H. C. Zhou, J. L. Song, Z. M. Liu and B. X. Han, Angew. Chem., Int. Ed., 2016, 55, 737–741 CrossRef CAS PubMed.
- (a) M. Yoshida and M. Ihara, Chem. – Eur. J., 2004, 10, 2886–2893 CrossRef CAS PubMed; (b) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed; (c) J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adan, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228–1231 CrossRef PubMed; (d) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC; (e) J. Ma, J. L. Song, H. Z. Liu, Z. F. Zhang, T. Jiang, H. L. Fan and B. X. Han, Green Chem., 2012, 14, 1743–1748 RSC; (f) P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC; (g) T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489–4491 RSC; (h) B. Schaffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef CAS PubMed.
- (a) A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS PubMed; (b) M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS PubMed; (c) X. B. Lu, B. Liang, Y. J. Zhang, Y. Z. Tian, Y. M. Wang, C. X. Bai, H. Wang and R. Zhang, J. Am. Chem. Soc., 2004, 126, 3732–3733 CrossRef CAS PubMed; (d) M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC; (e) H. Yasuda, L. N. He, T. Sakakura and C. Hu, J. Catal., 2005, 233, 119–122 CrossRef CAS; (f) T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed; (g) M. R. Kember, A. Buchhard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC; (h) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC.
- (a) K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526–4527 CrossRef CAS; (b) E. J. Doskocil, S. V. Bordawekar, B. C. Kaye and R. J. Davis, J. Phys. Chem. B, 1999, 103, 6277–6282 CrossRef CAS; (c) T. Yano, H. Matsui, T. Koike, H. Ishiguro, H. Fujihara, M. Yoshihara and T. Maeshima, Chem. Commun., 1997, 1129–1130 RSC; (d) H. Yasuda, L. N. He and T. Sakakura, J. Catal., 2002, 209, 547–550 CrossRef CAS; (e) Y. Xie, T. T. Wang, X. H. Liu, K. Zou and W. Q. Deng, Nat. Commun., 2013, 4, 1960–1966 Search PubMed.
- (a) B. Wang, A. P. Côté, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nature, 2008, 453, 207–211 CrossRef CAS PubMed; (b) H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ő. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed; (c) Y. B. He, W. Zhou, G. D. Qian and B. L. Chen, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC; (d) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC; (e) D. P. Broom and K. M. Thomas, MRS Bull., 2013, 38, 412–421 CrossRef CAS; (f) W. M. Xuan, C. F. Zhu, Y. Liu and Y. Cui, Chem. Soc. Rev., 2012, 41, 1677–1695 RSC.
- (a) W. Y. Gao and S. Q. Ma, Comments Inorg. Chem., 2014, 34, 125–141 CrossRef CAS; (b) J. W. Liu, L. F. Chen, H. Cui, J. Y. Zhang, L. Zhang and C. Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- (a) A. C. Kathalikkattil, D. W. Kim, J. Tharun, H. G. Soek, R. Roshan and D. W. Park, Green Chem., 2014, 16, 1607–1616 RSC; (b) D. A. Yang, H. Y. Cho, J. Kim, S. T. Yang and W. S. Ahn, Energy Environ. Sci., 2012, 5, 6465–6473 RSC; (c) W. Y. Gao, L. Wojtas and S. Q. Ma, Chem. Commun., 2014, 50, 5316–5318 RSC; (d) B. Zou, L. Hao, L. Y. Fan, Z. M. Gao, S. L. Chen, H. Li and C. W. Hu, J. Catal., 2015, 329, 119–129 CrossRef CAS; (e) Z. Zhou, C. He, J. H. Xiu, L. Yang and C. Y. Duan, J. Am. Chem. Soc., 2015, 137, 15066–15069 CrossRef CAS PubMed.
- (a) X. Q. Huang, Y. F. Chen, Z. G. Lin, X. Q. Ren, Y. N. Song, Z. Z. Xu, X. M. Dong, X. G. Li, C. W. Hu and B. Wang, Chem. Commun., 2014, 50, 2624–2627 RSC; (b) D. X. Ma, B. Y. Li, K. Liu, X. L. Zhang, W. J. Zou, Y. Q. Yang, G. H. Li, Z. Shi and S. H. Feng, J. Mater. Chem. A, 2015, 3, 23136–23142 RSC; (c) J. L. Song, Z. F. Zhang, S. Q. Hu, T. B. Wu, T. Jiang and B. X. Han, Green Chem., 2009, 11, 1031–1036 RSC; (d) O. V. Zalomaeva, A. M. Chibiryaev, K. A. Kovalenko, O. A. Kholdeeva, B. S. Balzhnimaev and V. P. Fedin, J. Catal., 2013, 298, 179–185 CrossRef CAS; (e) W. Kleist, F. Jutz, M. Maciejewski and A. Baiker, Eur. J. Inorg. Chem., 2009, 2009, 3552–3561 CrossRef; (f) H. Y. Cho, D. A. Yang, J. Kim, S. Y. Jeong and W. S. Ahn, Catal. Today, 2012, 185, 35–40 CrossRef CAS; (g) X. Zhou, Y. Zhang, X. G. Yang, L. Z. Zhao and G. Y. Wang, J. Mol. Catal. A: Chem., 2012, 361–362, 12–16 CrossRef CAS; (h) C. M. Miralda, E. E. Macias, M. Q. Zhu, P. Ratnasamy and M. A. Carreon, ACS Catal., 2012, 2, 180–183 CrossRef CAS.
- (a) W. Y. Gao, Y. Chen, Y. H. Niu, K. Williams, L. Cash, P. J. Perez, L. Wojtas, J. F. Cai, Y. S. Chen and S. Q. Ma, Angew. Chem., Int. Ed., 2014, 126, 2653–2657 CrossRef; (b) G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed; (c) J. J. Perry IV, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400–1417 RSC; (d) S. T. Zheng, T. Wu, B. Irfanoglu, F. Zuo, P. Y. Feng and X. H. Bu, Angew. Chem., 2011, 123, 8184–8187 ( Angew. Chem., Int. Ed. , 2011 , 50 , 8034–8037 ) CrossRef.
- (a) G. M. Sheldrick, SHELXL-97 Program for X-ray Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed; (b) A. L. Spek, PLATON-97, University of Utrecht, Utrecht, The Netherlands, 1997 Search PubMed.
- (a) S. T. Zheng, T. Wu, F. Zuo, C. S. Chou, P. Y. Feng and X. H. Bu, J. Am. Chem. Soc., 2012, 134, 1934–1937 CrossRef CAS PubMed; (b) X. S. Wang, L. Meng, Q. G. Cheng, C. Kim, L. Wojtas, M. Chrzanowski, Y. S. Chen, X. P. Zhang and S. Q. Ma, J. Am. Chem. Soc., 2011, 133, 16322–16325 CrossRef CAS PubMed.
- (a) X. Kuang, X. Wu, R. Yu, J. P. Donahue, J. Huang and C. Z. Lu, Nat. Chem., 2010, 2, 461–465 CrossRef CAS PubMed; (b) J. Heine, J. Schmedt auf der Günne and S. Dehnen, J. Am. Chem. Soc., 2011, 133, 10018–10021 CrossRef CAS PubMed; (c) L. Jiang, P. Ju, X. R. Meng, X. J. Kuang and T. B. Lu, Sci. Rep., 2012, 2, 668–772 Search PubMed; (d) S. Ma, X. S. Wang, D. Yuan and H. C. Zhou, Angew. Chem., Int. Ed., 2008, 47, 4130–4133 CrossRef CAS PubMed; (e) Y. Shen, H. B. Zhu, J. Hu and Y. Zhao, CrystEngComm, 2015, 17, 2080–2082 RSC; (f) Y. H. Han, C. B. Tian, P. Lin and S. W. Du, J. Mater. Chem. A, 2015, 3, 24525–24531 RSC.
- (a) A. Khutia and C. Janiak, Dalton Trans., 2014, 43, 1338–1347 RSC; (b) N. Planas, A. L. Dzubak, R. Poloni, L. C. Lin, A. McManus, T. M. McDonald, J. B. Neaton, J. R. Rong, B. Smit and L. Gagliardi, J. Am. Chem. Soc., 2013, 135, 7402–7405 CrossRef CAS PubMed; (c) S. Pal, A. Bhunia, P. P. Jana, S. Dey, J. Möllmer, C. Janiak and H. P. Nayek, Chem. – Eur. J., 2015, 21, 2789–2792 CrossRef CAS PubMed.
- (a) N. W. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176–182 CrossRef CAS PubMed.
- Y. Chen, V. Lykourinou, T. Hoang, L. J. Ming and S. Ma, Inorg. Chem., 2012, 51, 9156–9158 CrossRef CAS PubMed.
- (a) T. Lescouet, C. Chizallet and D. Farrusseng, ChemCatChem, 2012, 4, 1725–1728 CrossRef CAS; (b) Y. J. Kim and D. W. Park, J. Nanosci. Nanotechnol., 2013, 13, 2307–2312 CrossRef CAS PubMed; (c) R. Srivastava, D. Srinivas and P. Ratnasamy, Microporous Mesoporous Mater., 2006, 90, 314–326 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystallographic data, additional figures, PXRD, TGAs, and FT-IR. CCDC 1450479. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c6gc00413j |
| This journal is © The Royal Society of Chemistry 2016 |