 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assessing chemistry schemes and constraints in air quality models used to predict ozone in London against the detailed Master Chemical Mechanism
Tamsin L.
Malkin
a,
Dwayne E.
Heard
ab,
Christina
Hood
c,
Jenny
Stocker
c,
David
Carruthers
c,
Ian A.
MacKenzie
d,
Ruth M.
Doherty
d,
Massimo
Vieno
e,
James
Lee
fg,
Jörg
Kleffmann
h,
Sebastian
Laufs
h and
Lisa K.
Whalley
*ab
aSchool of Chemistry, University of Leeds, Leeds, UK. E-mail: L.K.Whalley@leeds.ac.uk
bNational Centre for Atmospheric Science, University of Leeds, Leeds, UK
cCambridge Environmental Research Consultants, 3 King's Parade, Cambridge, UK
dSchool of GeoSciences, University of Edinburgh, Edinburgh, UK
eCentre for Ecology & Hydrology, Bush Estate, Penicuik, UK
fDepartment of Chemistry, University of York, York, UK
gNational Centre for Atmospheric Science, Wolfson Atmospheric Chemistry Laboratory, York, UK
hPhysikalische und Theoretische Chemie/Fakultät Mathematik und Naturwissenschaften, Bergische Universität Wuppertal (BUW), Wuppertal, Germany
First published on 11th February 2016
Abstract
Air pollution is the environmental factor with the greatest impact on human health in Europe. Understanding the key processes driving air quality across the relevant spatial scales, especially during pollution exceedances and episodes, is essential to provide effective predictions for both policymakers and the public. It is particularly important for policy regulators to understand the drivers of local air quality that can be regulated by national policies versus the contribution from regional pollution transported from mainland Europe or elsewhere. One of the main objectives of the Coupled Urban and Regional processes: Effects on AIR quality (CUREAIR) project is to determine local and regional contributions to ozone events. A detailed zero-dimensional (0-D) box model run with the Master Chemical Mechanism (MCMv3.2) is used as the benchmark model against which the less explicit chemistry mechanisms of the Generic Reaction Set (GRS) and the Common Representative Intermediates (CRIv2-R5) schemes are evaluated. GRS and CRI are used by the Atmospheric Dispersion Modelling System (ADMS-Urban) and the regional chemistry transport model EMEP4UK, respectively. The MCM model uses a near-explicit chemical scheme for the oxidation of volatile organic compounds (VOCs) and is constrained to observations of VOCs, NOx, CO, HONO (nitrous acid), photolysis frequencies and meteorological parameters measured during the ClearfLo (Clean Air for London) campaign. The sensitivity of the less explicit chemistry schemes to different model inputs has been investigated: Constraining GRS to the total VOC observed during ClearfLo as opposed to VOC derived from ADMS-Urban dispersion calculations, including emissions and background concentrations, led to a significant increase (674% during winter) in modelled ozone. The inclusion of HONO chemistry in this mechanism, particularly during wintertime when other radical sources are limited, led to substantial increases in the ozone levels predicted (223%). When the GRS and CRIv2-R5 schemes are run with the equivalent model constraints to the MCM, they are able to reproduce the level of ozone predicted by the near-explicit MCM to within 40% and 20% respectively for the majority of the time. An exception to this trend was observed during pollution episodes experienced in the summer, when anticyclonic conditions favoured increased temperatures and elevated O3. The in situ O3 predicted by the MCM was heavily influenced by biogenic VOCs during these conditions and the low GRS [O3]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MCM [O3] ratio (and low CRIv2-R5 [O3]:MCM [O3] ratio) demonstrates that these less explicit schemes under-represent the full O3 creation potential of these VOCs. To fully assess the influence of the in situ O3 generated from local emissions versus O3 generated upwind of London and advected in, the time since emission (and, hence, how far the real atmosphere is from steady state) must be determined. From estimates of the mean transport time determined from the NOx:NOy ratio observed at North Kensington during the summer and comparison of the O3 predicted by the MCM model after this time, ∼60% of the median observed [O3] could be generated from local emissions. During the warmer conditions experienced during the easterly flows, however, the observed [O3] may be even more heavily influenced by London's emissions.
:MCM [O3] ratio (and low CRIv2-R5 [O3]:MCM [O3] ratio) demonstrates that these less explicit schemes under-represent the full O3 creation potential of these VOCs. To fully assess the influence of the in situ O3 generated from local emissions versus O3 generated upwind of London and advected in, the time since emission (and, hence, how far the real atmosphere is from steady state) must be determined. From estimates of the mean transport time determined from the NOx:NOy ratio observed at North Kensington during the summer and comparison of the O3 predicted by the MCM model after this time, ∼60% of the median observed [O3] could be generated from local emissions. During the warmer conditions experienced during the easterly flows, however, the observed [O3] may be even more heavily influenced by London's emissions.
1. Introduction
Air pollution is the environmental factor with the greatest impact on human health in Europe1. More than 65% of the population in Europe live in cities where levels of particulate matter (PM) exceed the WHO Air Quality Guidelines, and when considering exceedances in the secondary pollutant ozone (O3), this figure rises to 95%.1,2 PM2.5 (particulate matter less than 2.5 micrometres in diameter) and O3 are estimated to contribute to 458000 and 17400 premature deaths each year across Europe, respectively.1 Both PM and O3 concentrations are influenced by many factors including local emissions, chemistry and regional advection.3 Understanding the key processes driving air quality across the relevant spatial and temporal scales, especially during pollution episodes, is necessary to inform both policymakers and the public. For the development of effective mitigation policies, it is important to isolate the contribution of local emissions and production during peak events from that due to long-range transport of regional pollution.
Air pollution events in the UK are generally associated with stagnation events, which can occur at any time, but which in summer may be coincident with heatwaves.4 Large contributions of European pollution to the UK are often associated with slow-moving easterly air masses.3,5 Several studies have highlighted the downward entrainment of O3-rich air from Europe to ground level in the UK during the heatwave of 2003.6–8 Interactions between the weather conditions and chemistry processes can also contribute to elevated O3, for example, the role of enhanced biogenic emissions in the production of ozone in urban plumes in the UK was first considered by MacKenzie et al.9 Other processes including reduced dry deposition, and extensive forest fires in mainland Europe during heatwaves, can also influence UK O3.5–8
There remains some debate in the analysis of surface ozone levels as to the origin of the ozone and in particular the role of long-range transport.10 Coupled modelling studies allow regional and local processes affecting air quality across the UK and, specifically, in London to be examined. Recent intensive measurements from the NERC ClearfLo (Clean Air for London) and REPARTEE (Regent's Park and Tower Environmental Experiment) campaigns provide an opportunity to combine box models and measurements to gain an insight into the relative contributions of locally generated and transported O3. During the winter campaigns of REPARTEE (October 2006 and October/November 2007),11 O3 concentrations recorded at the BT Tower were always higher than at surface sites, consistent with the city acting as an efficient chemical sink for regional O3, which was confirmed by strong downward fluxes of O3 measured on the BT Tower.11
The ClearfLo project was a measurement programme in and around London lasting 2 years (2011–2012) including two month-long intensive observation periods (IOPs) in the summer and wintertime.3 The summer IOP, which coincided with the London 2012 Olympics, provides the opportunity to investigate factors controlling O3 when photochemical activity is high, in contrast to the earlier REPARTEE campaign which took place in winter. Understanding O3 production in summer is critical as this is when the exceedances typically occur. During the summer IOP, a number of high pollution events were observed where meteorological conditions favoured sustained, elevated O3 levels (peaking at ∼100 ppb3). In addition to the high O3 levels recorded during the summer IOP, much lower O3 concentrations were recorded during the winter IOP (January–February, 2012), with a peak concentration of ∼40 ppb recorded.
One of the main objectives of the Coupled Urban and Regional processes: Effects on AIR quality (CUREAIR) project is to determine local and regional contributions to ozone (O3) events. This paper presents a comparison of O3 concentrations predicted to be produced locally in London (both during the summertime and wintertime) using three different chemistry schemes. The level of chemical complexity between the three schemes investigated varies greatly from (1) a simple 7 step reaction scheme (the generic reaction scheme – GRS) which is used in the Atmospheric Dispersion Modelling System (ADMS-Urban)12 to (2) a lumped, 609 reaction, chemistry scheme (Common Representative Intermediates – CRI) used in some versions of the EMEP4UK regional transport model to (3) a near-explicit, 16940 reaction, chemistry scheme (the Master Chemical Mechanism – MCMv3.2) used in this paper as the benchmark scheme against which the smaller chemistry schemes are assessed.
The sensitivity of simulated O3 produced by the chemistry schemes to a variety of influences including the effects of VOC and NOx concentrations, and the photolysis frequency of NO2, as well as the impact of HONO are presented. Comparison of the O3 simulated by the three schemes when equivalent constraints are used is also presented.
This paper first describes the model methodology and is then divided into 4 results sections: (1) investigating the impact of VOCs, NOx, NO2 photolysis frequency and HONO during the summer ClearfLo IOP on the O3 concentration predicted by the GRS scheme, (2) investigating the influence of HONO and NOx during the summer ClearfLo IOP on the O3 concentration predicted by the CRIv2-R5 scheme, and (3) and (4) investigating the same impacts as (1) and (2) respectively but for the winter ClearfLo IOP. The paper then concludes with a summary of the results and discussion of the influence of chemistry and transport on O3 production in London.
2. Methodology
Models of atmospheric composition typically fall into one of several categories according to the spatial coverage (global, regional, urban, street canyon, single point (box)). Urban-scale dispersion models allow simulation at roadsides and hotspots but rely on background observations for their initialisation conditions whereas regional chemistry-climate models can be used for future climate simulations but have insufficient spatial resolution for direct comparison with urban monitoring sites. A combination of these two modelling strategies can be used to study regional and local drivers of urban air quality. In addition, chemical box models can provide a detailed representation of the chemical environment and may be used for evaluation of simpler chemical oxidation schemes represented in regional and urban-scale models.2.1 Master Chemical Mechanism
The Master Chemical Mechanism (MCMv3.2)13,14 is a near-explicit chemical mechanism which describes the detailed gas-phase chemical processes involved in the tropospheric degradation of a series of primary emitted VOCs. The entire MCM treats the degradation of methane and 142 non-methane VOCs and considers photolysis and oxidation by OH, Cl, O3 and NO3. In its entirety, the MCM (v3.2) contains 5734 species and 16940 gas-phase reactions. The MCM has been utilised using input data from numerous field campaigns to investigate the chemistry of the polluted urban boundary layer,15,16 marine boundary layer,17,18 continental low-NOx regions influenced by biogenic emissions19,20 and polar regions;21,22 a comprehensive review of these and more can be found in Stone et al., 2012.23
In this work, a 0-D box model was run using the MCMv3.2 and constrained to measurements from the ClearfLo project of NO, CO, CH4, HONO, 62 individual VOC species measured by GC-FID and also 2D-GC,24,25 PAN, HCHO, HNO3, water vapour, temperature, pressure and measured photolysis frequencies (including j(O1D), j(NO2), j(HONO), j(HCHO), j(CH3COCH3) and j(CH3CHO)) calculated using wavelength-resolved actinic flux measurements by a spectral radiometer. A constant H2 concentration of 500 ppb was assumed.26 The model constraints were updated hourly. For species measured more regularly, data were averaged to hourly intervals. The model was run unconstrained to O3 and NO2 and the predicted in situ (or locally produced) O3 was output once a steady state concentration had been reached (further details on how the box model was run is provided in Section 2.4).
For all model generated species (including O3 and NO2) a first order loss rate (k) was included. This loss rate varied as a function of the measured boundary layer height (h) (k = Vd/h), where Vd represents a deposition velocity which was taken to be equal to 1 cm s−1 for all of the model-generated species.
In addition to the MCM, two smaller chemistry schemes are used in this study and run within the same box model framework: the Generic Reaction Set (GRS) as used in the urban dispersion model, ADMS-Urban, and the Common Representative Intermediates (CRI) mechanism as used in the regional chemistry transport model, EMEP4UK. Both these models are described below. The scheme sizes and capabilities are summarised in Table 1.
| Species | Reactions | Chemistry includes oxidation of | Models used in | |
|---|---|---|---|---|
| a MCM (Master Chemical Mechanism), subset sizes in brackets. b GRS (Generic Reaction Set). c ADMS-Urban (Atmospheric Dispersion Modelling System). d CRI (Common Representative Intermediates). e EMEP4UK (European Monitoring and Evaluation Programme (for the UK)). | ||||
| MCMv3.2 | 5734 (3789) | 16940 (11410) |
143 VOCs including 22 ≤C12 alkanes, 16 ≤C12 alkenes, 9 aldehydes, 18 aromatics, isoprene, α- and β-pinene | Benchmark scheme |
| GRS | 7 | 7 | Lumped VOCs | ADMS-Urban |
| CRIv2-R5 | 220 | 609; 4% The size of the MCM | 22 VOCs including ≤C4 alkanes and alkenes, 9 oxygenated compounds, benzene, toluene, isoprene, o-xylene, α- and β-pinene | EMEP4UK |
2.2 ADMS-Urban dispersion model and GRS mechanism
ADMS-Urban27 is a quasi-Gaussian air dispersion modelling tool able to resolve the details of concentration fields within an urban area at high resolution (tens of metres) by explicitly representing the near-field features of the dispersion of emissions from all source types; specifically: point, line, area, volume, road and airport runway source types. ADMS-Urban performs well in comparison to measured data for a wide range of pollutants including NO2, PM10 and O3 and by explicitly modelling complex urban features such as street canyons, tunnels and noise barriers and including a relatively simple photolytic NOx chemistry scheme, the model is able to predict concentrations at kerbside locations as well as at urban background sites; consequently, the model is used worldwide for air quality management and assessment studies. ADMS-Urban can be used as a stand-alone system, where upwind measurements of meteorology and pollutants are used to drive the model. Alternatively, the model can be coupled to regional modelling systems, for both assessment12,28 and forecasting applications.29Fig. 1a shows an ADMS-Urban dispersion model contour plot of the annual mean O3 concentration in London for 2010. | ||
| Fig. 1 (a) Contour plot of London showing the annual mean O3 concentrations predicted by ADMS-Urban for 2010 constrained to upwind observations. (b) EMEP4UK hourly average surface O3 concentrations at 5 km resolution over central London at 12:00, 24/07/2012 (during ClearfLo summer IOP). The star represents the North Kensington site in each case. | ||
Within the ADMS-Urban domain the local NOx and O3 chemistry is represented by the GRS, initially developed at the Commonwealth Scientific and Industrial Research Organisation (CSIRO)30,31 in Australia. The GRS was developed by fitting model estimates of O3 concentrations to data obtained from an outdoor smog chamber using VOC and NO concentrations typical of urban areas.30,31 It is a semi-empirical photochemical mechanism which reduces the many thousands of chemical reactions involving NO, NO2, O3 and many hydrocarbons (which are treated explicitly in the MCM) to the following seven reactions involving a number of surrogate species:
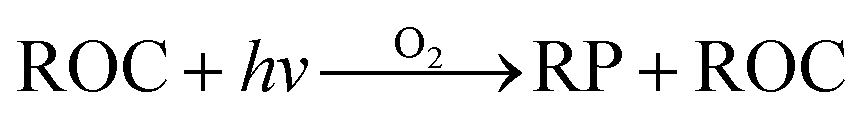 | (R1) |
| RP + NO → NO2 | (R2) |
 | (R3) |
| NO + O3 → NO2 | (R4) |
| RP + RP → RP | (R5) |
| RP + NO2 → SGN | (R6) |
| RP + NO2 → SNGN | (R7) |
Reactions (3) and (4) represent chemically exact mechanisms, while the rest of the reactions are only approximate representations of their chemical counterparts. Reaction (1) is a semi-empirical representation of all the processes that lead to radical production from VOCs through photo-oxidation. ROC is conserved in the reaction; thus, becomes a surrogate for the products of the initial oxidation of the emitted VOCs. Reaction (2) represents the conversion of NO to NO2 by reaction with radicals and leads to the termination of the radical, RP. Reaction (5) represents another sink for the radical pool. Reactions (6) and (7) lead to the formation of organic and inorganic nitrates. The rate of (R1) is a function of the photolysis frequency for NO2 (further details below), an empirically determined weighted mean reactivity coefficient of the ROC, and the temperature. By normalising the emitted VOCs by their rates of radical production, a composite ROC concentration is derived; in effect, the ROC concentration determined is weighted by the ability of the different VOCs used in the calculation to produce radicals.
Typically, the GRS chemistry scheme used in ADMS-Urban is run constrained to background measurements of NO, NO2, O3 and, if available, ROC. These background measurements are from a location approximately 10 km upstream from the point of interest and are representative of rural background concentrations. The reactions (R1)–(R7) are applied in the model in two steps: initially the rural background concentrations and sources undergo reaction, and then secondly the contribution of sources nearest to the receptor point (typically 72 s upwind, but dependent on local meteorology) undergo reaction (integrated for 72 s on average) generating the final modelled O3 concentration.
A 0-D box model was run using the GRS mechanism and (in the base model run) constrained to concentrations of ROC and NO which were also used as the near-field constraints in ADMS-Urban and which derive from ADMS-Urban dispersion calculations, including emissions and background concentrations,32 (further discussion on how ROC is determined is provided in Section 3.1.1.). Analogous to the MCM box model described above, the GRS box model was run unconstrained to O3 and NO2 and the predicted in situ (or locally produced) O3 was output once a steady state concentration had been reached. As for the MCM box model, the GRS model constraints were updated hourly. In the case of the GRS box model a constant loss rate of O3 and NO2, equal to 3 × 10−5 s−1, was used to represent deposition or loss of these species from the model box. The GRS predicted in situ O3 is particularly sensitive to this loss rate (owing to the lack of competing reactions in this mechanism). Despite this, the influence of the various model parameters on O3 reported in Sections 3.1.1 and 3.2.1 does not change even if this loss rate is varied and so it is still possible to assess the sensitivity of the GRS mechanism to various parameters.
2.3 EMEP4UK regional model and CRIv2-R5 chemistry mechanism
The EMEP MSC-W (Meteorological Synthesizing Centre-West) model33 is used by the United Nations Economic Commission for Europe Convention on Long Range Transboundary Air Pollution, at a grid resolution of 50 km to assess transboundary air pollution in Europe. EMEP4UK7,34 is a version of the EMEP4UK model targeted specifically at UK air quality and incorporating a nested sub domain with 5 km resolution over the British Isles. Fig. 1b shows the resolution and capability of the EMEP4UK model predicting the hourly average surface O3 concentration for London at 12:00 on 24/07/2012. As part of the CUREAIR project, EMEP4UK will be used to provide chemical boundary conditions for high resolution city-scale chemical dispersion modelling with ADMS-Urban.Within the CUREAIR project, the EMEP4UK regional model will be run with the CRIv2-R5 reduced chemistry mechanism as in a number of previous studies.7,34
Reduced chemistry mechanisms such as CRI are achieved either by (i) reducing the complexity of the chemistry for the considered suite of VOCs and/or (ii) by lumping emissions so that the chemistry for one VOC can be used to represent that of a number of VOCs. Method (i) was used to generate the CRIv2 mechanism which reduced the number of species and reactions used in the MCMv3.1 by 90% (i.e. to 434 species and 1183 reactions), whilst still adequately describing the degradation of methane and 115 non-methane VOCs.35 The non-methane VOC groups are defined by the Global Emissions Inventory Activity36 to impose a more severe level of reduction through emissions lumping. In this case, more limited selections of VOCs were considered to represent each VOC group, with the choice of species taking account of their photochemical ozone creation potentials (POCP) value, abundance in the detailed speciation, and the simplicity of the associated CRIv2 degradation mechanism. This allowed reductions of up to 55% in the numbers of reactions and species relative to CRIv2. The mechanism retains CRIv2 chemistry for isoprene, α-pinene and β-pinene and this contributes to the number of VOCs, species and reactions given. These reduced mechanisms display a degree of compromise in the O3-forming ability of the VOC sub-categories, but retain a good level of overall performance.37
By considering a series of emission lumping options for anthropogenic VOCs (method (ii)), a set of further reduced CRI mechanisms were developed by Watson et al.37 The smallest of these CRI schemes (known as CRIv2-R5) has 220 species and 609 reactions, with the suite of emitted non-methane VOCs represented by 22 compounds. This was a further 49% reduction in the numbers of reactions and species relative to CRIv2. The performances of CRIv2 and its reduced variants (including CRIv2-R5) were tested during development against that of MCMv3.1 for a wide range of ambient conditions, using box model scenarios and simulations of a major field campaign.35,37
A 0-D box model was run using CRIv2-R5 and (similar to the MCM box model) run constrained to measurements from the ClearfLo project of NO, CO, CH4, VOCs, PAN, HCHO, HNO3, water vapour, temperature, pressure and measured photolysis frequencies. The model was run unconstrained to O3 and NO2 (and unconstrained to observed HONO in the base run) and the predicted in situ (or locally produced) O3 was output once steady state conditions were reached. For all model generated species (including O3 and NO2) a first order loss rate (k) which varied with boundary depth was included as in the MCM box model. The CRIv2-R5 model constraints were updated hourly.
2.4 Running the box models to steady state conditions
In the three box models constrained to chemical mechanisms of varying complexity, each model point was initialised with zero [O3] and zero [NO2] and then run forward with O3 and NO2 concentrations allowed to vary until steady state concentrations were reached. This was found to take up to 168 hours (seven days) for the MCM model, but was quicker for the simpler GRS mechanism (<3 days typically). The O3 predicted by each of the three chemical mechanisms (run with equivalent NO and VOC constraints) for one model point on the 2nd August is presented in Fig. 2. All constraints including photolysis rates were held constant during each model point. By running to steady state in this way, the full O3 creation potential of the VOCs present under a particular NOx and radiation loading is determined. As can be seen in Fig. 2, this O3 creation potential is specific to the chemical mechanism employed. Given the length of time it can take before O3 reaches a steady state concentration, it is unlikely that the real atmosphere is in steady state; Lee et al.38 calculate a mean transport time to the North Kensington site since emission of ∼40 to 50 minutes during the summer from the NOx/NOy ratio. Furthermore, in the ADMS-Urban dispersion model the GRS scheme is run for just 72 s on average. Nevertheless, to evaluate the ability of different chemical mechanisms to produce O3 it is necessary to compare each under steady state conditions to prevent any bias caused by variability in the length of time it may take for [O3] to reach steady state when the complexity of the chemical mechanisms varies. | ||
| Fig. 2 Example of the modelled O3 concentration determined by the three chemistry schemes for a single model point run forward until steady state conditions are reached; [O3] output hourly for 7 model days. | ||
3. Results and discussion
3.1 Modelled O3 concentrations during the ClearfLo summer IOP
The majority of the summer measurement period was characterised by south-westerly winds, with the wind speed showing a diurnal cycle of less than 1 m s−1 at night to 4–6 m s−1 in late afternoon. The exception to this are two periods from 24–27 July and 8–10 August, during which the site was subjected to an easterly flow, with lower wind speed. Due to central London being to the east of the site, these periods are characterised by higher levels of NOx (up to 60 ppb of NO and 50 ppb of NO2), which has its source mainly from traffic.The measured O3 concentrations during the ClearfLo summer IOP are compared to the modelled O3 concentrations from the ADMS-Urban dispersion model and EMEP4UK regional model (Fig. 3a). Reasonable agreement is achieved, with both models able to reproduce the variability observed day to day. When compared as a median IOP diurnal cycle (Fig. 3c), the ADMS-Urban and EMEP4UK models under-predict the peak O3 concentrations by 30–40% during the day and this under-prediction is most evident on the days when the observed O3 was most elevated (e.g. on the 24th and 25th July). Fig. 3b compares the in situ O3 predicted by the three chemistry mechanisms. The MCM box model run to steady state generates significant concentrations of O3 each day (substantially more than was observed), with a peak concentration of 1240 ppb predicted on the 24th July during the polluted easterly flow. For reference, the [O3] predicted by the MCM after a model run time of 1 hour (which is similar to the estimated mean transport time to the North Kensington site since emission38) is also presented in Fig. 3b and d. The median predicted [O3] by the MCM model run for 1 hour peaks during the afternoon at 24 ppb (Fig. 3d) suggesting that ∼60% of the observed [O3] may be generated from local emissions. There is considerable variability day to day in the [O3] predicted by the MCM after a run time of 1 hour, however, with ∼290 ppb generated on the 24th July suggesting, under certain conditions, that the observed [O3] may be even more heavily influenced by London's emissions.
 | ||
| Fig. 3 (a) Full time series of ADMS-Urban, EMEP4UK modelled and measured O3 concentrations from ClearfLo summer IOP. (b) Time series of the ozone predicted by the chemistry schemes GRS, CRIv2-R5 and MCMv3.2 run in box models to steady state (MCM [O3] predicted after 1 h model run time also shown) from ClearfLo summer IOP (c) corresponding median diurnal cycle profile from the data in panel (a). (d) Corresponding median diurnal cycle from the data in panel (b). | ||
Elevated O3 concentrations are also predicted by the CRIv2-R5 model although the peak O3 predicted on the 24th and 25th July is roughly half that predicted by the MCM. During the south-westerly flows, when the daily maximum [O3] observed was lower, the CRIv2-R5 model is in reasonable agreement with the MCM. The GRS base model predicts significantly lower in situ O3, relative to the MCM model, with a peak concentration of 59 ppb simulated on the 24th July. It is evident from Fig. 3 that the GRS base model which is constrained with the same input parameters as ADMS-Urban is unable to generate as much O3 as the MCM model which is constrained to the observations made during ClearfLo. These differences may be due to differences in the model constraints or differences in the mechanisms themselves. In the following sections the sensitivity of the modelled O3 concentration from the GRS and CRIv2-R5 models to different variables (including HONO) is investigated. The ability of the GRS and CRIv2-R5 mechanisms to reproduce the level of O3 predicted by the explicit MCM when all models are constrained with equivalent inputs is also evaluated.
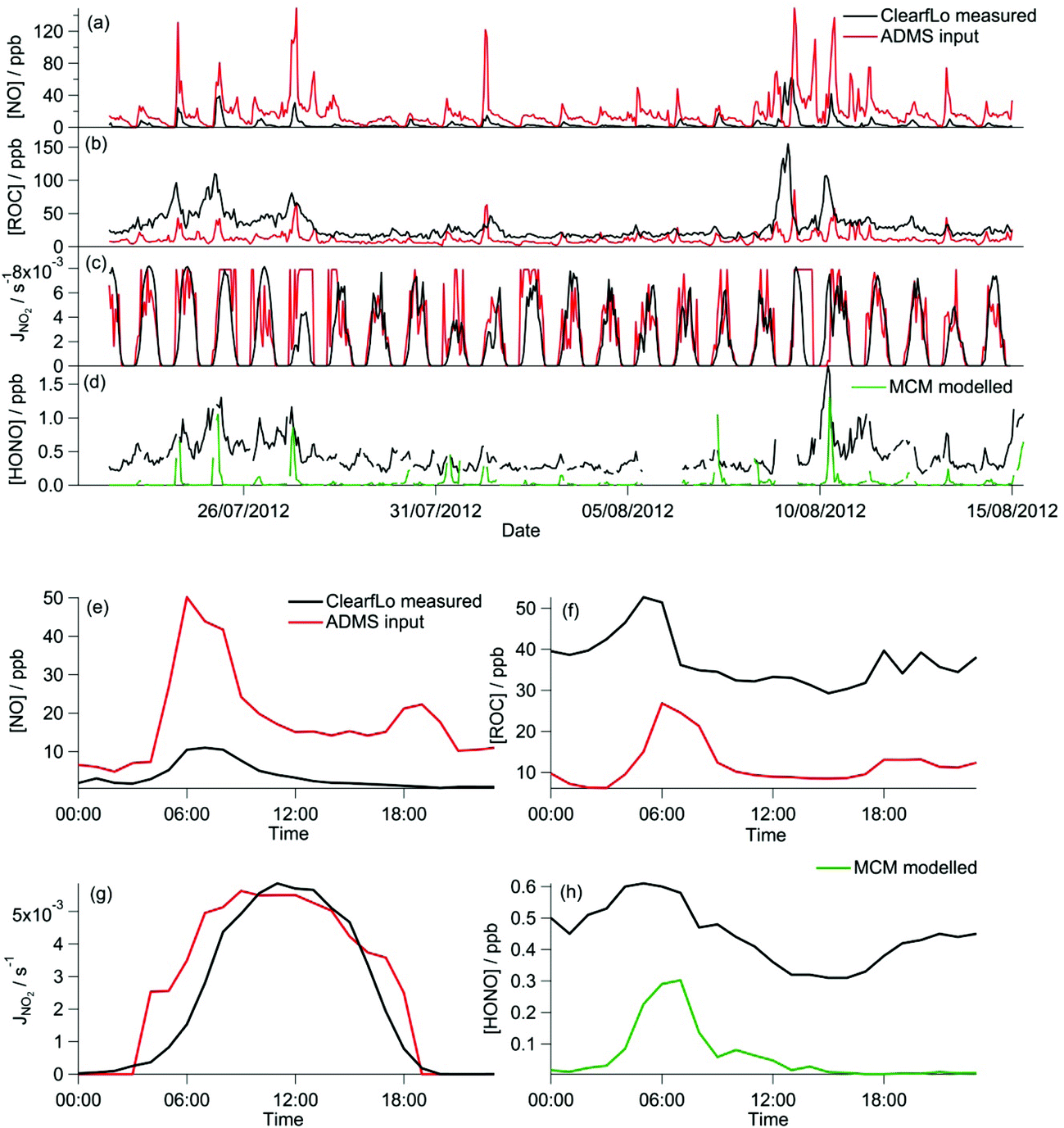 | ||
| Fig. 4 (a–c) Full time series of ADMS-Urban inputs and measured NO and VOC concentrations and NO2 photolysis frequency from ClearfLo summer IOP. (d) Full time series of MCM modelled HONO concentrations and ClearfLo measured HONO. (e–h) Average diurnal cycle of the time series in panels a–d. | ||
Further under-representation of ROC in the base run likely derives from the use of a single factor to convert VOC concentrations to ppb and the subsequent conversion to ROC. The ADMS-Urban model currently uses a factor of 0.31 (the value appropriate for benzene) to convert VOC concentrations from μg m−3 to ppb. This is likely to be too low in practice, hence underestimating the ppb concentrations of VOC. VOCs are converted to the composite ROC by their ability to produce radicals (RO2 or HO2). The concentration of ROC is defined as a reactivity coefficient multiplied by VOC concentration. For example, Johnson31 used [ROC] = 0.0067[VOC] for typical 1980s Australian urban air dominated by motor vehicles; ADMS-Urban currently uses 0.1. If measured rural concentrations of VOC are unavailable for input to ADMS-Urban, the increased reactivity coefficient partially compensates for the reduced concentration. Empirically determined reactivity coefficients for individual VOC species are available from smog chamber experiments, whilst numerically determined reactivity coefficients have been calculated using a comparison of the GRS mechanism with more complex mechanisms.31,40 A full description of this methodology for conversion can be found in Johnson31 or Venkatram et al.45The latter numerical approach is used to convert the comprehensive measurement suite of VOC concentrations from the ClearfLo campaigns to ROC for the GRS chemistry scheme.
Owing to a lack of long-term monitoring of solar actinic flux from which photolysis frequencies may be determined, the rate of photolysis of NO2 (ρ3) used in the GRS scheme is calculated from the background concentrations of O3 (CBGD(O3)), NO (CBGD(NO)) and NO2 (CBGD(NO2)):
 | (E1) |
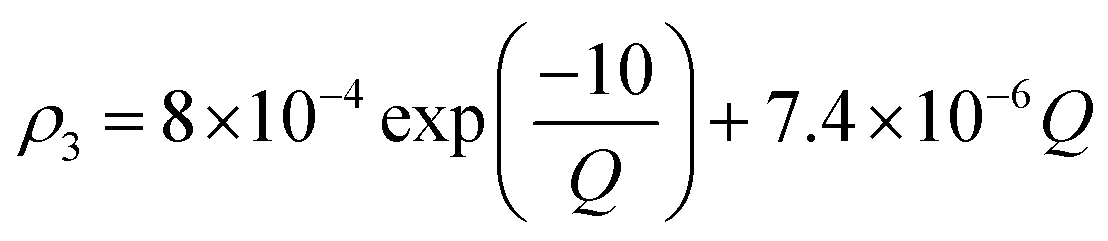 | (E2) |
 | (E3) |
 | (E4) |
 is taken to ensure that the reactions occur at a realistic rate. By utilising the measured j(NO2) from the ClearfLo IOP, the sensitivity of O3 to this parameterisation is investigated.
is taken to ensure that the reactions occur at a realistic rate. By utilising the measured j(NO2) from the ClearfLo IOP, the sensitivity of O3 to this parameterisation is investigated.
HONO can represent a major source of OH particularly in urban regions38 and has been shown to influence O3 and PM.41,42 The importance of HONO on the oxidation capacity during the ClearfLo campaign has recently been highlighted by Lee et al.38 From simultaneous measurements of HONO and OH during the summer ClearfLo IOP and subsequent modelling activities, Lee and co-authors determined an average 40% under-prediction in the daytime [OH] when the model was unconstrained to the measured [HONO] as opposed to constrained. HONO as a source of radicals (and NO) was included in the GRS scheme by the addition of the following reaction and using the measured photolysis frequency for HONO:
| HONO → RP + NO | (R8) |
This reaction assumes that the OH generated from HONO photolysis instantaneously converts to a peroxy radical (RP).
Fig. 5 presents the correlation of the MCM modelled O3 concentration from the summer ClearfLo campaign with the GRS-scheme modelled O3 for each scenario discussed above. As discussed above, the base GRS run predicts significantly less in situ O3 than the MCM model with a ratio of GRS O3:MCM O3 of 0.05. This ratio increases to 0.11 when only MCM-modelled O3 below 400 ppb is considered indicating that the base GRS model more closely reproduces the [O3] predicted by the MCM in the cleaner south-westerly flows than in the polluted easterlies. Nevertheless, the base GRS model predicts significantly lower O3 than the MCM model.
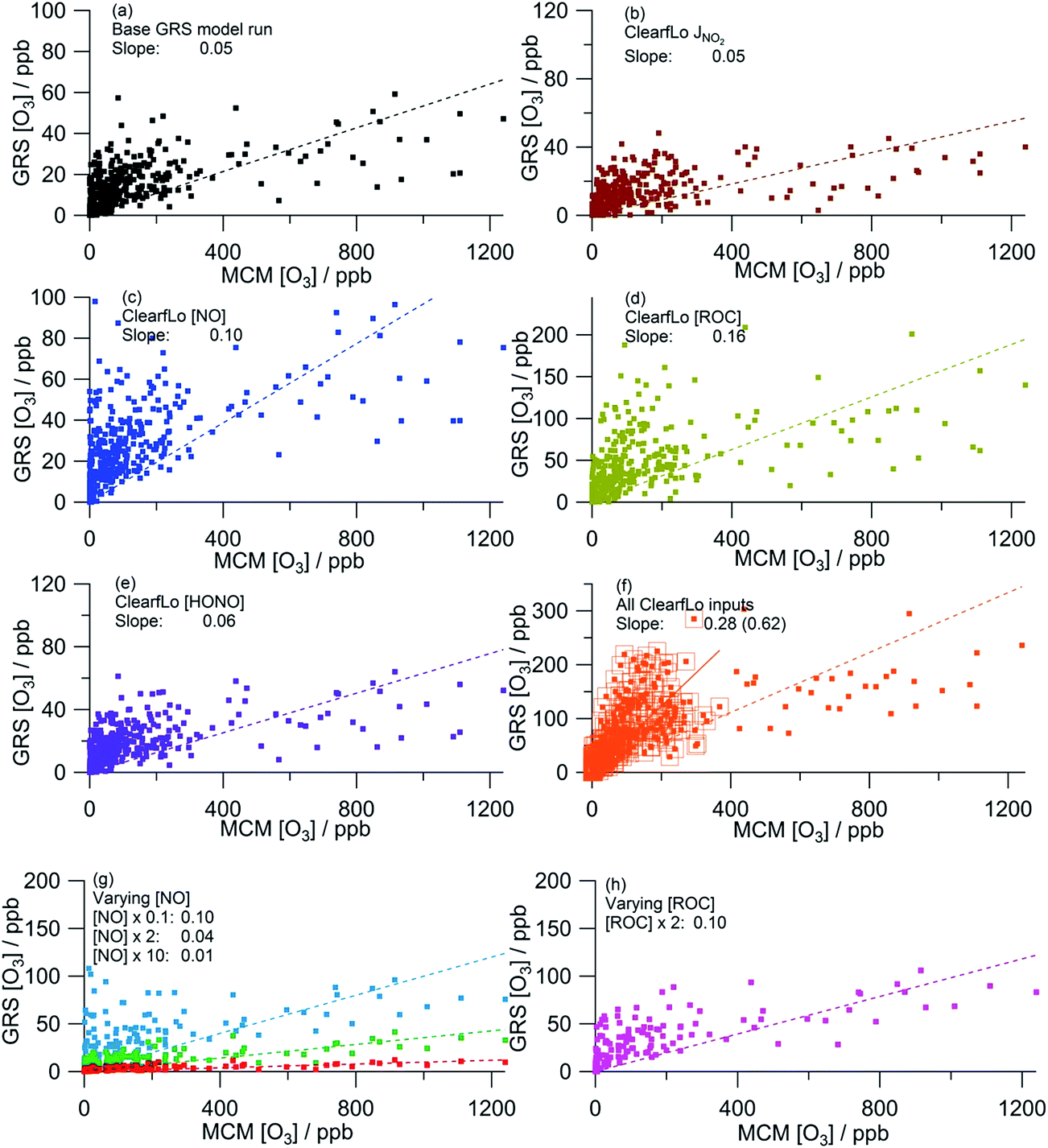 | ||
| Fig. 5 Correlation of MCM modelled [O3] from summer ClearfLo IOP against GRS modelled [O3]. (a) Base GRS scheme, (b) ClearfLo measured j(NO2), (c) measured ClearfLo [NO], (d) measured ClearfLo [ROC], (e) measured ClearfLo [HONO], (f) all ClearfLo measured data (value in parentheses = slope when MCM modelled [O3] is <400 ppb), (g) & (h) varying [NO] and [ROC]. All data can be found in Table 2. | ||
The sensitivity of O3 to j(NO2) (Fig. 5a and b) was investigated by replacing the calculated photolysis frequency (ρ3 and eqn (E1)) with the observed photolysis frequency. The level of agreement between the GRS model and the MCM remained very similar as did R2 (Table 2). It is apparent from Fig. 4c and g that the calculated NO2 photolysis frequency replicates the actual photolysis frequency on average over the time series (Fig. 4g) and was able to replicate the peak rate of NO2 photolysis. However, on a number of days (e.g. 25th, 27th) the calculated photolysis frequency is determined from ρ3(Qmax) and, as a consequence of this, on the 27th the correlation with the measured photolysis frequencies is poor. This difference likely contributes to the small decrease in GRS-predicted O3 that is seen (9%) when only [O3] <400 ppb is considered.
| Model |
|
R 2 | % change from base runa | |
|---|---|---|---|---|
| a Base GRS is the model run with the GRS emission inputs. b Values in parentheses derive from correlation of MCM modelled [O3] <400 ppb against GRS modelled [O3]. | ||||
| a | Base GRSa | 0.05 (0.11)b | 0.25 (0.36)b | — |
| b | Measured j(NO2) | 0.05 (0.10)b | 0.23 (0.42)b | 0 (−9)b |
| c | ClearfLo [NO] | 0.10 (0.20)b | 0.17 (0.28)b | +100 (+82)b |
| d | ClearfLo [VOC] | 0.16 (0.34)b | 0.23 (0.36)b | +220 (+209)b |
| e | ClearfLo [HONO] | 0.06 (0.13)b | 0.20 (0.37)b | +20 (+18)b |
| f | All ClearfLo inputs | 0.28 (0.62)b | 0.33 (0.57)b | +460 (+464)b |
| g | Varying [NO] × 0.1 | 0.10 (0.21)b | 0.07 (0.20)b | +100 (+91)b |
| Varying [NO] × 2 | 0.04 (0.07)b | 0.28 (0.38)b | −20 (−36)b | |
| Varying [NO] × 10 | 0.01 (0.02)b | 0.30 (0.38)b | −80 (−82)b | |
| h | Varying [ROC] × 2 | 0.10 (0.21)b | 0.22 (0.35)b | +100 (+91)b |

| Model |
|
R 2 | % increase from chemistry onlya | |
|---|---|---|---|---|
| a Base CRI, panel a: CRIv2-R5 run unconstrained to HONO. b Values in parentheses derive from correlation of MCM modelled [O3] <400 ppb against GRS modelled [O3]. | ||||
| a | Base CRI | 0.51 (0.85)b | 0.75 (0.83)b | — |
| b | ClearfLo HONO | 0.53 (0.91)b | 0.73 (0.83)b | +4 (+7)b |
| c | 2 × [NO] | 0.40 (0.57)b | 0.79 (0.81)b | −22 (−33)b |
| d | 10 × [NO] | 0.14 (0.17)b | 0.73 (0.71)b | −73 (−80)b |

Both decreasing the NO concentration by a factor of 10 (Fig. 5g) or using the measured NO concentration (Fig. 5c) improves the agreement between the GRS-predicted O3 and that of the MCM (GRS O3:MCM O3 ratio = 0.10). Increasing NO concentration from the base GRS run by a factor of 2 or 10 leads to reductions in the GRS-predicted O3 indicative of a NOx-saturated regime (Fig. 5g). The GRS O3:MCM O3 ratio falls to 0.04 when NO is doubled and to 0.01 when NO is increased by a factor of 10.
The sensitivity of the GRS-predicted O3 to the ROC concentration is significant (Fig. 5a, d and h): increasing the total ROC concentration by a factor of 2 leads to a 100% increase in [O3] relative to the base GRS run. The ClearfLo ROC, which is ∼3.5 times higher than the base ROC, improves the agreement between GRS and MCM modelled O3 concentrations further, with the GRS O3:MCM O3 ratio now equal to 0.16 (or 0.34 when MCM-modelled O3 is less than 400 ppb). A major cause of the improved agreement between the GRS and MCM modelled O3 concentrations is due to the significant concentration of VOCs that are missing from the ADMS-Urban dispersion calculations, which include VOCs that are based primarily on road transport (exhaust emissions).32 These lumped transport VOCs may account for a reasonable proportion of the total VOCs, but omits any biogenics e.g. isoprene, monoterpenes and also substituted aromatics such as the xylenes which are known to have a very high potential for O3 formation.43 Many biogenic VOCs are highly reactive and are readily oxidised by the OH radical to form peroxy radicals. It has been previously identified that the production of O3 in urban areas (including London) is highly sensitive to biogenic emissions.9,25,44
The sensitivity of GRS modelled O3 to HONO chemistry was investigated by including reaction (R8) in the GRS mechanism and constraining to the measured HONO concentration and HONO photolysis frequency from the ClearfLo IOP. The introduction of this additional radical (RP) source in the GRS chemistry scheme increased the GRS O3 concentration (+20% from the base run) and agreement with the MCM-predicted O3 improved marginally (Fig. 5a and e).
When the GRS model was constrained to all the measured ClearfLo parameters (j(NO2), ClearfLo ROC, NO and HONO) (Fig. 5f) agreement between the GRS-predicted O3 and MCM-predicted O3 increased to a ratio of 0.28 for the whole campaign and to 0.62 when only MCM-modelled O3 <400 ppb was considered. The correlation coefficient also increases (R2 = 0.33 or 0.57). Interestingly, the percentage increase from the base run (+460%) when all the ClearfLo parameters are included is greater than when the parameters are added into the GRS model individually, highlighting the non-linear dependence of the O3 chemistry on NOx and VOC concentrations.
Overall, when equivalent inputs are used as constraints in both the MCM and GRS schemes, the predicted O3 is significantly lower if the lumped, eight reaction GRS scheme (including HONO reaction) is used. This trend is most evident, however, on the days when the observed O3 was most elevated, i.e. during the easterly flows, typified by warm, stagnant conditions. If the modelled O3 is even more heavily filtered and only MCM-modelled O3 below 150 ppb is considered, the ratio GRS [O3]:MCM [O3] increases to 0.96 (R2 = 0.7) demonstrating that under certain conditions, the heavily lumped GRS scheme has the ability to reproduce the O3 predicted by the more explicit MCM scheme. Given that the ADMS-Urban dispersion model has a tendency to under-predict the peak [O3] observed during the easterly flows experienced at the start of the IOP (Fig. 3a), it becomes relevant to consider differences caused by the simplifications in the chemistry scheme employed. The MCM model generates a significant concentration of model-generated intermediate species which contribute ∼9 s−1 to the total OH reactivity during the polluted easterly flows.25 These intermediates (deriving largely from the biogenic species of alpha-pinene and limonene) increase the MCM-modelled peroxy radical concentrations and drive up the predicted O3.25 [ROC] is conserved in the GRS by reaction (R1) and this effectively increases the O3 creation potential of the GRS. It is apparent, however, that this single reaction under-represents the influence of the secondary chemistry of the MCM model intermediates at the start of the IOP which is dominated by the oxidation of the biogenics. Although biogenic species contribute only ∼1% to the total VOC loading (excluding methane), the contribution they make to [ROC] is ∼20% demonstrating their high radical (and O3) creation potential. From the comparison of the GRS constrained to ClearfLo ROC with the near-explicit MCM, however, it is evident that the conversion of these VOC types to ROC (i.e. the ability of the biogenics to generate radicals) is under-estimated by the methodology employed. In the [ROC] constraint used within the ADMS-Urban model the impact of biogenics is not included at all, yet in a warming atmosphere, in the presence of NOx, the impact of these species on [O3] is likely to become increasingly relevant.
The regional chemistry transport model EMEP4UK can be run with the CRIv2-R5 chemistry scheme and, hence, here the ability of this simplified (but still somewhat complex relative to GRS) chemistry scheme to predict O3, as compared to that simulated by the near-explicit MCM model, is assessed. Sensitivity tests investigating the impact of constraining to measured concentrations of HONO are presented. The impact of NO is also investigated (by doubling and increasing the NO concentration ten-fold) to contrast to the impact of NO in the GRS box model.
Fig. 6 shows the correlation of modelled CRIv2-R5 O3 concentrations with those from the MCM benchmark model. As discussed in Section 3.1, the base CRIv2-R5 run, unconstrained to HONO, predicts less O3 than the MCM box model with a ratio of CRI O3:MCM O3 = 0.51, although this increases to 0.85 if only MCM-modelled O3 which is less than 400 ppb is included. The correlation coefficient is significantly better (R2 = 0.75 (or 0.83)) than all the GRS–MCM correlations, likely reflecting the origin of the CRI scheme. The ratio CRI O3:MCM O3 increases (to 0.53 or 0.91) when the CRIv2-R5 model is run constrained to the observed HONO demonstrating the impact of HONO as a radical source and ultimately as a source of O3.
 | ||
| Fig. 6 Correlation of CRI modelled [O3] from summer ClearfLo IOP against MCM modelled [O3]. (a) Base CRI model run (value in parentheses = slope when MCM modelled [O3] is <400 ppb). (b) CRI constrained to ClearfLo measured HONO. (c) Base CRI model run, but with [NO] doubled. (d) Base CRI model run, but with [NO] increased ten-fold. All data can be found in Table 3. | ||
Increasing the [NO] by a factor of 10 (to concentrations similar to those used in the GRS base run) decreases the CRIv2-R5 modelled O3 by 73% relative to the base CRIv2-R5 run, demonstrating a slightly weaker NO dependence than observed in the GRS box model as NO concentrations were varied. This weaker NO dependence likely reflects the lower VOC:NOx ratios of the base GRS box model relative to the CRIv2-R5 box model.
The CRIv2-R5 when constrained to equivalent inputs to the MCM reproduces the MCM-predicted O3 well (ratio of CRIv2-R5:MCM = 0.91) at all times apart from during the polluted easterly conditions when elevated [O3] prevailed. As discussed in Section 3.1.1, the oxidation of monoterpenes significantly increases the MCM model intermediates which enhance modelled peroxy radical concentrations and O325 at these times. Although the concentration of individual monoterpenes can be represented explicitly in the CRIv2-R5 scheme (rather than as a single lumped biogenic) the oxidation scheme for these species differs from the MCM somewhat. Considering the ozonolysis of alpha pinene and the subsequent RO2 + NO reactions (proceeding along the dominant reaction pathways in both the MCM and CRIv2-R5), 10 molecules of NO2 can be generated using the MCM scheme relative to 8 molecules in CRIv2-R5. Furthermore, 3 OH radicals are consumed in the CRIv2-R5 scheme relative to 1 OH radical in the MCM, although this is offset somewhat by 4 HO2 radicals being generated along the CRIv2-R5 reaction pathway relative to 2 HO2 radicals in the MCM. Overall these differences lead to CRIv2-R5 predicting around twice as much HO2 as the MCM on the afternoon of the 25th July; however, the MCM predicts up to 4 times as much RO2 as CRIv2-R5 at this time and ultimately, as a consequence, more O3 is generated by the MCM scheme. EMEP4UK, run with CRIv2-R5, has a tendency to under-predict O3 under these conditions and this analysis suggests that the differences in the in situ chemistry of biogenic species in CRIv2-R5 could, in part, contribute to this under-prediction at these times.
3.2 Modelled O3 concentrations during the ClearfLo winter IOP (January–February 2012)
During the winter, the majority of the measurement period was characterised by south-westerly winds. Wind speeds were variable; early in the campaign (13–19 January) wind speeds dropped and extremely elevated levels of NOx and VOCs were observed (Fig. 8a and b). During this period O3 was almost entirely titrated away by NO. Similar stagnation events were also observed for shorter periods later in the campaign, e.g. on the 24th January. The ClearfLo measured O3 concentrations are compared to the modelled O3 concentrations from ADMS-Urban and EMEP4UK for the winter IOP (Fig. 7a). The ADMS-Urban and EMEP4UK models reproduce the measured winter O3 concentration time-series well, although the EMEP4UK model over-predicts the median O3 during the morning and under-predicts O3 concentrations during the afternoon (Fig. 7c). Both models capture the nighttime O3 trend reasonably well, with only a slight under-prediction observed from midnight to 6 am. The MCM box model (Fig. 7b) run to steady state predicts more O3 than the base CRIv2-R5 and the GRS models. The chemical O3 creation potential determined (by running the box models to steady state with respect to O3) is substantially lower than in summer and the daytime O3 concentrations predicted by the MCM are only slightly higher than observed, suggesting that the maximum O3 creation potential of the air-mass is close to being realised during the winter. It also takes less time to reach a steady state [O3] when running the winter MCM model than running the summer MCM model owing to the higher [NOx] in winter; this is most evident during the stagnation events, when wind speeds were extremely low. When wind speeds picked up (20–23 January), the MCM under-predicts the daytime O3 that was observed, highlighting that a fraction of the O3 must have been transported from local/regional/continental sources at these times, in agreement with results from REPARTEE.11 The MCM box model is unable to reproduce the observed O3 at night (Fig. 7b and d), again suggesting that local chemistry (even when the full O3 creation potential is determined by running to steady state conditions) is not controlling the observed O3 at these times. | ||
| Fig. 7 (a) Full time series of ADMS-Urban, EMEP4UK modelled and measured O3 concentrations from ClearfLo winter IOP. (b) Time series of the GRS, CRIv2-R5 and MCMv3.2 box model O3 concentrations from ClearfLo winter IOP. (c) Corresponding IOP median diurnal cycle profile from the data in panel (a). (d) Corresponding median diurnal cycle from the data in panel (b). | ||
The time series and median diurnal cycles for the box models can be seen in Fig. 7b and d. The GRS base model significantly under-predicts O3 relative to the MCM box model (and observations). The CRIv2-R5 model (unconstrained to observations of HONO) also under-predicts O3 compared to the MCM model albeit to a lesser extent. The sensitivity of the modelled O3 concentration to different variables in the GRS and CRIv2-R5 chemistry schemes is investigated below using a comparison with the MCM box model in Sections 3.2.1 and 3.2.2. The sensitivity of the GRS and CRIv2-R5 chemistry schemes during the winter IOP were subject to the same investigations as for summer.
 | ||
| Fig. 8 (a–c) Full time series of ADMS-Urban inputs and measured NO and VOC concentrations and NO2 photolysis frequency from ClearfLo winter IOP. (d) Full time series of MCM modelled HONO concentrations and ClearfLo measured HONO. (e–h) Average diurnal cycle of the time series in panels a–d. | ||
As discussed in Section 3.2, the GRS box model (Fig. 9a) significantly under-estimates O3 relative to the MCM box model, predicting ∼30 times less O3. In contrast to summer, the agreement with the MCM model improves modestly (6% increase relative to the base GRS run) when the calculated NO2 photolysis frequency is replaced with the observed NO2 photolysis frequency (Table 4, Fig. 9a and b), likely reflecting the faster photolysis frequency observed (on average) than calculated during the winter project (Fig. 8g).
 | ||
| Fig. 9 Correlation of MCM modelled [O3] from winter ClearfLo IOP against GRS modelled [O3], (a) Base GRS scheme, (b) ClearfLo measured j(NO2) , (c) measured ClearfLo [NO], (d) measured ClearfLo [ROC], (e) measured ClearfLo [HONO], (f) all ClearfLo measured data, (g) & (h) varying [NO] and [ROC]. | ||
| Model |
|
R 2 | % change from base runa | |
|---|---|---|---|---|
| a Base GRS is the model run with the GRS emission inputs. | ||||
| a | Base GRSa | 0.031 | 0.62 | — |
| b | Measured j(NO2) | 0.033 | 0.56 | +6 |
| c | ClearfLo [NO] | 0.069 | 0.61 | +123 |
| d | ClearfLo [VOC] | 0.24 | 0.52 | +674 |
| e | ClearfLo [HONO] | 0.10 | 0.67 | +223 |
| f | All ClearfLo inputs | 0.67 | 0.78 | +2061 |
| g | Varying [NO] × 0.1 | 0.101 | 0.50 | +226 |
| Varying [NO] × 2 | 0.019 | 0.59 | −39 | |
| Varying [NO] × 10 | 0.005 | 0.54 | −84 | |
| h | Varying [ROC] × 2 | 0.06 | 0.62 | +94 |

On average, the ClearfLo observed NO concentration was a factor of 2 lower than the NO constraint used in the GRS base run. This difference is less than observed during the summer, where the ClearfLo observations were a factor of ∼8.5 lower than the GRS base model constraint. Fig. 8a highlights reasonable agreement between the GRS base model NO constraint and the NO observed during the stagnation events and so the difference in the model constraints between the two IOPs likely reflects the ability of the ADMS-Urban dispersion calculations, including emissions and background concentrations, to capture these high NO episodes. Both decreasing the NO concentration by a factor of 10 (Fig. 9g) or using the measured NO concentration (Fig. 9c) improves the agreement between the GRS-predicted O3 and that of the MCM, with a 226% or 123% increase respectively in the GRS modelled O3 relative to the base run. Increasing NO concentration from the base GRS run by a factor of 2 or 10 leads to reductions in the GRS-predicted O3 indicative of a NOx-saturated regime (Fig. 9g); this is analogous to the summertime. The GRS O3:MCM O3 ratio falls to 0.019 when NO is doubled and to 0.005 when NO is increased by a factor of 10.
The ClearfLo-derived ROC concentration was approximately 9 times greater than the concentration used in the base GRS run (and used in ADMS-Urban). This difference is even greater than that observed during the summer comparisons where, in summer, the ClearfLo-derived ROC was ∼3.5 times greater than the concentration used to constrain the base GRS run. In winter, the concentration of substituted aromatics which are not considered in the base ROC constraint was ∼6 to 8 times greater than during the summer months and these species contribute to the ClearfLo-derived ROC significantly. Constraining the GRS box model to the ClearfLo-derived ROC increases the predicted O3 concentration by 674% relative to the base case and the GRS O3:MCM O3 ratio increases to 0.24.
The introduction of the additional gas-phase HONO chemistry reactions to the GRS chemistry scheme improved the correlation between the GRS and MCM modelled O3 concentrations (Fig. 9a and e). Inclusion of the observed HONO as a GRS-model constraint and RP source increases the GRS predicted O3 by 223% relative to the base run. The impact of HONO on O3 is much more significant during the wintertime than summertime, where, in summer, predicted O3 increased by just 20% relative to the base run. HONO concentrations were more elevated during the winter IOP, with peak HONO concentrations of 11.1 ppb observed during the stagnation event; in summer the peak HONO concentration observed was 1.77 ppb.
When the model is constrained to all the measured ClearfLo parameters (Fig. 9f), significantly more O3 is predicted relative to the base run and the GRS O3:MCM O3 ratio increases to 0.67. Analogous to summer, the percentage increase from the base run (+2061%) when all the ClearfLo parameters are included is greater than when the parameters are added into the GRS model individually, highlighting again the non-linear dependence of the O3 chemistry on NOx and VOC concentrations.
The GRS [O3]:MCM [O3] ratio in winter when each scheme is constrained with equivalent inputs is similar to the summer ratio when only MCM O3 <400 ppb is considered (winter ratio = 0.67, summer ratio = 0.62) demonstrating the ability of this simple scheme to replicate the near-explicit MCM reasonably well under a range of conditions including the higher NOx conditions experienced during the winter. The ADMS-Urban dispersion model is able to predict the observed O3 well throughout the winter IOP despite the very limited ability of the GRS, when constrained to the ADMS-Urban input parameters (Fig. 9a) to generate O3, indicating that the in situ chemistry is not controlling the [O3] observed in winter.
 | ||
| Fig. 10 Correlation of CRI modelled [O3] from winter ClearfLo IOP against MCM modelled [O3], (a) Base CRI model, (b) CRI constrained to ClearfLo measured HONO, (c) Base CRI model run, but with [NO] doubled, (d) Base CRI model run, but with [NO] increased ten-fold. All data can be found in Table 5. | ||

Constrained to the observed HONO, the CRI scheme is able to reproduce the MCM-predicted O3 for the winter ClearfLo IOP to within 20% and the R2 of 0.89 demonstrates the similarity of the O3 predicted by the two schemes throughout the IOP, during which a variety of chemical conditions were experienced.
4. Conclusions
This paper has analysed the ability of the reduced chemistry schemes of the GRS and CRIv2-R5 mechanisms to predict O3 compared to the near-explicit MCMv3.2 scheme constrained to measurements made in London, and has looked at the impact different model constraints have upon modelled O3. The main conclusions of these model comparisons and sensitivity analyses are as follows:1. The GRS semi-empirical chemistry mechanism utilised in the ADMS-urban dispersion model predicts lower O3 concentrations both during the summer and winter months compared to the MCM box model when constrained to the NO and ROC derived from dispersion calculations. The GRS scheme reproduces the in situ O3 predicted by the MCM box model to within 40% on most days in the summer and winter when the ROC constraint is increased to reflect the VOC observations during ClearfLo and the NO constraint is decreased to reflect observations. However, during the elevated O3 episodes experienced during the easterly flows in the summer, when temperatures increased and wind speeds dropped, the agreement between the GRS scheme and the MCM decreased. Similarly, agreement between CRIv2-R5- and MCM-predicted O3 also worsened during these periods. The in situ O3 predicted by the MCM was heavily influenced by biogenic VOCs during these conditions and the low GRS [O3]:MCM [O3] ratio (and low CRIv2-R5 [O3]:MCM [O3] ratio) demonstrates that the lumped schemes under-represent the full O3 creation potential of these species. Biogenics are not considered in the total [ROC] used to constrain the ADMS-Urban model, and even though they are only a small proportion of the total measured VOCs (1%), they have a high O3 creation potential. In a warming atmosphere, in the presence of NOx, the impact of these species on [O3] is likely to become increasingly relevant.
In winter, omission of substituted aromatic species such as xylenes in the total [ROC] constraint used by ADMS-Urban, which have high O3 creation potentials similar to the biogenic VOCs, leads to an under-representation of the total ROC and, similar to summer, lowers the O3 concentration predicted by the GRS scheme.
2. Despite the very limited ability of the GRS to generate O3, when constrained to ADMS-Urban input parameters in winter, the ADMS-Urban dispersion model is able to predict the observed O3 well indicating that the in situ chemistry is not controlling the [O3] in London in the winter. This is further reflected by the inability of the near-explicit MCM run to steady state (reflecting the full chemical O3 creation potential) to predict the observed [O3] on most days of the winter IOP other than during the stagnation episodes.
During the summer IOP, the near-explicit MCM run to steady state predicts extremely high O3 concentrations, up to 1240 ppb. The O3 observed was significantly lower, indicating that the real London atmosphere was far from steady state. To fully assess the influence of the in situ O3 generated from local emissions versus O3 generated upwind of London and advected in, knowledge of the time since emission (and, hence, how far the real atmosphere is from steady state) is needed. From estimates of the mean transport time determined from the NOx:NOy ratio observed at North Kensington during the summer and comparison of the O3 predicted by the MCM model after this time, ∼60% of the median observed [O3] could be generated from local emissions. During the warmer conditions experienced during the easterly flows, however, the observed [O3] may be even more heavily influenced by London's emissions.
3. The O3 predicted by the reduced chemistry mechanisms of GRS and CRIv2-R5 is highly sensitive to HONO chemistry in the wintertime; less so in the summer. An inclusion of a radical source from HONO and an accurate representation of the HONO concentrations observed in urban centres could improve the in situ O3 predicted by these reduced chemistry schemes. Long-term HONO measurements are not performed as standard and the contribution of different HONO sources, many of which are heterogeneous in nature, remains uncertain. HONO has been shown to correlate with NO2 at urban sites and so its concentration may be parameterised from NO2 observations as a first step to representing this radical source. HONO can be an important source of OH radicals in the urban environment so any underestimation has a significant impact on the oxidising capacity of the atmosphere and on secondary organic aerosol (SOA) production as well as influencing in situ O3 as demonstrated here.
4. One of the advantages of using reduced chemistry mechanisms is that computer processing time and hence the required computer power can be minimised. It can be seen from Table 6 that by replacing the explicit MCM with the chemistry mechanisms of CRIv2-R5 and GRS the run times can be significantly reduced by a factor of 1000 and 60000 respectively. This is an important consideration when investigating large time series (e.g. multiple years) or running chemistry in conjunction with other computationally intensive simulations, e.g. of transport and dispersion. The inclusion of the additional HONO chemistry caused little impact on these run times (Table 6).
ADMS-Urban and EMEP4UK contain simple chemical schemes and are constrained by incomplete predictions of source/sink species, e.g. volatile organic compounds and nitrous acid, and contain parameterisations for some inputs, e.g. photolysis frequencies. Despite these simplifications, these schemes are used widely to provide forecasts and assessments of O3 and other air quality markers for the community. In this paper we have provided an analysis of the conditions under which the simple schemes appear able to deliver reliable forecasts of a key pollution marker, O3, with the simple chemical schemes performing better (with respect to the explicit MCM) in winter and in summer under lower O3 conditions. In addition, we show the sensitivity towards O3 production of these simple schemes for variations of NOx and VOC loading/speciation. We also demonstrate the impact of the inclusion of nitrous acid (HONO) chemistry which is overlooked in these types of mechanism. HONO can play a dominant role in radical production particularly in winter when other sources of radicals which lead to the production of O3 are limited. Both the ADMS-Urban and EMEP4UK models have a tendency to under-predict peak daytime O3 concentrations in London, particularly in the summer during high O3 episodes. Increasing the contribution of the local chemistry, with ROC (VOC) and HONO concentrations representative of the levels observed during ClearfLo (and enhancing the full O3 creation potential of biogenic VOC in line with the near-explicit MCM) may help to reduce this under-prediction.
Acknowledgements
This work was funded by the UK NERC under grant NE/M002381/1. The authors would like to thank all participants of ClearfLo.References
- EEA Report No 5/2014, 2014 Search PubMed.
- WHO final technical report, Review of evidence on health aspects of air pollution – REVIHAAP project, 2013 Search PubMed.
- S. I. Bohnenstengel, S. E. Belcher, A. Aiken, J. D. Allan, G. Allen, A. Bacak, T. J. Bannan, J. F. Barlow, D. C. S. Beddows, W. J. Bloss, A. M. Booth, C. Chemel, O. Coceal, C. F. Di Marco, M. K. Dubey, K. H. Faloon, Z. L. Fleming, M. Furger, J. K. Gietl, R. R. Graves, D. C. Green, C. S. B. Grimmond, C. H. Halios, J. F. Hamilton, R. M. Harrison, M. R. Heal, D. E. Heard, C. Helfter, S. C. Herndon, R. E. Holmes, J. R. Hopkins, A. M. Jones, F. J. Kelly, S. Kotthaus, B. Langford, J. D. Lee, R. J. Leigh, A. C. Lewis, R. T. Lidster, F. D. Lopez-Hilfiker, J. B. McQuaid, C. Mohr, P. S. Monks, E. Nemitz, N. L. Ng, C. J. Percival, A. S. H. Prévôt, H. M. A. Ricketts, R. Sokhi, D. Stone, J. A. Thornton, A. H. Tremper, A. C. Valach, S. Visser, L. K. Whalley, L. R. Williams, L. Xu, D. E. Young and P. Zotter, Bull. Am. Meteorol. Soc., 2014, 96, 779–804 Search PubMed.
- A. M. Fiore, V. Naik, D. V. Spracklen, A. Steiner, N. Unger, M. Prather, D. Bergmann, P. J. Cameron-Smith, I. Cionni and W. J. Collins, Chem. Soc. Rev., 2012, 41, 6663–6683 RSC.
- S. Solberg, Ø. Hov, A. Søvde, I. S. A. Isaksen, P. Coddeville, H. De Backer, C. Forster, Y. Orsolini and K. Uhse, J. Geophys. Res.: Atmos., 2008, 113, D07307 CrossRef.
- X. V. Francis, C. Chemel, R. S. Sokhi, E. G. Norton, H. M. A. Ricketts and B. E. A. Fisher, Atmos. Environ., 2011, 45, 6880–6890 CrossRef CAS.
- M. Vieno, A. J. Dore, D. S. Stevenson, R. Doherty, M. R. Heal, S. Reis, S. Hallsworth, L. Tarrason, P. Wind, D. Fowler, D. Simpson and M. A. Sutton, Atmos. Chem. Phys., 2010, 10, 7963–7978 CrossRef CAS.
- J. D. Lee, A. C. Lewis, P. S. Monks, M. Jacob, J. F. Hamilton, J. R. Hopkins, N. M. Watson, J. E. Saxton, C. Ennis, L. J. Carpenter, N. Carslaw, Z. Fleming, B. J. Bandy, D. E. Oram, S. A. Penkett, J. Slemr, E. Norton, A. R. Rickard, L. K Whalley, D. E. Heard, W. J. Bloss, T. Gravestock, S. C. Smith, J. Stanton, M. J. Pilling and M. E. Jenkin, Atmos. Environ., 2006, 40, 7598–7613 CrossRef CAS.
- A. R. MacKenzie, R. M. Harrison, I. Colbeck and C. N. Hewitt, Atmos. Environ., Part A, 1991, 25, 351–359 CrossRef.
- J. M. Harris and S. J. Oltmans, J. Geophys. Res.: Atmos., 1997, 102, 8781–8791 CrossRef CAS.
- R. M. Harrison, M. Dall'Osto, D. C. S. Beddows, A. J. Thorpe, W. J. Bloss, J. D. Allan, H. Coe, J. R. Dorsey, M. Gallagher, C. Martin, J. Whitehead, P. I. Williams, R. L. Jones, J. M. Langridge, A. K. Benton, S. M. Ball, B. Langford, C. N. Hewitt, B. Davison, D. Martin, K. F. Petersson, S. J. Henshaw, I. R. White, D. E. Shallcross, J. F. Barlow, T. Dunbar, F. Davies, E. Nemitz, G. J. Phillips, C. Helfter, C. F. Di Marco and S. Smith, Atmos. Chem. Phys., 2012, 12, 3065–3114 CAS.
- J. Stocker, C. Hood, D. Carruthers and C. McHugh, Int. J. Environ. Pollut., 2012, 50, 308–316 CrossRef CAS.
- M. E. Jenkin, S. M. Saunders, V. Wagner and M. J. Pilling, Atmos. Chem. Phys., 2003, 3, 181–193 CrossRef CAS.
- S. M. Saunders, M. E. Jenkin, R. G. Derwent and M. J. Pilling, Atmos. Chem. Phys., 2003, 3, 161–180 CrossRef CAS.
- K. M. Emmerson, N. Carslaw, D. C. Carslaw, J. D. Lee, G. McFiggans, W. J. Bloss, T. Gravestock, D. E. Heard, J. Hopkins, T. Ingham, M. J. Pilling, S. C. Smith, M. Jacob and P. S. Monks, Atmos. Chem. Phys., 2007, 7, 167–181 CrossRef CAS.
- R. M. Harrison, J. Yin, R. M. Tilling, X. Cai, P. W. Seakins, J. R. Hopkins, D. L. Lansley, A. C. Lewis, M. C. Hunter, D. E. Heard, L. J. Carpenter, D. J. Creasey, J. D. Lee, M. J. Pilling, N. Carslaw, K. M. Emmerson, A. Redington, R. G. Derwent, D. Ryall, G. Mills and S. A. Penkett, Sci. Total Environ., 2006, 360, 5–25 CrossRef CAS PubMed.
- R. Sommariva, A. L. Haggerstone, L. J. Carpenter, N. Carslaw, D. J. Creasey, D. E. Heard, J. D. Lee, A. C. Lewis, M. J. Pilling and J. Zádor, Atmos. Chem. Phys., 2004, 4, 839–856 CrossRef CAS.
- R. Sommariva, W. J. Bloss, N. Brough, N. Carslaw, M. Flynn, A. L. Haggerstone, D. E. Heard, J. R. Hopkins, J. D. Lee, A. C. Lewis, G. McFiggans, P. S. Monks, S. A. Penkett, M. J. Pilling, J. M. C. Plane, K. A. Read, A. Saiz-Lopez, A. R. Rickard and P. I. Williams, Atmos. Chem. Phys., 2006, 6, 1135–1153 CrossRef CAS.
- P. M. Edwards, M. J. Evans, K. L. Furneaux, J. Hopkins, T. Ingham, C. Jones, J. D. Lee, A. C. Lewis, S. J. Moller, D. Stone, L. K. Whalley and D. E. Heard, Atmos. Chem. Phys., 2013, 13, 9497–9514 CrossRef.
- D. Stone, M. J. Evans, R. Commane, T. Ingham, C. F. A. Floquet, J. B. McQuaid, D. M. Brookes, P. S. Monks, R. Purvis, J. F. Hamilton, J. Hopkins, J. Lee, A. C. Lewis, D. Stewart, J. G. Murphy, G. Mills, D. Oram, C. E. Reeves and D. E. Heard, Atmos. Chem. Phys., 2010, 10, 9415–9429 CrossRef CAS.
- W. J. Bloss, M. Camredon, J. D. Lee, D. E. Heard, J. M. C. Plane, A. Saiz-Lopez, S. J. B. Bauguitte, R. A. Salmon and A. E. Jones, Atmos. Chem. Phys., 2010, 10, 10187–10209 CAS.
- P. Edwards, M. J. Evans, R. Commane, T. Ingham, D. Stone, A. S. Mahajan, H. Oetjen, J. R. Dorsey, J. R. Hopkins, J. D. Lee, S. J. Moller, R. Leigh, J. M. C. Plane, L. J. Carpenter and D. E. Heard, J. Geophys. Res.: Atmos., 2011, 116, D22306 Search PubMed.
- D. Stone, L. K. Whalley and D. E. Heard, Chem. Soc. Rev., 2012, 41, 6348–6404 RSC.
- R. E. Dunmore, J. R. Hopkins, R. T. Lidster, J. D. Lee, M. J. Evans, A. R. Rickard, A. C. Lewis and J. F. Hamilton, Atmos. Chem. Phys., 2015, 15, 9983–9996 CAS.
- L. K. Whalley, D. Stone, B. Bandy, R. Dunmore, J. F. Hamilton, J. Hopkins, J. D. Lee, A. C. Lewis and D. E. Heard, Atmos. Chem. Phys., 2016, 16, 2109–2122 CrossRef.
- G. L. Forster, W. T. Sturges, Z. L. Fleming, B. J. Bandy and S. Emeis, Tellus B, 2012, 64, 17771 CrossRef CAS.
- D. J. Carruthers, H. A. Edmunds, A. E. Lester, C. A. McHugh and R. J. Singles, Int. J. Environ. Pollut., 2000, 14, 364–374 CrossRef CAS.
- J. Stocker, C. Hood, D. Carruthers, M. Seaton, K. Johnson and J. Fung, 13th Annual CMAS Conference, Chapel Hill, NC, USA, 2014.
- http://www.airtext.info/ .
- M. Azzi, G. Johnson and M. Cope, Presented in part at the 11th International Conference of the Clean Air Society of Australia and New Zealand, 1992 Search PubMed.
- G. Johnson, presented in part at the 8th International Conference of the Clean Air Society of Australia and New Zealand, 1984 Search PubMed.
- Defra's Emission Factor Toolkit, EFTv6.0.2, 2014, http://laqm.defra.gov.uk/review-and-assessment/tools/emissions-factors-toolkit.html Search PubMed.
- D. Simpson, A. Benedictow, H. Berge, R. Bergström, L. D. Emberson, H. Fagerli, C. R. Flechard, G. D. Hayman, M. Gauss, J. E. Jonson, M. E. Jenkin, A. Nyíri, C. Richter, V. S. Semeena, S. Tsyro, J. P. Tuovinen, Á. Valdebenito and P. Wind, Atmos. Chem. Phys., 2012, 12, 7825–7865 CAS.
- M. Vieno, M. R. Heal, S. Hallsworth, D. Famulari, R. M. Doherty, A. J. Dore, Y. S. Tang, C. F. Braban, D. Leaver, M. A. Sutton and S. Reis, Atmos. Chem. Phys., 2014, 14, 8435–8447 CrossRef CAS.
- M. E. Jenkin, L. A. Watson, S. R. Utembe and D. E. Shallcross, Atmos. Environ., 2008, 42, 7185–7195 CrossRef CAS.
- http://www3.epa.gov/ttnchie1/conference/ei19/session5/granier2.pdf .
- L. A. Watson, D. E. Shallcross, S. R. Utembe and M. E. Jenkin, Atmos. Environ., 2008, 42, 7196–7204 CrossRef CAS.
- J. D. Lee, L. K. Whalley, D. E. Heard, D. Stone, R. E. Dunmore, J. F. Hamilton, D. E. Young, J. D. Allan, S. Laufs and J. Kleffmann, Atmos. Chem. Phys., 2016, 16, 2747–2764 Search PubMed.
- http://uk-air.defra.gov.uk/networks/network-info?view=hc .
- P. Hurley, TAPM V4. Part 1: technical description. CSIRO Marine and Atmospheric Research Internal Report No. 5, 2008 Search PubMed.
- M. Gonçalves, D. Dabdub, W. L. Chang, O. Jorba and J. M. Baldasano, Atmos. Environ., 2012, 54, 168–176 CrossRef.
- Y. F. Elshorbany, B. Steil, C. Brühl and J. Lelieveld, Atmos. Chem. Phys., 2012, 12, 9977–10000 CrossRef CAS.
- E. von Schneidemesser, P. S. Monks and C. Plass-Duelmer, Atmos. Environ., 2010, 44, 5053–5064 CrossRef CAS.
- B. K. Pun, S.-Y. Wu and C. Seigneur, Environ. Sci. Technol., 2002, 36, 3586–3596 CrossRef CAS PubMed.
- A. Venkatram, P. Karamchandani, P. Pai and R. Goldstein, Atmos. Environ., 1994, 28, 3665–3678 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |