
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of natural organic matter and sulfidation on the flocculation and filtration of silver nanoparticles†
Tongren
Zhu
a,
Desmond F.
Lawler
*a,
Yunqi
Chen
b and
Boris L. T.
Lau
*b
aDept. of Civil, Architectural & Environmental Engineering, University of Texas, One University Station C1786, Austin, TX, USA. E-mail: dlawler@mail.utexas.edu; Fax: +01 512 471 0592; Tel: +01 512 471 4595
bDept. of Civil & Environmental Engineering, University of Massachusetts, 130 Natural Resources Road, Amherst, MA 01003, USA. E-mail: borislau@umass.edu; Fax: +01 413 577 4940; Tel: +01 413 545 5423
First published on 19th October 2016
Abstract
Surface properties of engineered silver nanoparticles (AgNPs) are strongly affected by environmental transformation. The fate and transport of these transformed AgNPs is largely unknown and cannot be fully explained by the traditional Derjaguin–Landau–Verwey–Overbeek (DLVO) theory. The objective of this study was to investigate the changes in the composition and surface properties of polyvinylpyrrolidone (PVP) capped AgNPs after environmental transformation and their subsequent effects on the flocculation and filtration of these transformed AgNPs during water treatment processes. To study the aggregation and deposition behavior of the transformed particles, PVP-AgNPs exposed to humic acid (HA) and/or sulfidation were characterized, followed by separate flocculation, granular media filtration, and quartz crystal microgravimetry (QCM) experiments. X-ray photoelectron spectroscopy revealed that HA exposure modified the original PVP capping via adsorption and/or ligand exchange and that sulfidation stripped the PVP from the particle surface as a result of the formation of silver sulfide. Sulfidation thereby reduced the stability of PVP-AgNPs in self-aggregation but enhanced the mobility of AgNPs in granular media filtration and quartz collector deposition. Without unbound macromolecules in the background solution, polymers on the particle surface largely prevent self-aggregation but allow favorable clean bed deposition. This difference between the effects on self-aggregation and granular media filtration is in contrast to traditional DLVO theory. QCM yielded two types of results, the initial rate of deposition and the ultimate deposition, and both gave insights into expected filtration behavior.
Environmental significanceSilver nanoparticles (AgNPs) undergo natural organic matter (NOM) exposure and sulfidation upon their release into the environment. However, the subsequent fate and transport of transformed AgNPs is poorly studied. This paper demonstrated that NOM exposure and sulfidation modified the polyvinylpyrrolidone (PVP) capped AgNPs. Without unbound macromolecules in the background solution, polymers on the particle surface largely prevent self-aggregation but allow favorable clean bed deposition. Sulfidation stripped the PVP capping and reduced the stability of PVP-AgNPs in self-aggregation, but enhanced the mobility of AgNPs in granular media filtration and quartz collector deposition. This study broadens our knowledge on the particle–particle and particle–collector interaction by focusing on the effect of surface macromolecular capping. |
Introduction
The increasing use of engineered silver nanoparticles (AgNPs) due to their antimicrobial properties inevitably leads to their release into the aquatic environment, and much research has focused on the fate and transport of the AgNPs to evaluate their potential impact on the environment.1–3 While numerous studies on aggregation and deposition of engineered AgNPs have accounted for various environmental factors such as pH, ionic strength, electrolyte types, capping ligands, and natural organic matter (NOM),4–10 few have investigated environmental conditions where complex environmental transformation (e.g., photoreduction,11–13 oxidation,14 and sulfidation15,16) could alter the surface properties of AgNPs and influence their fate and transport. This study investigated the effect of NOM (specifically, humic acid (HA)) exposure and sulfidation on the change of surface properties as well as the subsequent fate and transport of engineered AgNPs in the conventional water treatment processes of flocculation and granular media filtration.NOM is ubiquitous in the environment and has been found to adsorb onto the AgNPs' surface and thereby to modify the stability and mobility of AgNPs by the electrosteric or hydrophobic effect.17–20 However, an explanation for the association of NOM and AgNPs is still limited, and the different effects of bound and unbound NOM require further clarification. In addition to their interaction with NOM, AgNPs also often undergo sulfidation upon their release into the aqueous environment through wastewater treatment facilities.21,22 Various laboratory and field studies have reported sulfidation of engineered AgNPs in a low redox state environment in the presence of sulfide,16,23 conditions which are common in sewer systems and in the anaerobic stage of wastewater treatment. The thermodynamic preference of metallic silver to sulfide (i.e., Ksp,Ag2S = 6.2 × 10−52)24 over either the original particle capping or NOM impacts the surface properties and reactivity of engineered AgNPs.25 The interaction with NOM and sulfidation affect the fate and transport of AgNPs in the environment, and pose uncertain challenges for their risk assessment;15,26 however, previous nanoparticle transport studies have mostly concentrated on untransformed particles.
Some recent studies25–29 have reported changes in aggregation state, ζ-potential, and capping composition after environmental transformation, but the subsequent different aggregation and deposition behavior has not yet been fully investigated. As NOM enhances the stability and mobility of AgNPs30–32 and sulfidized AgNPs are found to be stable and resistant to oxidation,33,34 it is conceivable that environmentally transformed engineered AgNPs (after both sulfidation and NOM adsorption) will emerge in a drinking water source after traveling through wastewater treatment facilities or an anaerobic aquatic environment. Release of engineered nanomaterials (ENMs) after a conventional coagulation process is also possible,35,36 so investigating granular media filtration for the removal of ENMs is vital. The majority of previous deposition studies have been limited to the subsurface scenario of groundwater flow. However, the filtration velocity of granular media filters in drinking water treatment is much higher than the Darcy velocity of groundwater transport, so that particles that would be well retained at low velocities could be mobile in rapid filtration.37 Therefore, it is necessary to validate the extensively used colloid filtration theory for assessing nanoparticle removal at high filtration velocity and in the presence of environmental transformation. Traditionally, column tests are used to mimic porous media transport and to investigate particle deposition with the help of colloidal filtration theory; recently, quartz crystal microgravimetry (QCM) has become widely employed as a powerful research tool to study the in situ particle–collector interaction.38–40 However, little focus has been placed on the comparison between the two techniques: whether the results from both systems can be used to interpret deposition behavior interchangeably remains largely unanswered.41,42
The objective of this study was to investigate whether environmentally transformed AgNPs could be effectively removed by conventional water treatment processes such as flocculation and rapid granular media filtration. First, the effects of sulfidation and of the exposure to HA on the surface properties of polyvinylpyrrolidone (PVP) coated AgNPs (PVP-AgNPs) were characterized. Next, to determine the impact of transformation on particle–particle interaction, the flocculation performance of the PVP-AgNPs after the environmental transformations was evaluated by measuring the particle number concentration and particle size distribution (PSD) over time as the AgNPs were kept in suspension in jar tests. Finally, filtration studies of transformed AgNPs were performed via both packed column tests and QCM to explore the effects of HA and sulfidation on particle–collector interaction and the transport of PVP-AgNPs in granular media. Results from both column tests and QCM were compared to validate the extension of results from one technique to the other.
Experimental
Chemicals
Spherical PVP-AgNPs were purchased (NanoComposix, CA) with a manufacturer-reported diameter of 54.8 nm by transmission electron microscopy (TEM). The PVP capping of these particles has a molecular weight of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da. ACS grade chemicals and ultrapure deionized water (DI) of 18.2 mΩ cm was used throughout the study for solution preparation. All flocculation and filtration experiments were performed at pH = 7.0 ± 0.2, buffered by either phosphate or carbonate. Predetermined ionic strengths for different experiments were controlled with the addition of NaNO3 or Ca(NO3)2 to explore the effects of both ionic strength and electrolyte types. Suwanee River HA (International Humic Substances Soc. MN) was purchased as the model NOM for the experiments, and Na2S solution was used to induce sulfidation. Laboratory glassware, stir-bars, and pipette tips were cleaned by soaking in 10% nitric acid overnight and kept in a particle-free room prior to each experiment.
000 Da. ACS grade chemicals and ultrapure deionized water (DI) of 18.2 mΩ cm was used throughout the study for solution preparation. All flocculation and filtration experiments were performed at pH = 7.0 ± 0.2, buffered by either phosphate or carbonate. Predetermined ionic strengths for different experiments were controlled with the addition of NaNO3 or Ca(NO3)2 to explore the effects of both ionic strength and electrolyte types. Suwanee River HA (International Humic Substances Soc. MN) was purchased as the model NOM for the experiments, and Na2S solution was used to induce sulfidation. Laboratory glassware, stir-bars, and pipette tips were cleaned by soaking in 10% nitric acid overnight and kept in a particle-free room prior to each experiment.
Transformation and characterization of AgNPs
The sulfidation process for PVP-AgNPs was adapted from reported literature.27 For the sulfidation process without the presence of HA, 100 mg L−1 PVP-AgNPs were exposed to 1 mM Na2S solution to yield a S/Ag molar ratio of 1.08 in 5 mM NaNO3 electrolyte. This ratio is higher than the stoichiometric S/Ag ratio for Ag2S to ensure sulfidation but lower than the sulfide concentration that would induce Ag2S precipitation. After 24 h, the suspension was centrifuged at 13200 g for 20 minutes, the supernatant (95% of the original volume) discarded, and the suspension reconstituted with DI water and sonication; this process was repeated three times to ensure minimal concentrations of the soluble materials (Ag+, PVP, sulfide, etc.) in the supernatant.
For the simultaneous NOM-sulfide exposure experiments, Na2S and HA solutions were added to the PVP-AgNPs suspension simultaneously to yield 10 mg L−1 HA concentration. All subsequent procedures were the same as the sulfidation only process described above. The same transformation procedures were also carried out in Ca(NO3)2 electrolyte at an ionic strength of 5 mM as a comparison. The silver concentrations of the final transformed AgNPs were determined by inductively coupled plasma-optical emission spectrometry (ICP-OES, Varian 710-ES) after 6% nitric acid dissolution.
Characterization of PVP-AgNPs was obtained from both the manufacturer certificate of analysis and laboratory analysis. Both the hydrodynamic diameter (HDD) distribution and the ζ-potential were measured by two different instruments. The HDD distribution was determined via nanoparticle tracking analysis (NTA) using a Nanosight LM 10 (Amesbury, UK) at 0.1 mg L−1 of silver in 5 mM NaNO3 electrolyte at pH = 7.0 ± 0.2. The ζ-potential was characterized using a ZetaCompact zetameter (CAD Instruments, FR) by converting the measured electrophoretic mobility (EPM) into ζ-potential using the Smoluchowski equation built into the instrument software. Aqueous AgNPs suspensions with 1.0 mg L−1 of silver were prepared at different pH values ranging from 2 to 10 with NaOH and HNO3 addition in 5 mM NaNO3 electrolyte. The HDD and ζ-potential at pH 7.0 were also determined by dynamic light scattering (DLS) and electrophoretic light scattering (ELS), respectively, using a Malvern Zetasizer NS (Worcestershire, U.K.). The light scattering patterns were recorded to yield both the diffusion coefficient (which is used to calculate HDD with the Stokes–Einstein equation) and the EPM (which is used to calculate the ζ-potential with the Smoluchowski equation). As shown subsequently, the different instruments with their different measurement methodologies resulted in some differences in both characteristics.
Absorbance scans of the PVP-AgNPs from 200 to 900 nm were measured with a 8453 ultraviolet-visible spectrophotometer (Agilent Technologies) through a 1 cm pathway quartz cuvette; samples were measured at least three times and blank corrected. The primary interest was whether the distinct absorption peaks of metallic silver at approximately 435 nm wavelength remained after sulfidation.29,43 X-ray photoelectron spectroscopy (XPS) was used for surface composition analysis; samples were deposited on aluminum discs and dried in air before measurement with a Kratos XPS-Axis Ultra DLD (Kratos Analytical Ltd, UK). The XPS spectra were corrected with the C 1s line at 284.6 eV.44 Curve fitting and analysis were performed assuming Gaussian–Lorentzian deconvolution following Shirley background subtraction using the Casa XPS 2.3.16 software.
Flocculation test
The flocculation experiments were performed under varying environmental transformations and electrolyte types to explore the effects of NOM and sulfidation on particle–particle interaction. For each experiment, the original PVP-AgNPs or the transformed AgNPs were diluted to an initial concentration of 0.5 mg L−1. NaNO3 with phosphate buffer or Ca(NO3)2 with NaHCO3 buffer was added to the particle suspension at time 0 to achieve the desired ionic strength of 10 mM and a pH value of 7.0 ± 0.2; the pH did not vary over the 120 minutes of the flocculation tests. After an initial rapid mixing, gentle mixing (with G value estimated as 10 s−1) was applied via a combined rocking and rolling motion in enclosed jars to avoid particle settling to simulate the flocculation process.45 The device to accomplish this mixing is a rotating cylinder (6 rpm) with a diameter of 11.5 cm, and the jars are attached non-axially at an angle of approximately 20° from the horizontal axis of rotation. Samples were collected at various times over 120 min and PSDs were measured immediately with NTA.Column filtration test
Column filtration experiments were performed to investigate the retention performance of the laboratory-scale granular media filter on environmentally transformed AgNPs and the underlying particle–collector interaction. The filtration setup and operation were adapted from the previously reported conditions.45 A 3.8 cm inner diameter cylindrical column was packed with 325 μm diameter glass beads (MO-SCI Co., MO) to a depth of 10 cm. Prior to use, the beads were washed by the following cleaning process: rinsing with DI water 10 times, sonication in 0.01 M NaOH solution for 10 min followed by rinsing with DI water 10 times, sonication in 1 M HNO3 solution overnight followed by rinsing with DI water 10 times, sonication in DI water for 10 min followed by a final rinsing with DI water 20 times, and complete drying in 105 °C oven.46The background solution and the AgNPs suspension were pumped separately at a ratio of 20:1 and mixed at the column top to yield an influent silver concentration of 0.1 mg L−1, an ionic strength of 5 mM (made with NaNO3), a pH of 7.0 ± 0.2 (buffered with phosphate), and a filtration velocity of 5 m h−1, the lower filtration velocity limit of rapid granular media filters in water treatment plants. At the beginning of each experiment, the filter was preconditioned in the background solution overnight and flushed with the background solution without the presence of particles for 30 min (equivalent to 75 pore volumes (PV) of the filter) before time zero. The filtration was then conducted with a 30 minute injection of AgNPs with the background solution followed by a 15 minute injection of the background solution without AgNPs. The effluent was collected every one to two minutes for total silver mass concentration measurement by ICP-OES after 3% nitric acid dissolution overnight. The effluent was also sampled at various times for pH and immediate particle size characterization by NTA to check that no aggregation occurred during the filtration period. The attachment efficiency (α) was calculated by.37

QCM and substrate preparation
Real-time deposition kinetics and extent for four types of AgNPs were quantitatively determined by QCM at 25 ± 0.02 °C. QCM resolves mass differences on the crystal surface with high sensitivity using the piezoelectric property of quartz.48 The mass change on the surface of the quartz (Δm, in ng cm−2) can be related to the change in oscillation frequency (Δf) according to the Sauerbrey relation,49| Δm = −CΔf/n |
Silica-coated quartz crystals (QSX 303, Q-Sense AB, Gothenburg, Sweden) were used as substrates for each experiment. Silica substrates were soaked in sodium dodecyl sulfate solution overnight, then rinsed with Milli-Q water, dried with nitrogen gas, and placed in an UV/ozone cleaner for 20 min to remove any trace organics before each experiment. The silica substrates were equilibrated with the background solution for 30 min to obtain baseline conditions. All solutions/suspensions were well mixed and injected into the QCM flow module at a rate of 0.1 mL min−1. The AgNP concentrations of the influent were maintained at 10 mg L−1 for all the QCM experiments. All QCM experiments were performed at an ionic strength of 5 mM (made with NaNO3) and buffered at pH 7.0 ± 0.2 with phosphate buffer.
The observed rate of AgNP deposition (dm/dt) was calculated from the time interval taken to reach 50% of the ultimate deposition; in this range, the relationship between deposition and time was linear (R2 > 0.98). This rate was divided by the influent particle mass rate to yield the normalized rate of deposition. The ultimate deposition was determined after the sensor electrode was washed with background electrolyte solution to remove unbound NPs and a new stable frequency reading (±0.05 Hz s−1) was reached. Because the third overtone exhibited the best signal-to-noise ratio, its Δf values are presented in this study.
Extended DLVO energy of interaction
The extended Derjaguin–Landau–Verwey–Overbeek (XDLVO) theory is widely used to calculate interparticle interactions of polymer capped nanoparticles.50 According to this approach, the total energy of interaction of particle–particle and particle–collector can be approximated as the sum of the electrostatic repulsion (VR), van der Waals attraction (VA) and steric hindrance (VS).51 Analytical expressions and values of parameters used in this study for the calculation of interactions are summarized in Tables S1 and S2.† It is worth mentioning that superposing steric hindrance over the classical DLVO energy of interaction is only semi-quantitative.52 Various inherent assumptions (e.g., Derjaguin approximation, linearized superposition, uniform polymer distribution, etc.) are used in the expressions listed in Table S1.52† Therefore, the calculated results of energy of interactions are for semi-quantitative comparison only. Nevertheless, the XDLVO calculations can highlight the influence on particle stability brought by the capping polymer.Results and discussion
Characterization of AgNPs after transformation
The HDD and the ζ-potential under different transformation conditions are shown in Fig. S1 and S2† and summarized in Table 1. All four types of particles were stable during the experimental time period as no aggregation was observed over 90 minutes (Fig. S3†). Exposure to HA did not lead to an apparent size increase of the PVP-AgNPs, either immediately or over time. This result can possibly be explained by the adsorption and/or ligand replacement of surface capping and the accompanying electrosteric hindrance as a result of the adsorbed HA.18,19,32 On the contrary, sulfidation led to a mean HDD increase from 87.3 nm for the pristine PVP-AgNPs to 93.6 nm after sulfidation using NTA and from 81.0 nm to 108.1 nm using DLS. The difference in the size change by these two measurements reflects the fundamental difference in the measurement methodology. The NTA tracks individual particles and calculates their sizes from the diffusive motion; the NTA makes no assumptions about the shape of the distribution. The DLS measures the light scattering from an ensemble of particles and fits a gaussian distribution (a mean size with a variance) that would give a similar light scattering. Because the intensity of scattered light is proportional to the sixth power of particle diameter and because of the assumption of the shape of the size distribution, the DLS intrinsically weights the larger particles to a greater extent than the NTA.53 Previous studies on the sulfidation process of AgNPs suggest the formation of Ag2S nanobridges between particles and a core–shell structure of a Ag2S shell surrounding the metallic silver core, both of which would lead to size increases.27,54 However, though sulfidation caused both a color change from the original yellow to black and the disappearance of the distinct surface plasmon resonance (SPR) absorption peak at 435 nm wavelength for spherical PVP-AgNPs (Fig. S4†), no significant aggregation during sulfidation as suggested by some literature27 was observed in this study. In preliminary experiments, pecipitates clearly formed and settled when the S/Ag ratio was greater than or equal to 50, but the sulfidized AgNPs remained in suspension at the stoichiometric S/Ag ratio of one used in this research.| PVP-AgNPs | HA-AgNPs | S-AgNPs | HA-S-AgNPs | |
|---|---|---|---|---|
| HDD (NTA) (nm) | 84.2 ± 25.0 | 84.5 ± 34.2 | 93.6 ± 24.8 | 94.6 ± 25.6 |
| HDD (DLS) (nm) | 81.0 ± 0.5 | 83.4 ± 1.0 | 108.1 ± 16.2 | 111.2 ± 14.3 |
| ζ-Potential (zetameter) (mV) | −25.3 ± 4.0 | −28.13 ± 6.3 | −38.9 ± 3.3 | −36.4 ± 3.2 |
| ζ-Potential (ELS) (mV) | −21.4 ± 2.4 | −20.1 ± 0.1 | −31.1 ± 8.1 | −29.2 ± 5.1 |
When HA was involved during sulfidation, the particle size increased after transformation very similarly to the increase with sulfidation alone, a finding consistent with other literature.28 Given the molar concentrations of sulfide and HA and the relative values of precipitation (Ksp,Ag2S = 6.2 × 10−52)24 and complexation constants (logKbinding,Ag-NOM = 9 − 9.2),55,56 we can expect that AgNPs associate with sulfide more favorably than with HA. Therefore, during sulfidation, a Ag2S shell would form at the surface of the pristine AgNPs core, and some fraction of the HA would adsorb on the Ag2S shell surface.
Although the data from the zetameter in Table 1 suggest that HA exposure alone led to a small increase in the negative ζ-potential, the difference is not statistically significant (p > 0.06). PVP was reported to bind more strongly with the Ag surface via direct bonding with oxygen or nitrogen,57 and was reportedly difficult to be displaced by NOM.58
Sulfidation led to substantially more negative ζ-potentials at pH 7. The more negative ζ-potential of sulfidized AgNPs compared to pristine PVP-AgNPs is thought to be caused by the stripping of the capping PVP polymer layer during sulfidation. In the presence of the PVP layer, the shear plane for the ζ-potential approximation was shifted towards the outer edge of the polymer layer from the particle core surface.59,60 As PVP is uncharged, a decreased potential is expected away from the silver core surface. After the PVP capping layer was removed by sulfidation, the determined potential of the sulfidized AgNPs was higher (more negative) than that of the original PVP-AgNPs. When both HA and sulfide were mixed with PVP-AgNPs, the more negative ζ-potential of the transformed AgNPs as compared to the pristine PVP-AgNPs again suggests a stripping of the PVP capping layer and an adsorption of HA on the particle surface. Compared to the transformation case where only sulfide was present, the presence of HA during sulfidation resulted in a surface charge closer to that of the HA only exposure. In the presence of both HA and sulfide at the concentrations present in this research, the formation of Ag2S on the particle surface is more favorable than the association of Ag-HA. Based on these results, a shell of Ag2S appears to first form around the particle core, followed by the binding of HA to the particle surface. With this interpretation, the ζ-potential is influenced by both the negatively charged core and the adsorbed HA on the surface.
The partial stripping of PVP and the adsorption of HA onto the particle surface during HA exposure and the complete stripping during sulfidation were confirmed by XPS analysis (Fig. 1 and 2, and Table 2). In Fig. 1a, after HA exposure, the intensity of the XPS nitrogen peak was greatly reduced; and after sulfidation, the nitrogen peak disappeared. Because the only source of nitrogen for PVP-AgNPs is the nitrogen in the hetero ring of PVP, the reduction or disappearance of the nitrogen peak in the XPS spectrum confirmed the covering or removal of the PVP during transformation. In addition, the appearance of a clear sulfur peak after sulfidation in Fig. 1b indicated the formation of Ag2S for the S-AgNPs and HA-S-AgNPs.
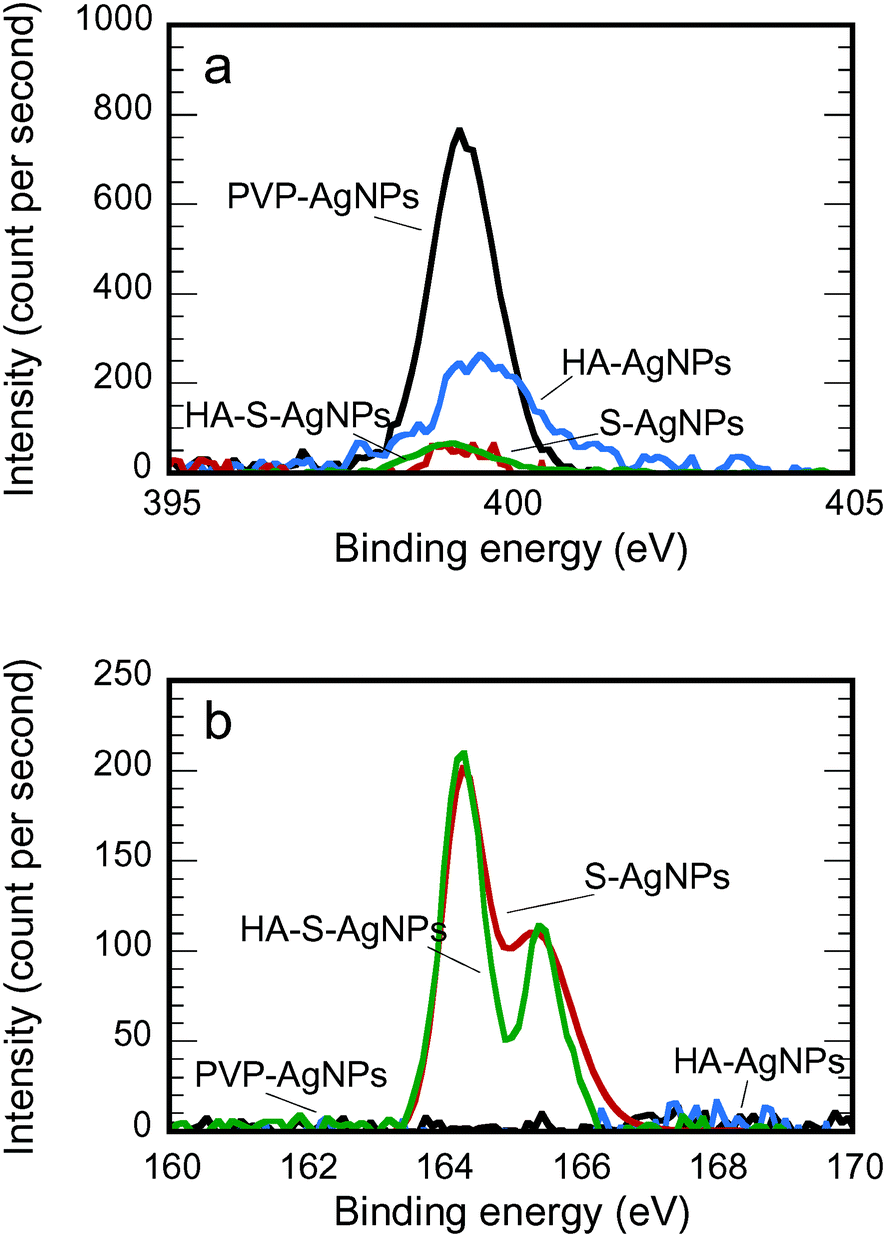 | ||
| Fig. 1 a) N 1s and b) S 2p XPS spectra for AgNPs with different environmental transformations. | ||
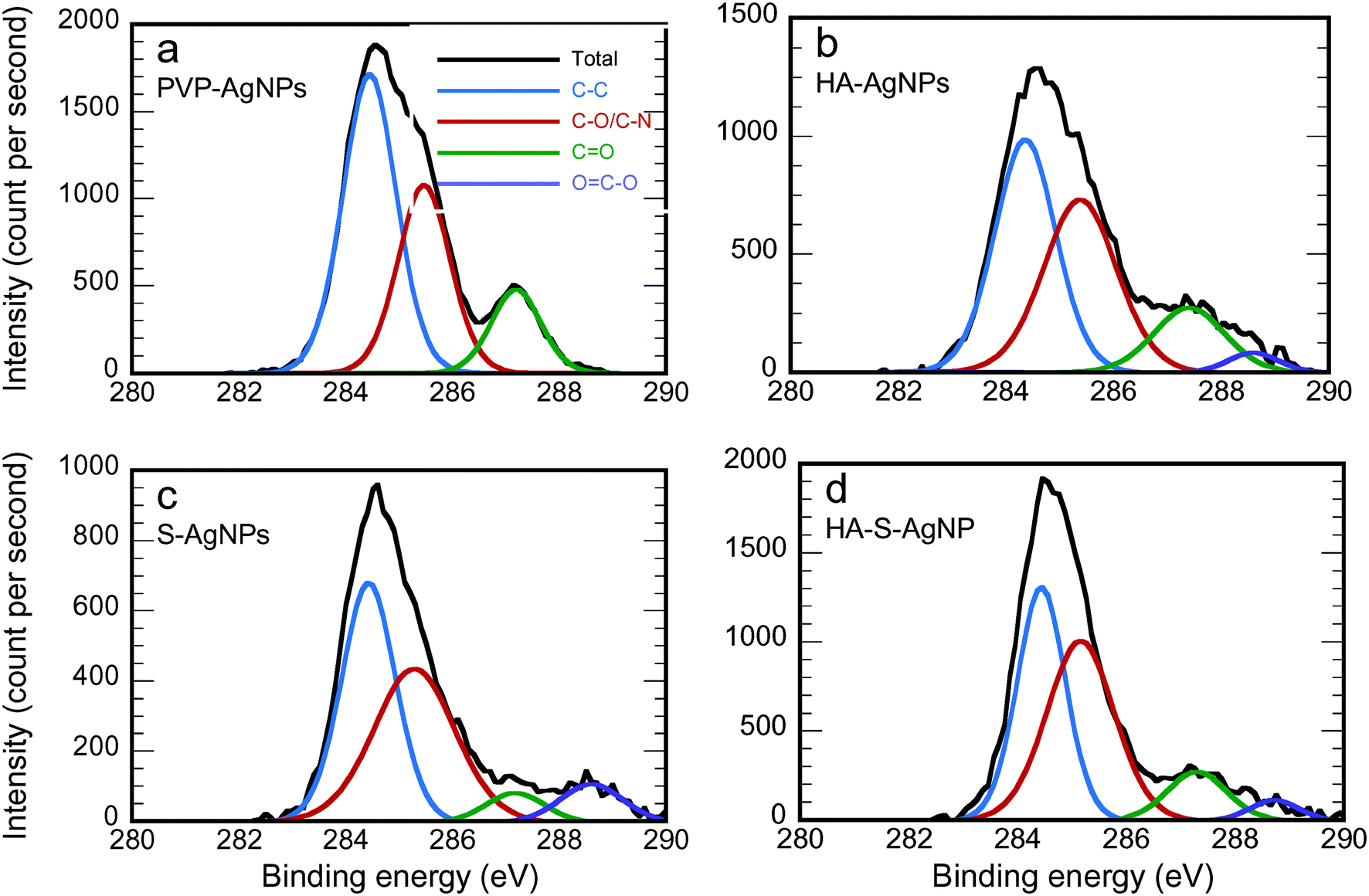 | ||
| Fig. 2 C 1s XPS experimental spectrum and the contribution of the fit of different transformed AgNPs (prepared in NaNO3 solution at pH = 7 with phosphate buffer): a) PVP-AgNPs, b) HA-AgNPs, c) S-AgNPs, and d) HA-S-AgNPs. | ||
| Functional group | Binding energy (eV) | PVP-AgNPs | HA-AgNPs | S-AgNPs | HA-S-AgNPs |
|---|---|---|---|---|---|
| C–C | 284.5 | 53.45 | 43.76 | 43.61 | 40.97 |
| C–N or C–O | 285.6 | 32.67 | 38.94 | 42.21 | 44.47 |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
287.5 | 13.89 | 14.19 | 5.94 | 10.80 |
| OC–O |
288.6 | 0 | 3.11 | 8.24 | 3.76 |
Fig. 2 shows the fitting of the C 1s XPS spectra by several Lorentzian and Gaussian functions representing different carbon bonds; the interpretation of these data is aided by the summary statistics in Table 2. The spectra in Fig. 2a for PVP-AgNPs match closely with what is expected for PVP; the data in Table 2 nearly equal the theoretical values for the percent of carbon in each type of bond, namely 50%, 33.3% and 16.7% for C–C, C–N, and CO, respectively. Considering the HA-AgNPs shown in Fig. 2b, there are two indications of the adsorption of HA onto the surface. First, the ratio of C–C to the combined C–N or C–O peak is closer to one than in the original PVP, and HA has been shown to have approximately that ratio.61 Second, the emergence of the peak for the carboxyl (OC–O) group stems from the carboxylic group of the HA.9,62 Considering both the data in Fig. 1a and 2b, the HA-AgNPs appear to have both PVP and HA on the surface. With sulfidation (Fig. 2c), the carbon is changed while the nitrogen is lost, as explained by the data in Fig. 1a. Finally, the spectra for the HA-S-AgNPs (Fig. 2d) is similar to that of the HA-AgNPs, indicating that the outer surface of the sulfidized particles exhibited adsorption of HA. A similar trend of surface composition change was also observed when the transformations were performed in Ca(NO3)2 electrolytes as shown in Fig. S5 and Table S3.†
Effects of transformation on AgNPs flocculation
In this research, flocculation was studied by measuring the particle size distribution via NTA on samples taken over time from the reaction vessels. Examples are shown in Fig. 3. The size distribution changed only slightly in 30 minutes when exposed to the NaNO3 solution (Fig. 3a) but the changes were much more substantial when exposed to the same ionic strength of a Ca(NO3)3 solution (Fig. 3b). Flocculation intrinsically reduces the particle number concentration (calculable as the area under the curves shown in Fig. 3) and shifts the distribution to larger sizes (to the right in Fig. 3); the reduction in particle number concentration is visible in both parts of Fig. 3 (albeit slight in Fig. 3a), but only the more extensive flocculation induced by the Ca(NO3)2 solution (Fig. 3b) leads to an obvious shift in the distribution. | ||
| Fig. 3 Changes in the particle size distribution brought about by flocculation for the non-transformed PVP AgNPs at an ionic strength of 10 mM induced by (a) NaNO3 and (b) Ca(NO3)2. | ||
The effects of environmental transformation on the flocculation of PVP-AgNPs are summarized from the particle size distributions via the loss in the total number concentration in Fig. 4. At I = 10 mM NaNO3 (Fig. 4a), the pristine PVP-AgNPs were quite stable. Though calculation of the classical DLVO interaction revealed no energy barrier (Fig. 5a), XDLVO calculation in Fig. 5b clearly indicates the contribution of steric hindrance to stability as the PVP polymer protrudes into the bulk solution and prevents aggregation when particles approach each other.5,63 In the same condition, the HA exposed particles were also stable. Though the protective PVP capping was partially removed, the adsorbed HA is thought to cause electrosteric hindrance.64
 | ||
| Fig. 4 Number fraction remaining during aggregation tests for PVP-AgNPs at pH 7 ± 0.2 and a) I = 10 mM with NaNO3 and b) I = 10 mM with Ca(NO3)2 with different environmental transformations. | ||
 | ||
| Fig. 5 Energy of interaction for self-aggregation at pH 7 ± 0.2 and I = 10 mM NaNO3 based on a) DLVO and b) XDLVO approaches. | ||
In the same NaNO3 solution, significant particle aggregation of both types of sulfidized AgNPs was observed by a decreased number fraction remaining. Loss of PVP reduced steric hindrance, the main stabilization mechanism for PVP-AgNPs. Although the sulfidized AgNPs had more negative ζ-potentials than the original PVP-AgNPs, the increased electrostatic repulsion was more than compensated for by the reduced steric hindrance: DLVO calculations reveal the lack of a repulsive energy barrier despite the charge due to the high Hamaker constant of silver (Fig. 5a and b). The PVP-AgNPs that were sulfidized with HA present showed less aggregation than the bare sulfidized AgNPs; the adsorbed HA on the particle surface apparently exerted some electrosteric hindrance in particle–particle interaction, thus partially recovering the stability of the AgNPs. According to the classical DLVO theory, the most negatively charged sulfidized AgNPs should be the most stable as the strongest electrostatic repulsion was expected. However, as shown by the experimental aggregation results in Fig. 4a and by the theoretical calculations in Fig. 5b, steric hindrance plays a more critical role than electrostatic repulsion during particle–particle interaction of macromolecule-coated particles, especially when the coating was intact (i.e., the pristine PVP-AgNPs and the HA-AgNPs).
Identical flocculation experiments were conducted at I = 10 mM with Ca(NO3)2 to determine the effects of divalent cation on particle–particle interaction. As shown in Fig. 4b, complexation between Ca2+ and surface functional groups resulted in stronger aggregation for PVP-AgNPs, HA-AgNPs and HA-S-AgNPs than in the sodium electrolyte. The PVP-AgNPs which were stable in NaNO3 showed an obvious particle number decrease over the experimental time period, probably as a result of the complexation between Ca2+ and the carbonyl group of the PVP on the particle surface.65,66 In contrast to the stable behavior of HA-AgNPs in the sodium electrolyte, HA-AgNPs showed strong aggregation when calcium electrolyte was used. Favorable inter-particle bridging aggregation through the Ca2+-carboxylic group complexation67–70 has been widely reported to explain the destabilization of HA coated particles in the presence of Ca2+. The HA-S-AgNPs also underwent extensive aggregation because of Ca2+-carboxylic group complexation.67–70 However, less aggregation was observed for HA-S-AgNPs than the HA-AgNPs, suggesting that less HA adsorbed onto the surface of S-AgNPs. Aggregation behavior of the bare S-AgNPs was similar to that in NaNO3, as expected from DLVO theory: the same diffuse layer thickness (i.e., the same inverse κ value) is expected under the same ionic strength regardless of electrolyte types, and the aggregation behavior should remain similar if only DLVO forces are considered. The bare S-AgNPs have no capping macromolecules on their surfaces, and only DLVO forces need to be included to evaluate their stability. Therefore, at the same ionic strength, similar aggregation behaviors were observed in both sodium and calcium electrolyte for these particles.
Effects of transformation on AgNPs deposition
The deposition behavior and the underlying particle–collector interaction of AgNPs under different transformation conditions were studied through both column tests and QCM. The column tests results indicated the affinity between the approaching particle and the collector, whereas QCM revealed both the initial rate of particle deposition and the ultimate deposition amount.As shown in Fig. 6a, PVP-AgNPs attached strongly to the collector surface (glass beads). The strong attachment of polymer coated particles has also been observed in previous research and was explained by “coating asymmetry”.6,43 We interpret this coating asymmetry to mean that hydrogen-bond bridging occurs between the amide carbonyl group of PVP and the silanol group on the glass surface.71 However, with the accumulation of PVP-AgNPs on the collectors' surface, the captured PVP-AgNPs will hinder the subsequent capture of PVP-AgNPs in the suspension as PVP molecules repulse each other sterically.5 Therefore, during the filtration experiment with PVP-AgNPs, the normalized effluent concentration increased continuously during column filtration, as shown in Fig. S7.† With more of the collector surface occupied by the attached PVP-AgNPs, the situation gradually changed from the favorable particle–collector interaction to the unfavorable particle–particle interaction. The rate of deposition from QCM exhibited the same strong initial attachment for PVP-AgNPs (Fig. 6b) as a result of the bridging effect of the PVP capping between the particle and the bare collector.
 | ||
| Fig. 6 a) Attachment efficiency (α) for the AgNPs transport in bare silica filter in column test, b) the normalized rate of AgNPs deposition during the first 50% deposition, and c) the ultimate deposition of AgNPs on bare silica at pH = 7.0 ± 0.2 and I = 5 mM NaNO3. | ||
In the filtration experiments with HA-AgNPs, the widely reported enhanced particle mobility due to HA exposure was not observed as a result of the different effects of unbound HA (in solution) and bound HA (on the particle surface). In those high particle mobility cases, the unbound HA molecules will surround the collector to induce electrosteric hindrance when the HA coated particles attempt to contact the collector surface.72,73 The current study excluded (to the extent possible) the influence of the unbound HA so only the bound HA on the particle surface can affect particle–collector interaction. Though the PVP capping was partially replaced or covered by the HA, the remaining PVP could still behave like a bridging agent between the particle and the collector. Moreover, as the original PVP capping was partially removed via HA adsorption and/or ligand exchange of HA for PVP, the unfavorable interaction between approaching particles and attached particles was reduced. Therefore, no climbing breakthrough curve as in the PVP-AgNPs case was observed for the filtration of the HA-AgNPs (Fig. S7†). The same trend was observed in the rate of deposition of QCM (Fig. 6b). HA-AgNPs also showed high deposition but with a higher variance due to the complexity of HA adsorption and/or ligand exchange.
Sulfidation was found to induce a different particle behavior in deposition from that in aggregation. Though sulfidized particles were less stable in the flocculation experiment, S-AgNPs showed much less attachment in column filtration (Fig. 6a) and a slower rate of deposition in QCM (Fig. 6b) than did the pristine PVP-AgNPs. The unfavorable particle–collector interaction can be explained from two perspectives. First, as the PVP for bridging was stripped, no particle–collector bridging effect remained to induce particle retention. Second, as shown in Table 1, sulfidation leads to a more negative ζ-potential for the particles than their pristine counterparts. As a result, more electrostatic repulsion is expected between the S-AgNPs and the collector surface and a clear DLVO energy barrier appears during the particle–collector interaction (Fig. S6a†). The increased particle mobility due to sulfidation was further evidenced when the S/Ag ratio was increased. As shown in Fig. S7,† when the S/Ag ratio was increased to 10 (as compared to the value of just greater than 1 in all of the other experiments), very few particles were captured by the collector because about 90% of the AgNPs injected into the filter penetrated to the effluent (Fig. S7†). In the case where the S/Ag ratio was as high as 10, stripping of PVP was more complete than in the case where S/Ag ≈ 1. Therefore, loss of PVP on the particle surface greatly reduced particle–collector affinity and enhanced particle mobility.
The filtration behavior in this study is different from the classical comparison between colloid aggregation and deposition where favorable chemical conditions for aggregation in jar tests generally lead to a strong deposition during granular media filtration.74 This contradiction to the classical particle–particle and particle–collector interaction theory also explains why ripening did not happen during the strong deposition condition of PVP-AgNPs filtration in this study. For ripening to occur, the collected particles act as additional media for particle capture, and the interaction between particles in the suspension and the captured particles is at least as favorable as the interaction between particles and the collector.75 However, in this study, the favorable surface condition is different for particle–particle interaction and particle–collector interaction.
In the column test, the HA-S-AgNPs showed similar deposition to the bare S-AgNPs, as shown in Fig. 6a. A similar trend in the deposition rate was observed in the QCM experiment with HA-S-AgNPs as the difference between S-AgNPs and HA-S-AgNPs are barely observable in Fig. 6b. However, it is clear that the deposition rate for both types of sulfidized particles (S-AgNPs and HA-S-AgNPs) is lower than the unsulfidized ones (PVP-AgNPs and HA-AgNPs) in both the column and QCM experiments. Considering the classical DLVO theory, the difference in deposition between the sulfidized and non-sulfidized particles could be attributed to the differences in their ζ-potentials. However, we believe that a stronger argument for the better deposition of the non-sulfidized particles is the strong affinity of PVP for the collector surfaces in both types of experiments.43,71
The different deposition of PVP-AgNPs, S-AgNPs and HA-S-AgNPs can be explained by the difference in their ζ-potentials; however, the strong deposition of HA-AgNPs cannot be explained from electrostatic repulsion alone. Non-DLVO forces have to be included to compare the interaction energies between particles and the surface in the presence of macromolecules.
Although the attachment efficiency in the column (Fig. 6a) and the rate of deposition in the QCM experiments (Fig. 6b) were similar, the ultimate deposition in the QCM measurements, shown in Fig. 6c, illustrated a quite different trend among the four types of AgNPs. The ultimate deposition of pristine PVP-AgNPs was quite low and far less than for the three modified AgNPs. PVP-AgNPs are sterically stabilized by the PVP molecules. Therefore, the PVP-AgNPs that first attached to the collector surface hindered the further attachment of approaching PVP-AgNPs sterically, resulting in the low ultimate deposition of PVP-AgNPs on substrate surfaces; this result from the QCM is also reflected in the time trend of the column deposition (Fig. S7†) in which only the PVP-AgNPs showed a substantial increase over time in the effluent concentration. For particles exposed to HA, the hindrance between the attached particles and the approaching particles was greatly reduced. As a result, no increasing effluent concentration profile was observed (Fig. S7†) and the ultimate deposition was greatly increased (Fig. 6c). Previous studies have shown that NOM can alter NP surfaces not only through adsorption but also by ligand exchange.76,77 The adsorption and/or ligand exchange of HA on the surface of AgNPs could potentially reduce steric repulsion by removing the PVP coating and/or altering its molecular orientation. The higher ultimate deposition of HA-AgNPs compared to PVP-AgNPs suggests that the adsorption and ligand-exchange of HA on the surface of AgNPs reduced steric repulsion and therefore enhanced the ultimate deposition of HA-AgNPs.
S-AgNPs also showed higher ultimate deposition than the original PVP-AgNPs because PVP was stripped during sulfidation. Attached particles would not exert steric repulsion to the approaching particles; therefore, the relative concentration curve in the column experiment remained stable and a higher ultimate deposition was observed. However, the ζ-potential of S-AgNPs was more negative than PVP-AgNPs. The QCM deposition and ζ-potential results indicated that the electrostatic force was less dominant than steric repulsion in controlling the ultimate deposition of S-AgNPs. With more particles deposited onto the collector surface, particle–collector interaction gradually became particle–particle interaction. HA-S-AgNPs showed the greatest ultimate deposition among the four types of AgNPs on the bare-silica substrate used in QCM. After some HA-S-AgNPs attached onto the collector surface, hydrophobic attraction exists between HA on the surface of both the attached particles and the approaching particles, leading to the increased deposition.18
Conclusions
In this study, the environmental transformations of HA adsorption and sulfidation were found to alter the surface composition and properties of PVP-AgNPs and thereby impacted the fate and transport of these particles in the water treatment processes of flocculation and granular media filtration. XPS revealed that HA alters the original PVP capping on the particle surface by adsorption and/or ligand exchange; and sulfidation stripped the surface PVP capping with the formation of Ag2S.Environmental transformations show different effects on aggregation and deposition of AgNPs. In experiments with NaNO3, the partial replacement of PVP by HA on the particle surface made little or no difference in either the stability in self-aggregation experiments or the mobility in clean bed deposition; as expected, the presence of calcium led to far greater self-aggregations of the HA-AgNPs than the PVP-AgNPs. When the filter media collector surface is clean, the remaining PVP on the HA-AgNPs exerts a bridging effect on the particle–collector interaction during filtration and allows effective removal to occur. Sulfidation stripped nearly all of the PVP from the surface and therefore rendered the AgNPs less stable during flocculation but more mobile during filtration.
The general idea that favorable flocculation conditions lead to favorable filtration conditions was not observed in the current study as a result of the changing role of the capping macromolecule as a bridging agent or a steric hindrance inducer. In addition, the bridging effect cannot be fully explained by the reduced electrostatic repulsion caused by the capping layer. Other forces need to be considered to clarify the difference between the bridging effect and the steric hindrance.
When comparing results from QCM and column tests, the initial rate of AgNPs deposition shows similar trend with the attachment efficiency calculated from colloidal filtration theory. The ultimate deposition in QCM indicates the final capacity of the collector and is more comparable to the interaction between attached particles and approaching particles. This study reveals the importance of clean surfaces and specific bridging to enhance the removal of AgNPs with macromolecules present.
Acknowledgements
The authors would like to thank Dr. Hugo Celio for technical support with the XPS measurements. This research was financially supported by the US National Science Foundation (CBET-1336139 and CBET 1336167).References
- G. Cornelis, Environ. Sci.: Nano, 2015, 2, 19–26 RSC.
- S. Yu, Y. Yin and J. Liu, Environ. Sci.: Processes Impacts, 2013, 15, 78–92 CAS.
- B. Nowack, J. F. Ranville, S. Diamond, J. A. Gallego-Urrea, C. Metcalfe, J. Rose, N. Horne, A. A. Koelmans and S. J. Klaine, Environ. Toxicol. Chem., 2012, 31, 50–59 CrossRef CAS PubMed.
- A. M. El Badawy, A. A. Hassan, K. G. Scheckel, M. T. Suidan and T. M. Tolaymat, Environ. Sci. Technol., 2013, 47, 4039–4045 CrossRef CAS PubMed.
- K. A. Huynh and K. L. Chen, Environ. Sci. Technol., 2011, 45, 5564–5571 CrossRef CAS PubMed.
- X. Yang, S. Lin and M. R. Wiesner, J. Hazard. Mater., 2014, 264, 161–168 CrossRef CAS PubMed.
- J. Flory, S. R. Kanel, L. Racz, C. A. Impellitteri, R. G. Silva and M. N. Goltz, J. Nanopart. Res., 2013, 15, 1–11 CrossRef.
- Y. Liang, S. A. Bradford, J. Simunek, H. Vereecken and E. Klumpp, Water Res., 2013, 47, 2572–2582 CrossRef CAS PubMed.
- M. Hoppe, R. Mikutta, J. Utermann, W. Duijnisveld and G. Guggenberger, Environ. Sci. Technol., 2014, 48, 12628–12635 CrossRef CAS PubMed.
- S. M. Louie, R. D. Tilton and G. V. Lowry, Environ. Sci.: Nano, 2016, 3, 283–310 RSC.
- J. M. Gorham, R. I. MacCuspie, K. L. Klein, D. H. Fairbrother and R. D. Holbrook, J. Nanopart. Res., 2012, 14, 1–16 CrossRef.
- Y. Yin, M. Shen, X. Zhou, S. Yu, J. Chao, J. Liu and G. Jiang, Environ. Sci. Technol., 2014, 48, 9366–9373 CrossRef CAS PubMed.
- X. Li and J. J. Lenhart, Environ. Sci. Technol., 2012, 46, 5378–5386 CrossRef CAS PubMed.
- A. L. Dale, E. A. Casman, G. V. Lowry, J. R. Lead, E. Viparelli and M. Baalousha, Environ. Sci. Technol., 2015, 49, 2587–2593 CrossRef CAS PubMed.
- C. Levard, E. M. Hotze, G. V. Lowry and G. E. Brown, Environ. Sci. Technol., 2012, 46, 6900–6914 CrossRef CAS PubMed.
- G. V. Lowry, B. P. Espinasse, A. R. Badireddy, C. J. Richardson, B. C. Reinsch, L. D. Bryant, A. J. Bone, A. Deonarine, S. Chae, M. Therezien, B. P. Colman, H. Hsu-Kim, E. S. Bernhardt, C. W. Matson and M. R. Wiesner, Environ. Sci. Technol., 2012, 46, 7027–7036 CrossRef CAS PubMed.
- G. R. Aiken, H. Hsu-Kim and J. N. Ryan, Environ. Sci. Technol., 2011, 45, 3196–3201 CrossRef CAS PubMed.
- O. Furman, S. Usenko and B. L. T. Lau, Environ. Sci. Technol., 2013, 47, 1349–1356 CAS.
- B. L. T. Lau, W. C. Hockaday, K. Ikuma, O. Furman and A. W. Decho, Colloids Surf., A, 2013, 435, 22–27 CrossRef CAS.
- A. Philippe and G. E. Schaumann, Environ. Sci. Technol., 2014, 48, 8946–8962 CrossRef CAS PubMed.
- L. Li, G. Hartmann, M. Döblinger and M. Schuster, Environ. Sci. Technol., 2013, 47, 7317–7323 CAS.
- S. M. King, H. P. Jarvie, M. J. Bowes, E. Gozzard, A. J. Lawlor and M. J. Lawrence, Environ. Sci.: Nano, 2015, 2, 177–190 RSC.
- R. Kaegi, A. Voegelin, C. Ort, B. Sinnet, B. Thalmann, J. Krismer, H. Hagendorfer, M. Elumelu and E. Mueller, Water Res., 2013, 47, 3866–3877 CrossRef CAS PubMed.
- J. R. Goates, A. G. Cole, E. L. Gray and N. D. Faux, J. Am. Chem. Soc., 1951, 73, 707–708 CrossRef CAS.
- M. Baalousha, K. P. Arkill, I. Romer, R. E. Palmer and J. R. Lead, Sci. Total Environ., 2015, 502, 344–353 CrossRef CAS PubMed.
- B. Thalmann, A. Voegelin, E. Morgenroth and R. Kaegi, Environ. Sci.: Nano, 2016, 3, 203–212 RSC.
- C. Levard, B. C. Reinsch, F. M. Michel, C. Oumahi, G. V. Lowry and G. E. Brown, Environ. Sci. Technol., 2011, 45, 5260–5266 CrossRef CAS PubMed.
- S.-W. Lee, S.-Y. Park, Y. Kim, H. Im and J. Choi, Sci. Total Environ., 2016, 553, 565–573 CrossRef CAS PubMed.
- B. Collin, O. V. Tsyusko, D. L. Starnes and J. M. Unrine, Environ. Sci.: Nano, 2016, 3, 728–736 RSC.
- J. Gao, K. Powers, Y. Wang, H. Zhou, S. M. Roberts, B. M. Moudgil, B. Koopman and D. S. Barber, Chemosphere, 2012, 89, 96–101 CrossRef CAS PubMed.
- M. Delay, T. Dolt, A. Woellhaf, R. Sembritzki and F. H. Frimmel, J. Chromatogr. A, 2011, 1218, 4206–4212 CrossRef CAS PubMed.
- Y. Yin, M. Shen, Z. Tan, S. Yu, J. Liu and G. Jiang, Environ. Sci. Technol., 2015, 49, 6581–6589 CrossRef CAS PubMed.
- E. Lombi, E. Donner, S. Taheri, E. Tavakkoli, Å. K. Jämting, S. McClure, R. Naidu, B. W. Miller, K. G. Scheckel and K. Vasilev, Environ. Pollut., 2013, 176, 193–197 CrossRef CAS PubMed.
- O. Choi, T. E. Clevenger, B. Deng, R. Y. Surampalli, L. Ross and Z. Hu, Water Res., 2009, 43, 1879–1886 CrossRef CAS PubMed.
- N. Kinsinger, R. Honda, V. Keene and S. L. Walker, Environ. Eng. Sci., 2015, 32, 292–300 CrossRef CAS.
- Q. Sun, Y. Li, T. Tang, Z. Yuan and C.-P. Yu, J. Hazard. Mater., 2013, 261, 414–420 CrossRef CAS PubMed.
- M. M. Benjamin and D. F. Lawler, Water Quality Engineering: Physical/Chemical Treatment Processes, John Wiley & Sons, Inc., Hoboken, New Jersey, 2013 Search PubMed.
- X. Liu, G. Chen and C. Su, Environ. Sci. Technol., 2012, 46, 6681–6688 CrossRef CAS PubMed.
- Q. Chen, S. Xu, Q. Liu, J. Masliyah and Z. Xu, Adv. Colloid Interface Sci., 2016, 233, 94–114 CrossRef CAS PubMed.
- K. Kubiak, Z. Adamczyk and M. Oćwieja, Langmuir, 2015, 31, 2988–2996 CrossRef CAS PubMed.
- M. Tong, J. Ding, Y. Shen and P. Zhu, Water Res., 2010, 44, 1094–1103 CrossRef CAS PubMed.
- X. Jiang, X. Wang, M. Tong and H. Kim, Environ. Pollut., 2013, 174, 38–49 CrossRef CAS PubMed.
- S. Lin, Y. Cheng, J. Liu and M. R. Wiesner, Langmuir, 2012, 28, 4178–4186 CrossRef CAS PubMed.
- P. Liu and M. Zhao, Appl. Surf. Sci., 2009, 255, 3989–3993 CrossRef CAS.
- D. F. Lawler, S. Youn, T. Zhu, I. Kim and B. L. T. Lau, Water Sci. Technol., 2015, 72, 2318–2324 CrossRef CAS PubMed.
- J. E. Tobiason and C. R. O'Melia, J. - Am. Water Works Assoc., 1988, 80, 54–64 CAS.
- N. Tufenkji and M. Elimelech, Environ. Sci. Technol., 2004, 38, 529–536 CrossRef CAS PubMed.
- A. A. Feiler, A. Sahlholm, T. Sandberg and K. D. Caldwell, J. Colloid Interface Sci., 2007, 315, 475–481 CrossRef CAS PubMed.
- M. Rodahl, F. Höök, A. Krozer, P. Brzezinski and B. Kasemo, Rev. Sci. Instrum., 1995, 66, 3924–3930 CrossRef CAS.
- A. M. Mikelonis, S. Youn and D. F. Lawler, Langmuir, 2016, 32, 1723–1731 CrossRef CAS PubMed.
- A. J. Worthen, V. Tran, K. A. Cornell, T. M. Truskett and K. P. Johnston, Soft Matter, 2016, 12, 2025–2039 RSC.
- C. A. S. Batista, R. G. Larson and N. A. Kotov, Science, 2015, 350, 1242477 CrossRef PubMed.
- V. Filipe, A. Hawe and W. Jiskoot, Pharm. Res., 2010, 27, 796–810 CrossRef CAS PubMed.
- R. D. Kent, J. G. Oser and P. J. Vikesland, Environ. Sci. Technol., 2014, 48, 8564–8572 CrossRef CAS PubMed.
- C. N. Glover, R. C. Playle and C. M. Wood, Environ. Toxicol. Chem., 2005, 24, 2934 CrossRef CAS PubMed.
- N. Janes and R. C. Playle, Environ. Toxicol. Chem., 1995, 14, 1847–1858 CrossRef CAS.
- W. A. Saidi, H. Feng and K. A. Fichthorn, J. Phys. Chem. C, 2013, 117, 1163–1171 CAS.
- I. L. Gunsolus, M. P. S. Mousavi, K. Hussein, P. Bühlmann and C. L. Haynes, Environ. Sci. Technol., 2015, 49, 8078–8086 CrossRef CAS PubMed.
- L. Kong, J. K. Beattie and R. J. Hunter, Phys. Chem. Chem. Phys., 2001, 3, 87–93 RSC.
- H. Ohshima, Adv. Colloid Interface Sci., 1995, 62, 189–235 CrossRef CAS.
- J.-L. Lin, C. Huang, B. Dempsey and J.-Y. Hu, Water Res., 2014, 56, 314–324 CrossRef CAS PubMed.
- S. Liang, G. Li and R. Tian, J. Mater. Sci., 2016, 51, 3513–3524 CrossRef CAS.
- K. M. Koczkur, S. Mourdikoudis, L. Polavarapu and S. E. Skrabalak, Dalton Trans., 2015, 44, 17883–17905 RSC.
- S. M. Louie, E. R. Spielman-Sun, M. J. Small, R. D. Tilton and G. V. Lowry, Environ. Sci. Technol., 2015, 49, 2188–2198 CrossRef CAS PubMed.
- V. Vao-soongnern, K. Merat and S. Horpibulsuk, J. Polym. Res., 2016, 23, 1–7 CrossRef CAS.
- R. Dluhy, D. G. Cameron, H. H. Mantsch and R. Mendelsohn, Biochemistry, 1983, 22, 6318–6325 CrossRef CAS.
- H. Dong and I. M. C. Lo, Water Res., 2013, 47, 2489–2496 CrossRef CAS PubMed.
- X. Liu, M. Wazne, T. Chou, R. Xiao and S. Xu, Water Res., 2011, 45, 105–112 CrossRef CAS PubMed.
- E. Topuz, J. Traber, L. Sigg and I. Talinli, Environ. Pollut., 2015, 204, 313–323 CrossRef CAS PubMed.
- A. M. El Badawy, T. P. Luxton, R. G. Silva, K. G. Scheckel, M. T. Suidan and T. M. Tolaymat, Environ. Sci. Technol., 2010, 44, 1260–1266 CrossRef CAS PubMed.
- M. Toki, T. Y. Chow, T. Ohnaka, H. Samura and T. Saegusa, Polym. Bull., 1992, 29, 653–660 CrossRef.
- G. Chen, X. Liu and C. Su, Environ. Sci. Technol., 2012, 46, 7142–7150 CrossRef CAS PubMed.
- L. Wang, Y. Huang, A. T. Kan, M. B. Tomson and W. Chen, Environ. Sci. Technol., 2012, 46, 5422–5429 CrossRef CAS PubMed.
- M. T. Habibian and C. R. O'Melia, J. Environ. Eng. Div., 1975, 101, 567–583 CAS.
- J. L. Darby and D. F. Lawler, Environ. Sci. Technol., 1990, 24, 1069–1079 CrossRef CAS.
- B. Gu, T. L. Mehlhorn, L. Liang and J. F. McCarthy, Geochim. Cosmochim. Acta, 1996, 60, 1943–1950 CrossRef CAS.
- A. P. Gondikas, A. Morris, B. C. Reinsch, S. M. Marinakos, G. V. Lowry and H. Hsu-Kim, Environ. Sci. Technol., 2012, 46, 7037–7045 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 7 figures and 3 tables showing the calculation methods and detailed experimental results. See DOI: 10.1039/c6en00266h |
| This journal is © The Royal Society of Chemistry 2016 |