
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFractal aggregation and disaggregation of newly formed iron(III) (hydr)oxide nanoparticles in the presence of natural organic matter and arsenic†
Chelsea W.
Neil‡
a,
Jessica R.
Ray§
a,
Byeongdu
Lee
b and
Young-Shin
Jun
*a
aDepartment of Energy, Environmental & Chemical Engineering, Washington University in St. Louis, St. Louis, MO 63130, USA. E-mail: ysjun@wustl.edu
bX-ray Science Division, Argonne National Laboratory, Argonne, IL 60439, USA
First published on 7th April 2016
Abstract
Water chemistry affects the nucleation kinetics, precipitate morphology, and quantity of iron(III) (hydr)oxide nanoparticles, directly impacting the reactive surface area of geomedia and fate of associated waterborne contaminants. In this study, we utilized in situ grazing-incidence small angle X-ray scattering (GISAXS) and complementary ex situ techniques to investigate heterogeneous iron(III) (hydr)oxide nucleation on quartz in the presence of natural organic matter (NOM) and arsenate. Results indicate unique fractal aggregation behavior in the systems containing NOM and precipitating iron(III) (hydr)oxide nanoparticles. Furthermore, the coexistence of arsenic and NOM lead to the formation of two distinct particle size ranges: larger particles dominated by arsenic effects, and smaller particles dominated by NOM effects. These new findings provide important implications for understanding the nucleation, growth, and aggregation of iron(III) (hydr)oxides in aqueous systems where NOM is present, such as natural surface waters and water and wastewater treatment plants. This study also offers new insight into how NOM-associated iron(III) (hydr)oxides can interact with aqueous contaminants such as arsenate.
Nano impactA better understanding of the fate and transport of iron(III) (hydr)oxide nanoparticles is crucial for controlling trace toxic contaminants in natural and engineered aquatic systems. Despite this, their nucleation, growth, and aggregation are not well understood. We studied the impacts of natural organic matter and arsenate on newly-forming iron(III) (hydr)oxide in situ and found that these constituents altered the nanoparticle size and aggregation state. Our findings allow us to elucidate how contaminants affect the aggregation or disaggregation of iron(III) (hydr)oxide nanoparticles in aqueous environments, where organic matter will be nearly ubiquitously present. Outcomes can also be informative in developing reactive transport models that better predict the ability of iron(III) (hydr)oxides to attenuate aqueous contaminants. |
Introduction
Iron(III) (hydr)oxide nanoparticles strongly influence contaminants' fate and transport in natural and engineered aquatic systems due to their high reactive surface area and capacity to adsorb waterborne trace contaminants.1–3 The ability of iron(III) (hydr)oxides to adsorb or co-precipitate with these contaminants depends largely on water chemistry, which affects the size, composition, surface charge, and aggregation state of these nanoparticles.4,5 In particular, common naturally-occurring water components, such as arsenic and natural organic matter (NOM), can have significant impacts on iron(III) (hydr)oxide nanoparticle precipitation kinetics and precipitate morphology.6–8In surface and groundwater environments, iron(III) (hydr)oxides form naturally due to the weathering of iron-containing minerals, such as pyrite.9 At high supersaturation, iron(III) (hydr)oxides can nucleate homogeneously, meaning that they will form in solution, while at low supersaturation, iron(III) (hydr)oxides will prefer to form heterogeneously on substrates due to lower activation energy barriers.10 The formation location of these iron(III) (hydr)oxides can impact their propensity to act as a sink for aqueous contaminants. When new iron(III) (hydr)oxide nanoparticles form homogeneously, they can continue to be transported downstream, while heterogeneously formed particles on substrates can be immobilized along with their associated contaminants. The fate and transport of newly formed nanoparticles can also be affected by their aggregation status.11,12 In natural environmental systems, various mineral surfaces can serve as nucleation substrates, including environmentally-abundant mineral surfaces13 and mineral surfaces coated by organics, such as biofilms.14 The organic coatings on minerals can alter heterogeneous iron(III) (hydr)oxide nucleation and growth kinetics from bare mineral surfaces by changing the hydrophilicity of the surface.14 To better characterize the fraction of contaminants which will be truly immobilized by iron(III) (hydr)oxide formation, it is crucial to quantitatively and qualitatively investigate heterogeneous precipitation in situ. To address this scientific need, in our previous study, we have established a new approach using in situ grazing incidence small angle X-ray scattering (GISAXS).10 Utilizing this new capability, the effects of arsenate on iron(III) (hydr)oxide nucleation and growth and their phase identity were studied.4 Interestingly, we found that the iron (hydr)oxides that formed in the presence of arsenate (As(V)) have larger heterogeneous particle sizes and higher water content.4 The results also indicated that arsenate was incorporated into nucleating iron(III) (hydr)oxides and acted as a bridge between iron tetrahedra, forming larger, less crystalline particles.
These strong interactions between iron(III) (hydr)oxides and arsenate can occur because iron(III) (hydr)oxides in the environment will frequently have a positive charge, attracting negatively charged arsenate oxyanions. For instance, the pHpzc values of ferrihydrite, goethite, and hematite are in the range of 8.5–9.5, sufficiently high to have a positive surface charge under the circumneutral pH conditions which dominate natural aqueous systems.15 These electrostatic forces can also promote iron(III) (hydr)oxide interactions with other negatively charged water components such as natural organic matter (NOM).4 In regions where arsenic contamination of ground and surface waters is a concern, such as Bangladesh,9 NOM is also ubiquitously present at concentrations ranging from 0–10 mg C (carbon) per L in surface water and 0–2 mg C per L in groundwater.16 Therefore, it is important to fully understand how the presence of arsenic, together with NOM, will impact iron(III) (hydr)oxide precipitation.
While there are limited studies which have looked at the precipitation of iron(III) (hydr)oxides in the co-presence of NOM and As(V), the studies which have examined sequential addition of these compounds can give some insight into this system. It has been reported that the adsorption of NOM on iron(III) (hydr)oxides inhibits arsenic adsorption due to steric and electrostatic effects.17–23 This occurs because NOM functional groups such as carboxylic acids and phenols bind directly to the iron(III) (hydr)oxide surface, or because they form outer sphere complexes.20,24–26 Studies have also focused on NOM effects on arsenic sorbed onto iron(III) (hydr)oxides. Bauer and Blodau27 found that a 25–50 mg L−1 dissolved organic matter (DOM) solution was able to mobilize arsenic from all iron(III) oxide solid phases, with a maximum mobilization of 53.3%. Sharma et al.28 found that 10–16% of sorbed As(V) could be desorbed from ferrihydrite in a soil column when flushed with a solution containing organic matter at concentrations ranging from 5 to 50 mg C per L.
In addition, because the functional groups of NOM deprotonate at different pH values, the system pH can impact the sorption efficiency of NOM onto iron(III) (hydr)oxides.29 The majority of these functional groups (e.g., carboxylic groups) will be negatively charged under neutral pH conditions; the humic fraction has been shown to have a negative surface charge at pH > 2.30 Studies of the fulvic fraction of NOM showed that it has the largest impact on arsenate sorption at a pH of 4, close to our system pH, and that this impact was decreased at pH 6 and 8 due to the change in deprotonation from carboxylic groups to phenolic groups.31 This phenomenon results from the formation of stronger, inner-sphere complexes for carboxylic groups, while phenolic groups formed outer-sphere complexes.20,31
While the previously discussed studies have focused on arsenic and NOM interactions with preformed iron(III) (hydr)oxides, only a few studies have also examined iron(III) hydroxide precipitation in the presence of NOM only and NOM and arsenic. With regards to As–Fe–NOM interactions, Liu et al. reported that As(III) can complex with stable Fe–NOM colloids.32 Chen et al. found that iron(III) (hydr)oxides formed in the presence of NOM contained more organic carbon, although arsenic was not present for their investigations.33 Currently, however, no studies have examined in situ the early stages of iron(III) (hydr)oxide formation in solutions containing both arsenic and NOM, which reflects natural systems more closely than using a sequential experimental approach.
The current study uses our established GISAXS experimental approach to study iron(III) (hydr)oxide nucleation and growth in the presence of NOM and in the presence of both NOM and arsenate, significantly advancing the available literature.21,32–34 In the system where arsenate and NOM coexist with precipitating iron(III) (hydr)oxides, our observations reveal unique behaviors that impact both the individual precipitate size and aggregate structure. These changes can in turn affect the fate and transport of iron(III) (hydr)oxides in the environment, and the consequential differences in the aggregate structure can further influence iron(III) (hydr)oxide sorption properties.35 Our findings thus have valuable implications for understanding and modeling iron(III) (hydr)oxide-associated contaminant fate and transport in aquatic systems containing arsenate and NOM.
Experimental methods
Sample preparation
For heterogeneous experiments, iron(III) (hydr)oxide nucleation was investigated on a cleaned (110) quartz surface. Quartz was chosen due to its abundance in natural aquatic systems. Because there is no natural cleavage plane for quartz, the abundance of the (110) surface is expected to be similar to the abundance of surfaces with similar energies.4 Single crystal wafers were purchased from MTI Corporation (Richmond, CA) with a surface roughness of less than 5 Å. Quartz wafers were cut to 0.5 cm squares and cleaned using Nochromix® and sulfuric acid prior to experiments.Reagent grade chemicals and ultrapure water were used to make all reaction solutions. All reaction solutions included 10−4 M Fe(III) from Fe(NO3)3·9H2O and 10 mM NaNO3. Systems for testing of arsenate and/or NOM effects also contained 10−5 M As(V) from Na2HAsO4·7H2O and 1.5 mg L−1 non-purgeable organic carbon (NPOC) from Suwanee River NOM (SRNOM). The pH of all experimental systems was 3.6 ± 0.2, and it did not vary over the course of the one hour reaction period.
The 10−5 M (∼750 ppb) concentration for arsenic was chosen as a worst case scenario for arsenic contamination. However, concentrations comparable to and even exceeding this value have been observed in the field, particularly for water sources impacted by mining activities. For example, well waters in the Pedro Dome–Cleary Summit area, Alaska, have measured arsenic concentrations as high as 1.7 × 10−5 M (1260 ppb) in stream waters.36 Gecol et al.37 also reported arsenic concentrations ranging from 5 × 10−9 M–1.3 × 10−5 M (0.38–1000 ppb) in lakes and 1.3 × 10−8 M–1.3 × 10−5 M (1–1000 ppb) in ground water. The concentration of 10−4 M for iron was chosen because this particular concentration is found frequently in the environment,38,39 and because it allows optimal conditions to observe the early nucleation and growth of iron(III) (hydr)oxides.
There are three reasons why a pH of 3.6 was used in this study: first, this pH can be relevant for water treatment using Fenton advanced oxidation processes (AOPs), which are more effective at pH 3,40 and for acid mine drainage systems, where there is also a particular interest in the interactions between aqueous iron and arsenic during or after the dissolution of arsenic-containing iron sulfide minerals. NOM can also be present in these natural and engineered aqueous systems. Second, a low pH is necessary to elucidate early nucleation processes. At high pH values, precipitation of iron(III) (hydr)oxides will occur too rapidly to be observed. Finally, the pH of 3.6 prevents introduction of any error due to additional pH adjustment.
Suwannee River NOM (SRNOM, 2R101N, RO isolation), purchased from the International Humic Substances Society, was used as the NOM source. Details on the preparation of SRNOM stock solutions can be found in Section S1 in the ESI.† Interactions between iron(III) (hydr)oxides and NOM occur primarily due to the carboxyl and phenolic acidic functional groups present on the NOM.41 Suwannee River NOM from the IHSS has been well-characterized. The quantification of carboxyl and phenolic acidic functional groups and their log K values can be found in Table S1 in the ESI.†
The pKa1 for our NOM is 4.16, thus the carboxylic portion of NOM will be at least partially deprotonated at the system pH. Our NOM also contains a large portion of carboxylic groups (11.21 meq g−1 C) relative to phenolic groups (2.47 meq g−1 C) according to characterization provided by the International Humic Substances Society (Table S1 in the ESI†). Furthermore, previous literature has shown that these carboxylic functional groups are key in forming strong complexes between NOM and oxide surfaces.41 This is especially true under acidic or slightly acidic conditions.42 Thus, we do expect some charge to be present which can impact interactions with highly positively charged iron(III) (hydr)oxides.
In situ GISAXS measurement and analysis procedures
For each GISAXS experiment, a clean piece of (110) quartz was placed in a specially designed GISAXS fluid cell. The quartz was aligned vertically and horizontally with respect to the X-ray beam and a background image was taken of the clean quartz surface in ultrapure water (resistivity > 18.2 MΩ cm). Then, water was removed and the freshly made reaction solution (Fe(III) only, Fe(III) + As(V), Fe(III) + NOM, or Fe(III) + As(V) + NOM) was injected into the cell. It is not likely for precipitates to form in the solution before injection. If any did form, these homogeneously formed precipitates would still be much larger than those observed to form on the quartz surface over the course of the 1 hour reaction, and thus did not affect our observation of heterogeneous nucleation and growth. The size of these homogeneous particles can range from 4 nm initially to greater than 20 nm after one hour of reaction.5Approximately two minutes lagged between when the solution was created and when the first GISAXS image was taken, which we counted in the reaction time. An incidence angle (αi) of 0.11° was chosen for GISAXS measurements to have 98% reflectivity on substrates at the 14 keV beam energy. The scattering vector range (e.g. q range) was 0.007–0.300 Å−1. All GISAXS experiments were conducted at beamline 12-ID-B at the Advanced Photon Source in Argonne National Laboratory (Argonne, IL).
GISAXS images were taken every 10 minutes for one hour to minimize any beam interactions with the systems. Scans of multiple locations on the sample were also taken to ensure no beam interactions took place. These time-resolved GISAXS images show how 2D scattering increases on the quartz surface as iron(III) (hydr)oxide nucleates and grows in situ. Cutting these images along the Yoneda wing reduces 2D information to 1D plots of scattering intensity (I) versus q. The bend position of this plot is inversely related to the particle size and the area under the plot, e.g., the integral, is proportional to the total particle volume. Scattering in the low q range, e.g., power law scattering, is related to particle aggregation. Fitting of the data gives estimations for the radius of gyration (Rg) of the newly formed nanoparticles as well as information about their aggregation state. Detailed information on data fitting can be found in Section S2 in the ESI† and in our previous publications.4,5,10,14 In addition, the invariant can be calculated from the scattering intensity using the following formula:4,5,10,14
 | (1) |
The invariant is proportional to the total nanoparticle volume, and thus these quantitative values can be used to compare heterogeneous precipitation amounts between the different reaction systems. Analysis of GISAXS data was carried out using the GISAXS-SHOP macro in Igor Pro (v 6.22 A, WaveMetrics, Inc., OR), which is available at APS beamline 12-ID-B.
Ex situ precipitate characterization
To examine precipitate morphology, homogeneously formed precipitates were characterized using transmission electron microscopy (TEM, FEI Tecnai Spirit, Hillsboro, OR) and heterogeneously formed precipitates were characterized using atomic force microscopy (AFM, Veeco Inc.). For TEM samples, a single drop of each reaction solution was placed on 300-mesh Cu Formvar-carbon grids after 1 hour of reaction and dried in a desiccator immediately prior to imaging. Particle size comparison with TEM images was not carried out because, while the size change trends can be similar, based on our previous studies, the homogeneously formed iron(III) (hydr)oxide particle size is much larger than the heterogeneously formed particle size.5,10 Thus, size comparison with AFM images will be more accurate.Tapping mode AFM was used to image the quartz substrates following one hour of reaction in the GISAXS fluid cell for all four reaction systems. In order to deduce the mechanism of NOM effects, sequential injection of reaction components for the Fe(III) + As(V) + NOM system was tested by first reacting Fe(III) and As(V) or NOM, and then feeding in the third component (either As(V) or NOM) after 30 minutes into the reaction period. Tapping mode probes (125 μm long with phosphorus (n) doped silicon tips, nominal tip radius of 10 nm, MPP-11100–10, Bruker probes) were used with a scanning rate of 0.988 Hz and the drive frequencies were between 312 and 320 kHz. Images were processed with Nanoscope 7.20 software.
A Zetasizer (Nano ZS, Malvern Instruments Ltd., Westborough, MA) was used to measure the zeta potential of the newly formed iron(III) (hydr)oxides. For these experiments, the reaction solutions were created as previously described, mixed with finely ground quartz powder, and reacted for one hour. Then, the solutions, which contained iron(III) (hydr)oxides precipitated on suspended quartz powder, were injected into a capillary cell to measure the zeta potential. More details on zeta potential measurement for the quartz powder can be found in the ESI† (S1). A thermal gravimetric analyzer (TGA, TA Instruments Q5000 IR Thermogravimetric Analyzer) was also used to quantify the water content of the precipitates. For TGA measurements, the nanoparticles were first concentrated using Millipore Amicon ultra-15 centrifugal filter units and dried overnight in a desiccator under ambient conditions. Then, the samples were placed in high temperature platinum TGA pans and the temperature was increased incrementally to 950 °C. It was assumed that after the temperature exceeded 107 °C, mass lost was due to water in the iron(III) (hydr)oxide crystal structure rather than from the nanoparticle slurry.4 The phase of homogeneous precipitates was determined using high-resolution X-ray diffraction (HRXRD). For this analysis, the homogeneously precipitated particles were first concentrated using Millipore Amicon ultra-15 centrifugal filter units and then dried overnight in a desiccator. The samples were packed in Kapton® polyimide capillary tubes and sent to sector 11-BM of the APS at ANL, where they were analyzed using HRXRD after approximately 7 days of particle aging. One caveat about ex situ phase identification using HRXRD is that drying of the particles and seven days of aging can result in a more crystalline phase than occurs in situ. Finally, a contact angle analyzer (Phoenix 300, SEO Corporation, Korea) was used to look at hydrophilicity/hydrophobicity changes of As(V) and NOM solutions. For these experiments, a clean (110) quartz substrate was utilized. The contact angle was measured between the substrate and solutions that contained 10 mM sodium nitrate, 10 mM sodium nitrate + 1.5 mg L−1 NPOC, and 10 mM sodium nitrate + 1.5 mg L−1 NPOC + 10−5 M As(V).
Details on additional characterization using Fourier transform infrared spectroscopy (FTIR) and X-ray absorption spectroscopy (XAS) can be found in the ESI† (S3).
Results & discussion
Fractal aggregation of NOM-containing precipitates
The in situ GISAXS results show obvious differences in the scattering pattern for systems with and without NOM present, particularly in the low q range (0.007–0.03 Å−1). Fig. 1 presents the 1D X-ray scattering for the four systems. The indicated radii show the growth of nanoparticles after a 1 h reaction (Fig. 1F). The particle size is larger for the system with arsenate when NOM is not present (e.g., radius of gyration (Rg) = 5.3 nm in Fig. 1B (Fe(III) + As(V)) vs. 2.1 nm in Fig. 1A (Fe(III) only), respectively), which is consistent with our previous publication.4 In the presence of NOM, the size of particles in the Fe(III) + NOM system decreases slightly to 1.8 nm (Fig. 1C). For the ternary system with Fe(III) + As(V) + NOM, there were differences between the in situ GISAXS replicate trials which were categorized into two distinct behaviors. In one case, smaller particles (Rg = 1.6 nm) similar in size to the Fe(III) + NOM system and fractal aggregation were observed on the surface (Fig. 1D). In another, larger particles (Rg = 8.6) more similar to the Fe(III) + As(V) system were observed (Fig. 1E). These two distinct trends were observed during multiple trials at different beamtime periods, indicating that for all samples reacted under ternary conditions, there were regions where particles were larger and regions where particles were smaller. | ||
| Fig. 1 GISAXS 1D scattering intensity for iron (III) hydroxide precipitation on quartz in the presence of (A) 10 mM sodium nitrate, (B) 10 mM sodium nitrate with 10−5 M As(V), (C) 10 mM sodium nitrate and 1.5 mg L−1 NPOC from NOM, and (D, E) 10 mM sodium nitrate, 10−5 M As(V), and 1.5 mg L−1 NPOC from NOM. The lack of dots in the low q range in plot A indicates that the background scattering for that region was higher than the scattering at the 2 min time point (low signal to noise ratio). The radius of gyration (Rg) of nanoparticles after 1 hour of reaction is indicated on the individual plots and the primary particle size evolution is shown in plot F. | ||
The relative total particle volumes were also calculated for these systems after a 1 hour reaction. However, the presence of arsenic can increase the electron density contrast of particles, increasing the scattering intensity in a manner independent of intensity change due to increased particle volume. Thus, the presence of arsenic in the Fe(III) + As(V) system and Fe(III) + As(V) + NOM systems complicates the direct comparison. The electron density contrast in the Fe(III) + As(V) system was calculated to be 1.2 times that of the Fe(III) only system.4 While this can prevent an absolute quantitative comparison, the scattering intensity difference between these two systems is several times larger than the contrast difference. Thus, we can conclude that the total volume of particles in the Fe(III) + As(V) system is larger than the Fe(III) only system.
For NOM-containing systems, the presence of NOM does not change the contrast. Thus, direct comparison between the Fe(III) only system and Fe(III) + NOM system indicates that the volume in the Fe(III) + NOM system was slightly higher than in the Fe(III) only system. This can occur because Fe(III) + NOM has a lower charge (Table 1), leading to more driving force for growth since the positively charged Fe(III) monomers in solution will be less repulsed by the nuclei in the Fe(III) + NOM system.
|
For the ternary system, the arsenic content was even higher than for the Fe(III) + As(V) system (14.5 mol% versus 8.1 mol%). Thus, the electron density contrast is even higher. However, for the ternary system the relative total particle volume was calculated to be less than half that of the Fe(III) only system. The true volume in these systems is, therefore, even lower, accounting for the contrast differences. In addition, the calculated relative particle volumes for the ternary system replicates were similar despite the difference in particle size. The relative total particle volumes calculated by comparison of invariants can be found in Fig. S1 in the ESI.† The primary particle size evolution was also calculated and is shown in Fig. 1F.
For both NOM-containing systems, there is strong power law scattering in the low q range (0.007–0.03 Å−1). This scattering is due to fractal aggregation of iron(III) (hydr)oxide precipitates, which can be characterized using the fractal dimension (d, where q−d represents the scattering from aggregates). This value is equal to the negative slope of the scattering in the low q region, i.e., the power law exponent.43,44 The fractal dimension gives insight into the density of the fractal aggregates forming in our experimental systems and can be used to distinguish between surface fractals and mass fractals.44 The fractal dimension also indicates how the rate of aggregation can alter the aggregate properties. If the aggregation is fast (d ≈ 1.8), it is characterized as diffusion limited cluster aggregation (DLCA), while slow aggregation (d ≈ 2.3) is characterized as reaction limited cluster aggregation (RLCA).45 In the DLCA regime, aggregates branch out more (i.e., fractal aggregation), while in the RLCA regime, aggregates are more compact. The changes in the aggregate structure can affect the surface area, reactivity, bioavailability, and transport of aggregates in the environment. For example, the surface area of fractal aggregates can be up to 30 times larger than if the aggregates were spherical.46
The extended linear region in the log–log plot for the Fe + NOM system and the Fe + As(V) + NOM (1) system allows us to calculate the fractal dimension in these cases. The d values were calculated to be 1.73 ± 0.09 and 1.87 ± 0.13 for the Fe + NOM system and the Fe + As(V) + NOM (1) system, respectively. In both systems, smaller values (d < 3) were observed, indicating mass fractal formation, i.e. less dense aggregates.47 This fractal dimension also indicates that the particles are forming in the DLCA regime. For the other particle size trend observed in the Fe + As(V) + NOM system (Fe + As(V) + NOM (2), Fig. 1E), the scattering was not linear due to the large particle size, and a fractal dimension could, therefore, not be calculated. The fractal dimension for the NOM-containing systems was also analyzed at ten minute intervals over the 1 hour reaction period. However, it did not change over the reaction period. This data can be found in Table S2 in the ESI.†
Chains of NOM tend to have a strong negative electric charge, and can, therefore, attract positively charged iron(III) (hydr)oxide particles. Due to their hydrophobicity, NOM chains and associated iron(III) (hydr)oxides will then aggregate. Fractal aggregation of NOM-associated nanoparticles has been observed previously.7,8,48,49 X-ray scattering for these systems thus reflects both the smallest individual primary particle sizes and the scattering of these particles with clusters of their neighbors along the NOM chains. For the Fe + NOM and Fe + As(V) + NOM (1) systems, there was no significant difference in the fractal dimension, indicating that the smaller particles in the ternary system had similar aggregation behavior around the NOM chains to the system with no As(V) present, i.e. the Fe + NOM system. However, the nucleation and growth behaviors differed greatly for the Fe(III) + As(V) + NOM (2) system, which has larger sized particles (Fig. 1E).
Exclusivity of arsenic and NOM interactions impacts iron(III) (hydr)oxide aggregation
Differences in aggregation between the ternary system and Fe(III) + NOM system were observed using ex situ TEM (Fig. 2). While the aggregation behavior in the Fe(III) only and Fe(III) + As(V) systems was similar (Fig. 2A and B, respectively), the morphology was dramatically different for systems with added NOM. For the Fe(III) + NOM system, large fractal aggregates were observed, which appeared to be coated by NOM (blue arrow in Fig. 2C). For the ternary, Fe(III) + As(V) + NOM system, much smaller fractal aggregates (red arrow in Fig. 2D) were observed along with individual particles. Furthermore, much fewer particles were observed compared to any of the other three systems. This is consistent with the calculated particle volumes from the GISAXS data. The caveat for ex situ TEM is that there may be changes in morphology due to drying effects. Drying during TEM sample preparation may also promote particle aggregation; however, the degree of aggregation should be the same in each sample. This is also supported by the fact that the in situ GISAXS observations of aggregation are consistent with TEM observations, despite drying prior to TEM observations. | ||
| Fig. 2 TEM images of homogeneous precipitations in the (A) Fe(III) only, (B) Fe(III) + As(V), (C) Fe(III) + NOM, and (D) Fe(III) + As(V) + NOM systems after 1 hour of reaction, showing different aggregation behaviors in the presence of different aqueous constituents. While some sodium nitrate crystallizes during the drying process, the particles imaged are iron(III) (hydr)oxides rather than salt crystals: They have a small size and exhibit weak diffraction, while sodium nitrate salts from drying have a much bigger size and a strong diffraction pattern. | ||
The mechanism of Fe–As–NOM interactions was investigated further using AFM and sequential addition of arsenate and NOM. All experiments were conducted in replicate tests to confirm the observed trends. First, all four systems were imaged following one hour of reaction (Fig. S2 in the ESI†). Next, in order to determine the sequence of Fe–As–NOM interactions, systems were investigated with Fe(III) + As(V) first added to solution, and then NOM added after 30 minutes, or with Fe(III) + NOM in solution initially, and As(V) added after 30 minutes (Fig. 3). Substrate morphology and height in the ternary system (Fig. 3A) were most similar to that of the system with Fe(III) + NOM initially and As(V) added later (Fig. 3B). Moreover, additional tests were run where the Fe(III) + NOM only system was run for 30 minutes and imaged. Large aggregates were observed in the Fe(III) + NOM only system at 30 minutes (Fig. 3D). However, when As(V) was added after 30 minutes and reacted for a total of 1 hour, these aggregates were not present in the system (Fig. 3B). This result indicates that in the early stages of iron(III) (hydr)oxide nucleation, precipitates may interact exclusively with NOM. However, further interactions with As(V) prevent aggregation or lead to disaggregation, thus resulting in a morphology which is dramatically different from the large aggregates observed in the Fe(III) + NOM system at 1 hour.
 | ||
| Fig. 3 AFM images from sequential addition tests. The morphology for the ternary Fe(III) + As(V) + NOM system (A) is most similar to the system with Fe(III) + NOM added initially and As(V) added after 30 minutes (B). Particles in the system with Fe(III) + As(V) added initially and NOM added after 30 minutes (C) had a more uniformly large particle size. Furthermore, large aggregates were observed in the Fe(III) + NOM system at 30 minutes (D) that were not observed in the system after As(V) was added (B), indicating that As(V) may lead to disaggregation of Fe(III)−NOM precipitates. | ||
The compositions of precipitates in the Fe(III) + As(V) + NOM systems and in the Fe(III) + NOM systems were investigated further for their total organic carbon and As(V) concentrations and water contents. The total organic carbon (i.e., NOM) content of the ternary system and Fe(III) + NOM system were found to be 2.05 ± 0.05 and 1.96 ± 0.10 mg C per μmol Fe, respectively, and the water content of these precipitates was 49 wt% water in the ternary system and 64 wt% water in the Fe(III) + NOM system (Fig. S3†). Thus, although the two NOM-containing systems have the same NOM content, the water content of the ternary system is less. This may be because particles which contain Fe(III) + As(V) without NOM have a water content of 22 wt%,4 and so the water content in the ternary system will be in between that of the Fe(III) + As(V) system and that of the Fe(III) + NOM system if the components are interacting with Fe(III) exclusively.
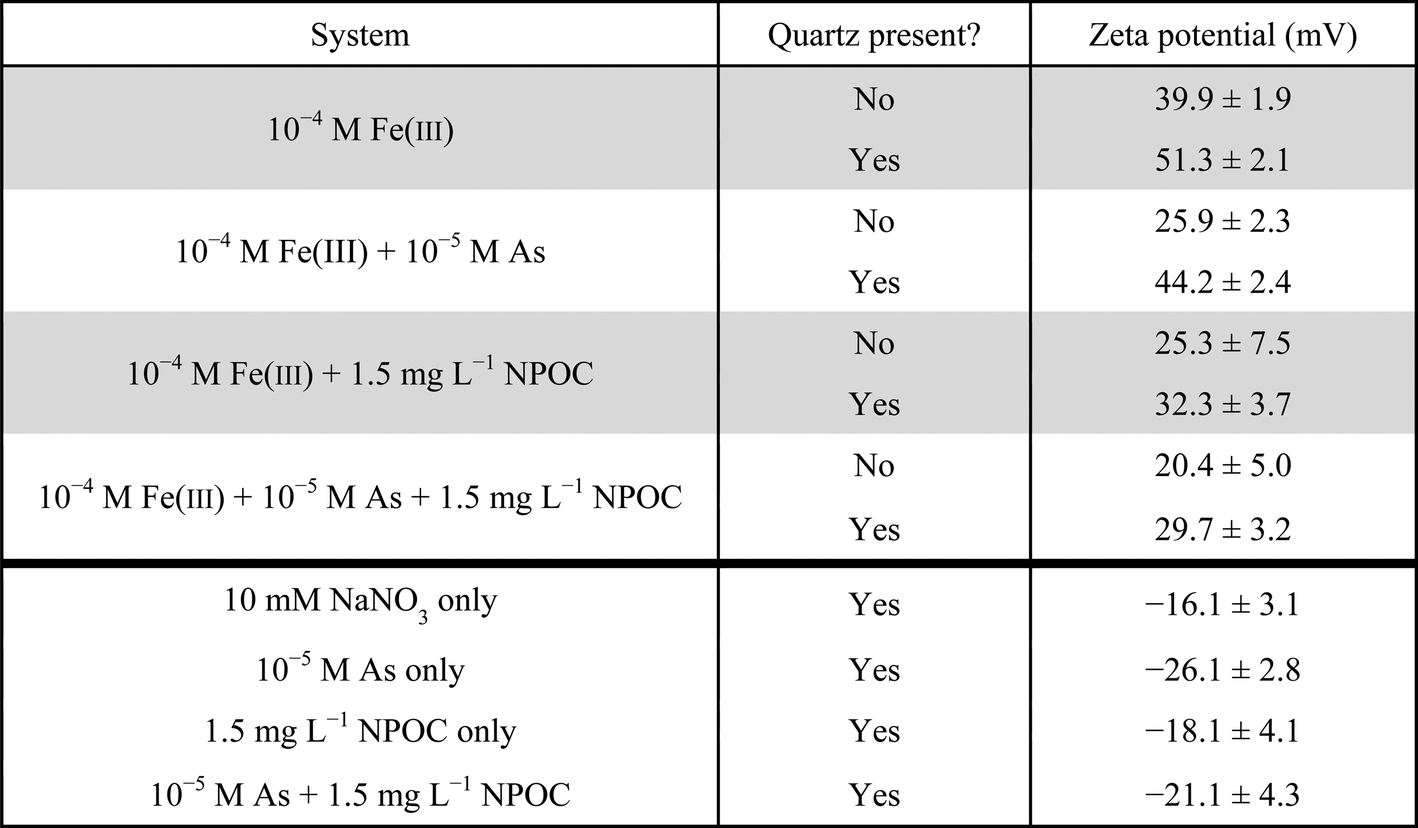
The arsenic content of the nanoparticles in the ternary system was measured to be 14.5 ± 1.5 mol% As, which is higher than the 8.1 ± 2.3 mol% As observed in the system without NOM.4 This composition is also reflected in the zeta potential measurements (Table 1). The zeta potential of iron(III) (hydr)oxide precipitates on quartz powder was 29.7 ± 3.2 mV for the system with As(V) + NOM, compared with 32.3 ± 3.7 mV for the system with NOM only. Without NOM, the zeta potential was 51.3 ± 2.1 mV for the Fe(III) only system and 44.2 ± 2.4 mV for the Fe(III) + As(V) system. Because NOM and As(V) are both negatively charged, the presence of both of these components together lowered the surface charge. By having As(V) incorporation, less positively charged iron(III) (hydr)oxide precipitates will form, resulting in a decreased tendency to adhere to the negatively charged NOM chains. Thus, this lower surface charge of As(V)-containing iron(III) (hydr)oxides may partially explain the smaller aggregate size observed in the ternary system.
Mechanism of interactions in As–Fe–NOM ternary systems
Sequential addition tests and observations suggest that As(V) can interact with Fe(III)-NOM aggregates, triggering disaggregation (Fig. 3). From the TEM images, we hypothesize that a large number of iron(III) (hydr)oxides in the sequential addition system will be coated with NOM prior to As(V) addition (Fig. 2). Thus, As(V) can interact with NOM directly to cause disaggregation. Previous studies have reported that phenolate groups in NOM can bind to the central arsenic atom of arsenate.26 In addition to arsenic–NOM interactions, FTIR and XAS results in this study (Fig. S4 and S5 in the ESI,† respectively) showed that bonding between iron and arsenic also occurs in the ternary system. This can happen when arsenic is added simultaneously with NOM, resulting in concurrent interactions with nucleating iron(III) (hydr)oxides. This can also result from the displacement of NOM on the iron(III) (hydr)oxide surface by arsenate, which is known to occur.21Furthermore, previous research has shown that electrostatic interactions play a significant role in NOM adsorption.50 Thus, the less positive zeta potentials of iron(III) (hydr)oxides in the ternary system (Table 1) can cause weaker attraction to NOM, preventing further aggregation of new particles. Iron(III) (hydr)oxides which remain associated primarily with NOM will act similarly to the particles observed in the Fe(III) + NOM only system, retaining a small particle size and high water content. On the other hand, iron(III) (hydr)oxides which are closely associated with arsenate in the ternary system, either through initial formation or displacement of NOM, will have a larger size and smaller water content more similar to the Fe(III) + As(V) system.
Although FTIR and XAS results suggest that arsenic which is bound to the iron(III) (hydr)oxide surface behaves similarly in the presence and absence of NOM, the HRXRD spectra for these systems show differences (Fig. 4). The broad peak between 2Theta = 10° and 25° in NOM-containing systems is indicative of NOM.51 For the ternary system, there was much less of a shift in the ferrihydrite peak positions, which indicates a larger extent of ferrihydrite that does not interact with As(V), but remains associated with NOM instead. While there appears to be less arsenate bound to iron(III) (hydr)oxides from the HRXRD results, there was still a high percentage of arsenate associated with precipitates according to ICP-MS results, indicating As(V) interactions with NOM. Therefore, we next investigated how As(V) can change the surface chemistry of NOM molecules by investigating the change in NOM hydrophilicity in the presence of As(V). We found when 10−5 M As(V) was added, the contact angle of the 1.5 mg L−1 NOM + 10 mM NaNO3 solution on quartz surfaces decreased from 23.9 ± 0.1° to 16.8 ± 0.1° (compared to 12.5 ± 0.2° for 10 mM NaNO3 only and 10.1 ± 0.5° for 10 mM NaNO3 + 10−5 M As(V)). This change demonstrates that the solution became more hydrophilic (Fig. 5). Thus, the hydrophobic–hydrophobic interactions which promote NOM aggregation are weaker in the ternary system. This can make the formation of large fractal aggregates, which were observed in the Fe(III) + NOM system, less favorable.
 | ||
| Fig. 4 HRXRD spectra for a ferrihydrite reference and systems containing Fe(III) only, Fe(III) + As(V), Fe(III) + NOM, and Fe(III) + As(V) + NOM. Spectra indicate that Fe(III) can form more ferrihydrite when NOM is present together with As(V) compared to the Fe(III) + As(V) system, thus indicating exclusive interactions. The broad peak between 2Theta = 10° and 25° in NOM-containing systems is indicative of NOM presence.51 | ||
 | ||
| Fig. 5 Contact angles on the (110) quartz surface for reaction systems. All systems were at a pH of 3.6. Systems with As(V) contained 10−5 M As(V) and systems with NOM contained 1.5 mg L−1 NPOC. | ||
In conclusion, the unique iron(III) (hydr)oxide nucleation and aggregation behaviors in the ternary systems result from a combination of physicochemical effects. Iron(III) (hydr)oxides can interact exclusively with As(V) and NOM, resulting in some particles with characteristics (e.g., size and water content) similar to that of the Fe(III) + As(V) system and some particles with characteristics similar to those of the Fe(III) + NOM systems. This alteration in aggregation behavior can be closely associated with less favorable electrostatic interactions as well as weaker hydrophobic–hydrophobic interactions, because As(V) interactions with NOM increased their hydrophilicity.
Conclusions
This study provides valuable, new insight into the nucleation and aggregation behaviors of iron(III) (hydr)oxides in systems containing NOM and arsenate. We found that in the ternary system (Fe(III) + As(V) + NOM), iron(III) (hydr)oxides interacted exclusively with both constituents, resulting in some particles with large sizes that interacted primarily with As(V) and other small fractal aggregates where iron(III) (hydr)oxides interacted primarily with NOM. In the ternary system, Fe(III) interacted first with NOM in solution, and later interactions with As(V) led to disaggregation of NOM-associated iron(III) (hydr)oxides. This change in aggregation and disaggregation behavior is an interesting finding that can have significant impacts on the fate of contaminants by influencing aggregate transport distances and times in natural and engineered aqueous environments. Furthermore, not all of the products formed are pure iron(III) (hydr)oxides. For example, ferric arsenate-like nanoparticles formed in the Fe(III) + As(V) system. It is important to consider other phases, such as ferric arsenate, when characterizing iron(III) (hydr)oxide formation because these phases can form in addition to or in place of iron(III) (hydr)oxides, affecting their reactivity in aqueous environments.These new findings contribute to the existing literature by thoroughly examining for the first time the scenario where iron(III) (hydr)oxides form in the presence of As(V) and NOM together. Because these observations were made in situ and in real-time using our GISAXS fluid cell setup, these findings give a clearer picture of the complex interactions among Fe(III), As(V), and NOM, which can occur when dissolved Fe(III) species precipitate to form iron (hydr)oxide or ferric arsenate-like nanoparticles in real aquatic systems. Using this advanced technique, the current study was able to capture iron(III) (hydr)oxide nucleation at its starting point, rather than after an elapsed time, as seen in the previous literature. Furthermore, the in situ study allowed us to observe the nucleation and growth of nanoparticles in a fully hydrated environment, allowing more accurate measurements of particle size, which can be useful in reactive transport modeling. In addition, the current study offers important clues to understand arsenic fate and transport in systems where NOM coexists with precipitating iron(III) (hydr)oxides. For instance, although precipitates in the ternary system contained significant quantities of As(V), there was a smaller volume of precipitates in this system. Therefore, it is likely that less arsenic can be attenuated in these systems.
These findings have also valuable implications for engineered aquatic processes, such as managed aquifer recharge, where ferrihydrite is frequently incorporated into reactive transport models as a sink for arsenic mobilized during arsenopyrite oxidative dissolution.52 The role of natural organic matter in limiting ferrihydrite formation when present with arsenic must be considered to develop more accurate models. It is also important to consider that, in natural systems, additional ions, such as phosphate and carbonate, can coexist with arsenic at higher concentrations. In one of our previous publications, we investigated the impact of arsenate and phosphate oxyanions on iron(III) (hydr)oxide precipitation and found that both oxyanions increase iron(III) (hydr)oxide heterogeneous precipitate size.4 However, for the arsenate system, the size and total volume of these particles was larger than for the phosphate system. Thus, arsenate can have some specific interactions compared to phosphate, despite the nearly identical structure of these oxyanions. Future studies should consider whether the specific interactions between iron(III) (hydr)oxides and arsenic can still occur in the presence of other environmentally abundant ions.
These new results advance our knowledge of early stage iron(III) (hydr)oxide formation mechanisms in natural and engineered aquatic systems and can help us to better evaluate the risk of arsenic contamination in complex aqueous environments.
Acknowledgements
We are grateful for support received from the National Science Foundation (EAR-1424927 and CHE-1214090). CWN acknowledges the generous support of the Mr. and Mrs. Spencer T. Olin Fellowship. We wish to thank the Environmental NanoChemistry Group members for valuable discussion. Use of the Advanced Photon Source (Sector 11-BM for HRXRD and Sector 12 ID-B for GISAXS) at the Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract no. DE-AC02-06CH11357.References
- G. A. Waychunas, C. S. Kim and J. F. Banfield, J. Nanopart. Res., 2005, 7, 409–433 CrossRef CAS.
- B. K. Theng and G. Yuan, Elements, 2008, 4, 395–399 CrossRef CAS.
- N. S. Wigginton, K. L. Haus and M. F. Hochella Jr, J. Environ. Monit., 2007, 9, 1306–1316 RSC.
- C. W. Neil, B. Lee and Y.-S. Jun, Environ. Sci. Technol., 2014, 48, 11883–11891 CrossRef CAS PubMed.
- Y. Hu, B. Lee, C. Bell and Y.-S. Jun, Langmuir, 2012, 28, 7737–7746 CrossRef CAS PubMed.
- P. H. Santschi, J. J. Lenhart and B. D. Honeyman, Mar. Chem., 1997, 58, 99–125 CrossRef CAS.
- M. Baalousha, Sci. Total Environ., 2009, 407, 2093–2101 CrossRef CAS PubMed.
- S. E. Mylon, K. L. Chen and M. Elimelech, Langmuir, 2004, 20, 9000–9006 CrossRef CAS PubMed.
- C. W. Neil, Y. J. Yang and Y.-S. Jun, J. Environ. Monit., 2012, 14, 1772–1788 RSC.
- Y.-S. Jun, B. Lee and G. A. Waychunas, Environ. Sci. Technol., 2010, 44, 8182–8189 CrossRef CAS PubMed.
- E. M. Hotze, T. Phenrat and G. V. Lowry, J. Environ. Qual., 2010, 39, 1909–1924 CrossRef CAS PubMed.
- A. A. Keller, H. Wang, D. Zhou, H. S. Lenihan, G. Cherr, B. J. Cardinale, R. Miller and Z. Ji, Environ. Sci. Technol., 2010, 44, 1962–1967 CrossRef CAS PubMed.
- Y. Hu, C. Neil, B. Lee and Y.-S. Jun, Environ. Sci. Technol., 2013, 47, 9198–9206 CrossRef CAS PubMed.
- J. R. Ray, B. Lee, J. Baltrusaitis and Y.-S. Jun, Environ. Sci. Technol., 2012, 46, 13167–13175 CrossRef CAS PubMed.
- H. P. Blume, G. W. Brummer, H. Fleige, R. Horn, E. Kandeler, I. Kogel-Knabner, R. Kretzschmar, K. Stahr and B. M. Wilke, Scheffer/Schachtschabel Soil Science, Springer, Berlin Heidelberg, 2015 Search PubMed.
- R. N. Carrow, R. R. Duncan and M. T. Huck, Turfgrass and landscape irrigation water quality: Assessment and management, CRC Press, 2008 Search PubMed.
- K. M. Campbell and D. K. Nordstrom, Rev. Mineral. Geochem., 2014, 79, 185–216 CrossRef.
- H. Xu, B. Allard and A. Grimvall, Water, Air, Soil Pollut., 1991, 57, 269–278 CrossRef.
- M. Grafe, M. Eick and P. Grossl, Soil Sci. Soc. Am. J., 2001, 65, 1680–1687 CrossRef CAS.
- M. Grafe, M. J. Eick, P. R. Grossl and A. M. Saunders, J. Environ. Qual., 2002, 31, 1115–1123 CrossRef CAS PubMed.
- A. D. Redman, D. L. Macalady and D. Ahmann, Environ. Sci. Technol., 2002, 36, 2889–2896 CrossRef CAS PubMed.
- I. Ko, J.-Y. Kim and K.-W. Kim, Colloids Surf., A, 2004, 234, 43–50 CrossRef CAS.
- M. Bauer and C. Blodau, Geochim. Cosmochim. Acta, 2009, 73, 529–542 CrossRef CAS.
- K. Kaiser, G. Guggenberger, L. Haumaier and W. Zech, Eur. J. Soil Sci., 1997, 48, 301–310 CrossRef.
- A. Saada, D. Breeze, C. Crouzet, S. Cornu and P. Baranger, Chemosphere, 2003, 51, 757–763 CrossRef CAS PubMed.
- J. Buschmann, A. Kappeler, U. Lindauer, D. Kistler, M. Berg and L. Sigg, Environ. Sci. Technol., 2006, 40, 6015–6020 CrossRef CAS PubMed.
- M. Bauer and C. Blodau, Sci. Total Environ., 2006, 354, 179–190 CrossRef CAS PubMed.
- P. Sharma, M. Rolle, B. Kocar, S. Fendorf and A. Kappler, Environ. Sci. Technol., 2010, 45, 546–553 CrossRef PubMed.
- T. Tuutijarvi, E. Repo, R. Vahala, M. Sillanpaa and G. Chen, Chin. J. Chem. Eng., 2012, 20, 505–514 CrossRef CAS.
- A. B. Giasuddin, S. R. Kanel and H. Choi, Environ. Sci. Technol., 2007, 41, 2022–2027 CrossRef CAS PubMed.
- M. A. Simeoni, B. D. Batts and C. McRae, Appl. Geochem., 2003, 18, 1507–1515 CrossRef CAS.
- G. Liu, A. Fernandez and Y. Cai, Environ. Sci. Technol., 2011, 45, 3210–3216 CrossRef CAS PubMed.
- C. Chen, J. J. Dynes, J. Wang and D. L. Sparks, Environ. Sci. Technol., 2014, 48, 13751–13759 CrossRef CAS PubMed.
- S. Wang and C. N. Mulligan, Environ. Geochem. Health, 2006, 28, 197–214 CrossRef CAS PubMed.
- B. Gilbert, R. K. Ono, K. A. Ching and C. S. Kim, J. Colloid Interface Sci., 2009, 339, 285–295 CrossRef CAS PubMed.
- F. Wilson and D. Hawkins, Environ. Geol., 1978, 2, 195–202 CrossRef CAS.
- H. Gecol, E. Ergican and A. Fuchs, J. Membr. Sci., 2004, 241, 105–119 CrossRef CAS.
- H. R. Diz, J. T. Novak and J. D. Rimstidt, Mine Water Environ., 1999, 18, 1–14 CrossRef CAS.
- C. A. J. Appelo and W. W. J. M. De Vet, in Arsenic in groundwater, ed. A. H. Welch and K. G. Stollenwerk, Kluwer Academ, Boston, Editon edn., 2003, pp. 381–401 Search PubMed.
- A. Mota, L. Albuquerque, L. C. Beltrame, O. Chiavone-Filho, A. Machulek Jr and C. Nascimento, Braz. J. Pet. Gas, 2009, 2(3), 122–142 Search PubMed.
- M. Edwards, M. M. Benjamin and J. N. Ryan, Colloids Surf., A, 1996, 107, 297–307 CrossRef CAS.
- B. Gu, J. Schmitt, Z. Chen, L. Liang and J. F. McCarthy, Adsorption and desorption of natural organic matter on iron oxide: mechanisms and models, Environ. Sci. Technol., 1994, 28(1), 38–46 CrossRef CAS PubMed.
- H. D. Bale and P. W. Schmidt, Phys. Rev. Lett., 1984, 53, 596 CrossRef CAS.
- J. Teixeira, J. Appl. Crystallogr., 1988, 21, 781–785 CrossRef.
- D. Robinson and J. Earnshaw, Phys. Rev. A: At., Mol., Opt. Phys., 1992, 46, 2065 CrossRef CAS.
- D. F. Blake and K. Kato, J. Geophys. Res.: Atmos., 1995, 100, 7195–7202 CrossRef CAS.
- B. Hammouda, Probing Nanostructures: The SANS Toolbox, Natl. Institute Standards Technology Center for Neutron Research, Gaithersburg, MD, 2008 Search PubMed.
- M. Baalousha, A. Manciulea, S. Cumberland, K. Kendall and J. R. Lead, Environ. Toxicol. Chem., 2008, 27, 1875–1882 CrossRef CAS PubMed.
- A. Vilge-Ritter, J. Rose, A. Masion, J.-Y. Bottero and J.-M. Laine, Colloids Surf., A, 1999, 147, 297–308 CrossRef CAS.
- A. Armanious, M. Aeppli and M. Sander, Environ. Sci. Technol., 2014, 48, 9420–9429 CrossRef CAS PubMed.
- P. H. de Carvalho, A. Jesus, V. M. Prata, D. S. Bezerra, L. P. Romao and S. Navickiene, J. Braz. Chem. Soc., 2010, 21, 659–664 CrossRef CAS.
- C. W. Neil, Y. J. Yang, D. Schupp and Y.-S. Jun, Environ. Sci. Technol., 2014, 48, 4395–4405 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5en00283d |
| ‡ Current address: Office of Research and Development, National Risk Management Research Laboratory, U.S. Environmental Protection Agency, Cincinnati, Ohio 45268, USA |
| § Current address: Civil & Environmental Engineering, University of California, Berkeley, California 94720, USA |
| This journal is © The Royal Society of Chemistry 2016 |