
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Environmental aging alters Al(OH)3 coating of TiO2 nanoparticles enhancing their photocatalytic and phototoxic activities†
Souhail R.
Al-Abed
a,
Jurate
Virkutyte
b,
Jayna N. R.
Ortenzio
c,
Robert M.
McCarrick
d,
Laura L.
Degn
e,
Robert
Zucker
e,
Najwa Haykal
Coates
e,
Kristin
Childs
f,
Hongbo
Ma
g,
Steve
Diamond
h,
Kevin
Dreher
e and
William K.
Boyes
*e
aNational Risk Management Research Laboratory, USEPA, Cincinnati, OH 45268, USA
bKeramida Inc, 5011 Kenwood Rd, Cincinnati, OH 45227, USA
cOak Ridge Institute for Science and Education (ORISE) fellow at the National Health and Environmental Effects Research Laboratory, USEPA, RTP, NC 27711, USA
dDepartment of Chemistry and Biochemistry, Miami University, Oxford, OH 45056, USA
eNational Health and Environmental Effects Research Laboratory, USEPA, RTP, NC 27711, USA. E-mail: boyes,william@epa.gov; Fax: (919) 541 4849; Tel: (919) 541 7538
fCenter for Human Disease Modeling, Duke University Health System, Durham, NC 27701, USA
gAssistant Professor in Environmental Health Science Zilber School of Public Health University of Wisconsin-Milwaukee Lapham Hall 442, Milwaukee, WI 53211, USA
hNanoSafe, Inc, Duluth, MN 55811, USA
First published on 18th May 2016
Abstract
As a component of sunscreen formulations, TiO2 engineered nanomaterials (ENM) are coated to prevent reactive oxygen species from causing damage to skin. We investigated the stability of an Al(OH)3 coating by exposing 25 nm Al(OH)3·TiO2 ENM to simulated swimming pool water (SPW) for 45 minutes, 1, 3, 10, or 14 days. Electron microscopy and spectroscopy indicated that exposure to SPW caused a redistribution of the Al(OH)3 coating allowing photocatalytic formation of hydroxyl radicals. Aged ENM showed significantly greater phototoxicity under UVA irradiation than un-aged ENM in a human-derived retinal pigment epithelium cell line (ARPE-19). Photocatalytic activity and phototoxicity of aged Al(OH)3·TiO2 was significantly less than that of the positive control—uncoated P25 TiO2. In summary, the aging of Al(OH)3·TiO2 ENM in SPW redistributed the coating and reduced its protective properties, thereby increasing reactivity and potential phototoxicity.
Nano impactMuch of the existing research in nanotoxicology involves the investigation of nanomaterials in their as-produced form. In environmental media, however, engineered nanomaterials may be transformed in a variety of ways, which can alter their toxicological properties and impact their risk assessment. In this study we demonstrate that the protective AlOH3 coating on TiO2 nanomaterials, which is intended to shunt photo-activated electrons and prevent the catalytic generation of reactive oxygen species, is degraded after exposure to simulated swimming pool water. This transformation caused a slight but measurable increase in both photocatalytic capability and phototoxic potential of these materials. This research illustrates the importance of studying environmentally transformed engineered nanomaterials as a component of assessing their potential environmental and health risks. |
1. Introduction
The conditions under which engineered nanomaterial (ENM) enabled products are used have the potential to cause transformations of ENMs resulting in subsequent changes in reactivity. Therefore, the entire ENM life cycle: manufacture, application, and disposal, is of concern when evaluating the potential for ENM to have unintended effects on human health or the environment.1Titania-based ENM are widely used in water and air treatment, clean energy production, and the fabrication of self-cleaning surfaces.2 Titanium dioxide (TiO2) is a semi-conductor that produces an electrochemical photolysis of water under UV irradiation,3 which in process creates intermediates including hydroxide, superoxide and singlet oxygen reactive oxygen species (ROS)4–7 In biological systems, ROS may cause extensive cell damage and ultimately lead to cell death.8–14
Due to their ability to absorb UV radiation, titania-based ENM are used in sunscreen formulations. For this application, the particle surface is coated with a material such as Al(OH)3 to shield the skin from ROS.5,15 Following dermal application of TiO2-containing sunscreen lotions, ENM may enter the aquatic environment.16–18 Widespread use has contributed to TiO2 detection in aquatic ecosystems and wastewater at concentrations ranging from 5 to 3000 μg L−1,19,20 and concentrations of sunscreen residuals detected in swimming pool water range from 21 to 60 μg L−1.21
One of the complexities of assessing potential environmental health consequences of ENM use is understanding ENM transformations in environmental media, and the consequences of those transformations.22 This study examines the integrity of the protective Al(OH)3 coating upon exposure to simulated environmental conditions relevant to product life-cycle. Previously, we and others23,24 have observed a redistribution and depletion of Al(OH)3 coating in TiO2-based sunscreens upon exposure to free chlorine in simulated swimming pool water (SPW).5 Here we extend those reports by evaluating the effect of SPW aging on three endpoints: condition of the Al(OH)3 coating; generation of ROS; and cellular uptake and phototoxicity. The hypothesis tested was that aging in SPW would degrade the protective Al(OH)3 coating leading to increased generation of ROS and increased potential for phototoxic reactions.
2. Materials and methods
2.1 Simulated swimming pool water
SPW was prepared by adding 0.3850 grams (g) of calcium sulfate dihydrate (CaSO4·2H2O; Acros Organics, 99%) and 0.150 g of sodium bicarbonate (NaHCO3; Fisher, ACS) to 1 liter (L) of Super-Q water and stirring overnight. Potassium bisulfate (KHSO4; Fisher, ACS) (1 N) was used to adjust the pH to 7.6. Clorox bleach (The Clorox Company, USA) containing 5.8% Cl2 was used as the free chlorine source.52.2 Aged ENM samples
Nanomaterials (TiO2 coated with Al(OH)3, MT100SA) were generously donated by Presperse Inc. (Somerset, NJ). A bulk suspension was prepared by adding 0.2056 g of MT100SA to 1 L of SPW. An aliquot of this initial suspension was removed to collect an un-aged ENM control sample. To start aging, 86 μL of commercial bleach was added to the bulk TiO2-SPW suspension while stirring (initial pH = 6.9). SPW in the TiO2-SPW mix was renewed every 12 hours by centrifuging at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g for 1 hour, removing the supernatant, and resuspending the pellet in 1 L of fresh SPW with 86 μL of commercial bleach. This 12 hour exchange cycle was chosen to mimic common practices as confirmed by local swimming pool operators.
000×g for 1 hour, removing the supernatant, and resuspending the pellet in 1 L of fresh SPW with 86 μL of commercial bleach. This 12 hour exchange cycle was chosen to mimic common practices as confirmed by local swimming pool operators.
To obtain samples of aged ENMs, 10 mL aliquots were collected from the bulk suspension at time points of 45 minutes and 1, 3, 10 and 14 days after the initial addition of free chlorine. After centrifuging at 10000×g for 1 hour and removing the supernatant, samples were resuspended in 10 mL of water. Each sample was washed by repeating this centrifugation process twice. After removing the supernatant at the end of the third cycle, samples were freeze-dried to a dry powder.
2.3 ENM characterization
ENM were analyzed using a FEI XL30 Environmental Scanning Electron Microscope (SEM) equipped with an EDAX Energy Dispersive Spectroscope (EDS) operating at 15 to 30 kV, and a high-resolution Transmission Electron Microscope (TEM, Jeol 2010F) equipped with an Oxford INCA EDS system operating at an accelerated voltage of 200 keV. Prior to SEM analyses, samples were degassed and completely dried in air to eliminate any impurities. Furthermore, samples were placed on SEM aluminum stubs gold-sputtered to ensure conductivity. The size of the analyzed area was 200 nm. Samples were selected from various locations across the sampling area to ensure more or less equal presentation of numerous analyses points.X-ray photoelectron spectroscopy (XPS) was performed using a QUANTERA II Spectrometer (PHI, USA). The samples were prepared and mounted for analysis using standard practices consistent with high vacuum surface analytical procedures. Powders were placed onto the XPS compatible mounts (using nonmagnetic screws and fixtures) prior to analysis. Calibration was performed using a clean Au/Cu sample and a pure Ag sample (99.99%). Measured values for electron binding energies (BE) were 84 ± 0.02 eV and 932 ± 0.04 eV. The samples were irradiated with monochromatic AlKα X-rays (hν = 1486.6 eV) using an X-ray spot size of 400 × 700 μm.2 Surface charging was compensated by means of a filament (I = 1.9 A, 3.6 V) inserted in a magnetic lens system, and all spectra were corrected by setting the C1s hydrocarbon component to 284.60 eV. For each sample, a survey spectra (0–1200 eV) were recorded at a pass energy of 20 and 160 eV to determine the surface chemical compositions as percentage. The data were processed using a PHI MultiPak data reduction software (Physical Electronics, USA). Sample compositions were obtained from the survey spectra after linear background subtraction using RSF (Relative Sensitivity Factors) derived from Scofield cross-sections. Curve fitting was carried out using the same initial parameters and inter-peak constraints to reduce scattering. The core level envelopes were fitted with Gaussian–Lorentzian function (G/L = 30) and variable full width half maximum.
The reactivity of the dried powder samples was evaluated with electron paramagnetic resonance (EPR) spectroscopy. For EPR measurements, 0.1 mM 5-(diethoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide (DEPMPO; ≥99%, Enzo Life Sciences) was added to 10 mL of the TiO2-SPW-bleach suspension. The samples were then irradiated with a UV lamp (Electron Microscopy Sciences, 6 W, 750 μm cm−2 at 6 inches (15.2 cm) for 7 minutes prior to EPR analysis. All experiments were conducted in triplicate. X-band EPR spectra were recorded at the Ohio Advanced EPR Laboratory on an EMX CW (Bruker, Germany) spectrometer with the following parameters: microwave power, 10 mW; modulation frequency, 100 kHz; modulation amplitude, 1G. Spectra simulations were performed with the “EasySpin” toolbox for MATLAB distributed by Stefan Stoll.25 Samples were mixed with the DEPMPO spin label and illuminated for 7 minutes using a UV lamp. Elemental analyses of the ENM were performed by acid digestion of the ENM (EPA Method 3051), filtering (0.45 μm), and analyzing for metals (EPA Method 6010B) using an inductively coupled plasma-atomic emission spectrophotometer (ICP-AES, IRIS Intrepid, Thermo Scientific, MA). Characterization of the positive control compound is described elsewhere.8
Statistical treatment of chemical characterization data was performed using Origin (OriginLab Corporation, Northampton, MA) utilizing the Anderson-Darling (AD) examination of distribution. Because the distribution of the data was not normal (e.g., using Kolmogorov–Smirnov test), non-parametric methods were used. A Wilcoxon signed rank test was run for each paired group to delineate whether the SPW-aging affected the coating distribution (expressed as Al/Ti%).
2.4 Photoreactivity assays
Two acellular photocatalytic ROS generation assays were used (see ESI† for methodological details). Briefly, the assays included photocatalytic generation of hydroxyl radicals (OH˙) using the fluorescence indicator 3′[p-aminophenyl] fluorescein (APF) and photocatalytic generation of thiobarbituric acid reactive substance (TBARS) as described previously.82.5 Characterization of ENM dispersion
Samples of untreated and SPW-aged Al(OH)3·TiO2 ENM were weighed into a 4 mL sterile glass vial and dispersed in saline (0.9% (w/v) NaCl in sterile H2O, pH 6.0; Hospira Inc., Lake Forest, IL) at 4 mg mL−1. The ENM were dispersed in saline to investigate suspension conditions as prepared for the TBARS assay. The suspensions were sonicated twice (6 watts; 2 min per sonication; TM 100 sonicator, TekMar, Cincinnati, OH) and placed in an ultrasonic bath (Model ME4.6; Mettler Electronic Corp., Anaheim, CA). Hydrodynamic size distributions were determined by dynamic light scattering (DLS) (Zetasizer Nano ZS; Malvern Instruments Ltd., Worcestershire, UK). Samples were diluted in saline (200 μg mL−1). Data collected included degree of agglomeration, polydispersity index (PDI) calculated from cumulants analysis, and count within the <100 nm, 100–500 nm, and >1000 nm size ranges.2.6 Phototoxicity experiments
A human derived cell line of retinal pigment epithelial cells was obtained from ATCC (Manassas, VA) and grown in culture conditions described previously and in the ESI.†8 Twenty four hours after plating, cells were exposed to Al(OH)3·TiO2 suspensions (un-aged or aged for 45 minutes, 1, 3, 10 or 14 days) at concentrations of 3, 10, 30, or 100 μg mL−1. Uncoated TiO2, Degussa P25 (30 μg mL−1) served as the positive control.8Twenty-four hours after ENM treatment, plates were exposed to either UVA radiation or dark conditions. A copper sulfate filter was used to exclude UVB wavelengths (295 to 315 nm). The emission spectrum of the UV bulbs (F20T12/BL/HO[PUVA], National Biological Corporation, Beachwood, OH) was measured with a spectroradiometer (RPS900, International Light, Newburyport, MA). Average UVA irradiance was 687.9 μW cm−2 when filtered through a cell culture plate lid and CuSO4 filter. During the 90 minute exposure time, plates were placed on heating blocks to maintain a temperature of 37 °C. Cells were exposed to approximately 3.7 J cm−2.
After 24 h viability was evaluated using calcein-AM and propidium iodide (PI) as a live/dead assay, described previously.8 Phototoxicity experiments were performed on three to five replicate plates for each of the SPW-aged ENM samples. Data were analyzed in SAS by three-way analysis of variance using the factors of UVA irradiance, nanoparticle age, and concentration of treatment suspension. The criterion for statistical significance was α = 0.05.
2.7 Cellular uptake
Cellular uptake was evaluated on a flow cytometer (BD FACSCalibur; BD Biosciences, San Jose, CA) 24 h after treatment and analyzed using a forward scatter (FSC) diode detector and side scatter (SSC) photomultiplier tube. Scatter results were expressed as a relative increase with respect to the negative control (no ENM treatment).
3. Results
3.1 Morphology assessment
The Al/Ti (%) distribution in the un-aged and SPW-aged ENM was measured using SEM-EDS. The ratio of Al/Ti was fairly constant across the sample field of the un-aged sample, but varied markedly in aged samples (Fig. 1). The range of Al/Ti ratio values expanded with aging, indicating a redistribution of Al(OH)3 protective layer. As SEM-EDS is only semi-quantitative, an Anderson-Darling (AD) examination of the distribution and a Wilcoxon signed rank test were used to interpret the statistical significance of differences due to aging from 45 minutes to 14 days.28 The Wilcoxon signed rank test was used to determine whether the median values of Al/Ti (%) distribution changed significantly when samples were subjected to 45 min – 14 days in SPW. The W values and their corresponding probabilities were calculated based on the Al/Ti (%) changes on the surface of the aged and un-aged particles. The probabilities for each aging time were tested for significance at alpha level of 0.05. Results in Table 1 indicate that there was no significant difference between un-aged and aged samples for the 45 minute and 1 day exposure times; however, the difference was statistically significant for samples that were aged for 3 days and longer.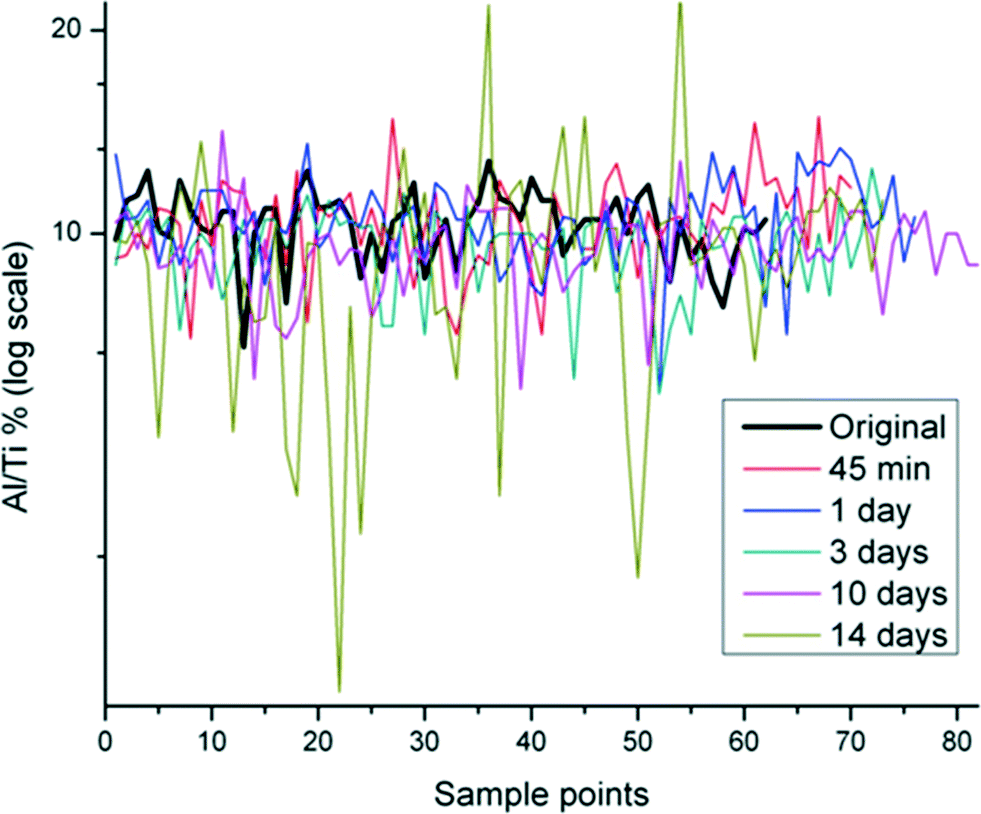 | ||
| Fig. 1 Distribution of Al/Ti% in Al(OH)3·TiO2 ENM aged in SPW from 45 min to 14 days in multiple scanning points using SEM-EDS. Original indicates the un-aged sample. | ||
| W | Z | Prob > |W| | |
|---|---|---|---|
| 45 min | 950 | 0.051 | 0.96 |
| 1 day | 930 | 0.084 | 0.93 |
| 3 days | 1400 | 3.9 | <0.0010 |
| 10 days | 1500 | 4.0 | <0.0010 |
| 14 days | 1300 | 2.8 | 0.0043 |
Electron micrographs indicated that ENM were slightly heterogeneous with a mean size of 60 nm and elongated to cuboidal-shaped up to 80–100 nm (Fig. S1 and S2†). The inter-reticular distance d (110) (∼3.3 Å) characteristic of rutile crystalline phase was maintained through 14 days of aging in SPW (Fig. S2†). In addition, EDS identified aluminum and copper in all samples (Fig. S3†), which can be attributed to the Al(OH)3 coating on the TiO2 with adventitious copper from the grid, respectively. The samples showed nearly similar mean percent atomic content of Al (2.5, 1.7, 2.1, 2.2, 2 and 2.6%) and Ti content (23.9, 17.2, 20.7, 23.2, 21.8 and 23.3%) in the un-aged, 45 min, 1, 3, 10 and 14 day aged samples, respectively.
The Ti 2p core level spectrum of the un-aged sample (Fig. 2a) showed a doublet at about 459.3 eV with a spin orbit splitting of 5.8 eV, which is compatible with the Ti4+ oxidation state.5,29 The shape of the peak excluded the presence of other oxidation states. When the samples were subjected to the SPW, the doublet shifted to 458.1, 458.4, 458.1, 458.2 and 458.1 eV for 45 min and 1, 3, 10 and 14 days, with a spin orbit splitting of 5.4, 5.7, 5.3, 6.1 and 5.7 eV, respectively (Fig. 2a). The small variation in doublet shifts are due to changes in the Al(OH)3 coating, not a change in the core material; doublet and splitting values for the aged samples remain consistent with that of a Ti4+ oxidation state. The peak intensities of Al (Fig. 2b) and Ti (Fig. 2a) varied throughout the aging process, which indicates the qualitative decrease (lower intensity after 45 min to 10 days of SPW treatment) or increase (higher intensity after 14 days of SPW treatment) in both Al and Ti concentration on the surface of the samples.
 | ||
| Fig. 2 High resolution XPS spectra of Ti (a) and Al (b) in Al(OH)3·TiO2 ENM aged in SPW from 45 min to 14 days. Ti and Al indicate the un-aged sample. | ||
3.2 Photoreactivity
EPR spectra were obtained for both aged and un-aged samples to determine the identity of the primary reactive oxygen species. A representative spectrum with a corresponding simulation is shown in Fig. 3. The simulated data contained a 70/30 ratio of two distinct sets of hyperfine couplings. In the predominant species, hyperfine couplings to three nuclei were observed (1H – 37 MHz, 14 N – 39 MHz, 31P – 132 MHz). These values were consistent with the formation of a DEPMPO-hydroxyl radical spin adduct.30 In the minor component, there was an additional coupling of 22 MHz to an I = 1/2 nucleus (perhaps an acidic proton in partial occupancy).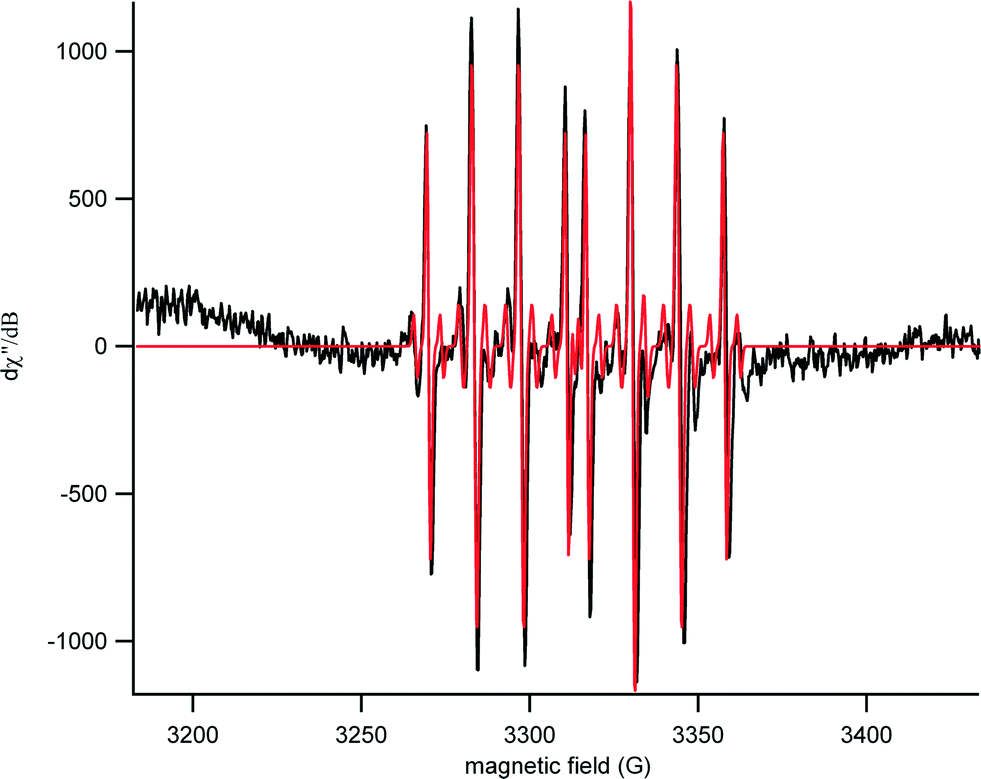 | ||
| Fig. 3 Representative EPR spectra for SPW-aged Al(OH)3·TiO2 ENM exposed to UV light for 7 minutes in the presence of DEPMPO spin trap (experimental spectrum in black and the corresponding simulation in red). The characteristic pair of 1:2:2:1 peaks is apparent with hyperfine couplings matching that of a DEPMPO-hydroxyl radical spin adduct. | ||
The APF assay showed a significant increase in photoreactivity under simulated solar radiation, relative to lab light controls, for particles aged 1 day or longer; however, the reactivity was substantially less than that of the P25 positive control (Fig. S4A†). In the TBARS assay, none of the samples were found to increase production of TBARS without UVA exposure (Fig. S4B†). With UVA, however, TBARS production was observed for un-aged Al(OH)3·TiO2, which increased with the duration of aging of the Al(OH)3·TiO2 in SPW (Fig. S4B†). The positive control, P25, had the greatest photoreactivity in all three assays, and the photocatalytic generation of ROS was less in all of the aged Al(OH)3·TiO2 conditions than for P25.
3.3 ENM dispersions
Dispersions of untreated and SPW treated Al(OH)3-coated TiO2 ENM had similar values for Z-average size and PDI, indicating that these dispersions had similar size distributions (Table 2). However, Al(OH)3-coated TiO2 ENM aged in SPW for 1 day had a higher particle count in the 500–1000 nm range, indicating this dispersion had the least agglomeration (Table 2). PDI values are similar to values reported for TiO2 throughout nanoparticle in vitro toxicology literature.31| Aging Time | % count by volume | % count by intensity | Z Average size (nm) | PDI | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| <100 nm | 100–500 nm | 500–1000 nm | >1000 nm | <100 nm | 100–500 nm | 500–1000 nm | >1000 nm | |||
| Untreated | 0 | 0 | 0.7 | 99.3 | 0 | 0 | 2 | 98 | 3074 | 0.434 |
| 45 min | 0 | 0 | 0.3 | 99.7 | 0 | 0 | 0.9 | 99.1 | 3048 | 0.433 |
| 1 day | 0 | 0 | 81.6 | 18.4 | 0 | 0 | 83.9 | 16.1 | 4453 | 0.454 |
| 3 days | 0 | 0 | 21.7 | 78.3 | 0 | 0 | 24.7 | 75.3 | 3589 | 0.334 |
| 14 days | 0 | 0 | 0 | 100 | 0 | 0 | 0 | 100 | 3302 | 0.317 |
3.4 Phototoxicity assays
The ocular tissues are sensitive to phototoxic damage and retinal pigment epithelial (ARPE-19) cell line is useful to assess phototoxic damage from TiO2 nanoparticles.8 The cell culture model provides an opportunity for comparative phototoxicity studies relating coated and aged Al(OH)3·TiO2 to other forms of TiO2.8 The RPE culture may also serve as a convenient test system to indicate potential phototoxicity to other tissues or aquatic species.12The ARPE-19 cells were treated with aged or un-aged Al(OH)3·TiO2 and then irradiated with UVA or kept in the dark. Decrease in viability at any particular dose was indicative of cytotoxicity (in the dark) or phototoxicity (under UVA irradiation compared to darkness). The results showed no difference in cell viability between irradiated and non-irradiated cells for un-aged Al(OH)3·TiO2, indicating a lack of phototoxicity (Fig. 4). In contrast, there was a significant difference in viability between irradiated and non-irradiated cells treated with aged ENM. Depending on aging condition and dose, the aged TiO2 particles under UVA irradiation showed up to approximately 30% greater reduction of cell viability than was seen in the dark. This was considered to be evidence of phototoxicity. However, the duration of aging did not affect cell viability, indicating that phototoxicity did not increase with duration of aging. The EDS data (Table 1) indicates a significant rearrangement of Al to Ti ratio after aging of 3 days or longer. The inability to see an effect of aging time on phototoxicity might be due to insufficient statistical power of the assay as opposed to a biophysical response. In all cases, the P25 positive control caused 100% cytotoxicity under UVA irradiation.
 | ||
| Fig. 4 Phototoxicity: viability of ARPE-19 cells under UVA or darkness after being treated with un-aged or SPW-aged Al(OH)3·TiO2 ENM. For each age group, the mean percent (±standard error) of viable cells is presented for cells exposed to darkness and for cells exposed to UVA radiation. Exposure to P25 TiO2 was used as a positive control. The dose axis in each panel is expressed on a log scale, which does not include a zero point, however untreated control cells (0 μg mL−1 Al(OH)3·TiO2) invariably showed 100% viability under either conditions of darkness or UVA exposure, and the data were indistinguishable from the lowest dose point plotted in the figures. | ||
3.5 Cellular uptake of ENM
Cells treated with both aged and un-aged Al(OH)3·TiO2 and Degussa P25 showed a clear increase in flow cytometry side scatter (Fig. 5; Table S1†) indicative of ENM internalization. Cells treated with Degussa P25 gave the highest mean SSC intensity value (6.67-fold increase over control). Treatment with the un-aged Al(OH)3·TiO2 showed a higher increase in SSC (3.29) than those treated with the aged ENM (ranging from 2.19 to 2.84). Duration of aging did not affect the SSC signal. | ||
| Fig. 5 Flow cytometry side scatter signals for ARPE-19 cells exposed to Degussa P25, un-aged and Al(OH)3·TiO2 ENM aged in SPW from 1 to 14 days. | ||
Darkfield/fluorescent images of ARPE-19 cells treated with P25 or aged Al(OH)3·TiO2 contained white, star-like reflective images of particles within the cytoplasm that congregated around the nucleus, co-localized with the endoplasmic reticulum (Fig. 6A). No obvious differences appeared among cells treated with aged or un-aged Al(OH)3·TiO2 (Fig. 6B–F). Agglomerates in cells treated with Al(OH)3·TiO2 appeared to be fewer in number but larger in size than agglomerates observed in cells treated with P25.
 | ||
| Fig. 6 Fluorescent and dark-field microscopy images (60×) for ARPE-19 cells treated with P25 (A), un-aged Al(OH)3·TiO2 ENM (B) and Al(OH)3·TiO2 ENM aged in SPW for 1 day (C), 3 days (D), 10 days (E), and 14 days (F). ENM appear white, mitochondria appear green (GFP), cell membrane appears red (CMO), and nuclei appear blue (DAPI). Images A–F show that internalized ENM are co-localized with the mitochondria and endoplasmic reticulum. ENM do not appear to enter the nuclei. | ||
4. Discussion
It is important to evaluate environmental transformations of ENM and determine if those transformations alter the activity of the material. In a previous publication5 we observed the redistribution of the Al(OH)3 coating on TiO2 ENM caused by aging in a chlorinated aqueous medium that simulated swimming pool water and discussed possible physicochemical mechanisms. Here we investigated effects of the coating transformation on the activity of the material. Coating thickness likely plays a significant role on the deterioration of the coating. The original thickness of the coating on the ENM as received from the manufacturer depends on synthesis methodology and quality control in production. For our study we assumed that all particles have a relatively uniform thickness of coating. The Al/Ti ratio across the particle surface, morphology, agglomeration, bioavailability, and reactivity were all altered by aging. The transformed particles were internalized by cells in culture and exhibited increased reactivity and potential phototoxicity under UVA irradiation.TEM (Fig. S2†) images indicated that aging of TiO2 ENM in SPW had no effect on the crystalline structure of the Al(OH)3 coating or of the rutile TiO2 core. This is consistent with X-ray diffraction reported in our previous publication5 as well as the findings of Fisichella et al.32 The XPS data showed that Ti was present in the Ti4+ oxidation state (doublet at about 459.3 eV with a spin orbit splitting of 5.8 eV (Fig. 2a)).5,29 Li & Fu observed a direct relation of concentration to XPS peak intensities when studying doping of TiO2 with heavy metals;33 our data show that the relative peak intensities of Ti (Fig. 2a) and Al (Fig. 2b) on the particle surfaces varied throughout the aging process, suggesting a general instability of the surface coating in SPW.
Dynamic light scattering characterization was performed as a qualitative assessment of the ENM suspensions. In particular it was important to determine whether any very small particles were generated as a result of the surrogate pool water treatment as this would have an impact on available surface area which in turn could potentially impact photoreactivity and cellular uptake associated with in vitro toxicity testing. As can be seen in Table 2, there were no detectable particles below 500 nm and the overall Z-average and PDI values were similar among the un-age and aged samples. This indicates that alteration of the coating, as opposed to variation in size distribution, was responsible for the effects induced by SPW treatment. Data from EPR spectra identified hydroxyl radical formation during UV irradiation. Results of the TBARS assay showed enhanced photocatalytic activity after SPW aging.
The uptake of TiO2 ENM into cells in vitro was evaluated with flow cytometry and darkfield microscopy. The uptake of reflective particles, including Degussa P25 and all Al(OH)3·TiO2 samples, resulted in dose-related increases in side scatter signal (greater amount of reflection away from the direct light path).8,26,27 In flow cytometry, the amount of side scatter light increases with the number of particles incorporated into a cell and/or the size or agglomeration of those particles. Under darkfield microscopy, the aged and un-aged ENM observed in the cytoplasm showed greater agglomeration than P25. When considered alone, increased agglomeration within the cells would correspond to greater side scatter intensity on the flow cytometer; however, cells treated with aged particles showed decreased side scatter signal relative to those treated with the un-aged samples. This suggests that cells treated with aged ENM internalized fewer particles than those treated with un-aged material. Overall, the dark-field images and flow cytometry indicate that cells treated with Al(OH)3·TiO2 incorporated particles into their cytoplasm; the incorporated ENM were fewer in number and larger in size than P25; and the aging of coated particles in SPW did not appear to influence the cellular uptake or distribution of the ENM.
Results of the phototoxicity experiments suggested the presence of a phototoxic reaction in that the aged Al(OH)3·TiO2 particles, despite lower uptake indicated by flow cytometry, showed up to about 30% greater toxicity under UVA irradiation than under darkness. Un-aged ENM were not phototoxic under the conditions tested. The analysis of cytotoxicity data showed an overall significant increase in toxicity following aging under UVA radiation, but not an interaction between the amount of aging and UVA irradiation. Inspection of Fig. 4 suggests that the lack of a significant aging-by-UVA interaction was related to insufficient statistical power in the assay to detect an significant interaction. The phototoxicity of the aged material was presumably caused by degradation of the Al(OH)3 protective coating which exposed the TiO2 particle surface to aqueous cellular media and, under photo-excitation, resulted in the catalytic generation of ROS that led to cytotoxicity. Possible mechanisms of underlying phototoxicity were discussed previously.8
Both the APF and TBARS assays of photoreactivity indicated a significant production of ROS under UV irradiation in the aged coated materials. It is well established that OH radicals are one of the important free radical species generated from UVA irradiation of TiO2, and our EPR results confirmed the formation of hydroxyl radicals in aged samples. It is possible that the phototoxicity of TiO2 in ARPE-19 cells involves the transformation of the OH radicals, produced by aged Al(OH)3·TiO2, into other free radical species.9
All of the coated samples tested here, aged or not, showed much less phototoxic potency that did the positive control P25. The low phototoxic potential of the aged Al(OH)3-coated particles, compared to P25, is likely attributed to a combination of shunting the free radical to the remaining intact coating, reduced cellular uptake of the agglomerated particles, and lower inherent photoreactivity of the rutile crystal structure than the mixed rutile/anatase structure of the P25. It is important to consider, however, that the exposures to UVA in these studies reflect only brief periods of time. In a real-world environment, the biological systems could be exposed to photo-catalytically active particles and UVA irradiation for longer periods of time, which might lead to cumulative free radical damage.
These experiments show that nanomaterials released into the environment might undergo physical and/or chemical transformations, altering their potential reactivity. This demonstrates the importance of considering environmental transformations when assessing risks across expected life cycles of engineered nanomaterials. In addition to the use of coated TiO2 materials in sunscreens, TiO2 has a broad range of industrial and commercial uses including as photo-catalysts, UV stabilizers, and pigments/colorants in paints, foods or other consumer products.34 Although the optimal particle size for coloration is in the range of 200–300 nm, these products may have a segment of the particle size distribution in the nanometer size range (i.e. between 1–100 nm).35 As particle size decreases and surface area to mass ratio increases, TiO2 becomes a more effective photo-catalyst, particularly in the anatase or mixed anatase/rutile crystal structures, as observed here for P25. Photo-catalyst applications of TiO2 nanomaterials include photovoltaics, photodynamic drug therapies, self-cleaning surfaces and photocatalytic degradation of environmental pollutants.36 Photocatalytic forms of TiO2, such as P25, are activated primarily by UV radiation. An active research area, however, is creation of modified TiO2 structures, which extend the activation spectrum into the visible wavelengths.37,38 These “doped” materials have potential applications to interior surfaces such as kitchen or bathroom self-cleaning surfaces where they become active under normal interior lighting. The widespread use of TiO2 in a variety of applications leads to the possibility of eventual release into the environment, and concerns about environmental contamination. Photo-activation of catalytic forms of TiO2 in surface waters, generating hydroxy and oxygen free radicals, can cause for phototoxic damage to aquatic species. For example, exposure to TiO2 in the presence of 48 hours of simulated solar radiation, including UV wavelengths, lowered the LC50 of TiO2 to Daphnia magna by up to four orders of magnitude in relation to the toxicity under normal laboratory lighting without UV wavelengths.12 This potentiation of TiO2 toxicity in UV wavelengths can make TiO2 toxic to marine species at concentration ranges that approach levels projected to result from environmental contamination.39 Recently, mammalian pharmacokinetic studies have observed that, although absorption of TiO2 is slow, its clearance may be even slower,40 raising the possibility of bioaccumulation after long-term exposure. The potential of long-term bioaccumulation of a photoactive material may produce of phototoxic effects at even lower environmental concentrations. A probabilistic risk assessment of five nanomaterials, including TiO2, compared probability distribution estimates of environmental exposures with those of toxic effects, including acute phototoxicity in aquatic species.41 They concluded that the probability distributions for TiO2 environmental exposures and sensitive effects were relatively close, with only about one order of magnitude separation. Assessments of potential environmental risks of nanomaterials to date, have not considered the combination of environmental transformations (demonstrated here), long-term bioaccumulation, and phototoxicity from co-exposure to UV wavelengths (or visible wavelengths in the case of doped TiO2 varieties).
Acknowledgements
This research was funded by and conducted at the National Risk Management Research Laboratory and the National Health and Environmental Effects Research Laboratory, of U.S. Environmental Protection Agency (EPA) under the EPA's Chemical Safety for Sustainability Research Program. Additionally, this research was supported in part by an appointment to the Research Participation Program for the U.S. Environmental Protection Agency, Office of Research and Development, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and EPA. This paper was subjected to the internal policy review of the U.S. EPA and approved for publication. Approval does not signify that the contents reflect the views of the Agency, nor does mention of trade names or commercial products constitute endorsement or recommendation for use. The authors wish to thank Dr. Gary Logan of Miami University (Oxford, OH) for the use of his facility for analyzing radical formation and for his insightful comments. The authors also thank John Havel for his work on the graphical abstract and Dr. Tim Shafer and Dr. Kim Rogers for review comments on an earlier version of the manuscript.References
- National Research Council (NRC). Research Progress on Environmental, Health, and Safety Aspects of Engineered Nanomaterials (The National Academies Press, Washington, District of Columbia, http://www.nap.edu/catalog.php?record_id=18475, 2015).
- J. Virkutyte and S. R. Al-Abed, J. Nanopart. Res., 2012, 14, 787–796 CrossRef.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- P. Tucci, G. Porta, M. Agostini, D. Dinsdale, I. Iavicoli, K. Cain, A. Finazzi-Agró, G. Melino and A. Willis, Cell Death Dis., 2013, 4, e549 CrossRef CAS PubMed.
- J. Virkutyte, S. R. Al-Abed and D. D. Dionysiou, Chem. Eng. J., 2012, 191, 95–103 CrossRef CAS.
- T. Ohno, K. Tokieda, S. Higashida and M. Matsumura, Appl. Catal., A, 2003, 244, 383–391 CrossRef CAS.
- Y. Li, W. Zhang, J. Niu and Y. Chen, ACS Nano, 2012, 6, 5164–5173 CrossRef CAS PubMed.
- K. Sanders, L. L. Degn, W. R. Mundy, R. M. Zucker, K. Dreher, B. Zhao, J. E. Roberts and W. K. Boyes, Toxicol. Appl. Pharmacol., 2012, 258, 226–236 CrossRef CAS PubMed.
- J.-J. Yin, J. Liu, M. Ehrenshaft, J. E. Roberts, P. P. Fu, R. P. Mason and B. Zhao, Toxicol. Appl. Pharmacol., 2012, 263, 81–88 CrossRef CAS PubMed.
- B. Balasubramanian, W. K. Pogozelski and T. D. Tullius, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9738–9743 CrossRef CAS.
- V. Brezová, S. Gabčová, D. Dvoranová and A. Staško, J. Photochem. Photobiol., B, 2005, 79, 121–134 CrossRef PubMed.
- H. Ma, A. Brennan and S. A. Diamond, Environ. Toxicol. Chem., 2012, 31, 2099–2107 CrossRef CAS PubMed.
- R. J. Miller, S. Bennett, A. A. Keller, S. Pease and H. S. Lenihan, PLoS One, 2012, 7, e303321 Search PubMed.
- H. Wang, R. L. Wick and B. Xing, Environ. Pollut., 2009, 157, 1171–1177 CrossRef CAS PubMed.
- M. Fouqueray, P. Noury, L. Dherret, P. Chaurand, K. Abbaci, J. Labille, J. Rose and J. Garric, Environ. Sci. Pollut. Res., 2013, 20, 3340–3350 CrossRef CAS PubMed.
- B. Faure, G. Salazar-Alvarez, A. Ahniyaz, I. Villaluenga, G. Berriozabal, Y. R. De Miguel and L. Bergström, Sci. Technol. Adv. Mater., 2013, 14, 023001–023023 CrossRef.
- Y. Ji, L. Zhou, C. Ferronato, A. Salvador, X. Yang and J. Chovelon, Appl. Catal., B, 2013, 140-141, 457–467 CrossRef CAS.
- T. Poiger, H.-R. Buser, M. E. Balmer, P.-A. Bergqvist and M. Müller, Chemosphere, 2004, 55, 951–963 CrossRef CAS PubMed.
- M. A. Kiser, P. Westerhoff, T. Benn, Y. Wang, J. Perez-Rivera and K. Hristovski, Environ. Sci. Technol., 2009, 43, 6757–6763 CrossRef CAS PubMed.
- C. Plagellat, T. Kupper, R. Furrer, L. F. de Alencastro, D. Grandjean and J. Tarradellas, Chemosphere, 2006, 62, 915–925 CrossRef CAS PubMed.
- D. R. Holbrook, D. Motabar, O. Quiñones, B. Stanford, B. Vanderford and D. Moss, Environ. Pollut., 2013, 181, 68–74 CrossRef PubMed.
- V. H. Grassian, A. J. Haes, I. A. Mudunkotuwa, P. Demokritou, A. B. Kane, C. J. Murphy, J. E. Hutchison, J. A. Isaacs, Y.-S. Jun, B. Karn, S. I. Khondaker, S. C. Larsen, B. L. T. Lau, J. M. Pettibone, O. A. Sadik, N. B. Saleh and C. Teague, Environ. Sci.: Nano, 2016, 3, 15–27 RSC.
- M. Auffan, M. Pedeutour, J. Rose, A. Masion, F. Ziarelli, D. Borschneck, C. Chaneac, C. Botta, P. Chaurand, J. Labille and J.-Y. Bottero, Environ. Sci. Technol., 2010, 44, 2689–2694 CrossRef CAS PubMed.
- J. Labille, J. Feng, C. Botta, D. Borschneck, M. Sammut, M. Cabie, M. Auffan, J. Rose and J.-Y. Bottero, Environ. Pollut., 2010, 158, 3482–3489 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- R. M. Zucker, E. J. Massaro, K. M. Sanders, L. L. Degn and W. K. Boyes, Cytometry, Part A, 2010, 77, 677–685 CrossRef CAS PubMed.
- R. M. Zucker, K. M. Daniel, E. J. Massaro, S. J. Karafas, L. L. Degn and W. K. Boyes, Cytometry, Part A, 2013, 83, 962–972 CAS.
- M. A. Stephens, J. Am. Stat. Assoc., 1974, 69, 730–737 CrossRef.
- S. Livraghi, I. Corazzari, M. Paganini, G. Ceccone, E. Giamello, B. Fubini and I. Fenoglio, Chem Commun., 2010, 46, 8478–8480 RSC.
- F. A. Villamena, C. M. Hadad and J. L. Zweier, J. Phys. Chem. A., 2003, 107, 4407–4414 CrossRef CAS.
- P. Bihari, M. Vippola, S. Schultes, M. Praetner, A. G. Khandoga, C. A. Reichel, C. Coester, T. Tuomi, M. Rehberg and F. Krombach, Part. Fibre Toxicol., 2008, 5, 1–14 CrossRef PubMed.
- M. Fisichella, F. Berenguer, G. Steinmetz, M. Auffan, J. Rose and O. Prat, Part. Fibre Toxicol., 2012, 9, 13 CrossRef PubMed.
- S. Li and J. Fu, Corros. Sci., 2013, 68, 101–110 CrossRef CAS.
- H. Shi, R. Magaye, V. Castranova and J. Zhao, Part. Fibre Toxicol., 2013, 10, 1–33 CrossRef CAS PubMed.
- A. Weir, P. Westerhoff, L. Fabricius, K. Hristovski and N. von Goetz, Environ. Sci. Technol., 2012, 46, 2242–2250 CrossRef CAS PubMed.
- S. Kwon, M. Fan, A. T. Cooper and H. Yang, Crit. Rev. Environ. Sci. Technol., 2008, 38, 197–226 CrossRef CAS.
- J. Virkutyte and R. S. Varma, New J. Chem., 2010, 34, 1094–1096 RSC.
- P. Wang, B. Huang, Y. Dai and M.-H. Whangbo, Phys. Chem. Chem. Phys., 2012, 14, 9813–9825 RSC.
- R. Kaegi, A. Ulrich, B. Sinnet, R. Vonbank, A. Wichser, S. Zuleeg, H. Simmler, S. Brunner, H. Vonmont, M. Burkhardt and M. Boller, Environ. Pollut., 2008, 156, 233–239 CrossRef CAS PubMed.
- G. Bachler, N. von Goetz and K. Hungerbuhler, Nanotoxicology, 2015, 9, 373–380 CrossRef CAS PubMed.
- C. Coll, D. Notter, F. Gottschalk, T. Sun, C. Som and B. Nowack, Nanotoxicology, 2016, 10, 436–444 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5en00250h |
| This journal is © The Royal Society of Chemistry 2016 |