
Non-covalent functionalization of high-surface area nanomaterials: a new class of sorbent materials†
Kara M.
Nell‡
a,
Sean A.
Fontenot‡
a,
Timothy G.
Carter
a,
Marvin G.
Warner
b,
Cynthia L.
Warner
b,
R. Shane
Addleman
*b and
Darren W.
Johnson
*a
aDepartment of Chemistry & Biochemistry and Materials Science Institute, University of Oregon, Eugene, OR 97403-1253, USA. E-mail: dwj@uoregon.edu
bPacific Northwest National Laboratory, P.O. Box 999, Richland, Washington DC 99352, USA. E-mail: Raymond.addleman@pnl.gov
First published on 27th October 2015
Abstract
A non-covalent approach to functionalizing nanostructured materials with high-specificity ligands is described. In this work a variety of thiol ligands were non-covalently attached to self-assembled phenyl monolayers on nanostructured materials by taking advantage of favorable aromatic interactions. The resulting sorbent materials, both mesoporous silica and magnetic nanoparticles, were found to be very effective at scavenging soft heavy metal cations, Cd(II), Hg(II), Pb(II) and Ag(I), from aqueous matrices, performing better than commercial sorbents and comparably to the best covalently functionalized thiol sorbents available. This approach can be extended to a variety of surface chemistries and has application to chemical functionalization of a broad range of support structures used for chemical separations and processing.
Introduction
Clean water is a health concern for the entire world population: the demand for clean drinking water continues to increase as a result of population growth as well as increased standards of living. At the same time, supply is decreasing due to contamination of fresh water sources.1,2 Thus, the demand for new sorbent materials and strategies for remediation of contaminated aqueous systems is on the rise. The capture of toxic heavy metals—whether occurring naturally in the water system or introduced by humankind—is essential for human health. Heavy metals, such as mercury (Hg), lead (Pb), or cadmium (Cd), have been shown to have adverse health effects at concentrations as low as part per billion levels.3,4The need for water purification methods extends to numerous industries that require purification of their aqueous waste streams.5 As a specific example, the mining industry seeks trace collection materials for both harmful contaminants and recovery of desirable precious metals from their waste streams.6 The present work focuses on fundamental science aimed at developing a novel, reversible (non-covalent) functionalization method for mesoporous silicas and related nanomaterials. While the present work is aimed at developing the initial basic research behind these new sorbents, these resulting materials offer long-term promise in advancing sorbent technologies by enabling: i) facile screening of surface ligands that can be attached through non-covalent interactions rather than stepwise synthesis; ii) possible new routes to sorbent regeneration that could avoid the harsh acid/base conditions required for current commercial, high-capacity sorbents; and iii) exceptionally high capacities and selectivities that are easily tunable through non-covalent functionalization even in these first generation materials. Gratifyingly, these materials feature comparable performance to commercial and specialty sorbents for trace collection of heavy metals.
A variety of methods exist that can be tapped as solutions to aqueous remediation, from liquid–liquid extraction to sorption.7,8 Solid phase sorbent materials serve as excellent filtration media due to their ease of use, selectivity, and trace collection capabilities.9 Mesoporous silica sorbents, such as MCM-41, exhibit many attractive properties, including well established surface chemistries, controllable “honey-comb”-like porosity, structural integrity, hydrothermal stability, and high surface areas approaching 1000 m2 g−1.10–12 Other materials, like polymer and resin-based sorbents, have good selectivity for target contaminants due to their diverse and easily modifiable surface chemistries, but suffer from low contaminant loading capacities as a result of limited surface area and low ligand densities. Alternatively, activated carbons have high surface area but limited selectivity.13 By functionalizing mesoporous materials with highly selective ligands, one can integrate desirable substrate properties such as a large surface area, rapid kinetics, and a high density of selective ligands to afford effective sorbent materials for trace level capture of contaminants in drinking water as well as other applications.
There are many examples of functionalized silicas as sorbents for aqueous remediation.14,15 A class of covalently functionalized silicas, self-assembled monolayers on mesoporous silica (SAMMS), have shown excellent performance as sorbent materials for a range of contaminants including toxic metals, metalloids, oxyanions as well as radioactive species.14,16–18 Thiol- and sulfur-functionalized materials have exceptional affinities for soft ions such as Hg(II), Ag(I), Cu(II), Cd(II), and Pb(II), and have been shown to enable enhanced collection and detection of trace metal contaminants.19–24 The materials discussed herein are a non-covalent complement to thiol-SAMMS. As such, we tested their performance for trace collection of heavy metals from natural waters for comparison.
Silica surfaces are typically chemically modified using established silane chemistries, such as co-condensation or post synthetic grafting, which result in a covalent connection.25 Unfortunately, covalent attachment requires optimization for each class of organic molecules; additionally, the use of bulky protecting groups is necessary when working with incompatible functional groups.26 Protecting groups hinder the ligand density, and the deprotection can be harmful to the base material. Utilizing our functionalization method, we would be able to bypass these steps reducing the amount of waste produced, and providing a material that would not suffer from partial deprotection. Herein, thiol functionalized ligands were employed directly without the use of protecting groups: simply using a supramolecular approach enables the synthesis of materials that can be directly functionalized with a variety of ligands using non-covalent aromatic interactions.
Results and discussion
Non-covalent functionalization method
In a previous communication, we demonstrated a non-covalently modified material featuring benzylmercaptan-functionalized Ph-SAMMS (BM, Fig. 1). This first-generation material, although very effective in toxic metal ion uptake, suffered from leaching of the secondary thiol layer into the aqueous matrices.27 In our previous report a third of the initial thiol surface leached into the aqueous media; this work has greatly improved upon these numbers by altering the hydrophobic nature of the secondary surface (Fig. 1).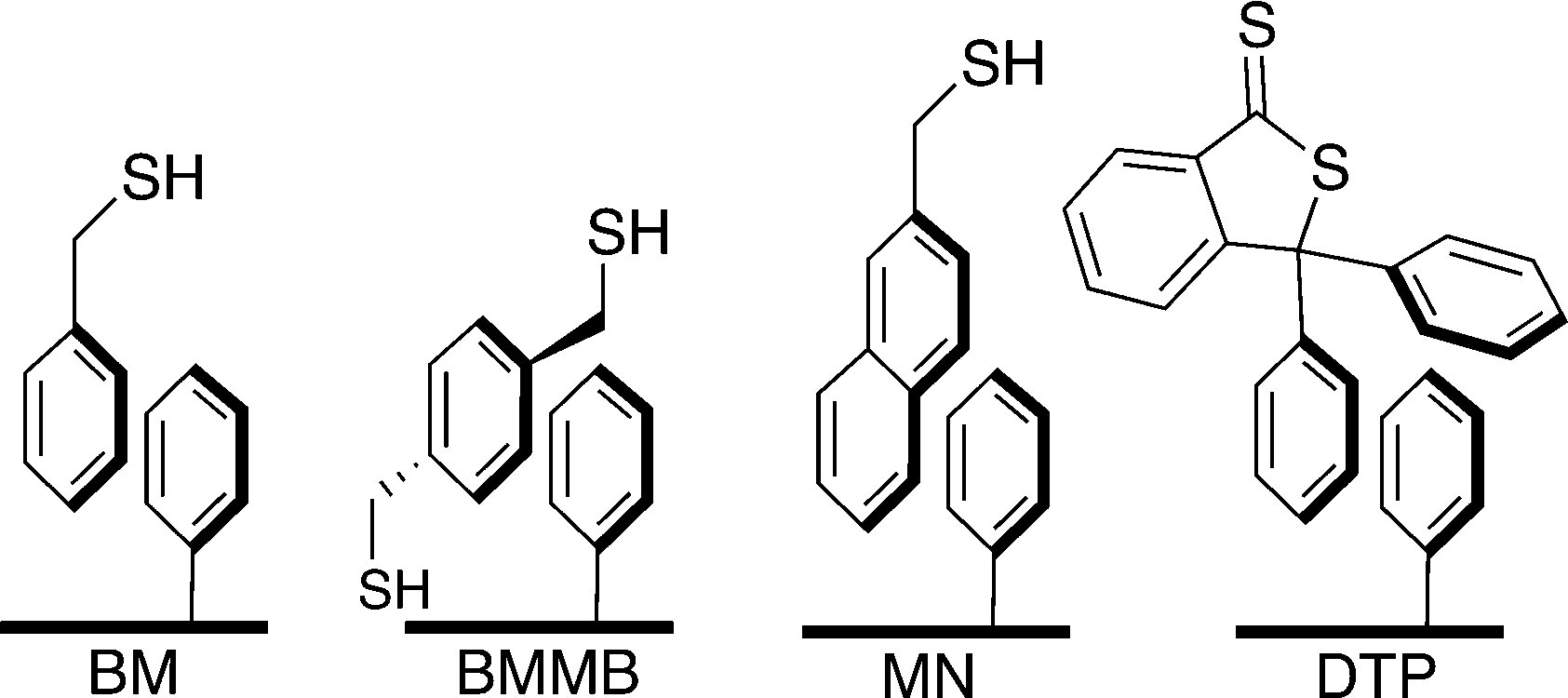 | ||
| Fig. 1 Depiction of idealized surface structure of materials created with varying ligands that were tested for stability. Benzyl mercaptan (BM), 1,4-bis(mercaptomethyl)benzene (BMMB), 2-(mercaptomethyl) naphthalene (MN), and 3,3-diphenylbenzo[c]thiophene-1(3H)-thione (DTP). | ||
Herein, Ph-SAMMS surfaces have been modified with several different thiol-bearing aromatic molecules (Fig. 1, Scheme 1). A primary surface of a phenyl-functionalized mesoporous silica material (Ph-SAMMS) serves as an anchor layer that supports a secondary surface layer of thiol-bearing aromatic ligands, which in turn, present a high density of thiol sites to the interior of the material. Fig. 1 depicts idealized representations of aromatic interactions between the ligands and Ph-SAMMS surface; in reality the attachment of these ligands is likely dominated by edge-to-face (CH–π) interactions anchoring these hydrophobic ligands to the hydrophobic phenyl substrate in aqueous streams.28–30 The hydrophobicity of each ligand was altered to optimize surface coverage and minimize ligand leaching. The resulting materials were evaluated for stability in aqueous matrices, and this functionalization motif was expanded to magnetic nanoparticles with the potential to span to a variety of form factors (vide infra).
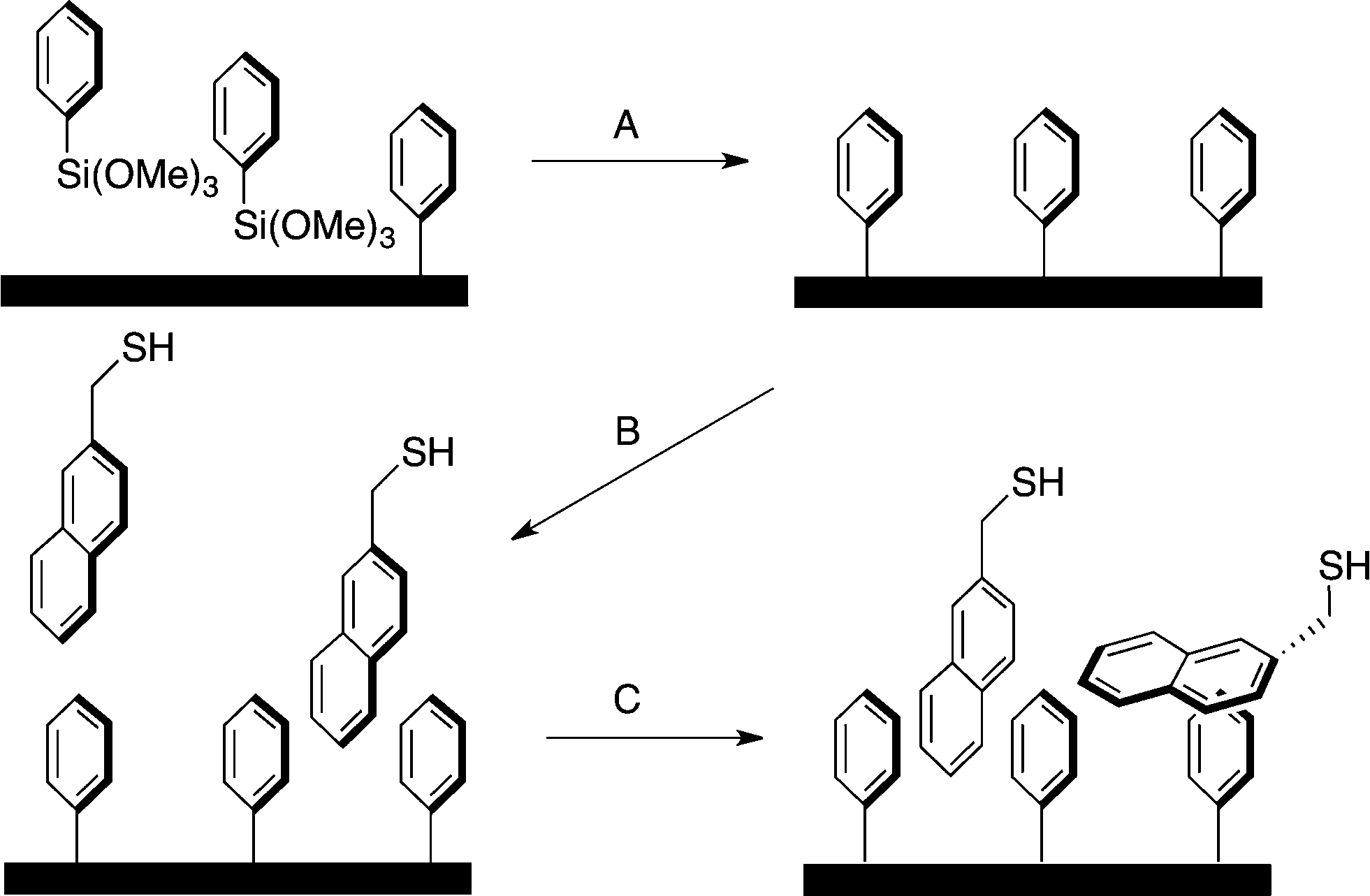 | ||
| Scheme 1 Preparation of Ph-SAMMS and supramolecular modification thereof. A) A phenyl layer is covalently attached to the MCM-41 surface yielding Ph-SAMMS. B) Ph-SAMMS is incubated with aromatic thiols. C) Slow evaporation yields a non-covalently functionalized thiol-bearing material, which is effective for capture of Cd, Hg, and Pb. The orientations of molecules in this illustration are idealized, but are shown in several probable orientations with ligands likely anchored by a variety of non-covalent interactions. | ||
Synthesis of functionalized mesoporous silica
All synthetic details and the crystal structure for 3,3-diphenylbenzo[c]thiophene-1(3H)-thione (DTP, ESI† Fig. S1) are contained in the ESI.†31The general material generation is depicted in Scheme 1. Ph-SAMMS (A) materials were prepared by stirring MCM-41 in dry toluene for 30 minutes to ensure that the silica has been solvated (see ESI† for additional details). A small amount of water (enough to coat the surface of the materials) was added to the reaction ensuring accessible surface hydroxyl groups. After an hour of stirring under argon, trimethoxyphenyl silane was added and the reaction was brought to reflux and left overnight. The methanol, a byproduct, was removed by evaporation at 120 °C leading to increased ligand density. To remove any excess trimethoxyphenyl silane, the reaction was hot filtered and washed with methanol. The resulting functionalized silica was dried at room temp then placed under vacuum for at least 6 hours.
The density of the phenyl anchor layer is easily varied by adjusting the stoichiometric addition of reagents (Table 1 and ESI†). The chosen thiol ligand was incubated with the Ph-SAMMS materials in dichloromethane (B) then evaporated overnight (C). During this time, the thiol ligands assembled on the surface due to favourable aromatic interactions with the primary phenyl surface. The materials were washed thoroughly with methanol, which removed any excess ligand that had simply precipitated as opposed to incorporating within the Ph-SAMMS material. The material was dried under vacuum for at least 6 hours to afford the final material.
| Phenyl silane coverage (phenyl molecules nm−2) | Surface area (m2 g−1) | Mean pore diameter (Å) |
|---|---|---|
| Surface area was measured using the Brunauer–Emmett–Teller (BET) method. As expected, surface area and mean pore diameter are lower for more completely functionalized materials. The pore diameter measurements indicated the reduced surface area is not due to obstruction of the pore by phenyl silane oligomers. | ||
| 2.43 | 390 | 17 |
| 2.22 | 400 | 17 |
| 1.60 | 550 | 19 |
| 0.72 | 730 | 27 |
| 0.34 | 770 | 31 |
| 0.06 | 875 | 34 |
| 0 | 1050 | 35 |
The ligand density was determined using thermal gravimetric analysis (TGA). Surface coverage was also confirmed by comparing pore sizes with increasing ligand coverage (Table 1). Pore size was determined by the Barrett–Joyner–Halenda (BJH) method,32 and it was found that the pore size decreases as the extent of silanation increases (Table 1). The surface area, determined by the Brunauer–Emmett–Teller (BET) method, was also found to decrease proportionally as phenyl silane loading increased, producing a material with a higher density but slightly reduced surface area/volume.33§ These results support that the surface is being functionalized while the pores remain unobstructed by oligomerization of the organosilanes (Table 1). High density functionalization of the surfaces can be seen to reduce the mean pore diameter from 35 to 17 Å. This 18 Å reduction in diameter is in good agreement with a phenyl silane monolayer (~9 Å) bound to each side of the pore wall.
Because X-ray and microscopy methods are not effective in analyzing interior surfaces of highly porous materials, characterizing the interior surface remains challenging. Ellman's test for thiols acts as a quick and effective characterization method in addition to TGA for quantification of surface thiols.34 Ellman's test relies on the use of DTNB (5,5′-dithiobis-(2-nitrobenzoic acid) or Ellman's Reagent), which releases the brightly-colored anion, 2-nitro-5-thiobenzoate (TNB) upon reaction with a free thiol. This reaction is stoichiometric: one molecule of TNB is produced per thiol. The TNB concentration may be measured by UV-vis spectroscopy and is a direct indication of thiol concentration. Ellman's test is ideal for measuring surface thiols since the indicator, TNB, is free of (not bound to) the thiol itself. Thus, the concentration of TNB in solution can be used to determine the number of accessible surface thiols. Using this method yielded a standard deviation across four measurements of 0.003 mmol SH per gram sorbent, proving to be highly precise. The DTP ligand density was measured with TGA and leaching was quantified with UV-vis spectroscopy using standard calibration curves. Additional material characterization method details are provided in the ESI.†
Impact of phenyl layer density on stability of secondary surface
For these non-covalently modified sorbent materials to be effective in water remediation applications, it is imperative that the interaction between the active surface layer and the anchoring base Ph-SAMMS substrate is robust enough to prevent the secondary surface layer from leaching into water. The density of the base/phenyl anchoring layer has a significant impact on the stability of the interaction and resulting functional material.Hydrophobic effects presumably drive the assembly of the secondary surface on to the Ph-SAMMS base layer, with specific attractive aromatic interactions anchoring the molecules to the Ph-substituted surface. We hypothesize that the surface density of the anchoring phenyl rings affects both the capacity of the Ph-SAMMS material for secondary surface molecules as well as the stability of that secondary surface (Fig. 2). In order to test the stability/capacity of the material, we varied the anchoring phenyl density from 0.1 nm−2 to 2.4 nm−2, functionalized the surfaces with mercaptomethyl naphthalene ligands (MN, Fig. 1), and then measured the loading coverage and leaching percentage (Fig. 2). The Ph-SAMMS capacity for loading aromatic thiol ligands increases with density of the phenyl anchor with an optimal range (for MN) of 1.3–1.9 nm−2. Similarly, data show the stability of the secondary surface is related to the density of the phenyl layer. Higher concentrations of phenyl groups provide more anchoring sites for the aromatic ligands, resulting in very little leaching for ~1–2 phenyl silanes per nm2. It can be seen in Fig. 2 that as the density of the anchor layer increases beyond ~2 phenyl silanes per nm2 the amount of thiol ligands bound to the surface decreases and leaching of the ligands begins to increase. Clearly steric crowding in the phenyl anchor layer can compromise surface ligand loading and stability.
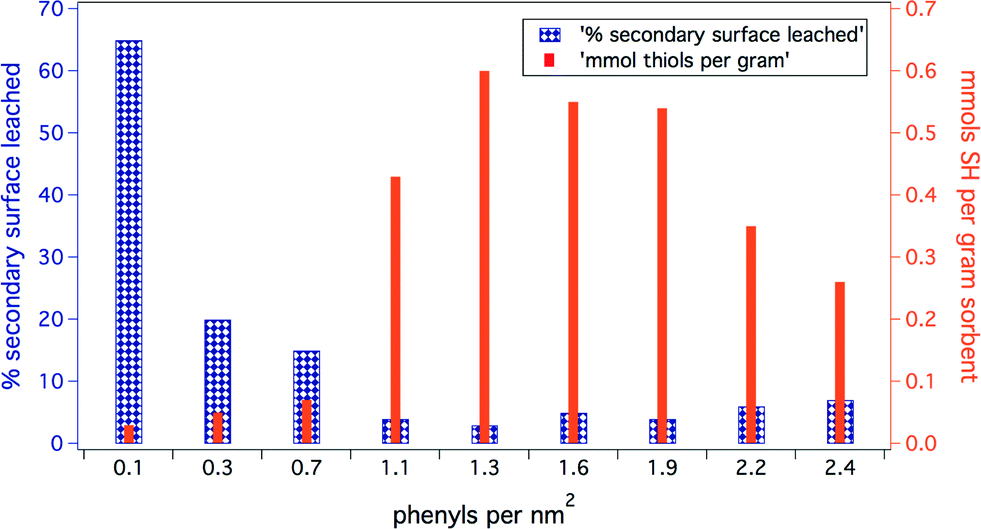 | ||
| Fig. 2 Comparative stability of mercaptomethyl naphthalene (MN) surface installed on Ph-SAMMS scaffolds of different anchoring densities. The lowest leaching and highest SH loading occurs for 1.3–1.9 anchoring phenyls per nm2. | ||
Impact of density and identity of secondary surface components
The identity of the ligands used to make up the secondary surface layer has a significant effect on material stability (Fig. 3). Benzyl mercaptan (BM) forms the least stable surface: 34% leached after 2 hours in filtered Columbia River water, which is not surprising given its relatively high water solubility. 1,4-bis(mercaptomethyl)benzene (BMMB) shows reduced leaching compared to BM, but still loses up to 10% of the thiol surface under identical conditions. Mercaptomethylnaphthalene (MN), which has very limited water solubility, forms the most robust secondary surface tested, leaching as little as 5%. 3,3-Diphenylbenzo[c]thiophene-1(3H)-thione (DTP) is the most hydrophobic ligand and did not show any detectable leaching. Unfortunately, DTP creates a surface layer that is too hydrophobic, and resists wetting, which causes ineffective solution contact and thus precludes effective metal uptake. Mercaptomethyl naphthalene was recognized as the preferred ligand for the secondary surface due to its ability to self-wet, its high thiol density and the low leaching of the secondary surface. All four non-covalently functionalized materials can be prepared with thiol surface densities similar to the covalently bound propyl thiol surface of SH-SAMMS.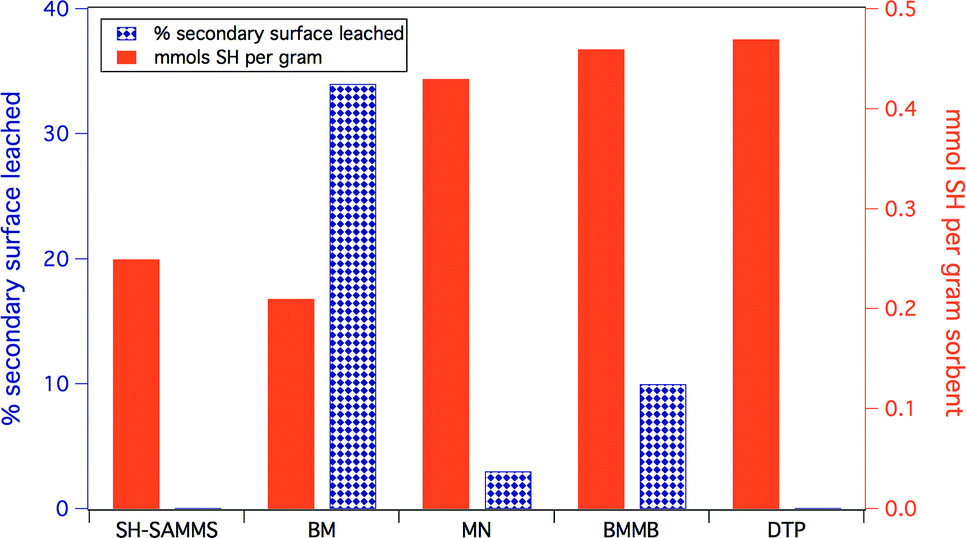 | ||
| Fig. 3 Comparative stability of materials based on secondary surface identity. Ligands are defined in Fig. 1 Both the covalently bound SAMMS as well as the most hydrophobic ligand, DTP, had no detectable leaching. Substrates had a phenyl silane anchor area density of 1.3 silanes per nm2. | ||
Because the density of the secondary surface also affects the stability of the final material to aqueous matrices, by varying the amount of thiol component one can increase the robustness of the secondary surface. Materials were prepared with less than 0.01 to 0.11 mmols thiol per gram on Ph-SAMMS that have 1.3 anchoring phenyls per nm2. The samples with 0.01 to 0.11 mmols thiol per gram leached ca. 34% and 5%, respectively. Presumably, highly dense and complete secondary surfaces contribute to the overall hydrophobicity of the surface environment, making the more densely covered surfaces more attractive for hydrophobic species.
Comparative sorbent performance of mesoporous materials
Sorbent performance was characterized by fundamental affinity of target analytes. A sorbent's affinity for a target species can be described by the distribution coefficient, Kd: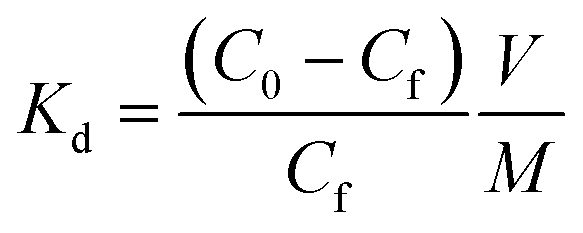
Here, C0 is the initial concentration of the target analyte, Cf is the final equilibrium concentration, V is the volume (mL) of the testing solution, and M is the mass (g) of sorbent used. The distribution coefficient is the solid phase equilibrium constant which acts as an empirical measurement of a sorbent's affinity (for the specified chemical in the specified matrix). A liquid to solid (L/S) ratio of 5000 was chosen and kept constant through all experiments. For these experiments this meant we had one milligram of material removing 50 ppb of target metals from 5 mL of water. Analytical methods details are provided in the ESI.†
High chemical affinity is essential when removing contaminants at trace levels. The material must selectively bind target species that are present in much lower concentrations than other ions. Kd values of 1 × 104 are good, and Kd values exceeding 1 × 105 are considered excellent and preferred for sorbents being used for trace collection applications.9 In addition to sorbent affinity, the percent metal removal percentage is shown from real water sources (Tables 2 and 3, respectively). We compared the uptake of the toxic metal ions, Cd(II), Hg(II), and Pb(II), with Ag(I) another soft thiophillic metal ion.
| Materials | Cd (%) | Hg (%) | Pb (%) | Ag (%) |
|---|---|---|---|---|
| Percent target removal of soft metals with selected materials. Noncovalently (NC) surface modified material loadings are given as mmol thiols (MN) per gram material, respectively. Experiments in Columbia River water (pH ~ 7.8) with a L/S of 5000. Analytical error ±2%. | ||||
| MCM-41 | 15 | 3 | 95 | 2 |
| Ph-SAMMS | 5 | 3 | 62 | 4 |
| GT-74 thiol resin | 62 | 57 | 66 | 84 |
| SH-SAMMS | >99 | 99 | >99 | >99 |
| NC 0.05 SH per g | 97 | 97 | 92 | >99 |
| NC 0.26 SH per g | 99 | 97 | >99 | >99 |
| NC 0.55 SH per g | 98 | 97 | 99 | >99 |
| Materials | Cd | Hg | Pb | Ag |
|---|---|---|---|---|
| K d values for selected materials. > indicate that the values were calculated based on limit of detection values since there was not enough metal left in solution to accurately measure. Noncovalently (NC) surface modified material loadings are given as mmol thiols (MN) per gram material, respectively. Experiments in Columbia River water (pH ~ 7.8) with a L/S of 5000. Analytical error ±2%. | ||||
| MCM-41 | 8.3 × 102 | 1.3 × 102 | 9.3 × 104 | 1.3 × 102 |
| Ph-SAMMS | 2.7 × 102 | 9.0 × 102 | 2.2 × 104 | 4.7 × 102 |
| GT-74 thiol resin | 8.2 × 103 | 6.6 × 103 | 9.8 × 103 | 2.7 × 104 |
| SH-SAMMS | 8.3 × 106 | 4.8 × 105 | 2.2 × 105 | >3.5 × 107 |
| NC 0.05 SH per g | 2.0 × 105 | 1.5 × 105 | 6.2 × 104 | >3.1 × 107 |
| NC 0.26 SH per g | 4.9 × 105 | 1.8 × 105 | >1.3 × 106 | >3.1 × 107 |
The thiol-functionalized SAMMS has been demonstrated repeatedly to be a superlative sorbent material for heavy metals19–24 and almost 100% removal of heavy metals from river water samples can be observed in Table 2. The non-covalently (NC) functionalized materials reported herein provide high performance that is on par with the covalent counterpart SH-SAMMS (Tables 2 and 3). By keeping the support material and ligand density constant we were able to compare and contrast performance of the covalent vs. non-covalent surface attachment. The NC functionalization motif provides similar performance to covalent grafting, and we were able to achieve almost identical thiol ligand densities for this comparison as well: 0.27 ligands per nm2 for SH-SAMMS versus 0.28 ligands per nm2 for the NC material (with equivalent SH per g of sorbent loadings).
Both surface modified nanostructured sorbents substantially outperformed commercial resin sorbent with similar thiol based surface chemistry. We compared the performance the noncovalently (NC) bound materials with three different ligand densities (0.05, 0.26, and 0.55 mmols thiol per gram) of mercaptomethylnapthalene surface chemistry. It can be observed that even at low ligand densities (0.05 SH g−1, equivalent to 0.06 ligands per nm2), the noncovalently bound material provides excellent performance, outperforming commercial sorbent materials. While Table 2 shows the different NC bound materials perform similarly at relatively low ligand loadings, one would expect that materials featuring higher ligand loadings would maintain high metal capture performance longer as the volume of water treated increases (i.e., larger L/S ratios).
For all non-covalently functionalized sorbents reported (as well as SH-SAMMS) the concentrations of Pb, Hg, and Cd were all reduced from 50 ppb to well below the Environmental Protection Agency's maximum contaminant levels for safe drinking water.3,4 Materials presented here exhibit ~100% capture of target contaminants when working with the L/S ratio of 5000; it is clear the sorbent material could successfully process much higher relative solution volumes to safe drinking water levels.
Raw MCM-41 silica and Ph-SAMMS (1.3 silanes per nm2) were also tested as control reference materials as shown in both Tables 2 and 3. Both materials show much lower metal capture and chemical affinity than the thiol functionalized materials. This confirms that the performance of our non-covalently functionalized materials is in fact due to the thiol component of the secondary layer.
As previously mentioned Kd is the solid phase distribution coefficient directly measures the chemical affinity of a sorbent (for the specified conditions). As shown in Table 3, all thiol-functionalized MCM-41 silica materials outperform GT-74, a commercially available macroporous resin with thiol functionality by orders of magnitude in affinity (Kd) for Hg(II), Cd(II), Ag(I) and Pb(II) (Table 3). For example, when looking at mercury affinity GT-74 has a Kd of 6.6 × 103 while the non-covalent functionalized material has Kd's > 105. Furthermore, the affinity of the non-covalently functionalized material for Hg(II), Cd(II), Ag(II) and Pb(II) is on par with that of their covalently functionalized counterpart, SH-SAMMS (Table 3). This supports that our non-covalent approach to surface modification is as effective as traditional covalent surface functionalization techniques. Further, the non-covalent surface modification approach offers some advantages over direct covalent linking. The active surface layer of the non-covalent attached ligands can be removed from the substrate with appropriate solvents, regenerated and replaced. Pragmatically, this enables the reuse and recycling of the expensive nanostructured sorbent (details will be reported in separate focused work). For fundamental studies the non-covalent attachment method also provides a rapid and practical way to screen many ligands for efficacy, thereby eliminating the need for stepwise covalent attachment (potentially with complicating protection and deprotection steps) for each new material-ligand combination.
Non-covalent functionalization of Fe3O4 nanoparticles
To demonstrate the potential and general applicability of our non-covalent surface functionalization method we applied it to a different sorbent form factor; magnetic nanoparticles. The magnetic Fe3O4 nanoparticles are nonporous with convex surfaces providing a different physical configuration and chemical composition than the mesoporous silica. Recently Fe3O4 nanoparticles have been shown to be effective sorbents for a variety of toxic metals from aqueous matrices.35–44 Unfortunately, it can be challenging to functionalize these particles with high affinity ligands since the complexing groups tend to bind to the iron surface of the particles rendering the installed surface chemistry inert. We were able to eliminate this problem by utilizing the two-stage non-covalent functionalization motif previously described.As a support structure, Fe3O4 nanoparticles (NPs) are advantageous due to their high surface area, dispersability, and susceptibility to magnetic fields. For application as a sorbent material, the NPs can be dispersed into solution and provide rapid kinetics resulting from the high surface area and good contact efficiency provided by the fine grain particles dispersed in solution. The sorbent material can then be retrieved magnetically as opposed to using filtration or packed bed configuration used with traditional sorbent materials.35–37 The use of dispersed magnetic media eliminates some of the problems typically encountered with filtration media such as backpressure and plugging due to fines and fouling.
The Fe3O4 nanoparticles were prepared as previously described.38 Magnetic Fe3O4 nanoparticles were modified to have a covalently bound phenyl surface (see ESI†). The phenyl-bearing surface on the nanoparticles is analogous to that of Ph-SAMMS and can act as an anchor layer for aromatic ligands to form the secondary chemically-functionalized surface (Fig. 4).
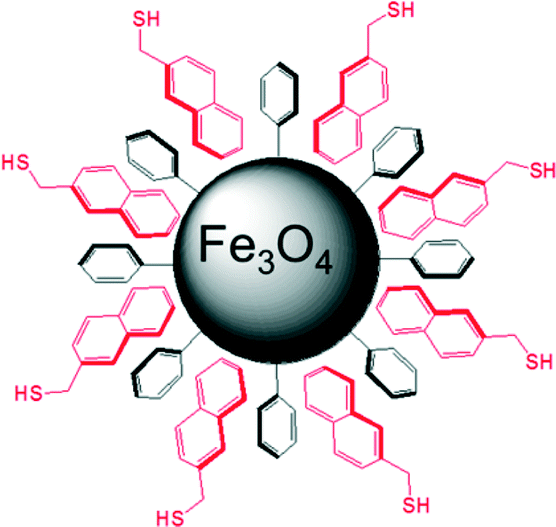 | ||
| Fig. 4 Illustration of a non-covalently functionalized Fe3O4 nanoparticle. The phenyl (black) ligand shell enables non-covalent attachment of the thiol-bearing layer in idealized orientation that is not to scale. | ||
Synthesis of modified Fe3O4 nanoparticles
The nanoparticles were prepared with lauric acid ligand shells.41 The lauric acid molecules are attached to the Fe3O4 surface through their carboxylate functionality, which were then exchanged for benzoic acid. As a result, the phenyl rings form the accessible surface of the benzoic acid Fe3O4 nanoparticles. The benzoic acid Fe3O4 NPs were then incubated with aromatic thiols in an organic solution and slow evaporation of the solvent yielded a non-covalently attached thiol-bearing surface.Fe3O4 NPs were characterized using BET, TGA, as well by scanning transmission electron microscopy coupled with energy-dispersive X-ray spectroscopy (STEM, EDX, respectively; see ESI†). The BET surface area of the benzoic acid NPs was 110 m2 per g while TGA indicated 0.8 mmol benzoic acid per g of material. These values are expected for the type and diameter (approximately 8 nm average) of these NPs. Fe3O4 materials are unfortunately not compatible with the Ellman's test for direct measurement of thiols, yielding a false positive. TGA analysis indicated approximately 0.04 mmols of BMMB per g material (0.22 ligands per nm2), 0.05 mmols g−1 MN (0.27 ligands per nm2), and 0.01 or less mmol per g BM (0.05 ligands per nm2). These values represent similar surface layer densities and approximately the same secondary ligand surface![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :phenyl anchor layer ratio (roughly 1:15) as most of our other non-covalently functionalized mesoporous silica materials. This suggests that the overall hydrophobic effect and specific favorable aromatic interactions between the phenyl anchor layer and secondary surface layer function similarly in both the convex and concave nanostructured materials reported herein.
:phenyl anchor layer ratio (roughly 1:15) as most of our other non-covalently functionalized mesoporous silica materials. This suggests that the overall hydrophobic effect and specific favorable aromatic interactions between the phenyl anchor layer and secondary surface layer function similarly in both the convex and concave nanostructured materials reported herein.
Comparative sorbent performance of magnetic nanoparticles
The addition of the thiol-bearing secondary surface on the benzoic acid Fe3O4 NPs yields a material with excellent sorption ability for toxic metals Cd(II) and Hg(II) (Table 4). The phenyl functionalized benzoic acid NPs were used as a comparative material for control, and mercaptopropionic acid functionalized NPs (MPA NP) were compared as the covalent counterpart to the new surface chemistry. As expected the thiol-modified surfaces provide very effective capture of heavy metals, particularly mercury. The NC functionalized NPs perform better than conventional resin materials while enabling faster kinetics, magnetic separation and renewable surface chemistry.| Sorbent material | Cd | Hg | ||
|---|---|---|---|---|
| % | K d | % | K d | |
| MPA NPs are covalently functionalized with mercaptopropionic acid (MPA). Noncovalently (NC) ligands bound to NP surfaces are defined in Fig. 1. Experiments in Columbia River water (pH ~ 7.8) with an L/S of 103. Analytical error ±2%. | ||||
| Benzoic acid NP | 76 | 3.5 × 103 | 28 | 3.6 × 102 |
| GT-74 thiol resin | 62 | 8.2 × 103 | 57 | 9.8 × 103 |
| MPA NP | 95 | 2.5 × 104 | 99 | 1.1 × 105 |
| NC BMMB + phenyl NP | 88 | 1.2 × 104 | 94 | 1.3 × 104 |
| NC MN + phenyl NP | 90 | 1.4 × 104 | 92 | 1.2 × 104 |
| NC BM + phenyl NP | 86 | 1.0 × 104 | 97 | 5.8 × 104 |
Conclusions
A simple non-covalent method for surface functionalization, utilizing an aromatic anchor layer to retain high affinity surface chemistry, has been generally demonstrated and optimized for selected nanomaterials. The surface functionalization method was shown to work for both nanoporous (silica) and nanoparticle (magnetic Fe3O4 NPs) structures. The binary surface chemistry with a phenyl anchor layer and an active layer of aromatic ligands is flexible, reversible and enables installation of high affinity ligands without many of the complications associated with covalent attachment. We show that altering the density of the primary phenyl scaffold and hydrophobicity of the secondary surface layer (aromatic thiols in this effort) affects the stability of the materials. We demonstrate these non-covalently functionalized sorbents performed on par to the best covalently functionalized materials and better than commercial resins with similar surface chemistry. The materials with non-covalently functionalized thiol surfaces showed excellent sorption capabilities for softer heavy metals such as Hg(II), Cd(II), Ag(I) and Pb(II). Results from this work show that this functionalization motif may be applied to a wide variety of support structures used for chemical separations and environmental remediation. The surface modification methods can be used for various applications, from remediation to immobilized catalysis, by expanding out from thiol containing arenes into a wide variety of aromatic ligands containing application relevant complexation moieties.Current efforts are focused on the regeneration of these materials in a sustainable closed-loop approach, making the use of these nanostructured sorbent supports more economically viable for remediation and trace collection purposes, as well as decreasing their environmental impact. Material stripping and regeneration methods for conventional sorbent materials are based on repetitive washes containing strong acids, bases, or competitive chelating ligands. The higher chemical affinity ligands that are particularly useful for trace collection and environmental remediation require more aggressive stripping methods, and sorbent materials can be significantly damaged by these processes. Utilizing the non-covalent interactions should enable the use of simple benign washes to remove the ligand-metal complex, and we are optimizing such processes to report in due course. This will allow the most expensive component—the sorbents support material—to be reused many times. Initial experiments suggest polar aprotic organic solvents can effectively strip the metal ligand complexes, however we are working towards a greener processing system.
Acknowledgements
The authors gratefully acknowledge the support of Pacific Northwest National Laboratory (PNNL) Laboratory-Directed Research & Development (LDRD) Program. PNNL is operated for the U.S. DOE by Battelle Memorial Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the DOE, PNNL or Battelle. Early work was supported by an NSF CAREER Award (CHE-0545206), and we also acknowledge the University of Oregon's Department of Chemistry & Biochemistry and Graduate Internship Program for generous support of this work. We gratefully acknowledge the use of UO CAMCOR facilities, which have been purchased with a combination of federal and state funding, and Dr. Lev N. Zakharov in CAMCOR for solving the crystal structure of DTP contained in the ESI.†Notes and references
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Mariñas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS PubMed.
- M. Hightower and S. A. Pierce, Nature, 2008, 452, 285–286 CrossRef CAS PubMed.
- US EPA, http://water.epa.gov/drink/info/lead/index.cfm, (accessed May 2015).
- US EPA, http://water.epa.gov/drink/info/chromium/index.cfm, (accessed May 2015).
- M. A. Barakat, Arabian J. Chem., 2011, 4, 361–377 CrossRef CAS.
- B. Lettermoser, Mine Wastes: Characterization, Treatment, and Environmental Impacts, Springer, Verlag Berlin Heidelberg, 2010 Search PubMed.
- D. Clifford, S. Subramonian and T. J. Sorg, Environ. Sci. Technol., 1986, 20, 1072–1080 CrossRef CAS.
- F. Fu and Q. Wang, J. Environ. Manage., 2011, 92, 407–418 CrossRef CAS PubMed.
- S. A. Fontenot, T. G. Carter, D. W. Johnson, R. S. Addleman, M. G. Warner, W. Yantasee, C. L. Warner, G. E. Fryxell and J. T. Bays, Trace Analysis with Nanomaterials, ed. D. T. Pierce and J. X. Zhao, Wiley-VCH Verlag GmbH &Co KGaA, Weinheim, 2010, p. 191 Search PubMed.
- I. Ali, Chem. Rev., 2012, 112, 5073–5091 CrossRef CAS PubMed.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson and E. W. Sheppard, J. Am. Chem. Soc., 1999, 114, 10834–10843 CrossRef.
- W. Yantasee, Y. Lin, G. E. Fryxell, K. L. Alford, A. Brad, J. Busche and C. D. Johnson, Ind. Eng. Chem. Res., 2004, 43, 2759–2764 CrossRef CAS.
- A. Walcarius and L. Mercier, J. Mater. Chem., 2010, 20, 4478–4511 RSC.
- A. Sayari, S. Hamoudi and Y. Yang, Chem. Mater., 2005, 17, 212–216 CrossRef CAS.
- B. E. Johnson, P. H. Santschi, C.-Y. Chuang, S. Otosaka, R. S. Addleman, M. Douglas, R. D. Rutledge, W. Chouyyok, J. D. Davidson, G. E. Fryxell and J. M. Schwantes, Environ. Sci. Technol., 2012, 46, 11251–11258 CrossRef CAS PubMed.
- G. E. Fryxell, Y. Lin, S. Fiskum, J. C. Birnbaum, H. Wu, K. Kemner and S. Kelly, Environ. Sci. Technol., 2005, 39, 1324–1331 CrossRef CAS PubMed.
- W. Yantasee, Y. Lin, G. E. Fryxell, B. J. Busche and J. C. Birnbaum, Sep. Sci. Technol., 2003, 38, 3809–3825 CrossRef CAS.
- X. Feng, G. E. Fryxell, L. Q. Wang, A. Y. Kim, J. Liu and K. M. Kemner, Science, 1997, 276, 923–926 CrossRef CAS.
- A. Walcarius, Chem. Soc. Rev., 2013, 42, 4098 RSC.
- X. Feng, Science, 1997, 276, 923–926 CrossRef CAS.
- T. Sangvanich, J. Morry, C. Fox, W. Ngamcherdtrakul, S. Goodyear, D. Castro, G. E. Fryxell, R. S. Addleman, A. O. Summers and W. Yantasee, ACS Appl. Mater. Interfaces, 2014, 6, 5483–5493 CAS.
- W. Yantasee, R. D. Rutledge, W. Chouyyok, V. Sukwarotwat, G. Orr, C. L. Warner, M. G. Warner, G. E. Fryxell, R. J. Wiacek, C. Timchalk and R. S. Addleman, ACS Appl. Mater. Interfaces, 2010, 2, 2749–2758 CAS.
- J. Liu, X. Feng, G. E. Fryxell, L.-Q. Wang, A. Y. Kim and M. Gong, Adv. Mater., 1998, 10, 161–165 CrossRef CAS.
- T. Asefa and Z. Tao, Can. J. Chem., 2012, 90, 1015–1031 CrossRef CAS.
- G. E. Fryxell, S. V. Mattigod, Y. Lin, H. Wu, S. Fiskum, K. Parker, F. Zheng, W. Yantasee, T. S. Zemanian, R. S. Addleman, J. Liu, K. Kemner, S. Kelly and X. Feng, J. Mater. Chem., 2007, 17, 2863–2874 RSC.
- T. G. Carter, W. Yantasee, T. Sangvanich, G. E. Fryxell, D. W. Johnson and R. S. Addleman, Chem. Commun., 2008, 5583 RSC.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- L. F. Newcomb, T. S. Haque and S. H. Gellman, J. Am. Chem. Soc., 1995, 117, 6509–6519 CrossRef CAS.
- T. J. Dickerson, N. N. Reed, J. J. LaClair and K. D. Janda, J. Am. Chem. Soc., 2004, 126, 16582–16586 CrossRef CAS PubMed.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
- C. L. Warner, R. S. Addleman, A. D. Cinson, T. C. Droubay, M. H. Engelhard, M. A. Nash, W. Yantasee and M. G. Warner, ChemSusChem, 2010, 3, 749–757 CrossRef CAS PubMed.
- W. Yantasee, C. L. Warner, T. Sangvanich, R. S. Addleman, T. G. Carter, R. J. Wiacek, G. E. Fryxell, A. C. Timchalk and M. G. Warner, Environ. Sci. Technol., 2007, 41, 5114–5119 CrossRef CAS PubMed.
- M. Hua, S. Zhang, B. Pan, W. Zhang, L. Lv and Q. Zhang, J. Hazard. Mater., 2012, 211–212, 317–331 CrossRef CAS PubMed.
- Y. Liu, G. Su, B. Zhang, G. Jiang and B. Yan, Analyst (Cambridge, U. K.), 2011, 136, 872 RSC.
- C. T. Yavuz, J. T. Mayo, W. W. Yu, A. Prakash, J. C. Flakner, S. Yean, L. Cong, H. Shipley, A. Kan, M. Tomson, D. Natelson and V. L. Colvin, Science, 2006, 314, 964–967 CrossRef PubMed.
- R. D. Ambashta and M. Sillanpaa, J. Hazard. Mater., 2010, 180, 38–39 CrossRef CAS PubMed.
- J. H. Jung, J. H. Lee and S. Shinkai, Chem. Soc. Rev., 2011, 40, 4464 RSC.
- P. Xu, G. M. Zeng, D. L. Huang, C. L. Feng, S. Hu, M. H. Zhao, C. Lai, Z. Wei, C. Huang, G. X. Xie and Z. F. Liu, Sci. Total Environ., 2012, 424, 1–10 CrossRef CAS PubMed.
- P. Z. Ray and H. J. Shipley, RSC Adv., 2015, 5, 29885–29907 RSC.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, A. Shan, X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures; RSDs for analytical measurements, TEM and EDX data for nanoparticles; crystal structure representation of DTP. CCDC 1408666. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5en00170f |
| ‡ These authors contributed equally to this work. |
| § It should be noted that Table 1 shows the surface area per gram material decreasing with increasing phenyl loading. Surface area per volume does not decrease drastically during the process rather the addition of phenyl silane increases the material density reducing the surface area/mass. |
| This journal is © The Royal Society of Chemistry 2016 |