
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of supported lipid bilayers containing phase-segregated domains and their interaction with gold nanoparticles†
Eric S.
Melby
ad,
Arielle C.
Mensch
b,
Samuel E.
Lohse‡
c,
Dehong
Hu
d,
Galya
Orr
d,
Catherine J.
Murphy
c,
Robert J.
Hamers
b and
Joel A.
Pedersen
*ab
aUniversity of Wisconsin-Madison, Environmental Chemistry and Technology Program, 1525 Observatory Dr., Madison, WI 53706, USA. E-mail: joelpedersen@wisc.edu
bUniversity of Wisconsin-Madison, Department of Chemistry, 1101 University Ave., Madison, WI 53706, USA
cUniversity of Illinois at Urbana-Champaign, Department of Chemistry, 600 S. Mathews Ave., Urbana, IL 61801, USA
dEnvironmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, WA 99352, USA
First published on 21st September 2015
Abstract
For nanoparticles that have been released into the environment, the cell membrane represents an initial site of interaction with eukaryotic cells. The encounter of nanoparticles with cellular membranes may alter membrane structure and function, lead to uptake into cells, or elicit adverse biological responses. Supported lipid bilayers have proven to be valuable ex vivo models for biological membranes, allowing investigation of their mechanisms of interaction with nanoparticles with a degree of control impossible in living cells. To date, the majority of research on nanoparticle interaction with supported lipid bilayers has employed membranes composed of single or binary mixtures of phospholipids. Cellular membranes contain a wide variety of lipids and exhibit lateral organization. Ordered membrane domains enriched in specific membrane components, also referred to as lipid rafts, have not been explored with respect to their interaction with nanoparticles. Here we develop model membranes containing segregated domains differing in fluidity that are amenable to investigation by a variety of surface-sensitive analytical techniques and demonstrate that these domains influence the extent of nanoparticle attachment to model membranes. We determined conditions that allow reliable formation of bilayers containing liquid-ordered domains enriched in sphingomyelin and cholesterol and confirmed their morphology by structured illumination and atomic force microscopies. We demonstrate that the presence of liquid-ordered domains increases attachment of cationic gold nanoparticles to model membranes relative to those lacking such domains under near physiological ionic strength conditions (0.1 M NaCl) at pH 7.4. We anticipate that these results will serve as the foundation for and motivate further study of nanoparticle interaction with phase-segregated domains.
Nano impactThe interaction of nanoparticles with cellular membranes can be a critical determinant in eliciting adverse outcomes in organisms. Model membranes composed of single or binary phospholipid mixtures have provided insight into the interaction with nanomaterials. Cytoplasmic membranes of living organisms, however, are made up of hundreds of components laterally organized into dynamic functional domains. We have established a platform to investigate the interaction of nanoparticles with model membranes containing ordered lipid domains with a variety of surface-sensitive analytical techniques. We demonstrate that the presence of these domains influences the interaction of cationic gold nanoparticles with model membranes under high ionic strength conditions. This study highlights the need to systematically investigate the interaction of nanoparticles with important membrane components and structures. |
Introduction
The cytoplasmic membrane serves as an initial point of contact between eukaryotic cells and nanomaterials that have been either intentionally or inadvertently released into the environment.1 Nanoparticle interactions at these nano-bio interfaces2 include attachment,3 passive penetration, endocytotic uptake,4 membrane permeabilization,5,6 and toxicological insult due to nanoparticle dissolution, generation of reactive oxygen species, or oxidative stress.7 If nanoparticles are internalized, they can also interact with the membranes that bound organelles including lysosomes, mitochondria, and the nucleus.8Mechanistic understanding of nanomaterial interaction with the cellular membranes of eukaryotes requires consideration of chemical composition, physical properties, and lateral organization of these critical structures. Cellular membranes are composed of a complex mixture of phospholipids, sphingolipids, sterols, and peripheral, integral and lipid-anchored proteins. Sphingolipids, phosphatidylinositol (a class of phospholipids) and membrane proteins can bear glycans, which can vary in composition and size. Cellular membranes typically contain several hundred lipid species when differences in headgroup, acyl chain length, the number and location of unsaturated bonds, and degree of glycosylation are taken into account.9,10 The lipid distribution between the inner and outer leaflet of biological membranes is typically asymmetric.11 The majority lipid components in the outer leaflet of animal cytoplasmic membranes are zwitterionic phosphatidylcholine and sphingomyelin, and neutral cholesterol.12 Minority lipid components in the outer leaflet of these membranes include zwitterionic phosphatidylethanolamine and the anionic phospholipids phosphatidylserine, (phosphorylated) phosphatidylinositol, and phosphatidic acid.13 Lipid molecules in cellular membranes not only provide a barrier function, but also participate in signaling processes and membrane trafficking.14
Cellular membranes are laterally organized, exhibiting domains differing in composition and lipid order. Domains of higher lipid order and enriched in specific membrane components are sometimes referred to as lipid rafts. These highly ordered domains are enriched in sphingolipids and cholesterol, and the acyl chains of majority of raft lipids are saturated.12 The degree of acyl chain saturation and preferential lipid association with sterols result in lipid segregation into a distinct liquid-ordered (Lo) phase, the components of which are, to a large degree, segregated from those of the liquid-disordered (Ld) phase.15 For the purposes of this paper we refer to segregated Lo domains in supported lipid bilayers as “domains”. The Lo phase is a highly ordered and tightly packed phase similar to the gel phase, but with much higher lateral mobility.12 The existence of lipid rafts in eukaryotic cellular membranes has been debated since the concept was introduced in 1997.16 The primary source of this debate is the small size and transient nature of lipid rafts within membranes,17,18 with typical length scales <50 nm and time scales on the order of milliseconds.19,20 A definition for these structures was developed at the Keystone Symposium on Lipid Rafts and Cell Function in 2006: “Membrane rafts are small (10–200 nm), heterogeneous, highly dynamic sterol- and sphingomyelin-enriched domains that compartmentalize cellular processes”.21 At the time of writing, the lateral organization of biological membranes is a topic of intense research. Further refinement of the lipid raft concept is expected as new techniques are brought to bear on understanding membrane structure.22 The rafts in cellular membranes are credited with a variety of functions, including preferential association by some membrane proteins,16 mediation of sorting in the trans-Golgi network,23 endocytotic pathway sorting,24 and signaling in hematopoietic cells.25 Membrane rafts appear to be ubiquitous features of cellular membranes; evidence exists for their presence in animal,12 plant,26 fungal,27 and bacterial28 cell membranes.
Supported lipid bilayers have proven to be useful models for biological membranes, allowing mechanisms of their interaction with nanomaterials to be studied with a degree of control impossible in living cells.29–31 Lipid bilayers on solid supports can be probed with a variety of optical,31 acoustic30–33 and scanning probe34–36 techniques. Among these techniques, quartz crystal microbalance with dissipation monitoring (QCM-D) and atomic force microscopy (AFM) allow label-free and in situ investigation of supported lipid bilayers. To date, most studies examining nanoparticle interaction with such model membranes have been restricted to supported lipid bilayers composed of single and binary mixtures of phospholipids.31,35–39 Despite the importance of lipid rafts in organismal membranes, investigation of nanoparticle interaction with these features of cellular membranes has not been previously reported. Prior studies have demonstrated successful formation of supported lipid bilayers containing phase-segregated domains on mica (by AFM)40 and optical glass (by fluorescence microscopy).41 Formation of domain-containing supported lipid bilayers on QCM-D sensors has been attempted,42 but success has not been demonstrated.
The objectives of this study were to develop a method to form model membranes containing phase-segregated domains on QCM-D sensor surfaces and to investigate the influence of such domains on nanoparticle attachment to model membranes. In this study, we employed a model membrane system comprising three components: the unsaturated phospholipid 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), the sphingolipid N-stearoyl-D-erythro-sphingosylphosphorylcholine (octadecanoyl sphingomyelin, SM), and the sterol lipid cholesterol (Chol). Sphingomyelin has a high affinity for Chol, which results in dense lipid packing and the formation of Lo regions (domains) within a Ld phase.43,44 To accomplish the first objective, we varied experimental parameters and the ratios of lipids in the ternary mixture to form supported lipid bilayers on planar SiO2 substrates by the vesicle fusion method.32,33 We confirmed the formation of supported lipid bilayers containing phase-segregated domains using a combination of QCM-D, AFM, and super-resolution fluorescence structured illumination microscopy (SIM). To achieve the second objective, we probed nanoparticle interactions with these domains using 4 nm gold nanoparticles (AuNPs) functionalized with anionic or cationic ligands by QCM-D.
Materials and methods
Materials
The following lipids were purchased from Avanti Polar Lipids (Alabaster, AL): 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC, 850375C), N-stearoyl-D-erythro-sphingosylphosphorylcholine (SM, 860586P), plant-derived cholesterol (Chol, 700100P), ganglioside GM1 (860065P), and TopFluor PC (810281C). We procured Atto647N DOPE from Atto-Tec (Siegen, Germany). Hydrogen tetrachloroaurate trihydrate (HAuCl4·3H2O), sodium borohydride, and mercaptopropanoic acid were purchased from Sigma-Aldrich. We obtained 3-amino propanethiol hydrochloride from MolPort Chemicals. Solid NaCl and Tris were obtained from Sigma-Aldrich, and all buffer solutions were made in ultrapure water (18.2 MΩ cm; Barnstead Nanopure) and filtered through a Millex GP 220 nm PES filter cartridge. PALL tangential flow filtration capsules (50 kDa pore size) were purchased from VWR. SiO/Cu mesh transmission electron microscopy grids were obtained from TedPella. All materials were used as received, unless otherwise noted.Formation and characterization of small unilamellar vesicles
Small unilamellar vesicles were prepared by the extrusion method.32,45 Just prior to use, SM and Chol were dissolved in chloroform (Sigma Aldrich). DOPC, SM, and Chol were mixed to the desired ratio, and the chloroform was evaporated under a stream of ultrapure N2 gas. For some fluorescent studies, 1 mol% ganglioside GM1 dissolved in 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :32:8 chloroform:ethanol:water (by volume), 0.1 mol% TopFluor PC, and/or 0.1 mol% Atto647N DOPE was included prior to evaporation under a stream of ultrapure N2 gas. The dried lipid films were held under vacuum for at least 1 h, rehydrated in 0.001 M NaCl, pH 7.4 (10 mM Tris) buffer to a stock concentration of 1.25 mg mL−1, subjected to three freeze (liquid N2)/thaw (bath sonicator) cycles to ensure complete sample mixing,46 and extruded 11 times through 50 nm polycarbonate membranes (Avanti Polar Lipids, 610003) with an extruder set (Avanti Polar Lipids, 610000). The resulting small unilamellar vesicles were stored at 4 °C and used within one week. A 0.03 mg mL−1 solution of the small unilamellar vesicles (diluted in either 0.001 or 0.1 M NaCl, pH 7.4 (0.01 M Tris)) was used to determine their hydrodynamic diameter (dh) by dynamic light scattering and zeta potential (ζ) by laser Doppler microelectrophoresis (Malvern Zetasizer Nano ZS). The literature is not unified in the definition of small unilamellar vesicles. For the purpose of this study we define small unilamellar vesicles as vesicles with diameters smaller than 100 nm.32 The size and ζ for small unilamellar vesicles containing SM and Chol (and GM1) and those composed solely of DOPC were similar (Table 1).
:SM:Chol molar ratios were 60:20:20. Ganglioside GM1 was incorporated at 1 mol%. Solutions were buffered to pH 7.4 with 0.01 M Tris and contained the indicated amount of NaCl
:32:8 chloroform:ethanol:water (by volume), 0.1 mol% TopFluor PC, and/or 0.1 mol% Atto647N DOPE was included prior to evaporation under a stream of ultrapure N2 gas. The dried lipid films were held under vacuum for at least 1 h, rehydrated in 0.001 M NaCl, pH 7.4 (10 mM Tris) buffer to a stock concentration of 1.25 mg mL−1, subjected to three freeze (liquid N2)/thaw (bath sonicator) cycles to ensure complete sample mixing,46 and extruded 11 times through 50 nm polycarbonate membranes (Avanti Polar Lipids, 610003) with an extruder set (Avanti Polar Lipids, 610000). The resulting small unilamellar vesicles were stored at 4 °C and used within one week. A 0.03 mg mL−1 solution of the small unilamellar vesicles (diluted in either 0.001 or 0.1 M NaCl, pH 7.4 (0.01 M Tris)) was used to determine their hydrodynamic diameter (dh) by dynamic light scattering and zeta potential (ζ) by laser Doppler microelectrophoresis (Malvern Zetasizer Nano ZS). The literature is not unified in the definition of small unilamellar vesicles. For the purpose of this study we define small unilamellar vesicles as vesicles with diameters smaller than 100 nm.32 The size and ζ for small unilamellar vesicles containing SM and Chol (and GM1) and those composed solely of DOPC were similar (Table 1).
:SM:Chol molar ratios were 60:20:20. Ganglioside GM1 was incorporated at 1 mol%. Solutions were buffered to pH 7.4 with 0.01 M Tris and contained the indicated amount of NaCl
| Small unilamellar vesicles | d h (nm) | ζ (mV) | ||
|---|---|---|---|---|
| 0.001 M NaCl | 0.1 M NaCl | 0.001 M NaCl | 0.1 M NaCl | |
| DOPC | 71 ± 3 | 67 ± 2 | −17 ± 1 | −1.6 ± 1.0 |
| DOPC/SM/Chol | 78 ± 4 | 80 ± 8 | −21 ± 1 | −1.6 ± 1.1 |
| DOPC/SM/Chol + GM1 | 79 ± 3 | 73 ± 2 | −23 ± 1 | −3.1 ± 0.7 |
Gold nanoparticle synthesis and characterization
Thiol-stabilized 4 nm AuNPs were synthesized using previously reported methods.47,48 In an aqua regia-cleaned 500 mL round bottom flask, 400 mL of ultrapure water, 1.5 mL of HAuCl4 (0.1 M), and either 200 μL of NaOH (1.0 M) and 1.5 mL of 0.1 M mercaptopropionic acid (MPA) or 1.8 mL of HCl (0.1 M) and 3.5 mL of 0.1 M mercaptopropylamine (MPNH2) were combined, and stirred at vortex for 10 min. These additions produced a clear, colorless solution. (Warning:aqua regia is a strong oxidant and highly corrosive.) After the initial stirring, either 5.0 mL of a 0.1 M aqueous sodium borohydride solution (for MPA–AuNPs) or 35 mL of ice–cold 0.01 M aqueous sodium borohydride solution (for MPNH2–AuNPs) was added to the solution. The solution rapidly turns brown, then red-orange. The resulting red-orange AuNP solution was stirred for 3 h. The crude AuNP solution was then purified by diafiltration.47 The 500 mL AuNP solution was concentrated to an initial volume of 50 mL and purified by passing 3.0 L of ultrapure water through the diafiltration system.Gold nanoparticle core size and concentration were determined from their UV-vis spectra.49 Core size was confirmed by transmission electron microscopy (Fig. S1†). A 10 nM solution of the synthesized AuNPs (diluted in either 0.001 or 0.1 M NaCl, pH 7.4 (0.01 M Tris)) was used to determine AuNP hydrodynamic diameter (dh) by dynamic light scattering and apparent zeta potential (ζapp) by laser Doppler microelectrophoresis (Malvern Zetasizer Nano ZS). All AuNP concentrations are expressed as number concentrations.
Quartz crystal microbalance with dissipation monitoring
All QCM-D experiments were conducted using the Q-Sense E4 system containing four silica-coated sensors (QSX 303) mounted in temperature-controlled, liquid flow cells (QFM 401; Biolin Scientific, Göteborg, Sweden). Quartz crystal microbalance with dissipation monitoring measures changes in the resonance frequency and energy dissipation induced by the interaction of an analyte with the surface of a coated piezoelectric quartz crystal. Frequency shifts (Δf) reflect changes in mass coupled to the sensor surface (analyte mass + hydrodynamically coupled water). Changes in the energy dissipation (ΔD) are related to the viscoelastic properties of laterally homogeneous adlayers or to the properties of the particle-surface contact region for films of discrete nanoscale objects.50The QCM-D silica sensor crystals were prepared for use by bath sonication in a 2% solution of sodium dodecyl sulfate for 20 min, rinsing consecutively with ethanol and ultrapure water three times, drying with ultrapure N2 gas, and UV/ozone treatment for 30 min (Bioforce Nanosciences UV/Ozone Procleaner). Supported lipid bilayers were formed by the vesicle fusion method.32 Briefly, after mounting the sensors in the flow cells, 0.1 M NaCl, pH 7.4 (0.01 M Tris) buffer was allowed to flow over the sensors until frequency and dissipation values stabilized. A suspension of small unilamellar vesicles (0.03 mg mL−1) was then allowed to flow across the sensor until stable values of Δf and ΔD were obtained after passing through a minimum and maximum, respectively, that indicate attainment of a critical adsorbed vesicle concentration, commencement of vesicle rupture, and supported lipid bilayer formation.51 The supported lipid bilayer was then rinsed with vesicle-free buffer for 15 min to remove any adhering vesicles. All experiments used a constant flow rate of 0.1 mL min−1 and temperature of 25 °C unless otherwise noted. Variables that were tested in arriving at conditions to form Lo domain-containing bilayers included vesicle concentration, solution ionic strength during vesicle deposition, the ionic strength of the rinsing solution, timing of the initiation of the rinsing step, and temperature. Specifically, we examined small unilamellar vesicle introduction in high ionic strength solutions (up to 0.15 M NaCl), temperature elevation to 35 °C after small unilamellar vesicle introduction, rinsing with low ionic strength (0.025 M NaCl) solutions, initiation of rinsing immediately after the rupture event, and vesicle introduction at temperatures up to 35 °C. The key modification allowing successful formation of Lo domain-containing supported lipid bilayers was the decrease in vesicle concentration relative to the previously published attempt to form such bilayers,42 which eliminated the adsorption of vesicles to the supported lipid bilayer.
After successful formation of supported lipid bilayers, for nanoparticle attachment experiments conducted at 0.1 M NaCl, pH 7.4 (0.01 mM Tris), nanoparticles were diluted to a concentration of 10 nM with the same buffer and flowed over the supported lipid bilayer for 20 min. The supported lipid bilayer was then rinsed with buffer for 20 min to assess the reversibility of nanoparticle attachment. Otherwise, for nanoparticle attachment experiments conducted at 0.001 M NaCl, pH 7.4 (0.01 mM Tris), after bilayer formation and rinsing the buffer was switched to 0.001 M NaCl for at least 10 min to allow a stable baseline to be attained, and the subsequent dilution of nanoparticles to 10 nM and rinsing were conducted in this buffer solution. We assessed the attachment of both MPNH2– and MPA–AuNPs to DOPC-only, 60/20/20 mol% DOPC/SM/Chol, and 60/20/20 mol% DOPC/SM/Chol with 1 mol% GM1 supported lipid bilayers. The initial rate of nanoparticle attachment to the supported lipid bilayers was determined as described previously.52 Briefly, the QCM-D Δf curve was subjected to locally weighted scatterplot smoothing (LOESS; α = 0.1),53 and the one minute moving Δf rate was averaged for the first 5 min of nanoparticle attachment to the supported lipid bilayer to determine the initial rate of nanoparticle attachment. All attachment experiments were conducted in triplicate. Data comparisons were made by one-way ANOVA with a Tukey post-hoc test at the p ≤ 0.05 level of significance.
Structured illumination microscopy
Imaging was carried out using a Zeiss Elyra fluorescence microscope with a 63× objective. Supported lipid bilayers were formed under batch conditions within 35/22 mm #1.5 glass bottom dishes (Willco Wells). Dishes were rinsed with ultrapure water (18.2 MΩ cm; MilliQ Advantage A10, Millipore), dried with N2, and cleaned in a UV/Ozone chamber (PSD Pro Series, Novascan) for 20 min. The 0.1 M NaCl, pH 7.4 (0.01 M Tris) buffer solution was introduced to the clean dishes at least 20 min prior to vesicle introduction. Suspensions of small unilamellar vesicles (0.06 mg mL−1 containing either 0.1 mol% TopFluor PC (λex = 488 nm; λem = 503 nm) or 0.1 mol% Atto647N DOPE (λex = 642 nm; λem = 667 nm) and sometimes 1 mol% ganglioside GM1 in the same buffered solution) were introduced to the dish, and bilayer formation was monitored by confocal fluorescence microscopy. We were able to distinguish unruptured vesicles from lipid bilayers using fluorescence recovery after photobleaching (FRAP; see below) because unruptured vesicles do not diffuse laterally, and as a result, they do not recover after photobleaching; in contrast, the lipids within supported lipid bilayers are able to diffuse in and out of the monitored region, resulting in recovery upon photobleaching. Upon bilayer formation, the solution contained within the dish was exchanged five times with 2 mL aliquots of the buffered solution. Two separate fluorescent phospholipids were used to ensure our observed results could not be attributed to the inclusion of the fluorophore. Additionally, these two fluorescent phospholipids were chosen because they differ in both the excitation/emission wavelengths of the fluorophore as well as the location of the fluorophore. TopFluor is covalently conjugated to one of the lipid acyl chains, while Atto647N is covalently conjugated to the lipid headgroup.Fluorescence recovery after photobleaching
To determine supported lipid bilayer formation and phospholipid diffusion coefficients, we conducted FRAP experiments using a Zeiss LSM 710 microscope with a scan rate of 240 ms. A minimum of eight FRAP curves from two different supported lipid bilayers were analyzed to obtain diffusion coefficients. Fluorescence intensities were normalized to that of the initial supported lipid bilayer. Data were analyzed using a nonlinear least-squares fitting method to determine the characteristic diffusion time, τD, using the equation:54,55where F(t) is the observed relative fluorescence as a function of time, F(0) is the relative fluorescence intensity immediately following the bleach pulse, F(∞) is the relative fluorescence intensity at infinite time following the bleach pulse, β is the bleach depth given by 1 − F(0), and τD is the characteristic diffusion time. The lateral diffusion coefficient, D, was then determined using
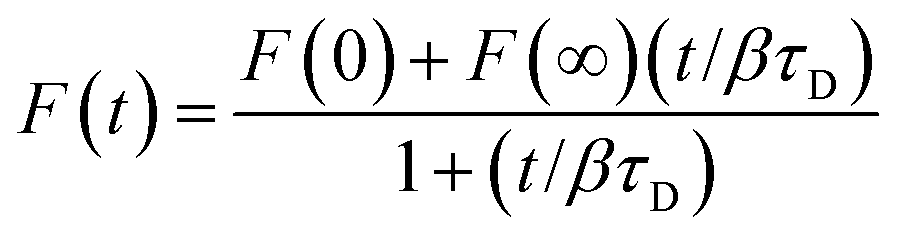
| D = ω2/4τD |
Atomic force microscopy
Images were collected in tapping mode using a Nanoscope IV controller (Digital Instruments) with a Type J scanner. Silicon nitride probes (Bruker, DNP) with a force constant of 0.35 N m−1 and a MTFML-V2 probe holder were used to collect images in fluid. Bilayers were formed on ultra-flat B-doped silicon wafers with a 200 nm thermal oxide layer (Ted Pella) under the same conditions as those described for QCM-D. To image under flow conditions (0.1 mL min−1, 25 °C), the wafer was first equilibrated in 0.1 M NaCl, pH 7.4 (0.01 M Tris) buffer for 20 min, vesicle solution (0.03 mg mL−1) was flowed for 20 min, the bilayer was rinsed with buffer for 20 min, and the bilayer was imaged. The Lo domains investigated here are readily observable in AFM as contiguous regions appearing higher than the surrounding regions. Initial image analysis was conducted with Gwyddion.56 Quantitative analysis of AFM data was performed using Igor Pro (Wavemetrics, Lake Oswego, OR, USA) to import the image data, calculate histograms of the height distributions, and then fit the histogram to two Gaussian distributions. The area of the peak corresponding to the higher height distribution (domains) was divided by the total area to give a fractional coverage of the Lo domains.Results and discussion
Formation of supported lipid bilayers containing phase-segregated domains
We first established a set of experimental conditions that allowed supported lipid bilayers to be formed on silica-coated QCM-D sensors from vesicles composed of 60/20/20 mol% DOPC/SM/Chol. Based on final stable Δf5/5 and ΔD5 values in QCM-D experiments,30,32,33 we determined that supported lipid bilayers could be formed from these small unilamellar vesicles at a vesicle concentration of 0.03 mg mL−1 in 0.1 M NaCl at pH 7.4 (0.01 M Tris) and 25 °C. Representative frequency and dissipation traces (Fig. 1) for formation of supported lipid bilayers under these conditions from vesicles composed of DOPC or 60/20/20 mol% DOPC/SM/Chol reflect the two-step process commonly observed in the formation of bilayers of zwitterionic lipids on silica substrates: attainment of a critical adsorbed vesicle concentration followed by initiation of vesicle rupture and formation of the supported bilayer.32,51 The final frequency and dissipation values for the DOPC bilayer were −24.9 ± 0.1 Hz and 0.17 (±0.02) × 10−6, respectively, consistent with prior reports of supported lipid bilayer formation from similar phospholipids.32 For supported bilayers formed from 60/20/20 mol% DOPC/SM/Chol the final Δf5/5 value was −26.8 ± 0.6 Hz, more negative than that for the pure DOPC bilayer. The more negative frequency value of 60/20/20 mol% DOPC/SM/Chol bilayer corresponds to higher bilayer mass than for the pure DOPC bilayer. We attribute the higher mass of the 60/20/20 mol% DOPC/SM/Chol bilayer to the interaction of sphingomyelin with cholesterol,57 increasing lipid packing density relative to pure DOPC. The final dissipation value for 60/20/20 mol% DOPC/SM/Chol bilayers was 0.51 (±0.29) × 10−6, higher than that for DOPC bilayers.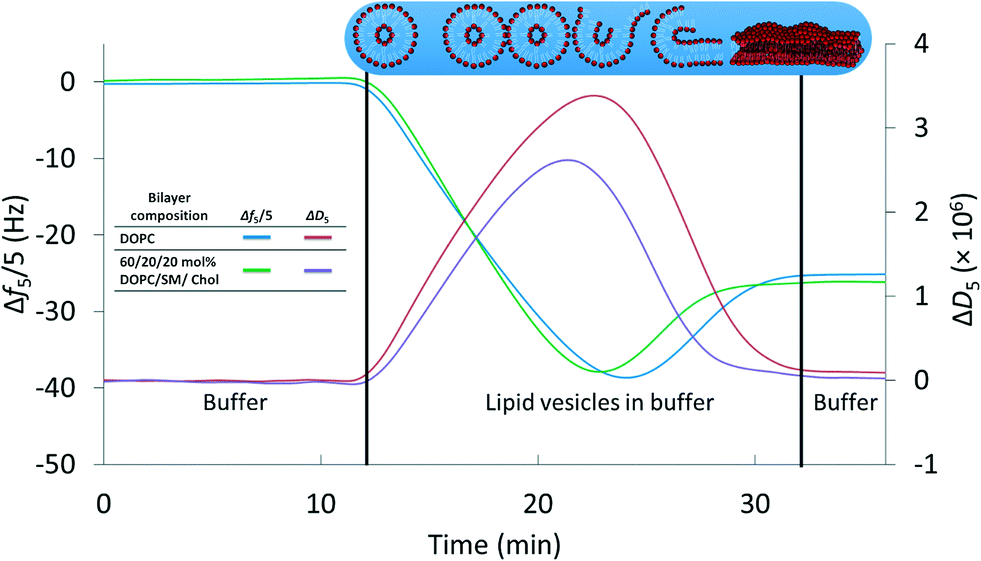 | ||
| Fig. 1 Formation of supported lipid bilayers from small unilamellar vesicles composed of DOPC or 60/20/20 mol% DOPC/SM/Chol as monitored by QCM-D. Final resonant frequency changes were −24.9 ± 0.1 Hz and −26.8 ± 0.6 Hz for the DOPC and DOPC/SM/Chol bilayers, respectively (n = 3). The corresponding dissipation values were 0.17 (±0.02) × 10−6 and 0.51 (±0.29) × 10−6. Overall supported lipid bilayer formation kinetics were similar for the two compositions. Data are shown from the 5th harmonic and were subjected to LOESS smoothing (α = 0.1).53 The buffer solution was 0.01 M Tris (pH 7.4) in 0.1 M NaCl. | ||
We employed super-resolution fluorescence structured illumination microscopy to determine whether the supported lipid bilayers formed from 60/20/20 mol% DOPC/SM/Chol vesicles contained phase-segregated domains with the expected morphology. Structured illumination microscopy allows fluorescence images to be obtained with sub-diffraction limit (~100 nm) resolution.58,59 Such super-resolution capabilities were critical because domains can have dimensions smaller than the diffraction limit of light, making these structures largely invisible to standard fluorescence microscopy techniques.60 Supported lipid bilayers were formed from vesicles containing one of two fluorescently labeled phospholipids, both of which were expected to segregate to the Ld regions with DOPC. We therefore expected to observe a fluorescent signal in the Ld regions and the absence of a fluorescent signal in the Lo domains. Fig. 2A and B show SIM images of 60/20/20 mol% DOPC/SM/Chol supported lipid bilayers that contain TopFluor PC and Atto647N DOPE, respectively. Both images display areas of fluorescent signal and patches of dark regions consistent with the presence of Lo domains that exclude the fluorescent phospholipids. To determine whether the dark regions were indeed Lo domains composed of SM and Chol, as opposed to being large defects in the bilayer, we formed supported lipid bilayers from small unilamellar vesicles with the same composition as above, but also containing 1 mol% ganglioside GM1. Ganglioside GM1 segregates to Lo domains40 and is the cell surface receptor for cholera toxin B (CTB) homopentamer, with each CTB pentamer binding five GM1 molecules.61,62 We used the binding of fluorescently labeled CTB (Alexa555-labeled CTB pentamer; Life Technologies) to GM1 to confirm that the dark regions were Lo domains. The SIM image in Fig. 2C displays the fluorescence of this cholera toxin B homopentamer in the dark regions that exclude the fluorescent phospholipids, confirming the existence of Lo domains.
 | ||
| Fig. 2 A–C. Structured illumination microscopy images displaying the exclusion of fluorescently-labeled (A) TopFluor PC (green) and (B) Atto647N DOPE (red) phospholipids from dark regions that are expected to be Lo domains in supported lipid bilayers composed of 60/20/20 mol% DOPC/SM/Chol with 0.1 mol% fluorescent phospholipid. To confirm the dark regions are Lo domains, 1 mol% GM1 was incorporated into the vesicles prior to formation supported lipid bilayers and is expected to segregate to the Lo domains.40 Alexa555-labeled cholera toxin subunit B protein (yellow) binds GM1, and its presence in the dark regions (C) confirms the dark regions are Lo domains. Scale bars are 2 μm. | ||
We further investigated the morphology and extent of Lo domains in these supported lipid bilayers by AFM. The majority of prior AFM studies of phase-segregated domains in planar bilayers has focused on model membranes supported on mica57 or glass coverslips.60 We formed Lo domain-containing bilayers on silica substrates to provide a direct connection with our QCM-D experiments. Fig. 3 displays an AFM image of a 60/20/20 mol% DOPC/SM/Chol supported lipid bilayer with a corresponding height profile and height distribution. These data confirm the presence of domains in the supported lipid bilayer that differ in height by ~1 nm. This result is consistent with prior studies that indicate that Lo domains are approximately 1 nm thicker than Ld regions due to the cholesterol-induced ordering of the unsaturated acyl chains of sphingomyelin.57 We note that the AFM data indicate that the bilayer is free from surface adsorbed vesicles, which would be evident due to their large size (70–80 nm; Table 1) relative to the height of the supported lipid bilayer. Therefore, the higher final dissipation value for bilayers composed of 60/20/20 mol% DOPC/SM/Chol relative to those composed of pure DOPC does not appear attributable to the presence of intact vesicles adhering to the bilayer.
 | ||
| Fig. 3 Atomic force microscopy confirms (A) the presence of distinct Lo and Ld phases with (B) the expected ~1 nm height difference between phases in a supported lipid bilayer consisting of 60/20/20 mol% DOPC/SM/Chol. (C) The coverage of Lo domains was determined by fitting the AFM image height distribution. Scale bar is 2 μm. AFM images of other compositions investigated are provided in Fig. S2.† | ||
To further characterize these Lo domain-containing supported lipid bilayers, we examined the lateral diffusivity of phospholipids within the Ld phase by FRAP. From these experiments we determined the lateral diffusion coefficient for TopFluor PC, which our SIM experiments confirmed to segregate to the Ld regions, to be 1.81 ± 0.16 μm2 s−1. This value agrees well with previous reports of fluorescent lipid diffusion in Ld regions ranging from 1.47 to 2.12 μm2 s−1.46,63–65
We next explored the compositional space bounded by the limits of sphingomyelin (0–30%) and cholesterol content (10–50%) in typical eukaryotic membranes.14 The ternary phase diagram in Fig. 4 displays the boundaries for the phases expected for various combinations of the three components used in our experiments based on prior work with intact giant unilamellar vesicles (10–100 μm).66 In this figure, we indicate the compositions of vesicles from which we successfully formed supported lipid bilayers based on the observation of the characteristic two-phase formation kinetics in QCM-D experiments and the final stable Δf5/5 and ΔD5 values (Table 2). The final Δf5/5 and ΔD5 values for bilayers formed from the ternary lipid mixtures were slightly more negative and more positive, respectively, than those formed from pure DOPC. The ternary lipid mixtures from which supported bilayers formed spanned a compositional range that included segregated Lo and Ld phases as well as a single continuous Ld phase in studies with giant unilamellar vesicles.66
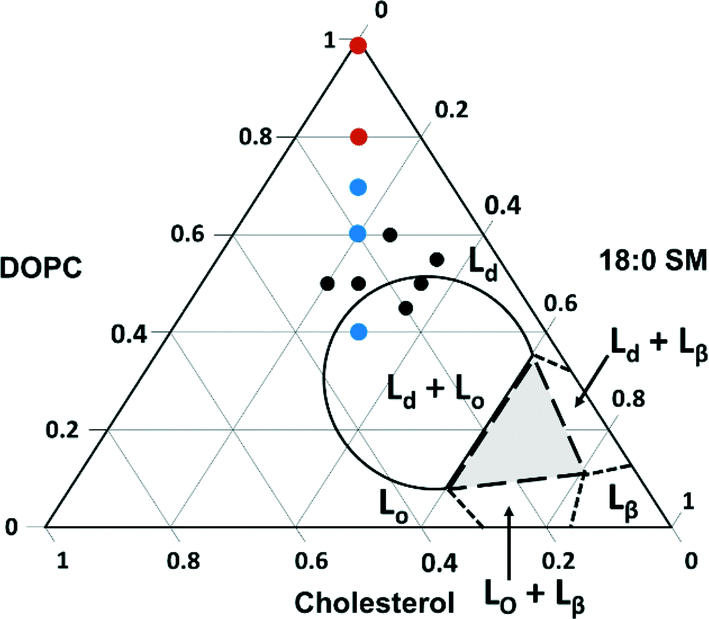 | ||
| Fig. 4 Ternary phase diagram showing the composition of vesicles from which supported lipid bilayers were successfully formed on SiO2 substrates as monitored by QCM-D. Black dots represent compositions investigated by QCM-D alone, and colored dots indicate those compositions (imaged by AFM) that resulted in segregated Lo domain formation (blue) and those that resulted in a single continuous phase (red). Lines indicate expected phase boundaries based on studies of giant unilamellar vesicles.66 | ||
| mol% DOPC/SM/Chol | Δf5/5final (Hz) | ΔD5 (×10−6) |
|---|---|---|
| 100/0/0 | −24.9 ± 0.1 | 0.17 ± 0.02 |
| 80/10/10 | −25.9 ± 0.4 | 0.20 ± 0.01 |
| 70/15/15 | −27.5 ± 1.3 | 0.46 ± 0.02 |
| 60/20/20 | −26.8 ± 0.6 | 0.51 ± 0.29 |
| 60/25/15 | −27.0 ± 0.5 | 0.35 ± 0.07 |
| 55/35/10 | −27.6 ± 0.2 | 0.24 ± 0.02 |
| 50/20/30 | −28.3 ± 0.3 | 0.47 ± 0.20 |
| 50/25/25 | −27.8 ± 0.6 | 0.43 ± 0.03 |
| 50/35/10 | −27.6 ± 0.2 | 0.24 ± 0.02 |
| 45/35/20 | −28.0 ± 0.3 | 0.54 ± 0.16 |
| 40/30/30 | −29.2 ± 0.6 | 0.52 ± 0.17 |
To determine the vesicle compositions that yielded domain-containing supported lipid bilayers, as well as to verify that intact vesicles were not adsorbed to the bilayer surfaces, we examined selected supported lipid bilayers by AFM (viz. those comprised of 40/30/30, 70/15/15, 80/10/10, and 100/0/0 mol% DOPC/SM/Chol; red and blue dots in Fig. 4). Representative images, height profiles, and Lo domain coverages are shown in Fig. S2.† All bilayers containing domains exhibit the expected ~1 nm height difference between Lo and Ld regions, and no intact vesicles were observed across the compositions investigated. These images indicate that within the compositional space examined, Lo domain formation is expected for supported lipid bilayers containing 15 mol% SM and Chol or more. Referring to the phase diagram boundaries (Fig. 4), phase-segregation does not occur in giant unilamellar vesicles at SM and Chol concentrations below ~27 mol%.66 Our results agree with a previous fluorescence microscopy study on domain formation in supported lipid bilayers that demonstrated phase segregation in bilayers containing ≥15 mol% Chol and sphingomyelin.67
To determine the impact of vesicle composition on supported lipid bilayer formation kinetics, we examined supported lipid bilayer formation time (defined as the time from vesicle adsorption to initial rupture) as a function of vesicle Chol content (Fig. 5). This analysis indicates that increasing Chol content in small unilamellar vesicles lengthens the time to initiation of vesicle rupture, consistent with a prior report.42 The lengthening of time to vesicle rupture is attributed to the ability of Chol to quickly flip-flop between the two bilayer leaflets.68 Vesicles flatten when adsorbed to silica surfaces,69 and the ability of Chol to flip-flop between leaflets reduces the stress of vesicle flattening. Often, the attraction of vesicles to a solid surface, and their subsequent flattening, is insufficient to promote supported lipid bilayer formation. Inter-vesicle interaction has an additional destabilizing effect on adsorbed vesicles that promotes vesicle fusion and ultimately supported lipid bilayer formation.70 The observation that supported lipid bilayer formation time increased with Chol content suggests that a larger inter-vesicle interaction force is required to overcome the vesicle stabilization caused by Chol flip-flop.
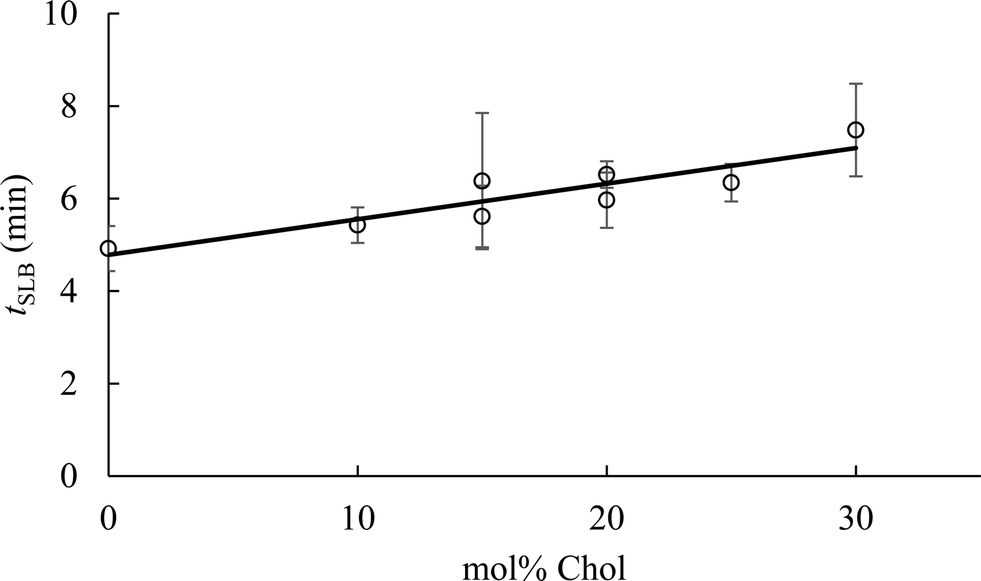 | ||
| Fig. 5 Supported lipid bilayer formation time (tSLB) as a function of vesicle cholesterol content as monitored by QCM-D. Supported lipid bilayer formation time was calculated from the time that vesicles were first observed adhering to the sensor surface to the time of initiation of vesicle rupture (as evidenced by the abrupt decrease in ΔD5). Fit (y = 0.077x + 4.782) was obtained using a standard least squares analysis (R2 = 0.82). | ||
Interaction of anionic and cationic gold nanoparticles with Lo domain-containing supported lipid bilayers
As a first step toward understanding the influence of phase-segregated domains on nanoparticle interaction with model membranes, we compared the attachment of cationic and anionic AuNPs to supported lipid bilayers containing Lo domains (60/20/20 mol% DOPC/SM/Chol ± 1 mol% GM1) with those composed of pure DOPC in QCM-D experiments. The AuNPs had 4 nm spherical cores (Fig. S1†) and were functionalized with cationic mercaptopropyl amine (MPNH2) or anionic mercaptopropionic acid (MPA). Attachment experiments were conducted in 0.001 M and 0.1 M NaCl at pH 7.4 (0.01 M Tris). The hydrodynamic diameters (dh) and apparent zeta potentials (ζapp) of the AuNPs under these solution conditions are provided in Table 3. At 0.1 M NaCl both types of AuNPs agglomerated to a considerable extent; however, nanoparticle agglomeration state remained stable over the course of QCM-D experiments.| AuNPs | d h (nm) | ζ app (mV) | ||
|---|---|---|---|---|
| 0.001 M NaCl | 0.1 M NaCl | 0.001 M NaCl | 0.1 M NaCl | |
| MPA–AuNP | 22 ± 10 | 160 ± 26 | −21 ± 5 | −28 ± 1 |
| MPNH2–AuNP | 6 ± 3 | 440 ± 120 | 26 ± 1 | 26 ± 1 |
Attachment of cationic MPNH2–AuNPs was observed for all supported lipid bilayers at both NaCl concentrations (Fig. 6) and was irreversible with respect to dilution (rinsing with AuNP-free buffer for 20 min). Attachment of the MPNH2–AuNPs to the bilayers studied can be attributed to Coulombic attraction of the positively charged nanoparticles to negative surface potential of the bilayers. The ζapp for the MPNH2-AuNPs was positive at both NaCl concentrations (26 ± 1 mV). Prior streaming current studies demonstrated that ζ values at pH 7.45 for pure DOPC bilayers on planar silica substrates were negative: −39 ± 1 mV for 0.001 M KCl and −17.5 ± 0.7 mV for 0.01 M KCl.71 Streaming current measurements of pure DOPC bilayers at 0.1 M KCl were not made in the referenced study, but ζ values at this salt concentration are expected to be somewhat less negative. (We note that at pH ~7.4 ζ is not expected to differ appreciably between solutions composed of KCl and NaCl.)72 The ζ of the DOPC/SM/Chol vesicles were similar to those composed of solely DOPC (identical at 0.1 M NaCl; Table 1). This comparison leads to the expectation that the DOPC/SM/Chol and pure DOPC supported lipid bilayers possessed similarly negative ζ values. Incorporation of the anionic glycosphingolipid GM1 at 1 mol% resulted in small unilamellar vesicles with slightly more negative ζ values than in its absence (Table 1). In the present study, solutions were buffered with Tris. At the pH of the solutions used, TrisH+ cations were present at 0.008 M, and these cations are present at the SiO2–water interface,31 likely leading to slightly less negative ζ values than indicated from the streaming current data. Second harmonic generation studies of phosphatidylcholine bilayers in the presence of Tris have shown that the surface potential remains negative at the solution conditions investigated here.31 For all bilayers investigated, the extent of attachment was much larger at 0.001 M than at 0.1 M NaCl. For example, Δf5/5 = −242 ± 23 Hz at 0.001 M NaCl and −10.0 ± 1.0 Hz at 0.1 M NaCl for attachment to supported lipid bilayers composed of 60/20/20 mol% DOPC/SM/Chol. The larger amount of MPNH2-AuNP attachment at 0.001 M relative to 0.1 M NaCl is due to the more negative ζ of the bilayer (vide supra) and the lower degree of nanoparticle aggregation (Table 3) at the lower ionic strength. At 0.001 M NaCl, 20 min exposure of the bilayers to MPNH2–AuNPs resulted in frequency changes corresponding to numbers of attached particles exceeding the jamming limit expected from random sequential adsorption of uniform, non-interacting spheres.73
 | ||
| Fig. 6 Attachment of MPNH2–AuNP to supported lipid bilayers of the indicated compositions. The molar ratio of DOPC to SM to Chol was 60:20:20. Ganglioside GM1 was incorporated into vesicles at 1 mol%. Solutions were buffered to pH 7.4 with 0.01 M Tris and contained the indicated amount of NaCl. The Δf values are for the 5th harmonic and represent the change in frequency from the supported lipid bilayer value after flowing 10 nM MPNH2–AuNPs for 20 min and rinsing with buffer for 20 min. Nanoparticle detachment was not observed upon rinsing with buffer. Letters indicate significant differences in attachment (p < 0.05) within each NaCl treatment. Bars represent mean values; error bars represent one standard deviation (n = 3). | ||
At 0.001 M NaCl, MPNH2–AuNP attachment to all supported lipid bilayers investigated, as well as initial rates of nanoparticle attachment (Fig. S3†), were statistically indistinguishable (p > 0.1). In contrast, at 0.1 M NaCl attachment to bilayers containing Lo domains was higher than that to pure DOPC bilayers (p < 0.01). For lipid bilayers containing Lo domains, Δf5/5 = −10.0 ± 1.0 Hz and −8.2 ± 2.6 Hz in the absence and presence of 1 mol% GM1. For pure DOPC bilayers, Δf5/5 = −1.1 ± 0.5 Hz. The extent of attachment to Lo domain-containing supported lipid bilayers was not affected by the presence of GM1 (p > 0.1).
The higher attachment of MPNH2-AuNPs to bilayers containing Lo domains relative to those lacking these structures may be attributable to differences in bilayer morphology. At 0.1 M NaCl, the DOPC and DOPC/SM/Chol vesicles possessed identical ζ values (Table 1), and the supported lipid bilayers are expected to exhibit similar ζ values as well. This argues against differences in bilayer ζ being responsible for the higher attachment to the DOPC/SM/Chol relative to DOPC bilayers. We hypothesize that differences in supported lipid bilayer morphology are responsible for the higher attachment to the Lo domain-containing bilayers. The Lo and Ld regions differ in height by approximately 1 nm (vide supra). This height difference occurs over a lateral distance of approximately 10–15 nm, and the altered water structure at this boundary may result in increased nanoparticle attachment.74 Alternatively, this boundary region may result in the exposure of sphingomyelin phosphate groups at the edges of Lo domains, providing local sites for increased electrostatic attraction with the cationic MPNH2–AuNPs. The GM1 molecule carries a negative charge; however, the ζ data for the vesicles show that incorporation of this glycosphingolipid into vesicles at 1 mol% produced a slight, but significant, reduction in ζ (p < 0.01; Table 1). In the QCM-D experiments, MPNH2–AuNP attachment to Lo domain-containing bilayers was insensitive to inclusion of 1 mol% GM1 (p > 0.1). This result provides further evidence that the presence of domains or their boundaries, not the overall surface potential of the bilayer, is responsible for the increase in attachment for Lo domain-containing supported lipid bilayers.
No attachment of anionic MPA-AuNPs was detectable at either NaCl concentration for the bilayers studied. This result can be rationalized by considering the negative ζapp of the nanoparticles (−21 ± 5 mV and −28 ± 1 mV for 0.001 and 0.1 M NaCl) and those expected for the bilayers (vide supra). We conclude that Coulombic repulsion prevented detectable attachment. The ζ for the MPA–AuNPs were large and negative under the solution conditions employed. We cannot exclude the attachment of MPA–AuNPs to the bilayers employed at amounts below the limit of QCM-D detection as we demonstrated in prior work for pure DOPC bilayers.31
Conclusions
Cellular membranes across all domains of life appear to exhibit lateral organization,12,26–28 and lipid rafts have been implicated as important membrane organizing structures.12,16,75 Knowledge of the diverse functions of lipid rafts in cellular membranes is expanding as the development and refinement of instrumentation allows these structures to be studied at minute length and time scales.18 At present strong evidence exists for the participation of lipid rafts in T-cell signaling,76 post-Golgi traffic to the cell surface,23 and glycosphingolipid-mediated endocytosis.24 Giant unilamellar vesicles formed from plasma membranes exhibit phase-segregated domains similar to those in model membranes.77–79 These similarities and the participation of lipid rafts in a large number of cellular processes justify the development of supported lipid bilayers to simulate these domains in cellular membranes, thereby enabling their study with a wider array of instrumentation. In this study we have demonstrated the ability to form Lo domain-containing supported lipid bilayers on silica substrates, permitting investigation of these domains by a variety of surface-sensitive techniques such as QCM-D, AFM, ellipsometry, electrochemical impedance spectroscopy, secondary ion mass spectrometry, and non-linear optical methods. The variety of techniques available to probe surfaces makes possible the investigation of dynamics occurring at the interface between phase-segregated regions, including protein incorporation,80,81 association of viruses,82,83 and interaction with nanoparticles (as demonstrated in this work). Furthermore, development of supported lipid bilayers that contain other domain-forming lipids (e.g., ceramides)84 and proteins that segregate to domains (e.g., glycophosphatidylinositol-anchored proteins)12 will enable investigation of the role such components play in the dynamic processes occurring at the lipid raft-solution interface.Observations of differential nanoparticle attachment to Lo domain-containing bilayers, relative to those containing one phospholipid in a continuous phase, both provide a foundation and highlight the need for further investigations of nanoparticle interaction with phase-segregated domains. Future investigation into the influence of nanoparticle size, shape, surface chemistry, and degree of aggregation on interaction with phase-segregated domains, as well as with domain-associating proteins, is warranted. Such studies would improve our understanding of the extent to which phase-segregated domains and their components govern nanoparticle interactions with cellular membranes.
Acknowledgements
This study was supported by the National Science Foundation (NSF) Center for Sustainable Nanotechnology, funded as a Center for Chemical Innovation (CHE-1240151). Part of the research was performed at the Environmental Molecular Sciences Laboratory, a Scientific User Facility sponsored by DOE-BER and located at the Pacific Northwest National Laboratory.Notes and references
- M. A. Maurer-Jones, I. L. Gunsolus, C. J. Murphy and C. L. Haynes, Anal. Chem., 2013, 85, 3036–3049 CrossRef CAS PubMed.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- C. C. Berry and A. S. Curtis, J. Phys. D: Appl. Phys., 2003, 36, 198–206 CrossRef.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS PubMed.
- P. R. Leroueil, S. Hong, A. Mecke, J. R. Baker, B. G. Orr and M. M. Banaszak Holl, Acc. Chem. Res., 2007, 40, 335–342 CrossRef CAS PubMed.
- J. Chen, J. A. Hessler, K. Putchakayala, B. K. Panama, D. P. Khan, S. Hong, D. G. Mullen, S. C. DiMaggio, A. Som, G. N. Tew, A. N. Lopatin, J. R. Baker, M. M. Banaszak Holl and B. G. Orr, J. Phys. Chem. B, 2009, 113, 11179–11185 CrossRef CAS PubMed.
- T. Xia, M. Kovochich, J. Brant, M. Hotze, J. Sempf, T. Oberley, C. Sioutas, J. I. Yeh, M. R. Wiesner and A. E. Nel, Nano Lett., 2006, 6, 1794–1807 CrossRef CAS PubMed.
- Z. Wang, N. Li, J. Zhao, J. C. White, P. Qu and B. Xing, Chem. Res. Toxicol., 2012, 25, 1512–1521 CrossRef CAS PubMed.
- M. Luckey, Membrane Structural Biology with Biochemical and Biophysical Foundations, 2nd edn, Cambridge University Press, Cambridge, UK, 2014 Search PubMed.
- O. Quehenberger, A. M. Armando, A. H. Brown, S. B. Milne, D. S. Myers, A. H. Merrill, S. Bandyopadhyay, K. N. Jones, S. Kelly, R. L. Shaner, C. M. Sullards, E. Wang, R. C. Murphy, R. M. Barkley, T. J. Leiker, C. R. H. Raetz, Z. Guan, G. M. Laird, D. A. Six, D. W. Russell, J. G. McDonald, S. Subramaniam, E. Fahy and E. A. Dennis, J. Lipid Res., 2010, 51, 3299–3305 CrossRef CAS PubMed.
- J. A. F. Op den Kamp, Annu. Rev. Biochem., 1979, 48, 47–71 CrossRef CAS PubMed.
- D. A. Brown and E. London, Annu. Rev. Cell Dev. Biol., 1998, 14, 111–136 CrossRef CAS PubMed.
- D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, 3rd edn, Worth Publishers, New York, 2000 Search PubMed.
- G. Van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112–124 CrossRef CAS PubMed.
- R. F. M. De Almeida, A. Fedorov and M. Prieto, Biophys. J., 2003, 85, 2406–2416 CrossRef CAS PubMed.
- K. Simons and E. Ikonen, Nature, 1997, 387, 569–572 CrossRef CAS PubMed.
- J. F. Hancock, Nat. Rev. Mol. Cell Biol., 2006, 7, 456–462 CrossRef CAS PubMed.
- K. Simons and M. J. Gerl, Nat. Rev. Mol. Cell Biol., 2010, 11, 688–699 CrossRef CAS PubMed.
- S. L. Veatch, P. Cicuta, P. Sengupta, A. Honerkamp-Smith, D. Holowka and B. Baird, ACS Chem. Biol., 2008, 3, 287–293 CrossRef CAS PubMed.
- P.-F. Lenne, L. Wawrezinieck, F. Conchonaud, O. Wurtz, A. Boned, X.-J. Guo, H. Rigneault, H.-T. He and D. Marguet, EMBO J., 2006, 25, 3245–3256 CrossRef CAS PubMed.
- L. J. Pike, J. Lipid Res., 2006, 47, 1597–1598 CrossRef CAS PubMed.
- D. M. Owen, A. Magenau, D. Williamson and K. Gaus, BioEssays, 2012, 34, 739–747 CrossRef CAS PubMed.
- T. Weimbs, S. Low, S. Chapin and K. Mostov, Trends Cell Biol., 1997, 8924, 393–399 CrossRef.
- S. Mukherjee, T. T. Soe and F. R. Maxfield, J. Cell Biol., 1999, 144, 1271–1284 CrossRef CAS PubMed.
- D. A. Brown and E. London, J. Biol. Chem., 2000, 275, 17221–17224 CrossRef CAS PubMed.
- J. L. Cacas, F. Furt, M. Le Guédard, J. M. Schmitter, C. Buré, P. Gerbeau-Pissot, P. Moreau, J. J. Bessoule, F. Simon-Plas and S. Mongrand, Prog. Lipid Res., 2012, 51, 272–299 CrossRef CAS PubMed.
- X. Xu, R. Bittman, G. Duportail, D. Heissler, C. Vilcheze and E. London, J. Biol. Chem., 2001, 276, 33540–33546 CrossRef CAS PubMed.
- M. Bramkamp and D. Lopez, Microbiol. Mol. Biol. Rev., 2015, 79, 81–100 CrossRef PubMed.
- K. Chen and G. Bothun, Environ. Sci. Technol., 2014, 48, 873–880 CrossRef CAS PubMed.
- P. Yi and K. L. Chen, Environ. Sci. Technol., 2013, 47, 5711–5719 CrossRef CAS PubMed.
- J. M. Troiano, L. L. Olenick, T. R. Kuech, E. S. Melby, D. Hu, S. E. Lohse, A. C. Mensch, M. Dogangun, A. M. Vartanian, M. D. Torelli, E. Ehimiaghe, S. R. Walter, L. Fu, C. R. Anderton, Z. Zhu, H. Wang, G. Orr, C. J. Murphy, R. J. Hamers, J. A. Pedersen and F. M. Geiger, J. Phys. Chem. C, 2015, 119, 534–546 CAS.
- N.-J. Cho, C. W. Frank, B. Kasemo and F. Höök, Nat. Protoc., 2010, 5, 1096–1106 CrossRef CAS PubMed.
- R. P. Richter, R. Berat and A. R. Brisson, Langmuir, 2006, 22, 3497–3505 CrossRef CAS PubMed.
- K. El Kirat, S. Morandat and Y. F. Dufrêne, Biochim. Biophys. Acta, 2010, 1798, 750–765 CrossRef CAS PubMed.
- A. Mecke, I. J. Majoros, A. K. Patri, J. R. Baker, M. M. Banaszak Holl and B. G. Orr, Langmuir, 2005, 21, 10348–10354 CrossRef CAS PubMed.
- A. Mecke, D. K. Lee, A. Ramamoorthy, B. G. Orr and M. M. Banaszak Holl, Langmuir, 2005, 21, 8588–8590 CrossRef CAS PubMed.
- P. R. Leroueil, S. A. Berry, K. Duthie, G. Han, V. M. Rotello, D. Q. McNerny, J. R. Baker, B. G. Orr and M. M. Banaszak Holl, Nano Lett., 2008, 8, 420–424 CrossRef CAS PubMed.
- S. Hong, P. R. Leroueil, E. K. Janus, J. L. Peters, M. M. Kober, M. T. Islam, B. G. Orr, J. R. Baker and M. M. Banaszak Holl, Bioconjugate Chem., 2006, 17, 728–734 CrossRef CAS PubMed.
- R. C. Van Lehn, M. Ricci, P. H. J. Silva, P. Andreozzi, J. Reguera, K. Voïtchovsky, F. Stellacci and A. Alexander-Katz, Nat. Commun., 2014, 5, 4482 CAS.
- C. Yuan, J. Furlong, P. Burgos and L. J. Johnston, Biophys. J., 2002, 82, 2526–2535 CrossRef CAS PubMed.
- C. Dietrich, L. A. Bagatolli, Z. N. Volovyk, N. L. Thompson, M. Levi, K. Jacobson and E. Gratton, Biophys. J., 2001, 80, 1417–1428 CrossRef CAS PubMed.
- M. Sundh, S. Svedhem and D. S. Sutherland, Phys. Chem. Chem. Phys., 2010, 12, 453–460 RSC.
- J. P. Slotte, Prog. Lipid Res., 2013, 52, 424–437 CrossRef CAS PubMed.
- P. J. Quinn, Langmuir, 2013, 29, 9447–9456 CrossRef CAS PubMed.
- R. Richter, A. Mukhopadhyay and A. Brisson, Biophys. J., 2003, 85, 3035–3047 CrossRef CAS PubMed.
- M. M. Lapinski, A. Castro-Forero, A. J. Greiner, R. Y. Ofoli and G. J. Blanchard, Langmuir, 2007, 23, 11677–11683 CrossRef CAS PubMed.
- S. F. Sweeney, G. H. Woehrle and J. E. Hutchison, J. Am. Chem. Soc., 2006, 128, 3190–3197 CrossRef CAS PubMed.
- L. Zhu, C. Zhang, C. Guo, X. Wang, P. Sun, D. Zhou, W. Chen and G. Xue, J. Phys. Chem. C, 2013, 117, 11399–11404 CAS.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- M. Rodahl, F. Höök, A. Krozer, P. Brzezinski and B. Kasemo, Rev. Sci. Instrum., 1995, 66, 3924–3930 CrossRef CAS.
- C. A. Keller and B. Kasemo, Biophys. J., 1998, 75, 1397–1402 CrossRef CAS PubMed.
- K. H. Jacobson, T. R. Kuech and J. A. Pedersen, Environ. Sci. Technol., 2013, 47, 6925–6934 CrossRef CAS PubMed.
- W. S. Cleveland and S. J. Devlin, J. Am. Stat. Assoc., 1988, 83, 596–610 CrossRef.
- S. Ladha, A. R. Mackie, L. J. Harvey, D. C. Clark, E. J. Lea, M. Brullemans and H. Duclohier, Biophys. J., 1996, 71, 1364–1373 CrossRef CAS PubMed.
- J. Yguerabide, J. A. Schmidt and E. E. Yguerabide, Biophys. J., 1982, 40, 69–75 CrossRef CAS PubMed.
- D. Nečas and P. Klapetek, Cent. Eur. J. Phys., 2011, 10, 181–188 Search PubMed.
- S. Chiantia, N. Kahya, J. Ries and P. Schwille, Biophys. J., 2006, 90, 4500–4508 CrossRef CAS PubMed.
- M. G. Gustafsson, J. Microsc., 2000, 198, 82–87 CrossRef CAS PubMed.
- B. Bailey, D. L. Farkas, D. L. Taylor and F. Lanni, Nature, 1993, 366, 44–48 CrossRef CAS PubMed.
- A. R. Burns, D. J. Frankel and T. Buranda, Biophys. J., 2005, 89, 1081–1093 CrossRef CAS PubMed.
- J. Holmgren, I. Lönnroth, J.-E. Månsson and L. Svennerholm, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 2520–2524 CrossRef CAS.
- W. I. Lencer, Am. J. Physiol., 2001, 280, G781–G786 CAS.
- N.-J. Cho, L. Hwang, J. Solandt and C. Frank, Materials, 2013, 6, 3294–3308 CrossRef CAS.
- C. Hamai, P. S. Cremer and S. M. Musser, Biophys. J., 2007, 92, 1988–1999 CrossRef CAS PubMed.
- C. Hamai, T. Yang, S. Kataoka, P. S. Cremer and S. M. Musser, Biophys. J., 2006, 90, 1241–1248 CrossRef CAS PubMed.
- N. Kahya, D. Scherfeld, K. Bacia, B. Poolman and P. Schwille, J. Biol. Chem., 2003, 278, 28109–28115 CrossRef CAS PubMed.
- A. V. Samsonov, I. Mihalyov and F. S. Cohen, Biophys. J., 2001, 81, 1486–1500 CrossRef CAS PubMed.
- Y. Lange, J. Dolde and T. L. Steck, J. Biol. Chem., 1981, 256, 5321–5323 CAS.
- E. Reimhult, F. Höök and B. Kasemo, J. Chem. Phys., 2002, 117, 7401–7404 CrossRef CAS.
- C. A. Keller, K. Glasmästar, V. P. Zhdanov and B. Kasemo, Phys. Rev. Lett., 2000, 84, 5443–5446 CrossRef CAS PubMed.
- R. Zimmermann, D. Küttner, L. Renner, M. Kaufmann, J. Zitzmann, M. Müller and C. Werner, Biointerphases, 2009, 4, 1–6 CrossRef PubMed.
- R. Zimmermann, D. Küttner, L. Renner, M. Kaufmann and C. Werner, J. Phys. Chem. A, 2012, 116, 6519–6525 CrossRef CAS PubMed.
- Z. Adamczyk, Particles at Interfaces: Interactions, Deposition, Structure, Academic Press, San Diego, CA, 2006 Search PubMed.
- K. H. Sheikh and S. P. Jarvis, J. Am. Chem. Soc., 2011, 133, 18296–18303 CrossRef CAS PubMed.
- R. G. W. Anderson and K. Jacobson, Science, 2002, 296, 1821–1825 CrossRef CAS PubMed.
- M. C. Seminario and S. C. Bunnell, Immunol. Rev., 2008, 221, 90–106 CrossRef CAS PubMed.
- T. Baumgart, A. T. Hammond, P. Sengupta, S. T. Hess, D. A. Holowka, B. A. Baird and W. W. Webb, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3165–3170 CrossRef CAS PubMed.
- I. Levental, F. J. Byfield, P. Chowdhury, F. Gai, T. Baumgart and P. A. Janmey, Biochem. J., 2009, 424, 163–167 CrossRef CAS PubMed.
- D. Lingwood, J. Ries, P. Schwille and K. Simons, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10005–10010 CrossRef CAS PubMed.
- P. Sharma, R. Varma, R. C. Sarasij, Ira, K. Gousset, G. Krishnamoorthy, M. Rao and S. Mayor, Cell, 2004, 116, 577–589 CrossRef CAS PubMed.
- N. Vyas, D. Goswami, A. Manonmani, P. Sharma, H. A. Ranganath, K. VijayRaghavan, L. S. Shashidhara, R. Sowdhamini and S. Mayor, Cell, 2008, 133, 1214–1227 CrossRef CAS PubMed.
- A. A. Waheed and E. O. Freed, Virus Res., 2009, 143, 162–176 CrossRef CAS PubMed.
- P. Scheiffele, A. Rietveld, K. Simons and T. Wilk, J. Biol. Chem., 1999, 274, 2038–2044 CrossRef CAS PubMed.
- B. M. Castro, M. Prieto and L. C. Silva, Prog. Lipid Res., 2014, 54, 53–67 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5en00098j |
| ‡ Present address: Colorado Mesa University, Department of Chemistry, 1100 North Ave, Grand Junction, CO, 81501. |
| This journal is © The Royal Society of Chemistry 2016 |