
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review of stereochemical implications in the generation of secondary organic aerosol from isoprene oxidation
James M.
Cash
ab,
Mathew R.
Heal
b,
Ben
Langford
a and
Julia
Drewer
*a
aNERC Centre for Ecology and Hydrology, Bush Estate, Penicuik, EH26 0QB, UK. E-mail: juew@ceh.ac.uk
bSchool of Chemistry, The University of Edinburgh, David Brewster Rd, Edinburgh EH9 3FJ, UK
First published on 12th October 2016
Abstract
The atmospheric reactions leading to the generation of secondary organic aerosol (SOA) from the oxidation of isoprene are generally assumed to produce only racemic mixtures, but aspects of the chemical reactions suggest this may not be the case. In this review, the stereochemical outcomes of published isoprene-degradation mechanisms contributing to high amounts of SOA are evaluated. Despite evidence suggesting isoprene first-generation oxidation products do not contribute to SOA directly, this review suggests the stereochemistry of first-generation products may be important because their stereochemical configurations may be retained through to the second-generation products which form SOA. Specifically, due to the stereochemistry of epoxide ring-opening mechanisms, the outcome of the reactions involving epoxydiols of isoprene (IEPOX), methacrylic acid epoxide (MAE) and hydroxymethylmethyl-α-lactone (HMML) are, in principle, stereospecific which indicates the stereochemistry is predefined from first-generation precursors. The products from these three epoxide intermediates oligomerise to form macromolecules which are proposed to form chiral structures within the aerosol and are considered to be the largest contributors to SOA. If conditions in the atmosphere such as pH, aerosol water content, relative humidity, pre-existing aerosol, aerosol coatings and aerosol cation/anion content (and other) variables acting on the reactions leading to SOA affect the tacticity (arrangement of chiral centres) in the SOA then they may influence its physical properties, for example its hygroscopicity. Chamber studies of SOA formation from isoprene encompass particular sets of controlled conditions of these variables. It may therefore be important to consider stereochemistry when upscaling from chamber study data to predictions of SOA yields across the range of ambient atmospheric conditions. Experiments analysing the stereochemistry of the reactions under varying conditions of the above variables would help elucidate whether there is stereoselectivity in SOA formation from isoprene and if the rates of SOA formation are affected.
Environmental impactThe predicted yield of isoprene-derived secondary organic aerosol (SOA) could be over- or underestimated when upscaling from chamber data due to the prevailing assumption that stereochemistry has no effect. Although studies have shown that one atmospherically-generated SOA product of isoprene is produced in a racemic mixture, the possibilities of a stereoselective influence have not been explored fully. Evidence suggests that heterogenous stereochemistry could influence the production of some of the highest-contributing isoprene-derived SOA components: oligomer chains created from reactions involving epoxydiols of isoprene (IEPOX), methacrylic acid epoxide (MAE) and hydroxymethylmethyl-α-lactone (HMML). Revealing the effects of stereochemistry on these reactions could considerably impact on the predicted properties and effects of isoprene-derived SOA, for example its hygroscopicity. |
Introduction
Isoprene (2-methyl-1,3-butadiene) is a volatile organic compound (VOC) emitted into the atmosphere predominantly from vegetation (and is hence often referred to as a biogenic VOC), although it is also emitted from some anthropogenic sources such as vehicle exhaust.1–3 Isoprene is observed in comparatively large amounts in the atmosphere and its global emissions are predicted by the Model of Gases and Aerosols from Nature (MEGAN2.1) to be as high as 535 Tg per year.4 Under the presumption that atmospheric oxidation of isoprene can lead to formation of secondary organic aerosol (SOA), even a small SOA product-yield would mean isoprene emissions make a significant contribution to global SOA production.5,6It was initially thought that isoprene photooxidation products were too volatile to condense to the aerosol phase and experiments showed almost zero aerosol growth.7 It was not until SOA was observed above the Amazonian rain forest that Claeys et al.8 showed that isoprene reacted in the atmosphere to form SOA. The observed SOA contained 2-methylthreitol and 2-methylerythritol (Fig. 1) which are named according to the diastereomers of four possible 2-methyltetrols. They have the C5 isoprene skeleton and were concluded to be atmospheric oxidation products from isoprene as the other biogenic sources of 2-methyltetrols were not present in this study.8 These diastereoisomers are now widely recognised as a source of, and tracer for, isoprene-derived SOA.9–11 However, the link between SOA production and the stereochemical effects of these diastereoisomers was not investigated until fairly recently.12
 | ||
| Fig. 1 The structures and names of the stereoisomers of 2-methyltetrol. The red arrows indicate the diastereomers of each structure, showing the two 2-methylerythritols on the left and the 2-methylthreitols on the right, and with their names in red. Names according to the R and S nomenclature are in black. The blue arrows indicate the enantiomer of each structure (adapted from Nozière et al.12). | ||
Until the 1980s and 1990s the stereochemical outcome of radical reactions was considered not to be relevant due to the reaction rates being too fast to create any stereoselectivity.13 (Stereoselectivity is the production of one enantiomer in larger quantities than another.) However, subsequent research on radical reactions showed that selectivity can be introduced under substrate control and chiral auxiliaries.13 It is therefore appropriate to consider whether the reactions leading to the first-generation products in isoprene oxidation are under stereoselective control. The first-generation products react further in the gas phase to create isoprene second-generation products. These species then react through a gas-to-aerosol-phase reaction pathway involving their uptake onto an aerosol surface, which creates further potential for stereochemical influence. A surface stereochemical effect has been seen in the atmospheric heterogeneous ozonolysis of quinuclidine diastereomers on a silica surface where an enantiomeric excess of isomer products was observed, together with a doubling of the rate of reaction.14 The study describes how stereoselectivity is introduced through the orientation of the reactive C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond on the solid surface. An analogous pathway in the formation of isoprene-derived SOA involves the uptake onto an aerosol surface of epoxydiols of isoprene (IEPOX) species. This suggests that stereoselectivity can be introduced into the mechanisms leading to SOA formation through the surface stereochemistry of the aerosol interacting with IEPOX. It also suggests that the rate of production of SOA could vary as a consequence of this.
C bond on the solid surface. An analogous pathway in the formation of isoprene-derived SOA involves the uptake onto an aerosol surface of epoxydiols of isoprene (IEPOX) species. This suggests that stereoselectivity can be introduced into the mechanisms leading to SOA formation through the surface stereochemistry of the aerosol interacting with IEPOX. It also suggests that the rate of production of SOA could vary as a consequence of this.
Where stereoselectivity is introduced, the varying tacticity of aerosol, which is the arrangement of stereocentres within its macrostructure, could change its properties such as melting point and water solubility.15 This could therefore influence its overall effect on the environment; for example, the probability of forming aerosol can vary with changes in these properties, which in turn leads to variation in the radiative forcing effects of the aerosol. That the composition and structure of an aerosol particle can affect its interaction with the environment is well documented.16 For example, the phase properties, hydrophilicity and melting properties of polymer systems have been linked to the tacticity of their amorphous and crystalline structures.17–19 Mikhailov et al.20 also described how interactions of water vapour varied with altering amorphous and crystalline structures of aerosol, whilst Baker et al.21 showed how water-solubility equilibrium dynamics differed by one order of magnitude when a supramolecular polymer structure was subjected to a change in the stereochemical orientation of a single methyl group.
Nozière et al.12 were one of the first to recognise that an enantiomeric excess of one enantiomer of the 2-methyltetrols is observed, when measuring SOA in Aspvreten, Sweden. However, they describe this enantiomeric excess as being due to biogenic sources which emit 2-methyltetrols directly rather than being due to atmospheric oxidation products. They supported this with chamber experimental data for the aqueous-phase oxidation of isoprene in H2O2 and the gas-phase photooxidation of isoprene in the presence of NOx. The quantities of the four isomers of 2-methyltetrol in both reactions gave racemic mixtures of the two sets of enantiomers. From this they developed a method of identifying the origin of 2-methyltetrols in ambient SOA by correlating its isomeric fractions (ratio of the concentration of 2-methylerythritol to the total concentration of the 2-methyltetrols) with those calculated for smog chamber measurements under set conditions.12 The method of comparing isomeric fractions between experimental and ambient SOA can be validated, in theory, through the study of the mechanisms which proceed to forming SOA. This method relies on two assumptions: one is that the stereochemical environments of both the chamber and the atmosphere are the same, and the second is that SOA produced in the atmosphere is racemic (i.e. an equal mixture of enantiomers). The second assumption has been accepted by a number of researchers12,22 but this has been based on chamber experiments and was accepted regardless of the mechanism which forms the SOA.
It can be established if stereochemistry is important from the reactions. For example, if a reaction is stereoselective, then the stereochemical environment can change the resultant quantities of enantiomers. However, if it is stereospecific (producing one enantiomer product only) the stereochemical environment will likely not have any effect. This kind of analysis can establish if atmospheric oxidation of isoprene produces only racemic mixtures of isomers. Using results from recent developments in SOA analysis and mechanistic predictions, this review analyses the pathways to SOA formation in the context of their stereochemistry. As this is a relatively new area of research, the focus here is on the pathways of most importance to SOA production. Using the reaction mechanisms which describe these pathways, the chirality of SOA is assessed, along with the possible implications for atmospheric SOA. It is hoped that findings from this review will help target future research on mechanistic areas where further understanding could yield major insights into the chirality of SOA created through atmospheric isoprene chemistry.
Isoprene first-generation products
The predominant initial reaction of isoprene in the atmosphere is with the OH radical, followed by O2 addition. The OH radical addition to one of the two CC double bonds in isoprene can, in principle, form four hydroxyalkyl radicals, which then rapidly react with O2 to form hydroxyperoxy radicals (Fig. 2).23,24
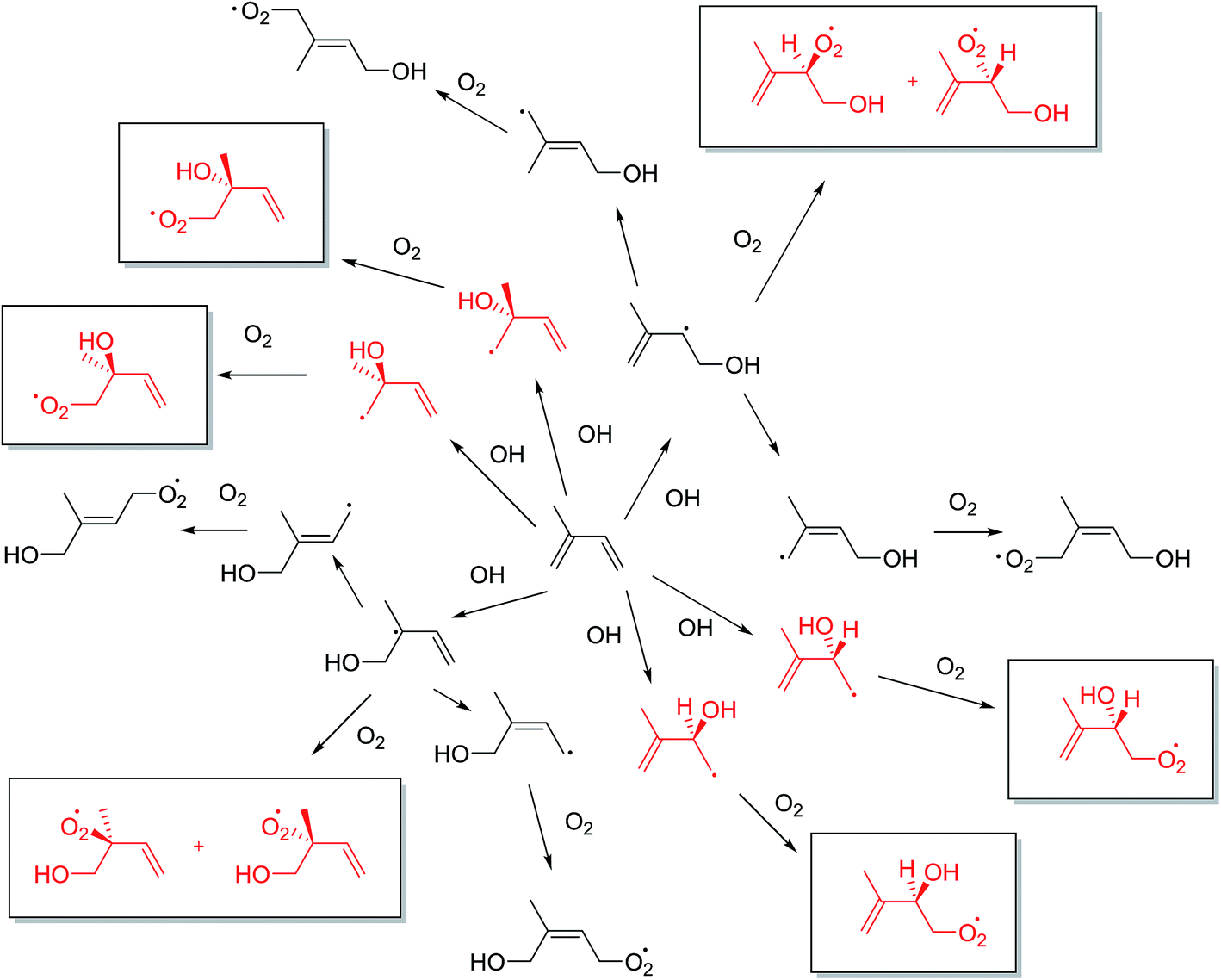 | ||
| Fig. 2 Generalised mechanism of isoprene photooxidation to produce 12 hydroxyperoxy radical isomers. The red structures not in boxes show where the enantiomers are first created and the boxed forms show the stereoisomers previously not reported. | ||
However, the eight hydroxyperoxy structural radical isomers generally written as products in this step are not the whole story as this does not take into account the possible enantiomers that can form. As Fig. 2 shows, 12 hydroxyperoxy radicals are possible. The omission of these four additional stereoisomers is relevant as the proceeding chemistry can involve a chiral environment which has the potential to change the outcome of the resultant SOA, especially as diastereoisomers are formed.
Isoprene first-generation products: high and low NOx conditions
The subsequent chemistry of the hydroxyperoxy radicals (RO2) can proceed along two pathways depending on the concentration of NOx in the air (Fig. 3). Under low NOx concentrations, which is associated with rural/tropical areas, the reaction pathways involving HO2 + RO2 or RO2 + RO2 are dominant. The reactions of RO2 with HO2 produce hydroxy-hydroperoxides, whilst the reactions with RO2 result in diols. Under high NOx concentrations (more polluted air), the reaction pathway involving NO + RO2 competes with the HO2 + RO2 reaction pathway. The products of the RO2 + NO reactions include hydroxynitrates and carbonyl products such as methyl vinyl ketone (MVK) and methacrolein (MACR) (Fig. 3).25 | ||
| Fig. 3 Generalised mechanism of hydroxyperoxy radicals reacting in high and low NOx conditions. | ||
The first-generation products such as hydroxy-hydroperoxides, diols and hydroxynitrates can give many possible diastereoisomers and enantiomers. The amount of enantiomer excess is unknown but the presence of thousands of volatile organic compounds (VOCs) emitted into the atmosphere, could cause substrate control and chiral auxiliary effects which may in turn create an enantiomer excess. A particular species which could be susceptible to stereoselectivity is the enantiomeric hydroxyperoxy radical. The cross-reaction of the hydroxyperoxy radicals with themselves or other hydroxyperoxy radicals (RO2 + RO2) could be a source of substrate control as the R group could cause interactions which lead to stereoselectivity. This pathway has been observed as a dominant source of SOA in the form of organic peroxides (ROOR) similar to that in high NOx conditions.26 Future experiments targeted on the formation of organic peroxides through this reaction would be beneficial to establish its stereochemical importance.
Another set of reaction pathways which involve RO2 radicals are the unimolecular isomerisations. The RO2 molecules undergo 1,6 and 1,4 H-atom-shift isomerisations which cause intramolecular interconversions and significant changes in molecular structures. The products of these reactions have been observed experimentally under low NOx conditions.27,28 Peeters et al.29 have formulated a theoretical mechanism called LIM1 which is an updated version of the original Leuven Isoprene Mechanism.30 Global models using LIM1 predict that 28% of isoprene peroxy radicals undergo the 1,6 H-atom-shift reaction which equates to 100–150 Tg C per year,29 which is significant in terms of isoprene oxidation pathways.
The fraction of isoprene-derived peroxy radicals which react via the 1,6 H-atom-shift is governed by the hydrogen bond strengths.29 This raises the question of whether the different chiral positions of substituents should be considered in this mechanism. It has been observed in structures of proteins that hydrogen bonding is preferential under certain conditions and that stereochemical orientations are preferred based on sterics.31 Considering the many different RO2 stereoisomers produced in isoprene photooxidation (see Fig. 2), it may be beneficial to incorporate isomerisation reaction stereochemistry into the LIM1 mechanism.
The full chemical mechanism of first to fourth generation hydroxynitrate formation from isoprene, as described by the Master Chemical Mechanism (a near-explicit chemical degradation mechanism), shows multi-step reactions with OH radicals and NOx species creating mainly di-nitrates.32 Studies have shown that this pathway leads to SOA production yields from isoprene of ∼14%.33 However, further laboratory and field studies of aerosol composition are required to assess its contribution to SOA.
Nonetheless, past compositional analyses of isoprene photooxidation chamber studies have indicated that under low NOx conditions, organic peroxides compose ∼61% and 25–30% of SOA produced with non-acidic and acidic seed, respectively.34 This indicates that the organic peroxides and hydroxyperoxy radicals are important intermediates in the production of SOA but the chamber conditions in the study by Surratt et al.34 may not be representative of atmospheric conditions. The RO2 + RO2 may be the dominant pathway for the conditions of this study which shuts down the HO2 + RO2 pathway, thus making organic peroxides the dominant SOA product. Given that atmospheric HO2 concentrations are much greater than RO2 concentrations,35 this suggests that the HO2 + RO2 reaction pathway will be the main source of SOA. More recent studies have shown that gas-phase low-volatility organic compounds (LVOC) produced from ISOPOOH (a major product of the HO2 + RO2 pathway) are a direct source of SOA.36 Nevertheless, the predicted contribution of 3.3% to global SOA is low.36
In summary, evidence suggests that organic peroxides and other direct sources of SOA from isoprene first-generation products make little contribution to SOA formation, which might imply that the possibility of stereochemistry impacting the production of SOA through gas-phase first-generation products is not relevant. However, stereochemical configurations may be retained through reactions to second-generation products which may mean that the reactions leading to first-generation products predefine the stereoisomers formed in SOA.
Isoprene second-generation products
Detailed mechanisms for the degradation of isoprene first-generation products have been established,32 and again the reaction pathways and products are strongly influenced by the level of NOx. The first-generation products are still relatively high volatility, but with further reactions in the atmosphere the products become less volatile and pass through to the aerosol phase. Claeys et al.37 first hypothesised that a multi-phase reaction occurs from experimental evidence. They found that the gas-phase photooxidation of isoprene with ozone and OH radicals forms MACR, and further reactions form 2-methylthreitol and 2-methylerythritol through an acid-catalysed condensed-phase reaction. These semi-volatile products then form SOA directly or react further to produce supramolecular structures.The relevance of stereochemistry becomes clearer for the formation of second-generation products. This is because the stereochemical outcomes of condensed-phase reactions are better understood compared to the radical chemistry of the first-generation products. Therefore in the following sections the assumption that atmospheric reactions produce only racemic mixtures is more closely examined.
Isoprene second-generation products: low NOx conditions
Epoxydiols of isoprene (IEPOX) are reactive intermediates to the reactions that result in 2-methylthreitol and 2-methylerythritol. This IEPOX-formation pathway dominates in low NOx conditions (i.e. when RO2 + HO2 ≫ RO2 + NO) and is considered a key route to producing a high percentage of aerosol from isoprene oxidation (Fig. 4).35 | ||
| Fig. 4 Generalised mechanism of epoxydiols of isoprene (IEPOX) formation from relevant hydroxy-hydroperoxides. | ||
The four IEPOX species shown in Fig. 4 are considered to be the only relevant species formed that proceed to yield SOA.38,39 The evidence for this is their observation by CI-MS techniques when isoprene is subjected to photooxidation. Paulot et al.38 postulated β-IEPOX species to be the most stable IEPOX species based on computational quantum chemical energies which is consistent with the observation that 97% of IEPOX species are β-IEPOX39 and the MCM prediction that 95% of IEPOX species are β-IEPOX.32
The IEPOX species then undergo acid-catalysed reactive uptake within acidic aerosol to produce low-volatility products. The mechanism of the reactive uptake of IEPOX species is understood to be an acid-catalysed ring opening of the epoxy group by a nucleophile (Fig. 5).35,40–42 The reaction itself is believed to take place within the aerosol following internal diffusion from the surface.43 Observations have shown the reaction to be driven by an increase in acidified sulfate seed when under low NOx conditions. For example, SOA mass yields are 28.6% using an acidic seed and 1.3% without.35 Previous field studies applied positive matrix factorization analysis of aerosol mass spectrometry data and resolved one factor as surrogate for SOA from the reactive uptake of IEPOX. This factor accounts for a large fraction of OA at multiple sampling sites, including Borneo,6 Amazon rain forest,44 and southeastern United States.45–47
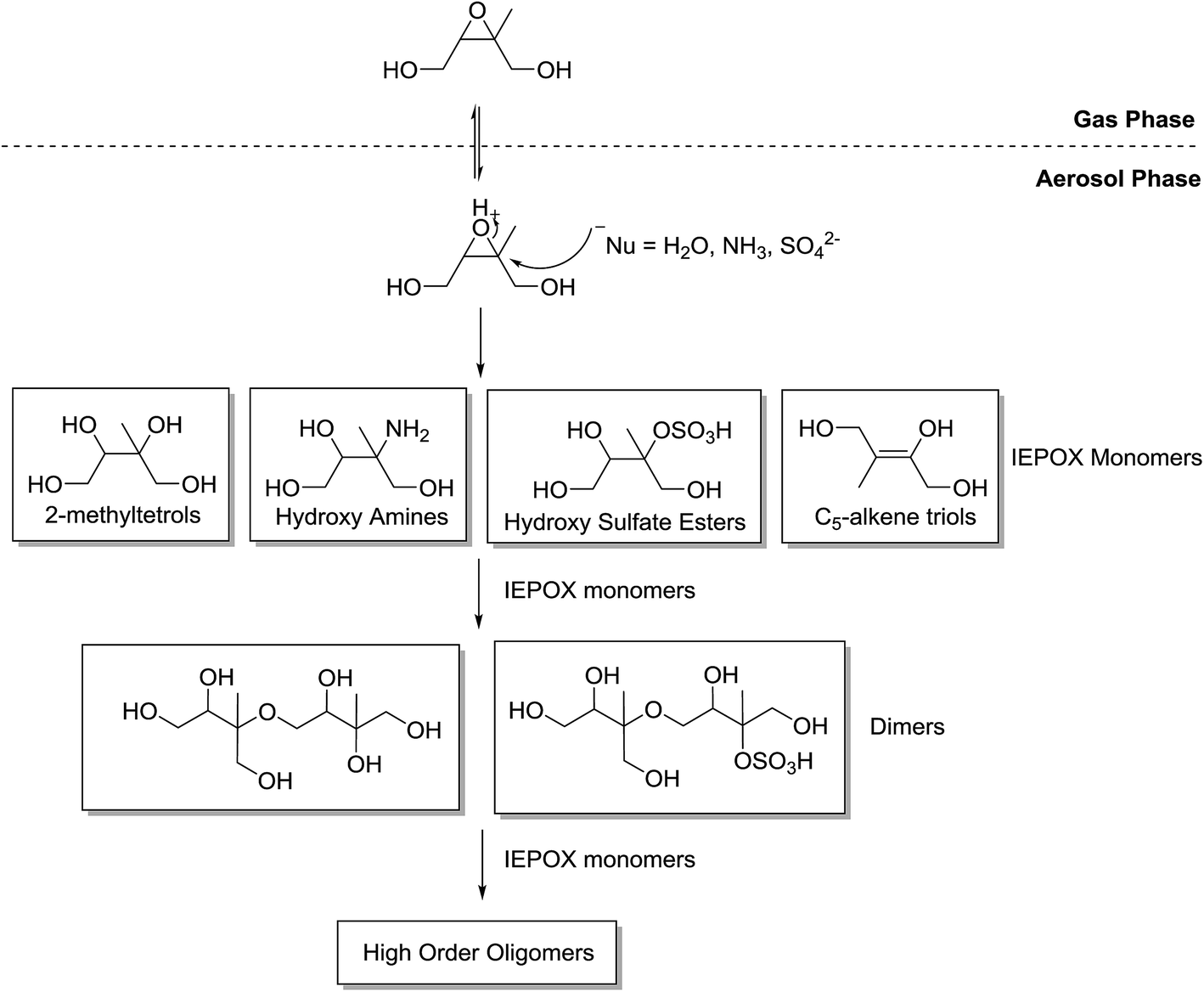 | ||
| Fig. 5 Generalised mechanism for the formation of aerosol species from β-IEPOX. The δ-IEPOX species is omitted for simplicity (adapted from Surratt et al.35). | ||
Studies argue differently about the contribution to SOA of the first generation of products from the IEPOX uptake reaction. Surratt et al.34 reported that IEPOX products such as 2-methyltetrols and C5-alkene triols make only small contribution to aerosol composition. Contributions were 3.91% and 0.6%, respectively, under low acidified-sulfate-seed concentrations, and 0.46% and 0.06%, respectively, under high acidic-seed concentrations. However, as described earlier, the conditions in the Surratt et al.34 study cause the RO2 + RO2 pathway to be dominant which prevents the pathway involving the IEPOX uptake reactions and is therefore considered not atmospherically-relevant. Riedel et al.48 determined the amount of SOA from the reactive uptake of β-IEPOX. They reported <5% of IEPOX-derived SOA is composed of 2-methyltetrols and C5-alkene triols under non-acidic seed conditions but ∼30% under acidic seed conditions. Despite this evidence of high contribution, a more recent study suggests that measured IEPOX organic aerosol tracers, such as 2-methyltetrols and C5-alkene triols, are thermal decomposition products of larger oligomer molecules.42 More evidence is clearly required to understand the direct contribution of these species to SOA composition.
Products of the IEPOX uptake reaction have been shown to polymerise to oligomer structures.35,40–42,49 The IEPOX product monomers combine through the IEPOX uptake reaction to form polymer chains in large structures (Fig. 5). The same studies show that the formation of oligomer species through the IEPOX uptake reaction produce significant amounts of SOA in chamber studies and has been considered the major component of IEPOX-SOA. The presence of oligomers in ambient IEPOX-derived SOA has also been observed in isoprene-rich locations in the field.40,50,51
In the acid-catalysed IEPOX uptake mechanism, the stereochemistry of the resultant products, such as the 2-methyltetrols (Fig. 6), is stereospecific because the epoxide functionality is acidified. The nucleophilic attack occurs on the more substituted carbon because a partial positive charge is created due to the activation of the epoxide ring by H+.52 The first products or IEPOX monomers will then undergo oligomerisation to form dimers and higher-order oligomers.35Fig. 6 illustrates an example where water is the nucleophile to produce two 2-methyltetrol diastereomers. In theory, this mechanism results in a single stereoisomer. This shows a key area where the stereochemistry of the precursors to aerosol formation may consequently predetermine the chiral configuration of the product aerosol because this reaction mechanism is not stereoselective.
 | ||
| Fig. 6 Mechanism for the nucleophilic ring-opening of trans- and cis-β-IEPOX by H2O. | ||
The ratio of quantities of trans- to cis-β-IEPOX found in chamber experiments has been observed as 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.39 Nozière et al.53 published data (in their supporting information) of the absolute concentrations of each isomer of the 2-methyltetrols analysed from ambient samples taken near Aspvreten, Sweden. If all the concentrations taken from the 12th of May to the 15th of December in that study are averaged, the quantities of (2S,3R)- and (2R,3R)-2-methyltetrol are found to be in a 1.82:1 ratio which is very similar to the reported 2:1 composition ratio of their precursors. This is assuming the reaction yields of (2S,3R)-2-methyltetrol from trans-β-IEPOX are similar to that of (2R,3R)-2-methyltetrol from cis-β-IEPOX. However, it does support the mechanism of H+-activated nucleophilic ring opening of β-IEPOX. The reason for the slight difference to the 2:1 ratio could also be due to the 5% of IEPOX species produced which are not β-IEPOX, as shown in the MCM.32 This 5% of other IEPOX species (such as δ-IEPOX) could therefore be producing the 2-methyltetrol isomers which cause this observed imbalance.
:1.39 Nozière et al.53 published data (in their supporting information) of the absolute concentrations of each isomer of the 2-methyltetrols analysed from ambient samples taken near Aspvreten, Sweden. If all the concentrations taken from the 12th of May to the 15th of December in that study are averaged, the quantities of (2S,3R)- and (2R,3R)-2-methyltetrol are found to be in a 1.82:1 ratio which is very similar to the reported 2:1 composition ratio of their precursors. This is assuming the reaction yields of (2S,3R)-2-methyltetrol from trans-β-IEPOX are similar to that of (2R,3R)-2-methyltetrol from cis-β-IEPOX. However, it does support the mechanism of H+-activated nucleophilic ring opening of β-IEPOX. The reason for the slight difference to the 2:1 ratio could also be due to the 5% of IEPOX species produced which are not β-IEPOX, as shown in the MCM.32 This 5% of other IEPOX species (such as δ-IEPOX) could therefore be producing the 2-methyltetrol isomers which cause this observed imbalance.
Taking into account that epoxides do not require to be activated by H+ and that no partial charge is created, a strong nucleophile could attack the least hindered (least substituted) carbon atom. Even under acid conditions, the attack at the least hindered carbon atom is likely to happen in epoxide reactions, especially where there is substantial steric hindrence.52 This mechanism is also stereospecific and produces the other two possible 2-methyltetrol isomers: (2R,3S)-2-methyltetrol is produced from trans-β-IEPOX and (2S,3S)-2-methyltetrol is produced from cis-β-IEPOX. Again, using the supplementary data published by Nozière et al.,12 the averaged quantities of (2S,3R)- and (2R,3R)-2-methyltetrol are 1.89:1 and is very similar to the 2:1 ratio of the precursors.
The influence of catalysts on the IEPOX reaction could also be a source of stereochemical influence. Through varying the cation and anion components of the aerosol formed in their chamber studies, Nguyen et al.54 showed that ammonium ions (NH4+) could be catalysing the IEPOX reaction along with H+ ions. Evidence suggests that NH4+ catalysis is likely to happen. A series of studies conducted by Noziere et al.53,55,56 showed how NH4+ could be a significant source of oligomer SOA in wet aerosol. This could cause a difference in the stereochemical outcome and in the rate of reaction. Ammonium cations have been shown to catalyse a number of ring-opening epoxide reactions57,58 and their activation of the epoxide ring can cause selectivity due to the substituents bonded to the nitrogen. There is the possibility, therefore, that NH4+ ions cause stereoselectivity and the reaction is no longer stereospecific. In addition, the isoprene oxidation products hydroxy amines (NH3R) could become protonated under acidic conditions to become ammonium cations (NH3R+) and their steric influence could lead to an alteration in the selectivity or rate of the reaction. The degree of their influence could relate to the difference in the R group, which will vary because hydroxy amines are produced from isoprene where potentially many different hydroxy amine chemical structures can be produced due to an oligomerisation reaction, as shown in Fig. 5. Nguyen et al.54 also suggest that activation of the epoxide ring may not only be limited to H+ and NH4+ ions. Future research involving epoxide ring activation of IEPOX may therefore be helpful for elucidating any hidden sources of influence on the composition and formation of SOA.
From the above possible factors that could cause stereoselectivity in the mechanism of nucleophilic ring opening of β-IEPOX it can be concluded that reaction conditions could play a key role. The chamber studies work carried out by Nozière et al.12 displayed racemic mixtures of the two sets of enantiomers of 2-methyltetrols. The reaction conditions however could be argued as not being representative throughout the atmosphere because aerosol seed, aerosol cationic and anionic content, pH and water availability were not varied. Future chamber studies involving aerosol growth where these parameters are changed, to mimic the varying conditions in the atmosphere, would help to establish if racemic mixtures are formed.
However, if there is no stereoselectivity involved and the mechanism shows only stereospecificity, it means that this part of the sequence of reactions leading from isoprene to SOA can be considered as insignificant in terms of stereochemistry. This is because the stereochemical configuration of the reactants is only inverted and is not disorganised into equal or unequal amounts of enantiomers. The reactions preceding this IEPOX intermediate reaction will therefore determine this disorganisation of enantiomers. Ebben et al.59 analysed the aerosol produced from another important biogenic VOC, α-pinene. They describe how mixtures of enantiomers of (+)-α-pinene and (−)-α-pinene strongly non-linearly correlate with chiral signals seen for the aerosol they produce in chamber studies. The aerosol is composed of oligomers which form chiral superstructures. This study suggests that a similar stereospecific reaction occurs for the formation of SOA from α-pinene, as it does for the formation of SOA from isoprene. The assumption that atmospheric reactions only produce racemic mixtures does not agree with these findings. If, for example, this was true then, for a ratio of 74:24 of (+)-α-pinene and (−)-α-pinene, a zero chiral signal (racemic mixture) should be produced. However, the study reports a chiral signal which non-linearly correlates with the 74:24 starting ratio. In comparing this with the IEPOX intermediate reactions, the stereoisomers of the hydroxy-hydroperoxides are analogous to the mixtures of α-pinene enantiomers. This supports the hypothesis that the stereochemistry is retained from early on in the first-generation products. Future work on discovering if mixtures of hydroxy-hydroperoxide enantiomers produce a non-linear chiral relationship with the oligomer structures produced could confirm whether this theory is valid.
The reaction rates of the IEPOX reaction could be influenced by heterogeneous stereochemistry. Stokes et al.14 have shown that surface-bound olefin diastereomers not only react to give stereoselectivity but also react twice as fast due to the orientation. These authors suggest that other heterogenous atmospheric reactions could be influenced by stereochemistry. To understand if the production of SOA is affected by stereochemistry, the IEPOX reaction itself can be considered. As discussed, this reaction governs the amount and composition of a large proportion of aerosol produced. Recent studies have shown that the reactive uptake coefficient (γ) correlates with SOA growth in chamber studies43,48,60,61 and it is being used to predict ambient aerosol in models.51,62 This parameter is calculated by determining the pseudo-first-order rate constant (khet) that describes the uptake of trans-β-IEPOX onto particles.60 The use of γ to measure the amount of aerosol growth has been beneficial in evaluating the effects of atmospheric conditions on the amount of SOA produced. Gaston et al.60 and Riva et al.43 both show how pre-existing coatings of aerosol significantly affects IEPOX uptake. It is therefore appropriate to consider if the pre-existing coating has a stereochemical influence on the IEPOX uptake reaction.
Furthermore, Gaston et al.60 describe how the morphology and composition of aerosol can affect reactive uptake. As discussed in the Introduction, the morphology of aerosol could be dependent on its tacticity, which could cause an indirect effect on aerosol production. Therefore studies in this area may show a dependency of IEPOX-SOA formation on stereochemistry.
Initial investigation could focus on whether the SOA framework produced from the IEPOX uptake reaction does indeed change in tacticity. A number of methods can be used and the simplest approach would be to create 2-methyltetrol enantiomers from IEPOX species using a similar chamber experimental method to Gaston et al.60 However, this method only considers the reactive uptake of trans-β-IEPOX whilst, despite the fact that the trans-β-IEPOX isomer is the most abundant of the possible isomers, the abundance of trans- to cis-IEPOX is 2:1, as discussed earlier.39 Without knowing the stereochemistry of each product from all the possible IEPOX species, the method should incorporate the reactive uptake of all atmospherically-relevant IEPOX species when forming aerosol directly from them in chamber studies. This would ensure that the tacticity of the aerosol is reflective of ambient aerosol which, if not, could significantly alter its morphology. For example, Shimizu et al.19 described how changes in physical properties (melting point, solubility and morphology) of a supramolecular structure were related to a stereochemical effect caused by a change in the arrangement of 1-glucosamide-headed bolaamphiphiles monomer units.
The amounts of the 2-methyltetrol species present in these chamber experiments can be quantified by GC-MS using electron-impact ionisation.12 The dependency of the 2-methyltetrol enantiomer compositions on different chamber conditions such as pH, ammonium sulfate seed aerosol, relative humidity, aerosol seed ion content and aerosol seed coatings, can then be determined. Using the same experimental procedure, the amount of chirality can be determined from spectra measured using vibrational sum-frequency-generation linear dichroism.59 In this technique the overall chirality of the aerosol is measured rather than individual components.
If the SOA framework does differ in stereoisomer quantities then the effect of altering the stereochemical SOA framework on the reactive uptake coefficient can be investigated. Experiments similar to those conducted by Gaston et al.60 and Reidel et al.48 could determine this. These authors measured the reactive uptake coefficient from the decay of trans-β-IEPOX against the interaction time with particles. Alternatively, for first-order kinetic conditions a pseudo-particle modulation method may be used.60 The uptake coefficient could then be measured for IEPOX species onto ammonium sulfate or bisulfate particles which are coated in media which differ in stereochemistry. The coatings could be a mixture of 2-methyltetrol enantiomers or, more realistically, oligomer molecules similar to those created in the production of aerosol under low-NOx conditions.
Another aspect of the reactive uptake of IEPOX is to consider the IEPOX species themselves and how their stereochemistry may affect the uptake coefficient. It is well known that diastereomers have different chemical properties and therefore react at different reaction rates (Kalsi, 2008). Indeed, two studies have shown that the hydroxy positioning on IEPOX species affects the rate of reaction for the opening of the epoxide ring.63,64 However, whether changing the hydroxy positioning also alters the uptake coefficient of the diastereomers of IEPOX species has not been investigated.
Isoprene second-generation products: high NOx conditions
2-Methyltetrols are reported to have not been observed under high NOx conditions in isoprene photooxidation chamber studies.34 However, the first-generation products of isoprene do have multiple chiral centres. Under high NOx conditions, MACR production is a dominant pathway and is formed following a reaction between hydroxyperoxy radicals and NO. Jenkin et al.32 quote MACR molar yields of 23.6% and 20.1% under concentrations of NO of 10 ppm and 100 ppt, respectively. However, MACR does not have any chiral centres but reacts further to produce condensable products in the form of organic acids such as pyruvic acid, and aldehyde derivatives such as methylglyoxal and hydroxyacetone.65 These products do contain chiral centres and their formation contributes to the overall SOA mass although further research is required to determine their significance, because there are contrasting views over their origin. The mechanism which has seen most interest, and is considered to be a large source of SOA, is the formation of the two-chiral-centred 2-methylglyceric acid (2-MG), which has been observed in chamber and field studies.10,34,66 Its formation from MACR has recently been theorised and the possible mechanisms are shown in Fig. 7. | ||
| Fig. 7 Generalised mechanism for the formation of aerosol from MACR through MPAN and MPAA intermediates (adapted from Surratt et al.35 and Lin et al.49). | ||
Two routes to forming 2-MG are considered and their relevance is still under debate. Methacryloylperoxynitrate (MPAN) is considered a key intermediate to forming SOA due to its high yields from MACR and high productivity of 2-MG.35,67 The mechanism through which 2-MG is formed is theorised to contain the reactive intermediates hydroxymethylmethyl-α-lactone (HMML) and methacrylic acid epoxide (MAE). The difference between the two stereochemically is that the MAE pathway is thought to involve an acid-catalysed ring opening of the epoxide group which, as shown previously, could theoretically be stereospecific. HMML, conversely, does not require H+ activation of the epoxide ring and therefore the reaction is stereospecific but the least hindered or substituted carbon is attacked through a nucleophilic SN2-like substitution reaction.52 Lin et al.68 suggest HMML may be insignificant compared to the yields of MAE. However, Kjaergaard et al.69 suggest that HMML is formed in significantly high yields and Nguyen et al.70 observe a ∼75% yield of HMML and a negligible (<2%) yield of MAE. Differences in experimental conditions could be the subject of the observed difference but further investigation is required to build a greater understanding.
Despite this, results of chamber studies have shown that SOA is composed of high percentages (22–34%) of 2-MG monomer-based oligomers34 and is produced in considerable yields (1.6–11.7%) from MACR.71 However, the relevance of this to atmospheric conditions has been questioned by Birdsall et al.72 In their study, oligomerisation of 2-MG is modelled as a Fischer esterification mechanism and the reaction calculated to be thermodynamically feasible only in dry conditions (RH < 20%), which is not normally encountered in the atmosphere. This is contradicted by MACR aqueous-phase photooxidation experiments by Liu et al.71 where 11 series of oligomer structures were observed. Each series amounted to a large number of different length oligomers with molecular weights that reached 1400 Da. These were obtained at a relative humidity of <45% and the yields produced were supported by previous studies.71 This suggests that the proposed Fischer esterification could be invalid and that there are other environmental conditions which drive the formation of oligomer aerosol.
Considering the HMML and MAE pathways to producing 2-MG and the observed oligomer SOA, there are two mechanisms. The mechanism for the pathway through which MAE exists is again predicted to be an acid-catalysed nucleophilic epoxide ring opening reaction68 and the HMML pathway is predicted to be a non-catalysed nucleophilic ring-opening reaction.68,70 These two pathways are predicted to produce very similar products of oligomer molecules comprising of 2-MG, 2-MG sulfate and 2-MG nitrate monomers (Fig. 7). However, MAE and HMML differ chemically and they react through different mechanisms. Therefore theoretically their products will differ stereochemically. If the two pathways react simultaneously, this could create an oligomer macromolecular structure with highly variable tacticity. As discussed for the low NOx IEPOX mechanism, the reaction conditions (e.g. aerosol cation/anion content, pH and water content) could play a crucial part in whether these reactions are stereospecific or stereoselective. If stereospecific, it supports the theory that the stereochemistry is conserved from isoprene first-generation products to isoprene-derived aerosol, similar to that seen for α-pinene in the study by Ebben et al.59 However, MACR does not contain any chiral centres and so the configuration of the chiral centres in the hydroxy-hydroperoxides which then react to form MACR are not significant. Nonetheless, in this situation the mechanisms to oligomer formation, although not understood, could still hold potential for causing stereoselectivity and creating non-racemic mixtures – especially if the two reaction pathways through HMML and MAE produce similar high amounts of SOA under atmospherically-relevant conditions. This would add complexity to the stereochemistry of the SOA framework and would most likely create non-racemic mixtures of stereoisomers. Thus, the SOA morphology and structures will differ and their properties, including lifetime and water solubility, may also be altered.
Conclusions and future directions
From the evidence of SOA composition studies, the first-generation products of isoprene oxidation do not produce aerosol directly in high amounts, although there are places in the chemistry where influence on stereoisomer outcomes is possible, such as the RO2 + RO2 cross-reaction or the 1,6 H-atom shift isomerisation reaction. Despite this, the evidence of a chiral supramolecular structure in oligomer aerosol correlating strongly with its precursors59 is supported by this review. Three mechanistic pathways of the epoxide intermediates, IEPOX, HMML and MACR, suggest a stereospecific product and could result in a chiral tacticity to the oligomer aerosol. This highlights the importance of stereochemistry in first-generation isoprene products. Therefore, this is potentially an important area of further research into atmospheric isoprene-derived SOA stereochemistry.To gain a better understanding of the stereochemical implications, studies on other supramolecular structures may be beneficial. Although it is a different system, it is worth noting that in one study a simple change in methyl group functionality caused a difference of one order of magnitude in the water-solubility equilibrium dynamics of a polymer.21 If stereoselectivity is involved in the production of atmospheric SOA from isoprene, the water-soluble properties of the SOA, which determines its formation and quantity, could theoretically be different with varying stereoselectivity. This results from the variance in the amounts of stereoisomers formed which, as shown previously, could oligomerise or form aerosol structures with different tacticity. Consequently, upscaling predictions from chamber studies may under- or over-estimate SOA production in different parts of the atmosphere if the ambient stereochemical environment is different.
It is well known that nature produces stereospecific compounds and structures, and some of the compounds which compose isoprene-derived SOA are also directly emitted from biogenic sources. The possibility of using the percentage of biogenic aerosol in ambient samples to give a stereochemical fingerprint to SOA could allow modellers to better quantify the SOA produced from different origins.12,22 However, with this method comes the assumption that all atmospheric reactions produce racemic mixtures. Mechanistic evaluation presented in this review has both supported and negated this assumption, in the latter case through the results for α-pinene from Ebben et al.59 It is clear that more experimental evidence is required in order to use this method with confidence.
Evidence of ammonium-catalysed ring opening of the epoxide intermediates suggests that chamber studies should monitor the cation and anion content more closely. The presence of ammonium cations (NH3R+) with varying substituents bonded to the nitrogen atom (R group) could theoretically cause selectivity and altered reaction rates in these reaction mechanisms. This could lead to a disordered stereochemical oligomer framework and a difference in the rate of formation of SOA.
The theoretical effects that stereochemistry may have over the rate of production of SOA from IEPOX, HMML and MAE reactions have been outlined. Past studies by Stokes et al.14 indicate that the rate of reaction can change due to heterogeneous stereochemistry in atmospheric aerosol. This direct effect on the rate of reaction has yet to be seen for the IEPOX reaction. A series of experimental suggestions have been described in order to determine if stereochemistry has an influence. Nonetheless, previous studies have shown that the reactive uptake coefficient is affected by pre-existing aerosol coatings.43,60 It is therefore suggested that if the tacticity of the aerosol changes the composition and morphology of the aerosol, the reactive uptake constant will be indirectly affected by stereochemistry.
From the evidence collected it is clear that, theoretically, SOA could display a varying tacticity due to differences in atmospheric stereochemical environments, but the influence this has on SOA production is still to be understood. If significant effects are observed, the accurate prediction of ambient SOA production from chamber studies would require stereochemistry to be considered. This review has highlighted the chemical mechanisms upon which future research efforts could focus so that the importance of aerosol stereochemistry in relation to SOA formation can be fully determined.
References
- A. Borbon, H. Fontaine, M. Veillerot, N. Locoge, J. Galloo and R. Guillermo, Atmos. Environ., 2001, 35, 3749–3760 CrossRef CAS.
- R. C. Hudman, L. T. Murray, D. J. Jacob, D. B. Millet, S. Turquety, S. Wu, D. R. Blake, A. H. Goldstein, J. Holloway and G. W. Sachse, Geophys. Res. Lett., 2008, 35 DOI:10.1029/2007gl032393.
- P. Wagner and W. Kuttler, Sci. Total Environ., 2014, 475, 104–115 CrossRef CAS PubMed.
- A. B. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl, L. K. Emmons and X. Wang, Geosci. Model Dev., 2012, 5, 1471–1492 CrossRef CAS.
- Y. Zhang, J.-P. Huang, D. K. Henze and J. H. Seinfeld, J. Geophys. Res.: Atmos., 2007, 112 DOI:10.1029/2007JD008675.
- N. Robinson, J. Hamilton, J. Allan, B. Langford, D. Oram, Q. Chen, K. Docherty, D. Farmer, J. Jimenez and M. Ward, Atmos. Chem. Phys., 2011, 11, 1039–1050 CAS.
- S. N. Pandis, S. E. Paulson, J. H. Seinfeld and R. C. Flagan, Atmos. Environ., Part A, 1991, 25, 997–1008 CrossRef.
- M. Claeys, B. Graham, G. Vas, W. Wang, R. Vermeylen, V. Pashynska, J. Cafmeyer, P. Guyon, M. O. Andreae and P. Artaxo, Science, 2004, 303, 1173–1176 CrossRef CAS PubMed.
- O. Böge, Y. Miao, A. Plewka and H. Herrmann, Atmos. Environ., 2006, 40, 2501–2509 CrossRef.
- E. O. Edney, T. E. Kleindienst, M. Jaoui, M. Lewandowski, J. H. Offenberg, W. Wang and M. Claeys, Atmos. Environ., 2005, 39, 5281–5289 CrossRef CAS.
- Q.-H. Hu, Z.-Q. Xie, X.-M. Wang, H. Kang, Q.-F. He and P. Zhang, Sci. Rep., 2013, 3, 2280 Search PubMed.
- B. Nozière, N. J. D. González, A.-K. Borg-Karlson, Y. Pei, J. P. Redeby, R. Krejci, J. Dommen, A. S. H. Prevot and T. Anthonsen, Geophys. Res. Lett., 2011, 38 DOI:10.1029/2011gl047323.
- D. P. Curran, N. A. Porter and B. Giese, Stereochemistry of radical reactions: concepts, guidelines, and synthetic applications, John Wiley & Sons, 2008 Search PubMed.
- G. Y. Stokes, E. H. Chen, A. M. Buchbinder, W. F. Paxton, A. Keeley and F. M. Geiger, J. Am. Chem. Soc., 2009, 131, 13733–13737 CrossRef CAS PubMed.
- I. S. Martinez, M. D. Peterson, C. J. Ebben, P. L. Hayes, P. Artaxo, S. T. Martin and F. M. Geiger, Phys. Chem. Chem. Phys., 2011, 13, 12114–12122 RSC.
- J. E. Penner, M. Andreae, H. Annegarn, L. Barrie, J. Feichter, D. Hegg, A. Jayaraman, R. Leaitch, D. Murphy and J. Nganga, in Climate Change 2001: The Scientific Basis. Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, 2001, pp. 289–348 Search PubMed.
- L. Chang and E. Woo, Polym. Chem., 2010, 1, 198–202 RSC.
- Y. Katsumoto, N. Kubosaki and T. Miyata, J. Phys. Chem. B, 2010, 114, 13312–13318 CrossRef CAS PubMed.
- T. Shimizu and M. Masuda, J. Am. Chem. Soc., 1997, 119, 2812–2818 CrossRef CAS.
- E. Mikhailov, S. Vlasenko, S. Martin, T. Koop and U. Pöschl, Atmos. Chem. Phys., 2009, 9, 9491–9522 CrossRef CAS.
- M. B. Baker, L. Albertazzi, I. K. Voets, C. M. Leenders, A. R. Palmans, G. M. Pavan and E. Meijer, Nat. Commun., 2015, 6 DOI:10.1038/ncomms7234.
- N. J. Gonzalez, A.-K. Borg-Karlson, P. Artaxo, A. Guenther, R. Krejci, B. Noziere and K. Noone, Environ. Sci.: Processes Impacts, 2014, 16, 1413–1421 CAS.
- M. Sprengnether, K. L. Demerjian, N. M. Donahue and J. G. Anderson, J. Geophys. Res.: Atmos., 2002, 107, ACH 8-1–ACH 8–13 CrossRef.
- J. Zhao, R. Zhang, E. C. Fortner and S. W. North, J. Am. Chem. Soc., 2004, 126, 2686–2687 CrossRef CAS PubMed.
- J. H. Kroll, N. L. Ng, S. M. Murphy, R. C. Flagan and J. H. Seinfeld, Environ. Sci. Technol., 2006, 40, 1869–1877 CrossRef CAS PubMed.
- N. L. Ng, A. J. Kwan, J. D. Surratt, A. W. H. Chan, P. S. Chhabra, A. Sorooshian, H. O. T. Pye, J. D. Crounse, P. O. Wennberg, R. C. Flagan and J. H. Seinfeld, Atmos. Chem. Phys., 2008, 8, 4117–4140 CrossRef CAS.
- J. D. Crounse, F. Paulot, H. G. Kjaergaard and P. O. Wennberg, Phys. Chem. Chem. Phys., 2011, 13, 13607–13613 RSC.
- T. Berndt, J. Atmos. Chem., 2012, 69, 253–272 CrossRef CAS.
- J. Peeters, J.-F. o. Müller, T. Stavrakou and V. S. Nguyen, J. Phys. Chem. A, 2014, 118, 8625–8643 CrossRef CAS PubMed.
- J. Peeters, T. L. Nguyen and L. Vereecken, Phys. Chem. Chem. Phys., 2009, 11, 5935–5939 RSC.
- J. A. Ippolito, R. S. Alexander and D. W. Christianson, J. Mol. Biol., 1990, 215, 457–471 CrossRef CAS PubMed.
- M. Jenkin, J. Young and A. Rickard, Atmos. Chem. Phys. Discuss., 2015, 15, 9709–9766 Search PubMed.
- A. W. Rollins, A. Kiendler-Scharr, J. Fry, T. Brauers, S. S. Brown, H.-P. Dorn, W. P. Dubé, H. Fuchs, A. Mensah and T. Mentel, Atmos. Chem. Phys., 2009, 9, 6685–6703 CrossRef CAS.
- J. D. Surratt, S. M. Murphy, J. H. Kroll, N. L. Ng, L. Hildebrandt, A. Sorooshian, R. Szmigielski, R. Vermeylen, W. Maenhaut and M. Claeys, J. Phys. Chem. A, 2006, 110, 9665–9690 CrossRef CAS PubMed.
- J. D. Surratt, A. W. Chan, N. C. Eddingsaas, M. Chan, C. L. Loza, A. J. Kwan, S. P. Hersey, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6640–6645 CrossRef CAS PubMed.
- J. E. Krechmer, M. M. Coggon, P. Massoli, T. B. Nguyen, J. D. Crounse, W. Hu, D. A. Day, G. S. Tyndall, D. K. Henze and J. C. Rivera-Rios, Environ. Sci. Technol., 2015, 49, 10330–10339 CrossRef CAS PubMed.
- M. Claeys, W. Wang, A. C. Ion, I. Kourtchev, A. Gelencsér and W. Maenhaut, Atmos. Environ., 2004, 38, 4093–4098 CrossRef CAS.
- F. Paulot, J. D. Crounse, H. G. Kjaergaard, A. Kürten, J. M. S. Clair, J. H. Seinfeld and P. O. Wennberg, Science, 2009, 325, 730–733 CrossRef CAS PubMed.
- K. H. Bates, J. D. Crounse, J. M. S. Clair, N. B. Bennett, T. B. Nguyen, J. H. Seinfeld, B. M. Stoltz and P. O. Wennberg, J. Phys. Chem. A, 2014, 118, 1237–1246 CrossRef CAS PubMed.
- Y.-H. Lin, Z. Zhang, K. S. Docherty, H. Zhang, S. H. Budisulistiorini, C. L. Rubitschun, S. L. Shaw, E. M. Knipping, E. S. Edgerton and T. E. Kleindienst, Environ. Sci. Technol., 2012, 46, 250–258 CrossRef CAS PubMed.
- Y.-H. Lin, S. H. Budisulistiorini, K. Chu, R. A. Siejack, H. Zhang, M. Riva, Z. Zhang, A. Gold, K. E. Kautzman and J. D. Surratt, Environ. Sci. Technol., 2014, 48, 12012–12021 CrossRef CAS PubMed.
- F. Lopez-Hilfiker, C. Mohr, E. L. D'Ambro, A. Lutz, T. P. Riedel, C. J. Gaston, S. Iyer, Z. Zhang, A. Gold and J. D. Surratt, Environ. Sci. Technol., 2016, 50, 2200–2209 CrossRef CAS PubMed.
- M. Riva, D. M. Bell, A.-M. K. Hansen, G. Drozd, Z. Zhang, A. Gold, D. Imre, J. D. Surratt, M. Glasius and A. Zelenyuk, Environ. Sci. Technol., 2016, 50, 5580–5588 CrossRef CAS PubMed.
- Q. Chen, D. Farmer, L. Rizzo, T. Pauliquevis, M. Kuwata, T. G. Karl, A. Guenther, J. D. Allan, H. Coe and M. Andreae, Atmos. Chem. Phys., 2015, 15, 3687–3701 CAS.
- S. H. Budisulistiorini, M. R. Canagaratna, P. L. Croteau, W. J. Marth, K. Baumann, E. S. Edgerton, S. L. Shaw, E. M. Knipping, D. R. Worsnop and J. T. Jayne, Environ. Sci. Technol., 2013, 47, 5686–5694 CrossRef CAS PubMed.
- L. Xu, H. Guo, C. M. Boyd, M. Klein, A. Bougiatioti, K. M. Cerully, J. R. Hite, G. Isaacman-VanWertz, N. M. Kreisberg and C. Knote, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 37–42 CrossRef CAS PubMed.
- L. Xu, A. M. Middlebrook, J. Liao, J. A. de Gouw, H. Guo, R. J. Weber, A. Nenes, F. D. Lopez-Hilfiker, B. H. Lee, J. A. Thornton, C. A. Brock, J. A. Neuman, J. B. Nowak, I. B. Pollack, A. Welti, M. Graus, C. Warneke and N. L. Ng, J. Geophys. Res.: Atmos., 2016 DOI:10.1002/2016jd025156.
- T. Riedel, Y.-H. Lin, Z. Zhang, K. Chu, J. Thornton, W. Vizuete, A. Gold and J. Surratt, Atmos. Chem. Phys., 2016, 16, 1245–1254 CrossRef CAS.
- Y.-H. Lin, E. Knipping, E. Edgerton, S. Shaw and J. Surratt, Atmos. Chem. Phys., 2013, 13, 8457–8470 Search PubMed.
- W. Hu, P. Campuzano-Jost, B. Palm, D. Day, A. Ortega, P. Hayes, J. Krechmer, Q. Chen, M. Kuwata and Y. Liu, Atmos. Chem. Phys., 2015, 15, 11807–11833 CAS.
- S. Budisulistiorini, X. Li, S. Bairai, J. Renfro, Y. Liu, Y. Liu, K. McKinney, S. Martin, V. McNeill and H. Pye, Atmos. Chem. Phys., 2015, 15, 8871–8888 CAS.
- J. Clayden, W. Warren, N. Greeves and P. Wothers, Organic Chemistry, Oxford University Press, 2001 Search PubMed.
- B. Nozière, P. Dziedzic and A. Córdova, Phys. Chem. Chem. Phys., 2010, 12, 3864–3872 RSC.
- T. B. Nguyen, M. M. Coggon, K. H. Bates, X. Zhang, R. H. Schwantes, K. A. Schilling, C. L. Loza, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Atmos. Chem. Phys., 2014, 14, 3497–3510 Search PubMed.
- B. Noziere, P. Dziedzic and A. Córdova, J. Phys. Chem. A, 2008, 113, 231–237 CrossRef PubMed.
- B. Nozière, P. Dziedzic and A. Córdova, Atmos. Chem. Phys. Discuss., 2009, 9, 1–21 CrossRef.
- Y. Du, J.-Q. Wang, J.-Y. Chen, F. Cai, J.-S. Tian, D.-L. Kong and L.-N. He, Tetrahedron Lett., 2006, 47, 1271–1275 CrossRef CAS.
- G. Fogassy, C. Pinel and G. Gelbard, Catal. Commun., 2009, 10, 557–560 CrossRef CAS.
- C. J. Ebben, S. R. Zorn, S.-B. Lee, P. Artaxo, S. T. Martin and F. M. Geiger, Geophys. Res. Lett., 2011, 38, L16807 CrossRef.
- C. J. Gaston, T. P. Riedel, Z. Zhang, A. Gold, J. D. Surratt and J. A. Thornton, Environ. Sci. Technol., 2014, 48, 11178–11186 CrossRef CAS PubMed.
- T. P. Riedel, Y.-H. Lin, S. H. Budisulistiorini, C. J. Gaston, J. A. Thornton, Z. Zhang, W. Vizuete, A. Gold and J. D. Surratt, Environ. Sci. Technol. Lett., 2015, 2, 38–42 CrossRef CAS.
- H. O. Pye, R. W. Pinder, I. R. Piletic, Y. Xie, S. L. Capps, Y.-H. Lin, J. D. Surratt, Z. Zhang, A. Gold and D. J. Luecken, Environ. Sci. Technol., 2013, 47, 11056–11064 CrossRef CAS PubMed.
- N. C. Eddingsaas, D. G. VanderVelde and P. O. Wennberg, J. Phys. Chem. A, 2010, 114, 8106–8113 CrossRef CAS PubMed.
- N. C. Cole-Filipiak, A. E. O'Connor and M. J. Elrod, Environ. Sci. Technol., 2010, 44, 6718–6723 CrossRef CAS PubMed.
- L. Schone, J. Schindelka, E. Szeremeta, T. Schaefer, D. Hoffmann, K. J. Rudzinski, R. Szmigielski and H. Herrmann, Phys. Chem. Chem. Phys., 2014, 16, 6257–6272 RSC.
- R. Szmigielski, J. D. Surratt, R. Vermeylen, K. Szmigielska, J. H. Kroll, N. L. Ng, S. M. Murphy, A. Sorooshian, J. H. Seinfeld and M. Claeys, J. Mass Spectrom., 2007, 42, 101–116 CrossRef CAS PubMed.
- A. Chan, M. Chan, J. D. Surratt, P. Chhabra, C. Loza, J. Crounse, L. Yee, R. Flagan, P. Wennberg and J. Seinfeld, Atmos. Chem. Phys., 2010, 10, 7169–7188 CAS.
- Y.-H. Lin, H. Zhang, H. O. T. Pye, Z. Zhang, W. J. Marth, S. Park, M. Arashiro, T. Cui, S. H. Budisulistiorini, K. G. Sexton, W. Vizuete, Y. Xie, D. J. Luecken, I. R. Piletic, E. O. Edney, L. J. Bartolotti, A. Gold and J. D. Surratt, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6718–6723 CrossRef CAS PubMed.
- H. G. Kjaergaard, H. C. Knap, K. B. Ørnsø, S. Jørgensen, J. D. Crounse, F. Paulot and P. O. Wennberg, J. Phys. Chem. A, 2012, 116, 5763–5768 CrossRef CAS PubMed.
- T. B. Nguyen, K. H. Bates, J. D. Crounse, R. H. Schwantes, X. Zhang, H. G. Kjaergaard, J. D. Surratt, P. Lin, A. Laskin and J. H. Seinfeld, Phys. Chem. Chem. Phys., 2015, 17, 17914–17926 RSC.
- Y. Liu, F. Siekmann, P. Renard, A. El Zein, G. Salque, I. El Haddad, B. Temime-Roussel, D. Voisin, R. Thissen and A. Monod, Atmos. Environ., 2012, 49, 123–129 CrossRef CAS.
- A. W. Birdsall, C. A. Zentner and M. J. Elrod, Atmos. Chem. Phys., 2013, 13, 3097–3109 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |