
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole of snow and cold environment in the fate and effects of nanoparticles and select organic pollutants from gasoline engine exhaust†
Yevgen
Nazarenko
a,
Uday
Kurien
a,
Oleg
Nepotchatykh
b,
Rodrigo B.
Rangel-Alvarado
c and
Parisa A.
Ariya
*ac
aDepartment of Atmospheric and Oceanic Sciences, McGill University, 805 Sherbrooke Street West, Montreal, QC H3A 0B9, Canada. E-mail: parisa.ariya@mcgill.ca; Fax: +1 514 398 3797; Tel: +1 514 398 6931 Tel: +1 514 398 3615
bPO-Laboratories, Inc., 609 McCaffrey Street, Saint-Laurent, QC H4T 1N3, Canada
cDepartment of Chemistry, McGill University, 801 Sherbrooke Street West, Montréal, QC H3A 2K6, Canada
First published on 21st December 2015
Abstract
Exposure to vehicle exhaust can drive up to 70 % of excess lifetime cancer incidences due to air pollution in urban environments. Little is known about how exhaust-derived particles and organic pollutants, implicated in adverse health effects, are affected by freezing ambient temperatures and the presence of snow. Airborne particles and (semi)volatile organic constituents in dilute exhaust were studied in a novel low-temperature environmental chamber system containing natural urban snow under controlled cold environmental conditions. The presence of snow altered the aerosol size distributions of dilute exhaust in the 10 nm to 10 μm range and decreased the number density of the nanoparticulate (<100 nm) fraction of exhaust aerosols, yet increased the 100–150 nm fraction. Upon 1 hour exhaust exposure, the total organic carbon increased in the natural snow from 0.218 ± 0.014 to 0.539 ± 0.009 mg L−1, and over 40 additional (semi)volatile organic compounds and a large number of exhaust-derived carbonaceous and likely organic particles were identified. The concentrations of benzene, toluene, ethylbenzene, and xylenes (BTEX) increased from near the detection limit to 52.48, 379.5, 242.7, and 238.1 μg kg−1 (± 10 %), respectively, indicating the absorption of exhaust-derived toxic organic compounds by snow. The alteration of exhaust aerosol size distributions at freezing temperatures and in the presence of snow, accompanied by changes of the organic pollutant content in snow, has potential to alter health effects of human exposure to vehicle exhaust.
Environmental impactThe study addresses the current lack of understanding of gasoline engine exhaust pollution behavior in cold environments. The study is first-of-its-kind, utilizing a novel snow chamber to decouple effects of exhaust from those of other interfering pollution sources to examine the effects of snow and freezing temperatures on the composition and fate of exhaust-derived air pollutants, exposure to which presents a health hazard. The findings are crucial for exposure assessment for exhaust-derived air pollutants in winter, which in turn is essential for the development of emissions and air quality regulations and technologies that would adequately protect public health in cold climates and wintertime. |
Introduction
The International Agency for Research on Cancer, IARC of the World Health Organization, WHO1 recently recognized inhalation exposure to air pollution as one of the leading environmental drivers of cancer deaths, in addition to cardiovascular and pulmonary diseases,2–6 estimated to be causing 1 in 8 premature deaths.6 Internal combustion engines (ICEs) are among the major sources of air pollution in urban areas,7 and their exhaust contains a variety of toxic and carcinogenic substances in gaseous and particulate (aerosol) forms.8 ICE exhaust as a contributor to hazardous air pollutants can cause up to 70 % of excess lifetime cancer incidences in urban areas.9 Moreover, in urban environments, gasoline-powered ICEs in vehicles are the biggest emitters of ozone-generating hydrocarbons and NOx,10 as well as nanoparticulate aerosols,11 which are important contributors to negative health effects.12Once released from the tailpipe as exhaust, internal combustion-derived gaseous and particulate air pollutants can spread over long distances13 or be trapped in urban canyons where air movement is hindered14 and undergo complex physicochemical transformations leading to the alteration of their toxic and carcinogenic properties.15,16 These transformations include alteration of the exhaust aerosol particle size distribution,2,15 interaction of exhaust constituents with particles and gases already present in the air,17 and with materials comprising the environment, namely, snow, soil, and vegetation. Research over the last decades suggests that snow and ice can interact with the atmosphere chemically and physically, including effects on the oxidation potential, as well as gas-to-particle partitioning.18–22 Notably, ice crystal and quasi-liquid water23 phases within snow provide a large surface area and a medium for adsorption and dissolution of pollutants and physicochemical reactions.22,24 Thereby, snowpack can serve as a reaction medium and a temporary or permanent sink for air pollutants, as well as their potential emission source.25–29
At low temperatures, condensation of gaseous water and organic substances onto particles is enhanced, while small particle motion leading to coagulation is reduced.30 Nucleation of aerosol nanoparticles from gaseous exhaust constituents is enhanced at lower temperatures.31 Additionally, exhaust passes through a cooler exhaust system and exits into a colder environment in close proximity to snow, if present. This results in thermophoresis, leading to increased aerosol removal by cold surfaces,32 such as cold roads or snow. Yet, the combined effects of a cold environment and the presence of snow on particulate and gaseous air pollution exposure from ICE are unknown, partly because observations of these effects in field studies are obscured by unevenness of exhaust dispersion due to the variability of wind direction and speed, which is also higher in wintertime,33 and other pollution sources, including vehicle and tire wear and tear.34 This challenge pointed to the need for an investigation that would examine the behavior of dilute exhaust at cold temperatures and in the presence of snow in a closed system, free from interference caused by other (non-exhaust-derived) air pollution components and where exhaust dilution and flow could be controlled. The data and findings were intended to be complementary to the results of prior field studies34–39 and provide evidence for the necessity to focus exposure and epidemiological research on the wintertime period of the year to further understand how freezing temperatures and the presence of snow influence health effects of exhaust-derived air pollution.
Methodology
Experimental setup
The experimental system was designed to mimic the real world conditions of exhaust release from a vehicle's tailpipe close to the ground surface. The system allows for dilution of released exhaust by cold air and simulates dispersion of exhaust constituents, including contact of dilute exhaust with the snowpack, as it happens on and near roads. The system presents a controlled enclosed environment, free from interference of unstable environmental factors, such as the presence of multiple pollution sources and instability of wind speed and direction, which are a challenge in field studies when trying to attribute observed concentrations of pollutants to specific sources and pollution-influencing factors.The experimental setup consisted of several modules: the dilution air control and conditioning module, the exhaust generation and diversion module, the environmental chamber with its cooling apparatus, the outflow heating module, and the sampling train. The schematic diagram of the setup is shown in Fig. 1. In the dilution air module, the dilution air was filtered with a high efficiency particulate air (HEPA) filter and scrubbed with activated carbon. For the cold temperature experiments only, the dilution air was also cooled using a heat exchanger cooling coil. To ensure the quality of measurements and ensure the absence of contamination of the airflow, multiple control experiments were performed, including clean air tests before each experimental run. For different experiments, the conditioned dilution air at room (warm) or cold temperatures, at a flow rate, adjusted around 4 SL min−1, was passed into a custom-made 12 L volume, borosilicate glass spherical snow exposure chamber, which, during cold-temperature experiments, was submerged in a standard cooling bath, described in the ESI.† Note that SL stands for standard liters at 273.15 K, 100 kPa. For the experiments with exhaust, the dilution air was combined with the exhaust stream, leading to dilution and cooling of the exhaust, similar to what happens when exhaust leaves a vehicle's tailpipe in the real world. The glass chamber was equipped with inflow and outflow glass ports, positioned one quarter of the diameter of the chamber from its bottom and top, respectively. The air or dilute exhaust entered via the top port and exited the chamber via the bottom port. It then passed through a 30 cm long 1/3′′ (1′′ = 2.54 cm) diameter stainless steel tube, wrapped in a thermo-controlled silicone heating tape that warmed the flow, if it was cold, to 23 ± 0.6 °C to match inflow temperature requirements of the sampling instruments. For additional details and operating parameters of the modules, see ESI.†
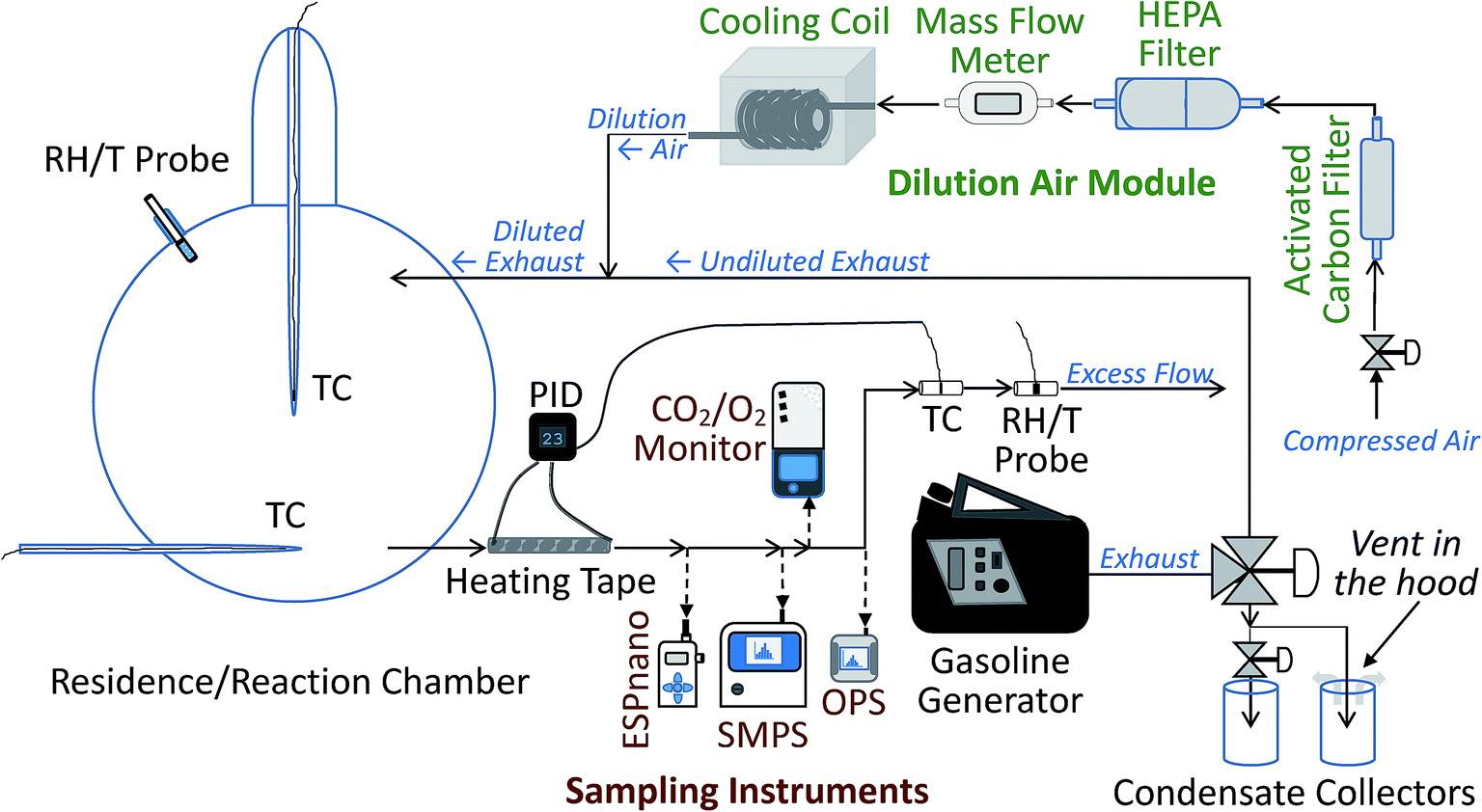 | ||
| Fig. 1 Simplified schematics of the environmental chamber setup. “RH/T Probe” stands for “relative humidity and temperature probe”. “TC” stands for “thermocouple”. “PID” stands for “proportional-integral-derivative controller”. “ESPnano” is the electrostatic precipitator. “SMPS” stands for “scanning mobility particle sizer”. “OPS” stands for “optical particle sizer”. “HEPA filter” stands for “high efficiency particulate air filter”. | ||
Generation of exhaust stream
To generate exhaust, we employed an ordinary portable gasoline generator rated at 450 W of output electrical power, model 500Wi manufactured by Powerhouse (Wilsonville, OR, USA). For full specifications of the generator, see ESI.† Up to three octane numbers of gasoline (87, 89 and 91) were compared in the experiments. The octane number in this article is expressed as anti-knock index (AKI), which is used in certain countries of North America, including the US and Canada. We diverted a portion of the total exhaust from the generator using a system of valves and tubes/hoses. The exhaust was then diluted with clean dry air at a ratio of approximately 6–15 times, depending on the experiment. This dilution resulted in mixing ratios of O2 in the dilute exhaust of approximately 18–19.5 % and ∼1–2 % of carbon dioxide (CO2). The method for calculating dilution ratios is standard and provided in the ESI.† The mixing ratios of oxygen (O2) and CO2 in the dilute exhaust were measured by using a CO2/O2 analyzer, model 902P (Quantek Instruments, Inc., Grafton, MA, USA), equipped with a data logger, model HOBO U12 (Onset Computer Corp., Cape Cod, MA, USA). The sampling flow rate of the CO2/O2 analyzer was 0.3 L min−1.Snow collection and storage
The snow collection and storage procedures have been previously described40–43 and are briefly outlined in the ESI.† The snow used in this work was collected using sterile equipment in an urban location (Montréal, Québec, Canada), in a wooded area of a public park (Park Mont-Royal (45°31′2′′ N 73°35′16′′ W)) during a light snowfall, immediately following a heavy night snowfall. The collection location receives fresh snow precipitation during a period of approximately 5 months per year. The collection occurred between 1:00 to 3:00 p.m. on 30 March 2014, and this snow was used for all the experiments reported herein. The weather conditions were: temperature −1 to 0 °C, light snow, wind NNE ∼43 km h−1, UV index 2 (low), relative humidity (RH) 87 %, pressure 1014.4 millibars, and visibility of ∼4 km. The collection site was not subject to human or pet/animal traffic.Aerosol size distribution analysis
To analyze aerosol size distributions, we used a NanoScan™ scanning mobility particle sizer (SMPS), model 3910 (TSI, Inc., Shoreview, MN, USA) and an optical particle sizer (OPS), model 3330 (TSI, Inc.), described in the ESI.† The sampling flow rate of the NanoScan™ SMPS was 0.75 L min−1; ± 20 % and that of the OPS was 1.0 L min−1; ± 5 %. The synergic operation of these two instruments provided continuous 10 nm to 10 μm aerosol size distributions every 1 min during the course of each operation cycle of the chamber system: (1) clean air passing through the empty chamber at warm temperature; (2) dilute exhaust passing through the chamber at warm temperature, (3) cold clean air passing through the cooled system, (4) cold dilute exhaust passing through the empty chamber, followed by chamber purging with cold clean air, (5) cold clean air passing over snow, and (6) cold dilute exhaust passing over snow.For the warm temperature 23 ± 0.6 °C and cold temperature −11 ± 0.6 to −7 ± 0.6 °C regimes with dilute exhaust, we measured number density-based aerosol size distributions upstream before the chamber inlet, as well as downstream, after the chamber outlet. Note that the number density of aerosol is also known as aerosol number concentration, and we use this term below. We note that the aerosol stream entering the sampling and measurement instruments was heated to 23 ± 0.6 °C in the heated line, controlled using a proportional-integral-derivative (PID) controller described in detail in the ESI,† and the instruments themselves operated at the matching ambient temperature in the laboratory. For each experimental situation, we collected and averaged at least three size distribution samples. The size distributions are reported as dN/d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logDp, cm−3, explained in the ESI.†
logDp, cm−3, explained in the ESI.†
Physical and chemical characterization of aerosol particles and snow samples
When collecting snow samples from the experimental chamber, we used the grid-shaped side of vertically held sterile HDPE sampling spatulas (Bel-Art Products, Inc., Wayne, NJ, USA). The samples were placed into sterile Falcon™ 50 mL Conical Centrifuge Tubes (Corning Life Sciences, Inc., Tewksbury MA, USA), which were frozen at −39 °C immediately after collection and weighing.For analyses by ion chromatography (IC) and for total organic carbon (TOC), the original unexposed snow from the environmental sample container (the blank) and exposed snow were melted at room temperature inside the original containers. The analysis procedures for melted snow by IC and TOC were performed using standard automated techniques outlined in the ESI.† For high-resolution transmission electron microscopy (HR-TEM), energy-dispersive spectroscopy (EDS) and selected area diffraction (SAED) of dried melted snow, we melted and placed on substrate grids small drops (∼20 μL) of snow, exposed to (1) only a cold clean airflow and (2) exhaust in the chamber. The grids used were mechanically strong FCF200-Cu-EB grids (Formvar Carbon Film on 200 Mesh Copper Extra Thick Option B grids, 25–50 nm Formvar & 3–4 nm Carbon), manufactured by SPI Supplies/Structure Probe, Inc., West Chester, PA, USA. The grids were then air-dried inside a biosafety cabinet (NuAire Inc., Plymouth, MN, USA) that provides a particle-free atmosphere. To collect airborne particles on the substrate grids for the HR-TEM, EDS and SAED from the chamber system outflow, we used an electrostatic precipitator, ESPnano Model 100 (ESPnano, Spokane, WA, USA), operating at a factory-set sampling flow rate of 0.1 L min−1. This instrument is described further in the ESI.† The same types of grids as used for melted snow analyses were utilized. The HR-TEM and EDS methodologies are described in the ESI.†
For gas chromatography with mass spectrometry detection (GC-MS), 10 ± 0.1 g of a snow sample, before and after exhaust exposure, were placed in a standard 20 mL headspace vial and closed with a PTFE butyl rubber septum and a crimp-closure. Subsequently, the sample temperature was raised to 23 °C, and a solid-phase microextraction (SPME) fiber was inserted into the liquid layer and kept there for 30 min while the vial was gently shaken in a lab rocker. Next, the SPME fiber was removed from the vial and injected into the GC-MS unit for analysis, which is described in detail in the ESI.†
Results and discussion
System characterization
The low-temperature experimental chamber system was designed and built specially for this study. It was therefore necessary to characterize the micro-environmental parameters inside it, as well as their dynamics for experimental timescales, including temperature and RH at different sites within the system. The system, equipped with cooling baths, successfully maintained snow in a frozen condition for the entire duration of each snow-exposure experiment. Specifically, the temperature at the top of the snowpack was close to −20.0 ± 0.6 °C after loading of the snow, and then slowly increased to around −1.0 ± 0.6 °C over the course of about 5 h. The length of time the system was able to maintain frozen snow was amply sufficient as each experimental routine was completed within 3 h. Temperature dynamics in the system during a typical experiment is shown in Fig. S1.† The inside of the system was completely isolated from the outside environment. This design provided a key advantage of being able to observe the effects of cold temperature and snowpack on the exhaust-derived aerosols without intrusion of natural environmental factors that interfere with field studies, including pollution from other sources and the variability of wind speed and direction44,45 that affect the stability of dilution and dispersion of pollutants. Moreover, using the enclosed design of the system allowed us to decouple the observed effects of exhaust interaction with snow from the second largest source of traffic-related particulate pollution: vehicle-derived aerosols, including those from wear and tear of the vehicle body, brakes and tires,34 which is important to obtain information specifically needed to decide on mitigation and regulation of internal combustion emissions.Aerosol analysis
Fig. 2 shows the aerosol size distributions of dilute exhaust. They are presented separately for warm temperature and cold temperature, as measured before and after the chamber. Also presented are the distributions of dilute exhaust passed over snow in the chamber. The NanoScan™ distributions are presented on a linear scale while the OPS distributions are on a logarithmic scale to better show the large drop of aerosol number concentrations with increasing particle size.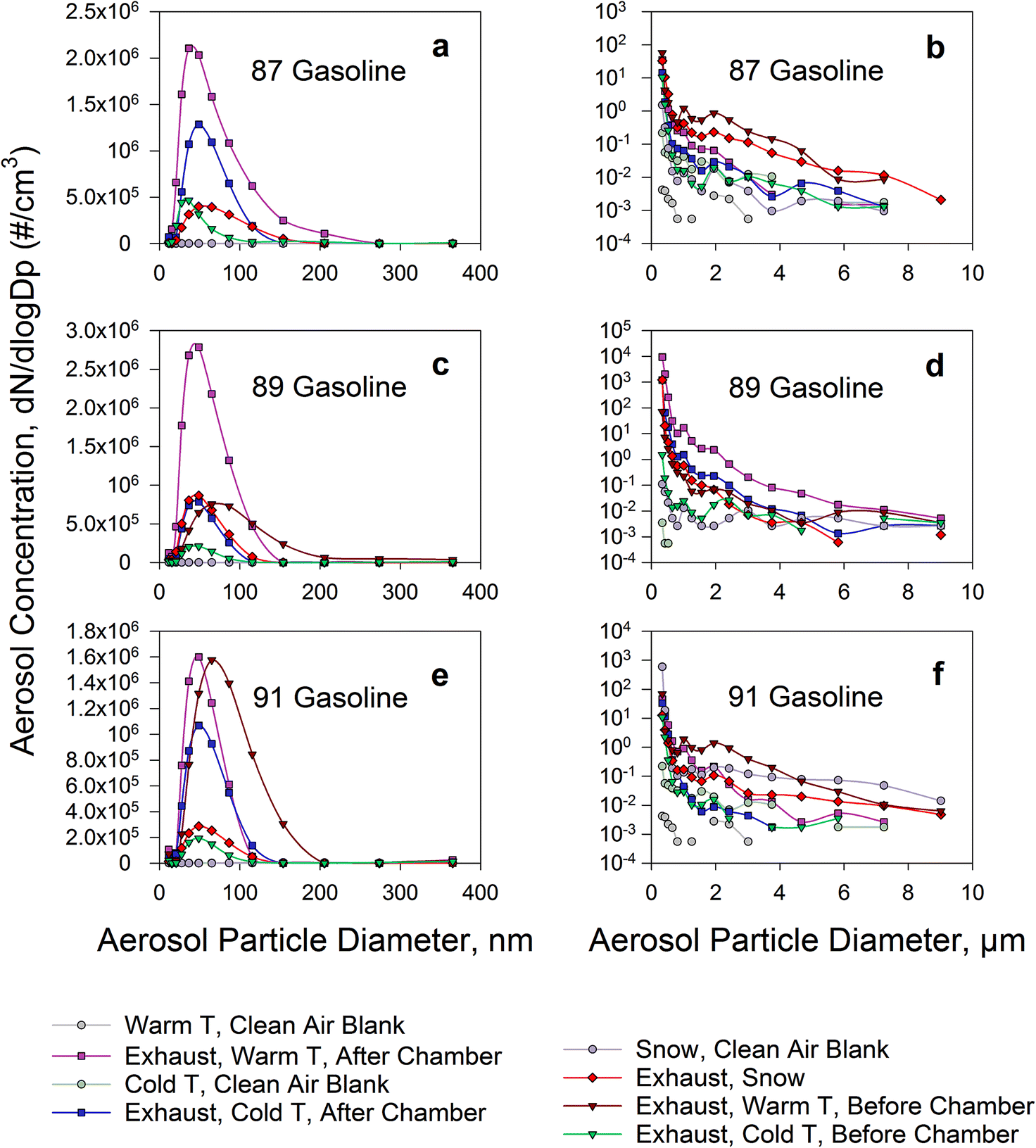 | ||
| Fig. 2 NanoScan™ (a, c, and e) and OPS (b, d, and f) aerosol size distributions. “87/89/91 Gasoline” refers to the octane number of gasoline used. The octane number is expressed as anti-knock index (AKI). “Warm/Cold T, clean Air Blank” refers to measurements at warm and cold temperature, performed with clean air only passed through the chamber. “Exhaust, Warm/Cold T, Before/After Chamber” refers to measurements at warm and cold temperature, performed with dilute exhaust before it entered the chamber and after it exited the chamber. “Warm” and “Cold” temperatures are defined in the methods. | ||
The mode diameters of the NanoScan™ aerosol size distributions for the three octane numbers of gasoline were ∼50 nm when passed through the chamber at both warm and cold temperature and in the presence of snow. This mode is generally consistent with measurements in previous studies,46–48 validating our system and measurement approach. The mode diameter was larger for the original dilute exhaust at warm temperature before it entered the chamber (60–90 nm). There seemed to be a tendency towards decreasing mode particle diameter with increasing gasoline octane number.
The evolution of the aerosol size distribution of dilute exhaust, as it passed through the chamber, indicated a marked alteration. Before the chamber, the mode was smaller for the exhaust diluted with cold (∼40 nm) compared to warm air (∼70 nm), but evolved to around 50 nm, for both cold and warm, after passing through the chamber. The observed differences in aerosol size distributions of dilute exhaust before and after the chamber support the important role aerosol aging plays in evolution of exhaust-derived air pollution. It is likely due to different mechanisms that the particle size increases at cold and decreases at warm temperatures, as dilute exhaust passes through the chamber, both leading to the same modal aerosol particle size of ∼50 nm at the exit from the chamber. At warm temperatures, particle evaporation49 is likely implicated, possibly dominating over coagulation of particles, which is more intense at higher temperatures.30 At cold temperatures, particles grow by condensation30 of volatile organic compounds and water, present in exhaust, on them while thermophoresis leads to a greater loss of smaller particles to cold surfaces.32 Despite the mode particle size being similar, these different mechanisms involved in aerosol dynamics likely lead to different chemical compositions of aerosol particles, including different contents of organic compounds.50 This is also supported by our observation of different carbon and oxygen abundance in particles collected from cold dilute exhaust vs. exhaust passed over snow (EDS data in Fig. S2f and h†). In turn, despite the similar size of particles, the different aerosol compositions may result in modifications of health effects due to differences in the quantities and bioavailability of toxic and carcinogenic compounds delivered into the respiratory system. The further fate of the particles in the atmosphere is also expected to depend on several factors including aerosol chemical compositions.51,52
The ∼50 nm mode diameter was also consistent between the experiments at cold temperature and in the presence of snow, yet the NanoScan™ distributions were stretched into larger sizes (100–150 nm) when snow was present. The OPS data showed a rapid decrease in aerosol number concentration with increasing particle size: from 10–80 cm−3 at 0.3 μm to 0.01–1 cm−3 at 1 μm, yet very low aerosol number concentrations were still observed at larger micron particle sizes (≥6–7 μm). At the cold temperature, the number concentration of particles larger than 0.3 μm dropped with increasing aerosol particle size at a greater rate than at the warm temperature. Thus, PM2.5 (mass-based regulatory metric) would be smaller at cold temperature. It was especially pronounced for 89 and 91 grade gasoline. Specifically, aerosol number concentrations for the 0.3 μm to 1 μm particle size range dropped at warm temperature from 35–57 down to 0.2–1.2 cm−3 (89 gasoline) and 45–69 down to 0.9–1.9 cm−3 (91 gasoline), and at cold temperature from 10–14 down to 0.02–0.06 cm−3 (89 gasoline) and 10–34 down to 0.03–0.05 cm−3 (91 gasoline), respectively. The number concentration of 1 μm particles decreased by approximately an order of magnitude at cold compared to warm temperature.
When fresh snow was present, there was less ultrafine aerosol: 2 × 106 cm−3 (snow) vs. 4 × 106 cm−3 (no snow); ultrafine defined as <115 nm, based on the available channels of NanoScan™, averaged for experiments with all three types of gasoline investigated. This observation suggests that under our experimental conditions, snow acted as a sink for the exhaust-derived ultrafine particles. In field studies, select anthropogenic particles, including sub-100 nm particles,38 were shown to accumulate in the roadside snowpack in association with other anthropogenic pollutants, such as heavy metals36,38 and organic compounds.37,53,54 However, further long-term research is required to evaluate the extent of pollutant accumulation and release, and the dynamics of such processes under different environmental conditions in polluted regions.
Snow analysis
The results from IC and TOC analyses are presented in Table 1. Halogen activations from salts are implicated in the formation of reactive radicals and photolabile molecules, which in turn leads to chemical transformations of (toxic) organic compounds.20,55,56 IC showed relatively low concentrations of salts in the snow collected at our sampling site, notably with chloride below the limit of detection. However, closer to roadways and sidewalks, where salt application is widespread, we expect higher salt concentrations accumulating in snow.35 Further, IC analysis did not show detectable alteration in the measured ion content of snow after exposure to dilute exhaust, within experimental uncertainties. Our results support the suggestion of Helmreich and colleagues36 that the likely source of certain ions in snow is not exhaust, but wear and tear of vehicles and road equipment, and application of de-icing agents and gravel to roads.35,36,38| Unexposed snow | Exposed snow | ||
|---|---|---|---|
| TOC, mg L−1 | 0.218 ± 0.028 | 0.539 ± 0.018 | |
| Organic pollutants, μg kg−1 | Benzene | 0.031 ± 3.1 × 10−3 | 52.48 ± 5.3 |
| Toluene | 0.174 ± 1.7 × 10−2 | 379.5 ± 38.0 | |
| Ethylbenzene | <0.01 | 242.7 ± 24.3 | |
| Xylenes | <0.01 | 238.1 ± 23.8 | |
| Ions, ppm | Sulfate | 0.11 ± 0.02 | 0.12 ± 0.02 |
| Phosphate | <0.1 | <0.1 | |
| Chloride | <0.1 | <0.1 | |
| Nitrate | <0.1 | <0.1 | |
| Sodium | 1.9 ± 0.60 | 2.1 ± 0.22 | |
| Potassium | <0.1 | <0.1 | |
| Magnesium | <0.1 | <0.1 | |
| Calcium | 0.84 ± 0.18 | 0.87 ± 0.06 | |
| Ammonium | <0.1 | <0.1 |
The TOC concentration in snow increased from 0.218 ± 0.014 to 0.539 ± 0.009 mg L−1 after a 1 hour exposure of snow to dilute exhaust (Table 1). The substantial increase of TOC in snow, exposed to exhaust, may be due to the deposition of a combination of both particulate and gaseous organic compounds into the snow, with uptake by ice crystal surfaces and the quasi-liquid water phase (present in snow above −20 °C).23 To confirm this supposition, we analyzed the melted original and the exhaust-exposed snow by TEM and EDS for particulate matter, as well as by SPME-GC-MS for organic compounds.
TEM and EDS analyses of the samples of dried melted snow, not exposed to exhaust, revealed particles containing such elements as carbon, oxygen, sulfur, silicon, iron, aluminum, calcium, titanium, sodium, potassium, and chlorine. Note that the two peaks for copper at ∼8 and 8.9 keV are attributed to the copper in the sample substrate (TEM-grid). Judging by the EDS spectra (Fig. 3 and S2†), particle compositions likely included salts, such as sodium and potassium sulfates (confirmed by IC analysis, described above), mineral particles, such as calcium carbonate, iron oxides, clay, silica, titanium-containing minerals, and more complex minerals containing these elements, as well as some carbonaceous and organic particles.
 | ||
| Fig. 3 Representative TEM micrographs and EDS spectra identifying the chemical elements in the particles: original snow (a and b), exhaust-exposed snow (c and d), aerosol particles captured from exhaust passed through an empty cold chamber (e and f), and from cold exhaust (gasoline with octane number 87 burned) passed over snow in the chamber (g and h). | ||
In the dried melted snow, exposed to exhaust, some particles expectedly exhibited similar elemental analysis to the original snow, since some particles had already been in the snow before exhaust exposure. The majority of identified particles were high-carbon-containing and high carbon-oxygen-containing particles that were similar to the exhaust particles we collected from the dilute exhaust by electrostatic precipitation (Fig. 3e–h, S2e–h and S4†) and to those previously observed.57
The presence of distinct crystalline structures within the amorphous phase in the exhaust particles was also observed, as illustrated in Fig. S3c.† TEM, EDS and SAED analyses of such particles from exhaust passed over snow confirm the presence of condensed particulate matter in the exhaust. From the EDS spectrum shown in Fig. S3d,† we conclude that these particles likely contain carbonaceous matter, which suggests that the crystalline phase may have condensed in the engine. Note that structures resembling bundles of nanotubes were also observed (Fig. S3b†), which merits further physicochemical research.
Exposure of snow to exhaust substantially increases carbonaceous and likely organic particles in the snow. The approximate doubling of TOC in the snow after 1 hour exposure to exhaust can be partially due to absorption of exhaust-derived particulate matter containing organic compounds. GC-MS further confirmed the transfer of organic pollutants from exhaust to snow that the TOC analysis initially showed. We observed a greater number of peaks and a higher baseline in the chromatogram of the exposed snow compared to the original snow. Specifically, the GC-MS analysis of the original unexposed snow showed a number of common biogenic58,59 (esters, lipids and fatty acids) and anthropogenic58,59 (long-chain hydrocarbons, thiazoles and other sulfur containing species) compounds (Fig. S5 and Table S2†). The species were diverse and ranged from a number of common plant-derived compounds,58,59 as well as several industrial chemicals,58,59 including a phthalate plasticizer and a pesticide. We cannot overrule, under experimental conditions, whether the identified plasticizer originated from the sampling equipment. In concurrence with previous studies,42 oxygenated species, such as ketones, aldehydes and esters, as well as saturated aliphatic compounds and a number of homo and heterocyclic aromatics were identified.
A substantial number (42) of additional compounds were identified in snow exposed to exhaust (Fig. S5 and Table S4†). Approximately half of these additional compounds (21) in exhaust-exposed snow were previously identified as components of engine exhaust in the United States Environmental Protection Agency's (U.S. EPA's) Master List of Compounds Emitted by Mobile Sources,8 which indicates that they were deposited into the snow from exhaust. The additional compounds identified in the exposed snow were dominated by long-chain alkanes, aromatic, polycyclic aromatic hydrocarbons, cyclic (including heterocyclic), as well as some esters, ketones, aldehydes, carboxylic acids, and phthalates.
We quantified benzene, toluene, ethylbenzene, and xylenes (BTEX), typical toxic exhaust constituents, before and after exhaust exposure (Table 1, Fig. S6†). While above-detection-limit amounts of benzene (0.031 μg kg−1) and toluene (0.174 μg kg−1) were found in unexposed snow, ethylbenzene and xylenes were undetectable (<0.01 ppb (μg kg−1)). After a 1 h exhaust exposure, BTEX concentrations increased dramatically and ranged between ∼50 and ∼380 μg kg−1.
Our results confirm that organic exhaust constituents are absorbed into the snowpack, consistent with several previous field studies,35–39 which discovered roadside snowpack to contain certain organic compounds found in vehicle exhaust.
Implications for emission regulations and air pollution research
Discovering the fate and effects of exhaust-derived air pollutants at cold temperatures and their interactions with snow is crucial for understanding the health and environmental impacts of exhaust in wintertime moderate and mountainous climates, polar and subpolar regions. The current vehicle emission regulations and air pollution standards are based on warm temperature research that does not consider the effects of subzero temperatures and snow on air pollution. The lack of adequate understanding of the environmental fate and effects of exhaust in winter leads to northern regions of the world missing evidence-based emission control measures and air pollution standards that would effectively protect human health during the cold months. Research of the fate and effects of air pollutants in cold environments will serve policy-makers to develop better air pollution regulations to protect public health. It will also contribute to climate research and modeling by providing information on the subzero temperature behavior and composition of exhaust-derived aerosols and gases involved in cloud interactions.We presented the novel results of the considerable impact cold temperatures and snow have on the aerosol size distribution of simulated exhaust pollution that is a public health hazard. We showed that snow provides a sink for exhaust-derived nanosized aerosols and certain organic constituents of exhaust, notably the BTEX. The effect on the exhaust aerosol size distribution is very important due to great differences in deposition fractions of different particle sizes in the different regions of the respiratory system30 and the key role of aerosols in the chemical transformations of gaseous air pollutants,60 with which they are in dynamic interaction.2 In the context of the growing evidence of the airborne nanoparticles' substantial health impacts,12,61 it is important to consider the significant decrease of the nanoparticle fraction of the exhaust aerosol emissions through interaction with snow. In view of these results, further development of nanoparticle number concentration emission limits within the European Union vehicle emissions standards and the corresponding potential for such regulations in North America and elsewhere in the world should take into consideration the effects of low temperature and snowpack in wintertime. Importantly, considering that ambient concentrations of nanoparticles, but not larger particles tend to be higher in wintertime, compared to summer,33 the contribution of other natural and anthropogenic sources of ambient nanoparticles and interaction of exhaust emissions with them should be investigated. Epidemiological studies should attempt to decouple health effect contributions of exhaust-derived emissions and products of their transformations and health effect contributions of similarly sized particulate air pollutants from other sources.
Since vehicle exhaust is such an important contributor to toxic and carcinogenic aerosols and gaseous constituents in urban environments,52,62–64 further research on the specific mechanisms of snow-aerosol physicochemical interactions at subzero temperatures is needed to inform exposure modelers, health scientists and, ultimately, regulators to adequately protect public health.
In summary, the results of the study point to the need for: (1) vehicle manufacturers to study and develop strategies for better operation of engine and exhaust aftertreatment technologies at freezing temperatures with a specific focus on the minimization of gaseous and particulate pollution from aged exhaust and its components in snow; modification of operating regimes depending on temperature may be considered, (2) additional field studies and systematic monitoring of air pollution in connection with traffic, temperature and snow cover, combined with epidemiological studies, to gain additional insights into the fate and the environmental and health effects of exhaust-derived air pollution in wintertime, (3) systematic monitoring of exhaust-derived pollutants and products of their chemical transformations in the runoff from melting snow, done separately for snow alongside major vs. minor roadways and away from roads, (4) search for correlations of pollutants' concentrations in snow with traffic, snowpack age, use of road surface treatment, and weather.
Conclusions
This study provides evidence that the presence of snow and freezing temperatures alters physical characteristics of particles, especially nano and fine particles, emitted in gasoline engine exhaust. The findings suggest that the snowpack is a sink for exhaust-derived nanoparticles, but its presence increases the smallest fine particles (100–150 nm). This is of high importance due to the known substantial dependence of health effects associated with inhalation exposure of airborne particulate matter on particle size. Notably, the present study uniquely demonstrates the cold temperature and snowpack-caused alteration of exhaust-derived aerosol in a completely enclosed controlled environment of the snow chamber, where interferences from various natural environmental factors are eliminated. Therefore, attributing observed changes in aerosol size distributions to cold temperature and the snowpack is not obscured by other sources of pollution, changes of wind speed and direction, and other factors that greatly influence particulate air pollution in the natural environment. Complementary to the limited number of previous studies that looked at deposition of traffic-related pollutants into the natural roadside snowpack,35,37,39 we also demonstrate that the chemical composition of snow is considerably changed upon exhaust exposure. Snowmelt in an urban environment leads to a rapid release of accumulated or transformed exhaust-derived organic pollutants into both runoff water53,65 and air.66 Our findings demonstrate the important role of exhaust as a source of pollutants in urban snow, decoupled from other sources of urban pollution. Therefore, the accumulation and transfer of pollutants from exhaust – to snow – to meltwater need to be considered by regulators and policy makers as an important area of focus for mitigation with the aim to protect public health and the environment. In summary, these findings demonstrate that the interaction of gasoline internal combustion exhaust with snow and the effect of cold temperature have a potential to influence human health and environmental effects associated with exposure to exhaust-derived air pollution. Further studies in this domain are required to provide quantitative data suitable for use in modeling studies.Conflict of interest
The authors declare no conflict of interest.Acknowledgements
We would like to thank Dr Chitra Narayanan for her assistance in performing the experiments, Mr Georges Kopp for making the glass chamber, the probe wells and in-flow probe fittings, Mr Jean-Philippe Guay for helping to build the engine exhaust distribution system, Mr Richard Rossi for tuning the proportional-integral-derivative controller, Ms Katherine Velghe for conducting the TOC analysis, Ms Monique Riendeau for conducting ion chromatography analysis, Mr Alexander Wahba for help with the analysis of the GC-MS data, and Mr Jim (Avik) Ghoshdastidar for help in proofreading the manuscript. We would like to express special thanks to TSI, Inc. and personally to Ms Sherrie Elzie and Ms Sarah Sakamoto for providing the NanoScan™ and the OPS instruments. We also kindly thank the anonymous reviewers for their valuable feedback that helped improve the final manuscript. The study is jointly funded by the Natural Science and Engineering Research Council of Canada, Canada Foundation for Innovation, and Fonds de recherche du Québec – Nature et technologies. Dr Yevgen Nazarenko is supported by Fonds de recherche du Québec – Nature et technologies. The views expressed in the manuscript are solely of the authors and do not necessarily reflect those of the funding agencies.References
- IARC (International Agency for Research on Cancer), IARC Scientific Publication No. 161, 2013, 1–169.
- R. Hetland, F. Cassee, M. Refsnes, P. Schwarze, M. Låg, A. Boere and E. Dybing, Toxicol. in Vitro, 2004, 18, 203–212 CrossRef CAS PubMed.
- M. Kampa and E. Castanas, Environ. Pollut., 2008, 151, 362–367 CrossRef CAS PubMed.
- H. A. Scott, L. N. Michelle, S. A. Umme, R. Neeraj, U. Bruce, S. S. Frances, C. Chung-Wai, J. E. Greg and A. Jeremy, Inhalation Toxicol., 2012, 24, 161–171 CrossRef PubMed.
- WHO (World Health Organication), Outdoor air pollution a leading environmental cause of cancer deaths, http://www.euro.who.int/en/health-topics/environment-and-health/urban-health/news/news/2013/10/outdoor-air-pollution-a-leading-environmental-cause-of-cancer-deaths, accessed September 28, 2014.
- WHO (World Health Organication), 7 million premature deaths annually linked to air pollution, http://www.who.int/mediacentre/news/releases/2014/air-pollution/en/, (accessed September 28, 2014).
- B. R. Gurjar, A. Nagpure, T. Singh and H. Hanson, Air Quality in Megacities, http://www.eoearth.org/view/article/149934/, accessed October 14, 2014 Search PubMed.
- U.S. Environmental Protection Agency, The Master List of Compounds Emitted by Mobile Sources, Report EPA420-b-06–002, National Service Center for Environmental Publications (NSCEP), Research Triangle Park, NC, 2006 Search PubMed.
- R. Morello-Frosch, M. Pastor and J. Sadd, Urban Aff. Rev., 2001, 36, 551–578 CrossRef.
- C.-C. Chang, J.-G. Lo and J.-L. Wang, Atmos. Environ., 2001, 35, 6201–6211 CrossRef CAS.
- H. Tsang, R. Kwok and A. H. Miguel, Aerosol Air Qual. Res., 2008, 8, 19–27 Search PubMed.
- K. Slezakova, S. Morais and M. do Carmo Pereira, in Current Topics in Public Health Atmospheric Nanoparticles and Their Impacts on Public Health, ed. A. J. Rodriguez-Morales, InTech, Rijeka, Croatia, 2013, p. 742, DOI:10.5772/54775.
- M. De Sario, K. Katsouyanni and P. Michelozzi, Eur. Respir. J., 2013, 42, 826–843 CrossRef CAS PubMed.
- K. Nazridoust and G. Ahmadi, J. Wind Eng. Ind. Aerod., 2006, 94, 491–522 CrossRef.
- J. S. Lighty, J. M. Veranth and A. F. Sarofim, J. Air Waste Manage. Assoc., 2000, 50, 1565–1618 CAS.
- J. S. Gaffney and N. A. Marley, Sci. World J., 2003, 3, 199–234 CrossRef PubMed.
- B. J. Finlayson-Pitts and J. N. Pitts Jr, Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Academic press, 1999 Search PubMed.
- V. F. McNeill, A. M. Grannas, J. P. D. Abbatt, M. Ammann, P. Ariya, T. Bartels-Rausch, F. Domine, D. J. Donaldson, M. I. Guzman, D. Heger, T. F. Kahan, P. Klán, S. Masclin, C. Toubin and D. Voisin, Atmos. Chem. Phys., 2012, 12, 9653–9678 CrossRef CAS.
- A. M. Grannas, A. R. Bausch and K. M. Mahanna, J. Phys. Chem. A, 2007, 111, 11043–11049 CrossRef CAS PubMed.
- P. A. Ariya, H. Niki, G. W. Harris, K. G. Anlauf and D. E. Worthy, Atmos. Environ., 1999, 33, 931–938 CrossRef CAS.
- P. Ariya, J. Hopper and G. Harris, J. Atmos. Chem., 1999, 34, 55–64 CrossRef CAS.
- P. A. Ariya, F. Domine, G. Kos, M. Amyot, V. Côté, H. Vali, T. Lauzier, W. F. Kuhs, K. Techmer, T. Heinrichs and R. Mortazavi, Environ. Chem., 2011, 8, 62–73 CrossRef CAS.
- A. M. Grannas, in Encyclopedia of Snow, Ice and Glaciers, Springer, 2014, pp. 138–139 Search PubMed.
- F. Dominé and P. B. Shepson, Science, 2002, 297, 1506–1510 CrossRef PubMed.
- M. Błaś, K. Cichała-Kamrowska, M. Sobik, Ż. Polkowska and J. Namieśnik, Environ. Rev., 2010, 18, 87–114 CrossRef.
- F. Cereceda-Balic, M. R. Palomo-Marín, E. Bernalte, V. Vidal, J. Christie, X. Fadic, J. L. Guevara, C. Miro and E. Pinilla Gil, Atmos. Environ., 2012, 47, 51–57 CrossRef CAS.
- A. Elik, Int. J. Environ. Anal. Chem., 2002, 82, 37 CrossRef CAS.
- K. Lee, S. D. Hur, S. Hou, S. Hong, X. Qin, J. Ren, Y. Liu, K. J. R. Rosman, C. Barbante and C. F. Boutron, Sci. Total Environ., 2008, 404, 171–181 CrossRef CAS PubMed.
- C. M. Roth, K.-U. Goss and R. P. Schwarzenbach, Environ. Sci. Technol., 2004, 38, 4078–4084 CrossRef CAS PubMed.
- W. C. Hinds, Aerosol Technology: Properties, Behavior, and Measurement of Airborne Particles, John Wiley & Sons, Inc., New York, NY, 2nd edn, 1999 Search PubMed.
- J. P. Shi and R. M. Harrison, Environ. Sci. Technol., 1999, 33, 3730–3736 CrossRef CAS.
- L. Isella, B. Giechaskiel and Y. Drossinos, J. Aerosol Sci., 2008, 39, 737–758 CrossRef CAS.
- Y. Wang, P. K. Hopke, D. C. Chalupa and M. J. Utell, Atmos. Environ., 2011, 45, 7672–7680 CrossRef CAS.
- M. S. Bućko, T. Magiera, B. Johanson, E. Petrovský and L. J. Pesonen, Environ. Pollut., 2011, 159, 1266–1276 CrossRef PubMed.
- E. L. Hautala, R. Rekilä, J. Tarhanen and J. Ruuskanen, Environ. Pollut., 1995, 87, 45–49 CrossRef CAS PubMed.
- B. Helmreich, R. Hilliges, A. Schriewer and H. Horn, Chemosphere, 2010, 80, 991–997 CrossRef CAS PubMed.
- C. Leuenberger, J. Czuczwa, E. Heyerdahl and W. Giger, Atmos. Environ., 1967, 1988(22), 695–705 Search PubMed.
- M. V. Vasić, A. Mihailović, U. Kozmidis-Luburić, T. Nemes, J. Ninkov, T. Zeremski-Škorić and B. Antić, Chemosphere, 2012, 86, 585–592 CrossRef PubMed.
- K. Kuoppamäki, H. Setälä, A.-L. Rantalainen and D. J. Kotze, Environ. Pollut., 2014, 195, 56–63 CrossRef PubMed.
- G. Kos and P. A. Ariya, Anal. Bioanal. Chem., 2006, 385, 57–66 CrossRef CAS PubMed.
- R. Mortazavi, C. T. Hayes and P. A. Ariya, Ice nucleation activity of bacteria isolated from snow compared with organic and inorganic substrates, Environ. Chem., 2008, 5(6), 373–381 CrossRef CAS.
- G. Kos and P. A. Ariya, J. Geophys. Res.: Atmos., 2010, 115, 2156–2202 CrossRef.
- G. Kos, V. Kanthasami, N. Adechina and P. A. Ariya, Environ. Sci.: Processes Impacts, 2014, 16, 2592–2603 CAS.
- D. Brugge, J. L. Durant and C. Rioux, Environ. Health, 2007, 6, 23 CrossRef PubMed.
- J. Hitchins, L. Morawska, R. Wolff and D. Gilbert, Atmos. Environ., 2000, 34, 51–59 CrossRef CAS.
- M. M. Maricq, R. E. Chase, D. H. Podsiadlik and R. Vogt, Vehicle Exhaust Particle Size Distributions: a Comparison of Tailpipe and Dilution Tunnel Measurements, SAE Technical Paper, 1999 Search PubMed.
- M. M. Maricq, D. H. Podsiadlik and R. E. Chase, Aerosol Sci. Technol., 2000, 33, 239–260 CrossRef CAS.
- S. J. Greenwood, J. E. Coxon, T. Biddulph and J. Bennett, An Investigation to Determine the Exhaust Particulate Size Distributions for Diesel, Petrol, and Compressed Natural Gas Fuelled Vehicles, SAE Technical Paper, 1996 Search PubMed.
- M. Jacobson, D. Kittelson and W. Watts, Environ. Sci. Technol., 2005, 39, 9486–9492 CrossRef CAS PubMed.
- S. Fuzzi, M. O. Andreae, B. J. Huebert, M. Kulmala, T. C. Bond, M. Boy, S. J. Doherty, A. Guenther, M. Kanakidou, K. Kawamura, V. M. Kerminen, U. Lohmann, L. M. Russell and U. Pöschl, Atmos. Chem. Phys., 2006, 6, 2017–2038 CrossRef CAS.
- K. Tsigaridis and M. Kanakidou, Atmos. Environ., 2007, 41, 4682–4692 CrossRef CAS.
- A. Calvo, C. Alves, A. Castro, V. Pont, A. Vicente and R. Fraile, Atmos. Res., 2013, 120, 1–28 CrossRef.
- T. Meyer and F. Wania, Water Res., 2008, 42, 1847–1865 CrossRef CAS PubMed.
- F. Wania, Chemosphere, 1997, 35, 2345–2363 CrossRef CAS.
- P. Ariya, B. Jobson, R. Sander, H. Niki, G. Harris, J. Hopper and K. Anlauf, J. Geophys. Res.: Atmos., 1984–2012, 1998(103), 13169–13180 Search PubMed.
- W. R. Simpson, R. von Glasow, K. Riedel, P. Anderson, P. Ariya, J. Bottenheim, J. Burrows, L. J. Carpenter, U. Frieß, M. E. Goodsite, D. Heard, M. Hutterli, H. W. Jacobi, L. Kaleschke, B. Neff, J. Plane, U. Platt, A. Richter, H. Roscoe, R. Sander, P. Shepson, J. Sodeau, A. Steffen, T. Wagner and E. Wolff, Atmos. Chem. Phys., 2007, 7, 4375–4418 CAS.
- A. Kocbach, B. V. Johansen, P. E. Schwarze and E. Namork, Sci. Total Environ., 2005, 346, 231–243 CrossRef CAS PubMed.
- AroKor Holdings Inc., Chemicalland21, http://www.chemicalland21.com/, accessed April 5, 2015, 2015.
- LookChem, Look for Chemicals, http://www.lookchem.com/, accessed April 5, 2015, 2015.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: from Air Pollution to Climate Change, John Wiley & Sons, 2nd edn, 2012 Search PubMed.
- N. Pieters, G. Koppen, M. van Poppel, S. De Prins, B. Cox, E. Dons, V. Nelen, L. I. Panis, M. Plusquin, G. Schoeters and T. S. Nawrot, Environ. Health Perspect., 2015, 123, 737–742 Search PubMed.
- K. Donaldson, L. Tran, L. Jimenez, R. Duffin, D. Newby, N. Mills, W. MacNee and V. Stone, Part. Fibre Toxicol., 2005, 2, 10 CrossRef PubMed.
- D. E. Newby, P. M. Mannucci, G. S. Tell, A. A. Baccarelli, R. D. Brook, K. Donaldson, F. Forastiere, M. Franchini, O. H. Franco, I. Graham, G. Hoek, B. Hoffmann, M. F. Hoylaerts, N. Künzli, N. Mills, J. Pekkanen, A. Peters, M. F. Piepoli, S. Rajagopalan and R. F. Storey, Eur. Heart J., 2014, 36, 83–93 CrossRef PubMed.
- E. Velasco, P. Siegmann and H. C. Siegmann, Atmos. Environ., 2004, 38, 4957–4968 CrossRef CAS.
- T. Meyer and F. Wania, Water Res., 2011, 45, 3627–3637 CrossRef CAS PubMed.
- J. Ma, H. Hung, C. Tian and R. Kallenborn, Nat. Clim. Change, 2011, 1, 255–260 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional details of methods and supplemental data, tables and figures are provided in the ESI. See DOI: 10.1039/c5em00616c |
| This journal is © The Royal Society of Chemistry 2016 |