
The severity of sediment desiccation affects the adsorption characteristics and speciation of phosphorus†
Nirmala W.
Attygalla
a,
Darren S.
Baldwin
*b,
Ewen
Silvester
a,
Peter
Kappen
cd and
Kerry L.
Whitworth
a
aDepartment of Ecology, Environment and Evolution, La Trobe University, Wodonga, Victoria 3689, Australia
bCSIRO Land and Water and the Murray-Darling Freshwater Research Centre, PO Box 821, Wodonga, Victoria 3689, Australia. E-mail: Darren.Baldwin@csiro.au
cAustralian Synchrotron, 800 Blackburn Rd, Clayton, Victoria 3168, Australia
dDepartment of Chemistry and Physics, La Trobe University, Melbourne, Victoria 3086, Australia
First published on 7th December 2015
Abstract
Phosphorus is an important nutrient for plants and algae, and can be the limiting nutrient in aquatic ecosystems. However, oversupply can lead to significant water quality issues. The largest source and sink of P in most aquatic systems is the sediment. As a consequence of drought, in many places sediments that normally would have remained inundated are now being desiccated. Based on previous studies, it is often difficult to predict what impact drying will have on the cycling of P. This is because most of these studies have looked at drying across a chronosequence in the field, where there may be differences in sediment composition or microbial community structure. In this paper we present the results of a study where sediment was exposed to progressively more severe drying in the laboratory – starting with wet sediment, followed by air drying and then sequential oven drying at 30, 50 and 85 °C. Drying resulted in a shift in P speciation, notably with an increase in NaHCO3-extractable reactive P and a decline in NaHCO3-extractable unreactive P, likely indicating an increase in bioavailable, easily exchangeable P. Drying also resulted in a decline in the microbial-P fraction. Drying significantly affected the P adsorption characteristics of the sediment. The total amount of P adsorbed by the sediment and the linear adsorption co-efficient both declined, while the amount of native P adsorbed to the sediment and the equilibrium P concentration both increased. Drying also affected iron speciation with a shift from more reactive oxalate-extractable Fe to more recalcitrant citrate–dithionate–bicarbonate-extractable Fe, suggesting an increase in iron crystallinity and hence decrease in P adsorption capacity. The increase in crystallinity is consistent with Fe EXAFS results, which showed that drying resulted in an increase in edge-sharing neighbours. We hypothesise that the shifts in P speciation, the decline in P adsorption capacity, the increase in the equilibrium P concentration, as well as the death of micro-organisms (as evidenced by a decline in microbial P) on drying all contribute to the Birch effect – the initial pulse of P and/or N upon inundation of dried soils or sediments.
Environmental impactDrying significantly affects sedimentary P dynamics, changing both P speciation and P adsorption capacity. Therefore sediment drying has the potential to alter the dynamics of P following re-inundation. |
1. Introduction
Drought has been a feature of the climate across most continents in the last few decades, including extended periods of drought in Sahelian Africa,1 south-western United States,2 South America and south-eastern Australia.3 One of the features of drought is the exposure to air and subsequent desiccation of sediments that normally would be inundated and, therefore, potentially anoxic. Water level drawdown, sediment oxidation and subsequent desiccation can have a significant impact on sediment chemistry,4 mineralogy4–6 and sediment microbial ecology.7 However the specific response to desiccation seems to be highly variable – especially with respect to phosphorus biogeochemistry. Phosphorus is an important nutrient for plants and algae, and can be the limiting nutrient in aquatic ecosystems. However, in oversupply it can lead to significant water quality issues. The largest source and sink of P in most aquatic systems is the sediment.8 The effect of drying of sediments on P dynamics is complex and includes various interdependent physical and biogeochemical changes. Previous field studies have shown that drying of soils and sediments can result in a decrease,5,6,9–12 an increase,13–16 or have no effect17 on orthophosphate binding capacity. Similarly, the effect of desiccation on P speciation seems to be inconsistent. For example, Kerr et al.18 reported a shift towards more reactive P species in river sediments on drying, while de Vincente et al.12 reported a shift towards less bioavailable pools in lake sediments as a consequence of desiccation.One of the possible reasons for the discrepancies in the role that desiccation plays in sedimentary P dynamics is that most of this research is based on field studies; the extent of desiccation is based on studying a chronosequence, usually including inundated, damp and dry sediments at the edge of a river, lake or wetland; i.e. not under controlled conditions. In the current study we examine P speciation, using a modified SEDEX extraction scheme,19 and sediment P adsorption characteristics, based on adsorption isotherms, of sediment from a floodplain wetland that had been exposed to progressively more severe drying regimes in the laboratory. Phosphorus dynamics are often closely linked to iron dynamics in sediments. Therefore we also explore shifts in iron mineralogy in response to drying using both sequential extraction and X-ray absorption spectroscopy.
2. Materials and methods
2.1 Sampling and initial handling
Inundated sediment was sampled to a depth of 10 cm using a soil auger from randomly selected sites from an unnamed wetland on the Kiewa River floodplain in north-eastern Victoria, Australia (36°34′S, 147°5′E). The wetland was quite shallow (<0.5 m in depth) and would most likely have periodically dried out during the period of drought this region experienced from about 1996 to 2010.3 Immediately on arrival at the laboratory (<1 h), the sediment was sieved (10 mm) to remove coarse woody debris, homogenised, and randomly placed into 25 polystyrene trays. Five trays (hereinafter Twet) were randomly selected, their content transferred individually to polycarbonate centrifuge tubes and then centrifuged at an average relative centrifugal field (RCFav) of 5900 g for 20 min at 5 °C (Beckman Avanti centrifuge) to remove pore-water before immediately undergoing analysis. The remaining sub-samples were subjected to a ‘sequential’ drying treatment. Firstly, all the remaining sediment was air-dried at ambient temperature (approximately 20 °C) for 7 days. Five sub-samples of air-dried sediment (hereinafter Tair) were then set aside for subsequent analysis. The remaining sediment was transferred into an oven (Thermocentre) and dried at 30 °C for 7 days. Five sub-samples were then removed for analysis (hereinafter T30). The remaining sediment was further oven-dried at 50 °C for 7 days, and a further five sub-samples removed (hereinafter T50). The remaining five sediment samples were then dried at 85 °C for 7 days before being analysed (T85).2.2 Chemical analyses
All results are expressed on a dry weight (DW) basis by taking into account the moisture content, which was determined by drying a sub-sample from each treatment at 105 °C for 24 h.Microbial biomass phosphorus was determined by the fumigation–extraction method22,23 with certain modifications. Sediment samples (10 g DW or equivalent) were fumigated with chloroform (CHCl3) vapour in a desiccator for 24 h. Non-fumigated samples were treated similarly, except that the desiccator contained no CHCl3. After residual CHCl3 was removed by evacuation, both fumigated and non-fumigated samples were shaken with 200 mL of 0.5 M NaHCO3 (pH 8.5) (at a 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution-to-soil ratio) in 250 mL Thermo Scientific Nalgene (HDPE) centrifuge bottles on an orbital shaker for 30 min at 20 °C. The extracts were centrifuged (RCFav = 1600 g, 10 min, 5 °C; Allegra X-15R Beckman Coulter) and the supernatants were passed through 0.45 μm cellulose acetate filters (Millipore). CHCl3-released inorganic P (Pi) was calculated as the difference between fumigated and non-fumigated samples. Microbial biomass P was calculated from CHCl3-released Pi by dividing by 0.4.22
:1 solution-to-soil ratio) in 250 mL Thermo Scientific Nalgene (HDPE) centrifuge bottles on an orbital shaker for 30 min at 20 °C. The extracts were centrifuged (RCFav = 1600 g, 10 min, 5 °C; Allegra X-15R Beckman Coulter) and the supernatants were passed through 0.45 μm cellulose acetate filters (Millipore). CHCl3-released inorganic P (Pi) was calculated as the difference between fumigated and non-fumigated samples. Microbial biomass P was calculated from CHCl3-released Pi by dividing by 0.4.22
Sediment phosphorus speciation was determined using a modified SEDEX sequential extraction procedure6,19 (Table 1). Briefly, well-mixed soil samples (0.5 g DW or equivalent) were placed into 50 mL acid-washed centrifuge tubes (polycarbonate). For each extraction step, 25 mL of extractant solution was added and the tubes were shaken on an orbital shaker for 16 h in the dark at 20 ± 1 °C. The tubes were then centrifuged (RCFav = 5900 g, 20 min, 5 °C; Beckman Avanti) and the supernatant filtered through 0.45 μm cellulose acetate membranes (Millipore). As proposed by Ruttenberg,19 each extraction by the principal extractants in steps I, II, III and IV of the sequence (Table 1) was followed by successive MgCl2 and H2O washes in order to prevent problems associated with secondary adsorption onto residual solid surfaces. The rP concentrations were determined on all extracts after first neutralizing the sample with concentrated acid or base as required. Because of reactivity issues, rP in the citrate–dithionite–bicarbonate (CDB) extract was determined on isobutyl alcohol extracts using the tin chloride assay24 after first diluting the supernatant by 1 in 10 with Milli-Q water (Millipore – SuperQ) to overcome interferences. Total P was also determined on the NaHCO3 and NaOH extracts.
| Step | Extractant | Operationally defined target phase | Reported as |
|---|---|---|---|
| I | 1.0 M MgCl2 | Exchangeable/loosely sorbed P | MgCl2 rP |
| IIa | 1.0 M NaHCO3 | Reactive P associated with labile organic matter | NaHCO3 rP |
| IIb | Non-reactive P associated with labile organic matter | NaHCO3 uP | |
| III | 0.3 M Na3–citrate, 0.56 g Na2S2O4, 1.0 M NaHCO3 | Easily reducible/reactive P associated with Fe or Mn phases | CDB P |
| IVa | 1.0 M NaOH | Reactive P associated with Al phases | NaOH rP |
| IVb | Non-reactive P associated with Al phases | ||
| V | 1.0 M HCl | Reactive P associated with Ca phases | HCl rP |
| VI | Ashed at 550 °C then extracted with 1.0 M HCl | Refractory P | Residual P |
Phosphorus L2,3-edge X-ray absorption near edge structural (XANES) spectra were acquired on the Soft X-ray Beamline at the Australian synchrotron. Spectra were collected in the energy range 125–155 eV in total electron yield mode. All spectra were normalised against incident beam intensity (I0) and energy calibrated against Au 4f5/2,7/2 absorption lines. Phosphorus-containing standards used included: phytic acid, sodium dihydrogen phosphate and iron(III) phosphate. As will be discussed, total P levels in the samples were too low to allow usable spectra to be acquired using this technique.
Phosphorus sorption characteristics were determined using phosphate adsorption isotherms based on a batch equilibration procedure developed by Nair et al.25 For each replicate, 1 g (DW) of sediment was placed in 50 mL acid-washed polycarbonate centrifuge tubes and suspended in 25 mL of standard orthophosphate solutions with P concentrations of: 0, 100, 250, 500, 1000, 2500, 5000, 10000, 20000 μg P l−1, spiked with 2 drops of chloroform. Stock phosphate solution was prepared by dissolving analytical grade anhydrous KH2PO4 in a 0.01 M CaCl2 matrix. The tubes were shaken on an orbital shaker for 24 h at 20 ± 1 °C in the dark. The tubes were then centrifuged (RCFav = 5900 g; 10 min; 5 °C; Beckman Avanti) and the supernatant passed through 0.45 μm cellulose acetate filters (Millipore) prior to rP analysis. The P adsorption isotherms were determined by measuring the amount of orthophosphate remaining in the solution (Ct) and the amount of orthophosphate retained by the sediment (S′) after 24 h; S′ was calculated as the difference between the amount of P in solution at 24 h and at 0 h.6 The linear adsorption co-efficient (Kd), the amount of native phosphate adsorbed to the sediment (S0) and the equilibrium phosphate concentration (EPC0) were calculated in the linear portion of the isotherm using the best fit of the data to the relationship:26,27
| S′ = Kd × Ct − S0 |
EPC0 is the same as Ct when S′ is 0.
Iron X-ray absorption fine structure (EXAFS) spectra at the Fe K-edge were collected at the Australian National Beamline Facility at the Photon Factory synchrotron, Tsukuba, Japan. All samples were stored under nitrogen in 120 mL serum bottles for transport to the facility. The X-ray energy was selected using a Si (111) double-crystal monochromator, with spectra recorded at the Fe K-edge (6.79–7.89 keV). The beam spot size was set to 5 mm (horizontal) × 1 mm (vertical). Data were acquired in fluorescence mode using a 36-element solid state Ge detector. For the determination of atomic distances and numbers of neighbours, individual peaks from the radial distribution function were Fourier filtered and fitted using Viper (v11), using a basis file developed from a goethite (α-FeOOH) standard.
2.3 Statistical analysis
All statistical analyses except analysis of similarity (ANOSIM) were performed using Sigmaplot (v12). All samples were assessed for normality and homogeneity of variance. Differences between sediment drying treatments were investigated using one-way analysis of variance (ANOVA) coupled with Tukey's post hoc test on absolute values. Errors are quoted as ±1 standard deviation from the mean. The ANOSIM analysis was performed using Primer (v6) on SEDEX speciation data for each individual sample that had first been standardised to total P concentration.3. Results
3.1 Modified-SEDEX P speciation
Drying caused a substantial change in sedimentary P speciation (Fig. 1). The ANOSIM showed that there were significant differences in overall P speciation between all treatments (global R = 0.914, p = 0.008) except T30 and T50 (p = 0.14).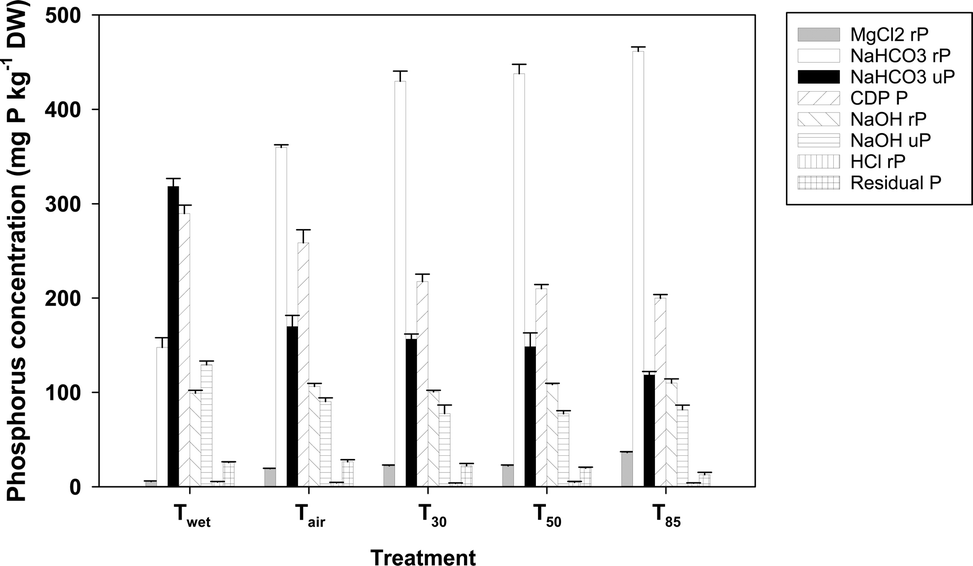 | ||
| Fig. 1 P speciation of sediment following different degrees of desiccation, as determined by sequential extraction. Error bars represent one standard deviation; rP = reactive P, uP = unreactive P. | ||
There was a statistically significant (p < 0.001) increase in MgCl2 P following air drying (from 5.5 ± 0.6 to 19.2 ± 0.3 mg P kg−1 DW), and again after air drying followed by oven drying at 30 °C (to 22.2 ± 0.8 mg P kg−1 DW). There was no statistically significant difference in MgCl2 rP between the T30 and T50 treatments (p = 0.79), but it was higher again (p < 0.001) in the T85 treatment. Reactive P concentrations in the NaHCO3 extract followed a similar pattern to that observed for the MgCl2 extract. Reactive P was significantly higher in the Tair treatment (360 ± 3 mg P kg−1 DW) than in the Twet treatment (148 ± 10 mg P kg−1 DW; p < 0.001) and was higher again in the T30 treatment (430 ± 11 mg P kg−1 DW; p < 0.001). There was no significant difference between T30 and T50 (p = 0.144), but rP in the T85 treatment was once again significantly higher (460 ± 4 mg P kg−1 DW; p < 0.001). Unlike rP, uP in the NaHCO3 extracts declined with drying; NaHCO3 uP was significantly lower (p < 0.001) in sediments from the Tair treatment (170 ± 11 mg P kg−1 DW) than in the Twet treatment (318 ± 8 mg P kg−1 DW). There was no significant difference in NaHCO3 uP between the Tair and T30 treatments (p = 0.08), nor between the T30 and T50 treatments (p = 0.195), but there was between Tair and T50 (p = 0.006). The T85 treatment had a lower NaHCO3 uP (118 ± 4 mg P kg−1 DW) than all the other treatments (p < 0.001). The CDB P also declined with drying, falling from 290 ± 9 mg P kg−1 DW for Twet, to 259 ± 14 mg P kg−1 DW for Tair and 218 ± 8 mg P kg−1 DW for T30; all of which were statistically significant (p < 0.001). There was no significant difference between CDB P in the T30 and T50 treatments (p = 0.178) or the T50 and T85 treatments (p = 0.153) but there was a difference between the T30 and T85 treatments (p = 0.012). Reactive P in the NaOH extract essentially remained constant, varying from 99 ± 3 mg P kg−1 DW in the Twet treatment to 110 ± 4 mg P kg−1 DW in the T85 treatment; noting however that the difference in NaOH rP between Twet and Tair (106 ± 3 mg P kg−1 DW) was statistically significant (p = 0.007). Unreactive P concentrations in the NaOH extracts fell in response to initial drying, going from 129 ± 4 mg P kg−1 DW in the Twet treatment to 90 ± 4 mg P kg−1 DW for the Tair treatment. Unreactive P was significantly higher in the Tair treatment than in the T30 treatment (78 ± 9 mg P kg−1 DW; p = 0.006) and the T50 treatment (77 ± 3 mg P kg−1 DW; p = 0.006) but not the T85 treatment (81 ± 5 mg P kg−1 DW; p = 0.07). The HCl rP concentrations were all low (<5 mg P kg−1 DW). There was no change in the residual P pool between the Twet and Tair treatments (p = 0.48) but there were differences between Tair and T30 (p = 0.01) and Tair and T50 (p < 0.001). Residual P in the T85 treatment (13 ± 2 mg P kg−1 DW) was lower than in all the other treatments (p < 0.001).
3.2 Microbial biomass P
Microbial biomass P declined with drying, falling from 20.3 ± 1.8 mg P kg−1 DW in the Twet treatment to 7.7 ± 1 mg P kg−1 DW in the Tair treatment and to 3.4 ± 0.8 mg P kg−1 DW in the T30 treatment. The microbial biomass P in the T30 and T50 treatments were similar (T50 = 3.1 ± 0.1 mg P kg−1 DW) but was lower again in the T85 treatment (1.0 ± 0.6 mg P kg−1 DW). Apart from T30/T50 couple, all differences were statistically significant (p < 0.05).3.3 Phosphorus L2,3-edge XANES analysis
High-quality spectra were obtained for reference P-containing compounds, similar to those reported elsewhere.30 However spectra for sediment samples from all treatments were featureless, most likely due to the low total P concentrations (example spectra are presented in the ESI†). Using phytic acid as an example, we demonstrate that rather higher total P levels (in excess of 2500 ppm P) would be required to allow useable spectra to be obtained for these type of samples (see ESI†).3.4 Phosphate adsorption isotherms
The phosphate adsorption isotherms are presented in Fig. 2. While air drying had some effect on the overall shape of the adsorption curves, the most dramatic changes occurred following oven drying at 30 °C (Fig. 2). The maximum amounts of P adsorbed in the T30, T50 and T80 treatments were all similar. | ||
| Fig. 2 Phosphate adsorption isotherms for sediment following different degrees of desiccation. Error bars represent one standard deviation. | ||
The values of Kd, S0 and EPC0 were also affected by drying (Table 2). Values of Kd were significantly lower in the Tair treatment than the Twet treatment (p < 0.001) and were lower again in the T30 treatment (p < 0.001). While the value for Kd was lower in the T85 treatment than in either the T30 or T50 treatments, the difference was not statistically significant (p = 0.87 and 0.85 respectively). The S0 increased with increasing severity of drying and each treatment was significantly different from the others (p < 0.001 except for the Tair/T30 couple where p = 0.04 and the T30/T50 couple where p = 0.002). Like S0, EPC0 also increased as the severity of drying increased and all treatments were significantly different from each other (p < 0.001 except for the Twet/Tair couple where p = 0.04).
| Treatment | K d (l kg−1) | S 0 (mg P kg−1) | EPC0 (μg l−1) |
|---|---|---|---|
| Twet | 1580 ± 330 | 2.2 ± 1.3 | 1.3 ± 0.6 |
| Tair | 627 ± 32 | 7.1 ± 1.1 | 11.3 ± 1.3 |
| T30 | 125 ± 5 | 8.5 ± 0.6 | 67.6 ± 2.1 |
| T50 | 108 ± 3 | 10.9 ± 0.8 | 101.5 ± 6.4 |
| T85 | 60 ± 1 | 21.2 ± 1.1 | 355 ± 14.5 |
3.5 Iron speciation
There was substantially more Feox than FeCDB in the sediments from the Twet treatment (Fig. 3). On drying the concentration of Feox declined while the concentration of FeCDB increased. There was significantly more Feox and FeCDB in the T30 treatment than in any of the other treatments where the sediment was dried. | ||
| Fig. 3 Fe speciation of sediment following different degrees of desiccation determined by sequential extraction with oxalate (Feox) followed by citrate–dithionite–bicarbonate (FeCDB). Error bars represent one standard deviation. | ||
3.6 Fe EXAFS
In all samples, the Fe K-edge showed evidence of two Fe–Fe distances; one at ∼3.05 Å and the second at 3.45 Å. There was a general increase in edge-sharing neighbours at 3.05 Å across the series Twet–T85 (Table 3). This increase is consistent with the building of extended chains of FeO6 octahedra, typical of many Fe(oxy)hydroxide minerals (e.g. goethite, lepidocrocite), and similar to that observed for the transformation of hydrous ferric oxides to more crystalline Fe(oxy)hydroxides.31 The number of neighbours at 3.45 Å initially decreased between Twet and Tair, and then increased across the series. However, it must be noted that in all samples, there was a strong background Fe mineralogy that diminishes the changes in EXAFS spectra across the drying series; this background is likely to be from Fe-containing clay minerals that are largely unaffected by the drying and heating treatments. As a consequence of this background, the changes in the numbers of neighbours are small, and similar in magnitude to the uncertainty in the fitted number of neighbours.| Treatment | Atomic pair | Distance (Å) | Number of neighbors | Debye–Waller (Å) | ΔE (eV) |
|---|---|---|---|---|---|
| Twet | Fe–Fe (1) | 3.10 | 0.77 | 2.3 × 10−3 | 0.62 |
| Fe–Fe (2) | 3.44 | 0.78 | 2.3 × 10−3 | ||
| Tair | Fe–Fe (1) | 3.05 | 0.94 | 2.3 × 10−3 | −0.36 |
| Fe–Fe (2) | 3.44 | 0.60 | 2.3 × 10−3 | ||
| T30 | Fe–Fe (1) | 3.04 | 0.94 | 2.3 × 10−3 | −0.36 |
| Fe–Fe (2) | 3.43 | 0.69 | 2.3 × 10−3 | ||
| T50 | Fe–Fe (1) | 3.04 | 0.99 | 2.3 × 10−3 | −0.41 |
| Fe–Fe (2) | 3.43 | 0.81 | 2.3 × 10−3 | ||
| T85 | Fe–Fe (1) | 3.04 | 1.03 | 2.3 × 10−3 | −0.67 |
| Fe–Fe (2) | 3.43 | 0.83 | 2.3 × 10−3 |
4. Discussion
This study examines how a wet–dry transitional phase could affect P speciation and adsorption characteristics in a floodplain wetland sediment. In the proceeding discussion we assume that the P and Fe speciation of the Twet samples are essentially the same as in the wetland – i.e. transportation back to the laboratory (<1 h) and sieving did not change the speciation. Oxidation of sulfidic materials (including pyrite) has been shown to shift P speciation in marine sediments from calcium phosphate mineral phases to iron phases through acidic dissolution during storage.32,33 While we cannot unequivocally discount that this has occurred in our sediments during sample processing, a number of studies of the regional distribution of sulfidic sediments (inland acid sulfate soils) in south-eastern Australia have shown that wetland sediments in the study area do not contain significant levels of reduced-sulfur compounds,34,35 which would suggest that the likelihood of this occurring during our experiment is low.The temperature treatments were chosen to reflect potential summer sediment temperatures encountered in many semi-arid and arid floodplains. While 85 °C is obviously an extreme temperature and one not encountered in these types of environments, soil temperatures approaching 50 °C are not unknown in these type of ecosystems (Dr Jessica McGregor, La Trobe University, pers. comm.).
Severity of desiccation had a substantial impact on P speciation in the sediments. Almost all operationally defined P fractions responded to the drying treatments. However the change in P speciation in response to desiccation appears to occur in phases. The largest change in P speciation occurs on initial drying. There was a statistically significant difference between Twet and Tair in the concentrations of P in all fractions with the exception of HCl rP and residual P; combined these latter two fractions represented only about 3% of the sedimentary pool. There were also statistically significant differences between Tair and T30 for many of the P species, although the magnitude of change was generally less than the change from Twet to Tair. For the most part there was little difference in P speciation between T30 and T50. However the P speciation in the T85 treatment was generally different to that found in T50 and/or T30, although the extent of change was not as large as seen in the Twet/Tair and Tair/T30 transitions.
Desiccation predominantly led to a shift in the P speciation to potentially more available pools. This is consistent with previous studies that have shown that desiccation leads to a shift away from non-reactive to more reactive pools in both lake6 and river18 sediments. In the current study MgCl2 P, which corresponds to easily exchanged P, increased with air drying. This increase in exchangeable P was also reflected by an increase in the amount of ‘native P’ adsorbed to the sediments (S0). The largest change in P speciation on drying was in the NaHCO3 P fraction. Reactive P in this fraction increased by about 200 mg P kg−1 DW on drying while non-reactive P fell by about 150 mg P kg−1 DW. The separate bicarbonate fraction was incorporated into the SEDEX procedure because the subsequent CDB extraction, where the target phase was P bound to reducible iron, also extracted substantial amounts of organic carbon when the procedure was applied to freshwater sediments.8 In the modified SEDEX extraction scheme the organic matter is pre-extracted using bicarbonate alone. Although the bicarbonate fraction co-extracts organic carbon and phosphorus, there is a shift from non-reactive to reactive P. Without confirmatory analyses (e.g.31P nuclear magnetic resonance (nmr)) it is not possible to unequivocally tell whether this represents a shift from organic to inorganic phosphorus phases.36 However given the strong relationship between reactive P and bioavailability37 the shift from uP to rP suggests a shift in the potential bioavailability of this fraction. A decrease was also observed in NaOH uP between Twet and Tdry. Previously a 31P nmr study has shown that in the modified SEDEX procedure NaOH can extract poly-phosphates, which are usually unreactive to the molybdenum blue assay.8 Poly-phosphates are used by bacteria as P storage compounds, which may, in part, explain the strong correlation between the overall decline in NaOH uP and the overall decline in microbial P with increasing severity of drying (Pearson's r = 0.92, n = 25).
The CDB extraction targets rP bound to Fe phases. Desiccation leads to a decrease in CDB rP, an effect previously observed for lake sediments.12 Generally, P bound to ferric iron is not readily available because of strong complexation. Typically, release of P from ferric minerals only occurs under anaerobic conditions, when microbial activity either directly38 or indirectly39 leads to the reductive dissolution of ferric phases with the concomitant release of P. While anaerobic conditions are common in inundated wetland sediments, when the sediment is exposed to the air, or prior to the onset of anoxia, P bound to iron phases will not generally be available. Therefore, the decline of CDB P with desiccation contributes to P availability, at least under oxic conditions.
One of the potential contributors to the decrease in iron-bound P on drying is the apparent shift to more crystalline iron phases, which would have a lower P-binding capacity.40 Acidic oxalate usually solubilises short-range (i.e. amorphous and poorly crystalline) iron minerals, while CDB solubilises both short-range and longer-range (more crystalline) phases.41 Therefore, in a sequential extraction Feox will represent less crystalline phases while FeCDB will represent more crystalline Fe minerals. On drying, there was a significant shift from Feox to FeCDB, indicating an increase in crystallinity on drying.
The sequential extraction data are supported by the Fe K-edge EXAFS data. It should be noted that due to the small contribution of Fe–Fe scattering to the total EXAFS spectrum we used a highly constrained fitting approach to determine the Fe–Fe distances and number of neighbours. These constraints included fixing the Fe–O number of neighbours (6) and the Debye–Waller factor for all Fe–Fe distances (0.0023 Å) across all samples. As a consequence, the uncertainty in the fitted number of neighbours at these Fe–Fe distances is high relative to the differences between samples. This is an inherent problem with such natural samples where the contribution of Fe-oxide minerals to total Fe mineralogy is small. Notwithstanding this caveat, the EXAFS study suggests that there was a general increase in edge-sharing neighbours at 3.05 Å across the series Twet–T85 (Table 3). This increase is consistent with the building of extended chains of FeO6 octahedra, typical of many Fe-(oxy)hydroxide minerals (e.g. goethite, lepidocrocite), and similar to that observed for the transformation of hydrous ferric oxides to more crystalline Fe-(oxy)hydroxides.31 An increase in crystallinity with desiccation explains the observed shifts in the P adsorption isotherms. As minerals become more crystalline both their surface area and the number of binding sites will decrease. Because they have fewer binding sites, more crystalline phases will have a lower maximum amount of P that they can bind, hence explaining the differences in the adsorption maxima in the P adsorption isotherms with extended drying. It also helps explain the shift to larger values of EPC0 with drying. The EPC0 represents the concentration above which P will adsorb to the sediment and hence indicates whether a sediment will act as a source or sink on re-inundation. The shift to higher EPC0 values on drying means that on re-inundation, at least initially, the concentration of P in the overlying water will be higher than it would have been if the sediments had not dried out. It is interesting to note that, unlike P speciation, EPC0, as well as S0 and Kd, continued to change quite substantially with the severity of desiccation. Increased EPC0 because of desiccation has also been reported for river sediments.18
The results of the current study are in agreement with a previous hypothesis proposed by Baldwin6 on the effect of increasing desiccation on P dynamics, but raise some interesting questions about the role of desiccation in shaping P speciation. Baldwin6 proposed that shifts in P adsorption in response to desiccation are due in large part to changes in iron mineralogy. Freshly exposed sediment has a high affinity for P because reduced iron in the sediment is rapidly oxidised when the sediment contacts air. As the sediment dries, dehydration of iron minerals leads to a transformation from amorphous oxyhydroxides to more crystalline forms – specifically goethite and/or haematite – with a concomitant loss of adsorption capacity and increase in EPC0. The more crystalline forms are harder to reduce once the sediment is re-inundated and becomes anoxic; hence repeated wetting and drying cycles can result in a substantial decrease in the sediment's affinity for P,17 with a potential concomitant increase in the concentration of P in the overlying water.
This study also contributes to our understanding of the mechanisms underlying the Birch effect.17 The Birch effect, first described in the 1950s and early 1960s,42 is the phenomenon of an initial pulse release of nutrients from dried soils or sediments upon re-wetting. It is often assumed that this pulse comes from microbial cells that have been killed either during the drying phase or by osmotic shock on re-wetting.17 This study suggests that drying does result in a decline in sediment microbial biomass, inferred from a reduction in microbial P, with an approximately corresponding increase in native adsorbed P (S0), suggesting that microbial necromass may indeed be a source of P for the Birch effect. However abiotic factors may also contribute to the effect. In particular the increase in EPC0 with drying means that on initial inundation, P would desorb from the sediment to the overlying water if the P concentration in the overlying water is less than the EPC0. Therefore drying and subsequent re-wetting of sediments has the potential to be a significant source of internal loading43 of P to water bodies. This has ramifications for the on-going management of water bodies previously subjected to nutrient pollution; in particular permanent water bodies that may start to undergo periodic desiccation in a changing climate.
The actual changes in molecular structure of P-containing compounds as a consequence of drying have yet to be elucidated. Sequential extraction is a rather crude technique for exploring the structural changes in P speciation, principally because it cannot unequivocally differentiate between organic and inorganic P species.32 As part of this study we attempted to further explore the sediment P speciation using phosphorus L2,3-edge XANES, but the native P concentrations appear to be below that required for useful spectra to be acquired (see ESI†). Further studies using more sophisticated techniques, in particular 31P nmr spectroscopy, are needed in order to explore both the extent and mechanisms of changing P speciation through desiccation.
5. Conclusions
This study has shown that drying has a significant effect on sedimentary P dynamics, changing both P speciation and P adsorption capacity. The latter is associated with increases in crystallinity of iron phases. Therefore sediment drying has the potential to alter the dynamics of P following re-inundation. This would include a pulse release of P immediately upon re-flooding, as well as the potential for less P being adsorbed to the sediment, leading to a higher concentration of P in the overlying water column. The results of this study also suggest that repeated short wetting and drying cycles may lead to continually decreasing sediment affinity for P.Acknowledgements
NWA was supported by a La Trobe University Postgraduate Research Scholarship. Additional funding was through the Australian Government's Commonwealth Environmental Research Facilities (CERF) Significant Projects Program. We also thank the Australian Synchrotron for access to the Soft X-ray Beamline at the Australian Synchrotron and funding to use the Australian National Beamline Facility at the Photon Factory, Tsukuba, Japan.References
- A. Dai, P. J. Lamb, K. E. Trenbeth, M. Hulme, P. D. Jones and P. P. Xie, Int. J. Climatol., 2004, 24, 1323–1331, DOI:10.1002/joc.1083.
- C. A. Woodhouse, D. M. Meko, G. M. MacDonald, D. W. Stahle and E. R. Cooke, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21283–21288, DOI:10.1073/pnas.0911197107.
- F. H. S. Chiew, W. J. Young, W. Cai and J. Teng, Stoch. Environ. Res. Risk Assess., 2011, 25, 601–612, DOI:10.1007/s00477-010-0424-x.
- J. C. de Groot and C. van Wijck, Hydrobiologia, 1993, 252, 83–94 CrossRef.
- R. N. Sah and D. S. Mikkelsen, Soil Sci., 1986, 142, 346–351, DOI:10.1097/00010694-198611000-00004.
- D. S. Baldwin, Limnol. Oceanogr., 1996, 41, 1725–1732 CrossRef.
- D. S. Baldwin and A. M. Mitchell, Regulated Rivers: Research & Management, 2000, 16, 457–467 Search PubMed.
- D. S. Baldwin, Hydrobiologia, 1996, 335, 63–73, DOI:10.1007/bf00013684.
- A. J. Twinch, Water Res., 1987, 21, 1225–1230, DOI:10.1016/0043-1354(87)90174-6.
- S. Qiu and A. J. McComb, Aust. J. Mar. Freshwater Res., 1994, 45, 1319–1328 CrossRef CAS.
- C. J. Watts, Hydrobiologia, 2000, 431, 27–39, DOI:10.1023/a:1004098120517.
- I. de Vincente, F. O. Anderson, H. C. B. Hansen, L. Cruz-Pizzaro and H. S. Jensen, Hydrobiologia, 2010, 651, 253–264, DOI:10.1007/s10750-010-0304-x.
- N. Barrow and T. C. Shaw, Commun. Soil Sci. Plant Anal., 1980, 11, 347–353 CrossRef CAS.
- R. Olsen and M. N. Court, J. Soil Sci., 1982, 33, 709–717 CrossRef CAS.
- J. M. Jacoby, D. D. Lynch, E. B. Welch and M. A. Perkins, Water Res., 1982, 16, 911–919 CrossRef CAS.
- R. J. Haynes and R. S. Swift, Geoderma, 1985, 35, 145–157 CrossRef CAS.
- D. S. Baldwin, A. M. Mitchell and G. N. Rees, Hydrobiologia, 2000, 431, 3–12, DOI:10.1023/a:1004015019608.
- J. G. Kerr, M. Burford, J. Olley and J. Udy, Mar. Freshwater Res., 2010, 61, 928–935, DOI:10.1071/mf09124.
- K. Ruttenberg, Limnol. Oceanogr., 1992, 37, 1460–1482 CrossRef CAS.
- APHA, Standard methods for the examination of water and wastewater, American Public Health Association, Washington, 20th edn, 1998 Search PubMed.
- M. Hosomi and R. Sudo, Int. J. Environ. Stud., 1986, 27, 267–275 CrossRef CAS.
- P. C. Brookes, D. S. Powlson and D. S. Jenkinson, Soil Biol. Biochem., 1982, 14, 319–329 CrossRef CAS.
- M. J. McLaughlin, A. M. Alston and J. K. Martin, Soil Biol. Biochem., 1986, 18, 437–443, DOI:10.1016/0038-0717(86)90050-7.
- F. S. Watanabe and S. R. Olsen, Soil Sci., 1962, 93, 183–188 CrossRef CAS.
- P. Nair, T. Logan, A. N. Sharpley, L. E. Sommers, M. A. Tabatabai and T. L. Yuan, J. Environ. Qual., 1984, 13, 591–595 CrossRef.
- K. R. Reddy, G. A. O'Connor and P. M. Gale, J. Environ. Qual., 1988, 27, 438–447 CrossRef.
- H. K. Pant and K. R. Reddy, J. Environ. Qual., 2001, 30, 1474–1480 CrossRef CAS.
- Y. S. Zhang, X. Lin and W. Werner, J. Plant Nutr. Soil Sci., 2003, 166, 68–75, DOI:10.1002/jpln.200390014.
- G. W. Kunze and J. B. Dixon, Pretreatment for mineralogical analysis, in Methods of soil analysis, Part 1: Physical and mineralogical methods, ed. A. Klute, American Society of Agronomy, Madison, 2nd edn, 1986, pp. 91–100 Search PubMed.
- J. Kruse, P. Leinweber, K. Eckhardt, F. Godlinski, Y. Hu and L. Zuin, J. Synchrotron Radiat., 2009, 16, 247–259, DOI:10.1107/s0909049509000211.
- A. Manceau and V. A. Drits, Clay Miner., 1993, 28, 165–165, DOI:10.1180/claymin.1993.028.2.01.
- P. Kraal, C. P. Slomp, A. Forster and M. M. M. Kupyers, Geochim. Cosmochim. Acta, 2009, 73, 3277–3290 CrossRef CAS.
- P. Kraal and C. P. Slomp, PLoS One, 2014, 9, e96859 Search PubMed.
- K. Hall, D. S. Baldwin, G. Rees and A. Richardson, Sci. Total Environ., 2006, 370, 235–244 CrossRef CAS PubMed.
- Murray–Darling Basin Ministerial Council, 2011, Acid Sulfate Soils in the Murray–Darling Basin, 81 pp, Murray–Darling Basin Commission, Canberra, http://www.mdba.gov.au/media-pubs/publications/acid-sulfate-soils-murray-darling-basin, accessed 13 November 2015.
- D. S. Baldwin, Environ. Chem., 2013, 10, 439–454, DOI:10.1071/en13151.
- For example, see: D. S. Baldwin, A. M. Mitchell and J. M. Olley, Pollutant–sediment interactions: Sorption, reactivity and transport of phosphorus, in Agriculture, hydrology and water quality, ed. P. Haygarth and S. Jarvis, CAB International Press, New York, 2002, pp. 265–279 Search PubMed.
- R. Gächter, J. S. Meyer and A. Mares, Limnol. Oceanogr., 1988, 33, 1542–1558 CrossRef.
- E. E. Roden and J. W. Edmonds, Arch. Hydrobiol., 1997, 139, 1618–1628 Search PubMed.
- L. Lijklema, Environ. Sci. Technol., 1980, 14, 537–541 CrossRef CAS.
- J. A. McKeague and D. H. Day, Can. J. Soil Sci., 1966, 46, 13–22 CrossRef CAS.
- For example, see: H. F. Birch, Plant Soil, 1960, XII, 81–96 CrossRef.
- D. W. Schindler, Limnol. Oceanogr., 2006, 51, 356–363 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5em00523j |
| This journal is © The Royal Society of Chemistry 2016 |