
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The critical role of lithium nitrate in the gas evolution of lithium–sulfur batteries†
Anna
Jozwiuk
*a,
Balázs B.
Berkes
*a,
Thomas
Weiß
b,
Heino
Sommer
ab,
Jürgen
Janek
ac and
Torsten
Brezesinski
*a
aBattery and Electrochemistry Laboratory, Institute of Nanotechnology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: anna.jozwiuk@kit.edu; balazs.berkes@kit.edu; torsten.brezesinski@kit.edu
bBASF SE, 67056 Ludwigshafen, Germany
cInstitute of Physical Chemistry, Justus-Liebig-University Gießen, Heinrich-Buff-Ring 17, 35392 Gießen, Germany
First published on 13th June 2016
Abstract
Sulfur–carbon composites are promising next generation cathode materials for high energy density lithium batteries and thus, their discharge and charge properties have been studied with increasing intensity in recent years. While the sulfur-based redox reactions are reasonably well understood, the knowledge of deleterious side reactions in lithium–sulfur batteries is still limited. In particular, the gassing behavior has not yet been investigated, although it is known that lithium metal readily reacts with the commonly used ethereal electrolytes. Herein, we describe, for the first time, gas evolution in operating lithium–sulfur cells with a diglyme-based electrolyte and evaluate the effect of the polysulfide shuttle-suppressing additive LiNO3. The use of the combination of two operando techniques (pressure measurements and online continuous flow differential electrochemical mass spectrometry coupled with infrared spectroscopy) demonstrates that the additive dramatically reduces, but does not completely eliminate gassing. The major increase in pressure occurs during charge, immediately after fresh lithium is deposited, but there are differences in gas generation during cycling depending on the addition of LiNO3. Cells with LiNO3 show evolution of N2 and N2O in addition to CH4 and H2, the latter being the main volatile decomposition products. Collectively, these results provide novel insight into the important function of LiNO3 as a stabilizing additive in lithium–sulfur batteries.
Broader contextThe lithium–sulfur system is without doubt one of the most promising next generation battery technologies. However, there are still many open questions and issues related to both its stability and safety that need to be addressed before full commercialization of lithium–sulfur batteries can be achieved. Major issues are fast capacity degradation and gas generation due to the polysulfide shuttle and decomposition of electrolyte components, respectively, the latter being strongly related to the lithium metal anode. Lithium nitrate is typically used as a stabilizing additive, but its function in operating lithium–sulfur batteries is largely unknown. Here, we present data on the gassing behavior of practical lithium–sulfur cells from in operando pressure measurements as well as online continuous flow differential electrochemical mass spectrometry/infrared spectroscopy. The results help to better understand failure mechanisms and thus to develop new electrolyte components and additives to improve the safety and performance of lithium–sulfur batteries. |
1 Introduction
The lithium–sulfur (Li–S) system forms the chemical basis for one of the most promising next generation battery technologies with a high theoretical energy density of 2500 W h kg−1 (or 2800 W h L−1) and with sulfur as a cheap and abundant cathode material.1 It relies on the stepwise reaction of S8 through intermediate lithium polysulfides of different chain lengths (Li2Sx with 2 ≤ x ≤ 8) to Li2S during discharge and back to elemental sulfur during charge.2 Simultaneously, lithium is dissolved and re-deposited at the anode, respectively. In the past decade, research has been conducted mainly to improve the cathode design, e.g., by the synthesis of novel carbons and composite structures, with the goal to better trap the lithium polysulfide species and improve their interactions with the host material.3–8 This leads to higher sulfur utilization and therefore, higher specific capacities and longer battery life. In addition, investigations have been performed to further understand reaction and conversion mechanisms of lithium polysulfides during cycling through advanced in situ and operando techniques.2,9 Major challenges of Li–S batteries still are fast capacity fading caused by the polysulfide shuttle and decomposition of the ethereal electrolyte solvent(s),10–12 which are mainly related to the lithium metal anode. The most commonly used solvents are 1,3-dioxolane (DOL) and 1,2-dimethoxyethane (DME) as a mixture or longer chain glymes.13 The shuttle reaction, briefly, is a typical problem of conversion-type batteries where oxidized/reduced species diffuse to the opposite electrode site and are re-oxidized/re-reduced. Lithium nitrate (LiNO3) was found to be the most effective shuttle suppressor when used as an additive. Its degradation products form a protective passivation layer on lithium which results in higher Coulombic efficiencies.14,15 Unfortunately, LiNO3 is continuously consumed during cycling.16 Typically, the problems related to the anode are compensated by use of excess electrolyte. This, however, decreases the gravimetric energy density considerably. The majority of publications also report low sulfur loadings resulting in improved cycling performance by not stressing the anode as much. To reach energy densities comparable to or higher than the state of the art lithium-ion batteries, high sulfur loadings (≥5 mg cm−2) and low amounts of electrolyte (electrolyte/sulfur ratio ≤4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by weight) are needed.17,18 However, under such conditions, Li–S batteries usually fail below a hundred cycles because of severe anode corrosion.19
:1 by weight) are needed.17,18 However, under such conditions, Li–S batteries usually fail below a hundred cycles because of severe anode corrosion.19
In recent years, a few ex situ studies have been performed to gain a better understanding of failure mechanisms with respect to decomposition reactions at the anode. For example, Aurbach and coworkers investigated the decomposition products on lithium in the presence of DOL/LiTFSI/LiNO3 through Fourier transform infrared (FTIR) spectroscopy and X-ray photoelectron spectroscopy (XPS) and found poly-DOL-oligomers as well as LixNOy and LixSOy on the surface.14 The generation of the latter inorganic products was confirmed by a different study using XPS.20 This study also revealed that only in the presence of both lithium polysulfides and LiNO3 a relatively stable and compact solid-electrolyte interphase (SEI) on lithium is formed. Overall, the ex situ examination of reactive electrode surfaces is a major challenge (e.g., with respect to contamination from the atmosphere before and during analysis) and thus, interpretation of data should be carried out with care. To the best of our knowledge, no in situ study on electrolyte decomposition reactions in Li–S batteries has been reported in the literature so far. Particularly, monitoring of gas formation should provide a better insight into degradation processes. Only if decomposition products are clearly identified can a better understanding of failure mechanisms in Li–S cells be gained and with that, novel additives suggested or better electrolyte components developed to protect the lithium metal anode.
Herein, we investigate, for the first time, the gassing processes in operating Li–S cells using diglyme/LiTFSI electrolyte (with and without LiNO3) by means of pressure measurements as well as simultaneous online analysis of the gases by mass spectrometry (MS) and FTIR spectroscopy. For the gas analysis, we have designed a special battery cell, in which a typical anode/separator/cathode setup is implemented and have established a novel and unique flow-through differential electrochemical mass spectrometry (DEMS) system.21 Our approach to add a gas FTIR cell to this system—referred to as differential electrochemical infrared spectroscopy (DEIRS) in the following—helped to facilitate the identification of gas molecules.22 First, we show the gassing of Li–S cells with and without LiNO3 in general. Then, we focus on the nature of the gaseous products and on their generation patterns during cycling and open circuit voltage (OCV) periods. Isotopically labeled nitrate was used to correctly identify the evolution of N2 and N2O.
2 Results and discussion
To obtain full information on the gassing behavior of conventional Li–S batteries, we have used the combination of in operando DEMS-DEIRS and pressure measurements. The electrolyte solvent was chosen to be diglyme instead of the more commonly used mixture of DOL and DME. The shorter chain ethers are too volatile to be used in a flow through DEMS-DEIRS setup, like that established in our laboratory (diglyme has only 1/16th or 1/23rd of the vapor pressure of DME or DOL, respectively).21,22 However, various glymes have been used before and they give similar voltage profiles during charge and discharge.132.1 Pressure measurements
To investigate how much gas is formed during cycling, pressure measurements were performed first. Fig. 1 shows the pressure trends with respect to the individual cycling profile in diglyme using LiTFSI as the sole supporting salt and additionally containing LiNO3 as an additive.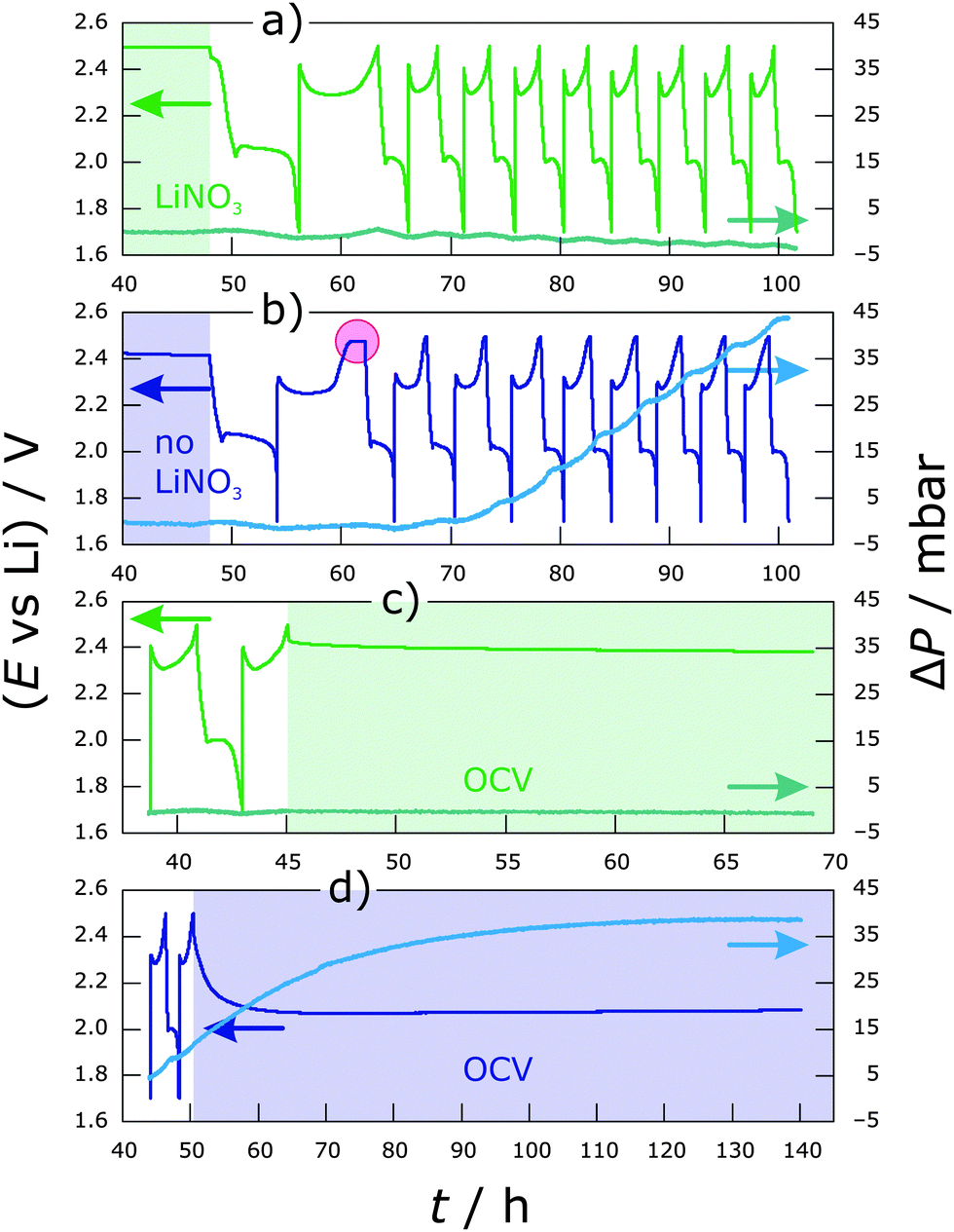 | ||
| Fig. 1 Pressure trends with dependence on the charge/discharge profile for Li–S cells using the diglyme/LiTFSI electrolyte with (a and c) and without LiNO3 (b and d). The red circle indicates an infinite charge region due to the polysulfide shuttle (charging is terminated by a time limit) and shaded areas represent OCV periods before and after cycling. | ||
After assembling, each system was allowed to equilibrate for 48 h prior to cycling to guarantee a stable pressure background (only the last 8 h of the OCV period are shown for clarity in Fig. 1a and b). ΔP was calculated from the absolute pressure according to: ΔP(t) = P(t) − P(40 h). This means the pressure after 40 h was subtracted from the measured values of P. Thus, for the sake of clarity, ΔP represents a relative value. Both cells show about the same pressure trend in the formation (first) cycle at C/10, with a small increase at the beginning of discharge as well as during the entire charge by about 2 mbar. Major differences can be seen upon further cycling at C/5. For the cell with LiNO3 (Fig. 1a), pressure always increases during charging (lithium deposition on the anode) by <1 mbar, while a decrease (>1 mbar) can be observed on discharge (see also Fig. S1, ESI† with a smaller ΔP range for more details). This results in an overall slight decrease of a few mbar after multiple cycles. A possible explanation for that could be that gases are produced spontaneously as soon as the electrolyte comes into contact with the fresh lithium anode, i.e., before the pressure sensor is connected to the cell. A decrease thereafter can only be explained by either dissolution of the gases within the electrolyte or consumption of certain gaseous products by reactions to form e.g., solid components or by the volume change of the electrodes. Another possibility is that sulfur from the cathode is being dissolved in the form of anionic species. This induces the dissolution of the lithium anode, causing a decrease in its volume and with that a decrease of the total pressure in the cell. The effect of volume change of the lithium anode on the pressure change in the system is shown in Fig. S2 (ESI†). In contrast, the slope of the pressure–time curve is always positive for the cell without LiNO3 from the 4th cycle onwards (Fig. 1b). The pressure increases significantly by 2–4 mbar (increasing with the cycle number) during a single charge step, and there is further pressure increase until the middle of discharge. Overall, an increase of >40 mbar is observed after 8 cycles—from the 4th cycle onwards it increases linearly with a rate of 1.5 mbar h−1. Applying the ideal gas law, an increase in pressure by 10 mbar in our cell (dead volume of ∼6 mL) corresponds to an amount of about 2.5 × 10−3 mmol or a gas volume of about 55 μL. Assuming a 1-electron process for gas evolution, this means a charge of 67 μA h. Fig. 1c and d show the pressure trend with respect to the voltage change during OCV after cycling. The pressure does not change significantly in the cell with LiNO3 and a stable voltage of 2.4 V is reached within less than 1 h (Fig. 1c and Fig. S1, ESI†). This suggests that a relatively stable SEI has been formed on lithium, thereby preventing shuttling or other decomposition reactions. On the other hand, when no LiNO3 is added, a further increase in pressure by ∼30 mbar is observed for >70 h, while the cell potential drops over time to 2.1 V (Fig. 1d). This indicates ongoing decomposition reactions.
2.2 DEMS-DEIRS measurements
To identify the gases formed during cycling, DEMS-DEIRS was conducted on nitrate-containing and nitrate-free Li–S batteries. Fig. 2 shows the ion currents for m/z = 2, 15 and 16 with respect to the voltage profile measured in cells with LiNO3. The m/z = 2 channel corresponds to H2 (beneath the H2 fragmented from the volatile diglyme in the MS). It is generated already in the initial OCV period (7 h for the DEMS-DEIRS experiments) due to the reaction of the electrolyte with lithium, later at the very beginning of the first discharge and during each subsequent charge cycle. It seems that less H2 is produced as the cell is cycled. During charge, a fresh lithium surface is created which could cause the reductive formation of H2 before the formation of the SEI layer. Another gaseous compound we were able to identify during cycling, though in small quantity, is CH4. The signals for m/z = 15 and 16 indicate its existence, and the characteristic FTIR vibrations (Fig. 4, inset) unequivocally identify it. Both the MS and FTIR data show very similar behavior. The dramatic change of the MS signals during the first OCV period and in the first cycle is caused by electrolyte signal fluctuation (m/z = 45 and 59, also fragments of diglyme, follow the very same trend) and the presence of O2 that is gradually purged out.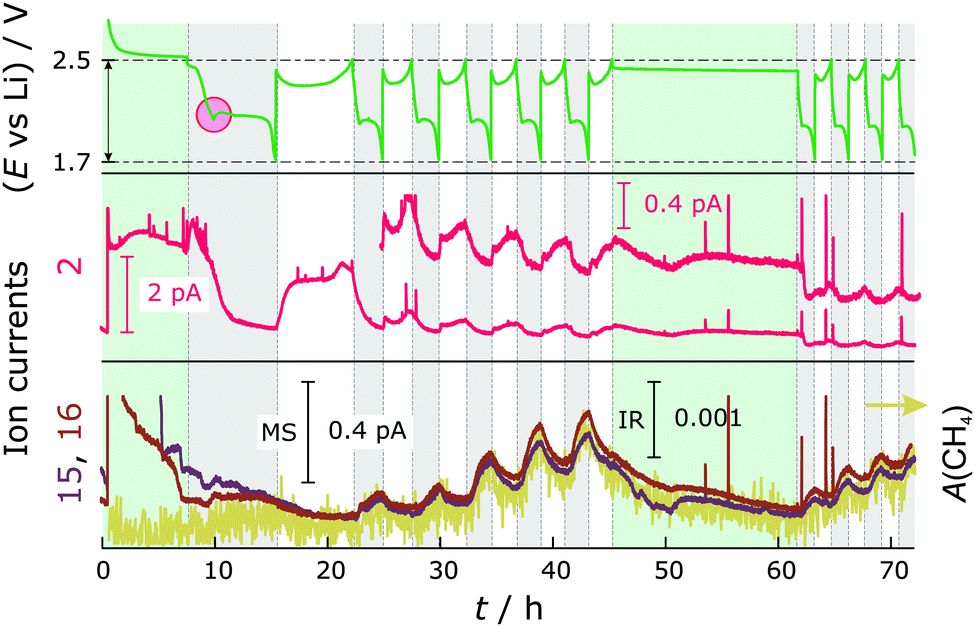 | ||
| Fig. 2 Top: Voltage profile of a Li–S cell with LiNO3. Middle: MS ion current for m/z = 2. Bottom: MS ion current for m/z = 15 and 16 as well as the integrated FTIR signal (A(CH4)) of the Q-branch of CH4 (yellow) at 3015 cm−1. The red circle indicates a voltage dip between the discharge plateaus. Green shaded areas represent OCV periods, while discharge and charge regions are highlighted in grey and white, respectively. | ||
There is virtually no generation of CH4 until the second cycle and this is further supported by FTIR, making the simultaneous use of vibrational spectroscopy even more powerful. CH4 evolves in an alternating fashion to H2 in each discharge, but not from the beginning (strong increase from the voltage dip between the discharge plateaus), and increases with the cycle number. The voltage dip indicates supersaturation of S2− and an increased electrolyte resistance.23,24 It is likely that the reactive Li is protected by an SEI layer formed through reactions with LiNO3 since the cell without LiNO3 shows more pronounced gassing. Therefore, the reductive decomposition of the electrolyte solvent (diglyme) occurs at the cathode side. The cathode still shows a quite negative potential even in a fully charged state with reference to the standard hydrogen electrode (ca. −500 mV). However, the most intense CH4 evolution can be observed with decreasing cathode potential, also supporting this assumption. At the beginning, the electrocatalytically active carbon surface is blocked by the impregnated sulfur that gradually dissolves during cycling, making reactive surfaces available. The generation of H2 could be either due to remaining traces of H2O in the cell components/electrolyte or a decomposition product of diglyme. However, H2O contamination presumably contributes only initially to the H2 evolution. We hypothesize that in addition to the formation of CH4 an alcohol (e.g., methanol or longer chain alcohol) is formed in the reduction process of diglyme during discharge. This “reaction product” can be reduced to form H2 in the next charge cycle. We note that the SEI might not only be Li+ conducting, but also H+ conducting. CH4 is most likely generated from the methyl group of diglyme. Similarly, CH4 should be generated from DME in DOL/DME-based electrolytes. To test whether the SEI on lithium really prevents further decomposition reactions, an OCV period of 16 h was added after cycling. As in the pressure measurements, the cell potential stabilized quickly at 2.4 V and the signals for H2 and CH4 decreased. Thereafter, the battery was cycled for a few more hours and the trends for CH4 and H2 gas evolution are very similar to the previous cycles.
We also observe some trends for ions with m/z = 28 and 44 with Li–S cells containing LiNO3 during cycling. The corresponding gases could be N2 or CO (m/z = 28) and N2O or CO2 (m/z = 44). To reveal the identity and origin of the gases concealed by these masses, experiments with isotopically labeled nitrate (Li15NO3 and Li15N18O3) were conducted (Fig. 3). The use of labeled nitrate identified the gases as N2 (m/z = 28 for N2 or 30 for 15N2) and N2O (m/z = 44 for N2O, 46 for 15N2O or 48 for 15N218O) and at the same time proves their origin from the added LiNO3 and not from contamination, e.g., air leaks or H2O in the electrolyte. These results should help to further understand the role and mechanism of the polysulfide shuttle-suppressing additive in Li–S batteries. During each charge, especially increasing towards the end of the charge cycle, N2 and N2O are generated, meaning that the nitrogen from nitrate is reduced. Their formation overlaps exactly with that of H2 gas during cycling. A feasible side product could be Li2O, which deposits on lithium and is a part of the forming SEI. It is possible that N2 and N2O further react with lithium to create Li3N and LixNOy species.14,20 This could explain why the pressure decreases during discharge in the nitrate-containing system.
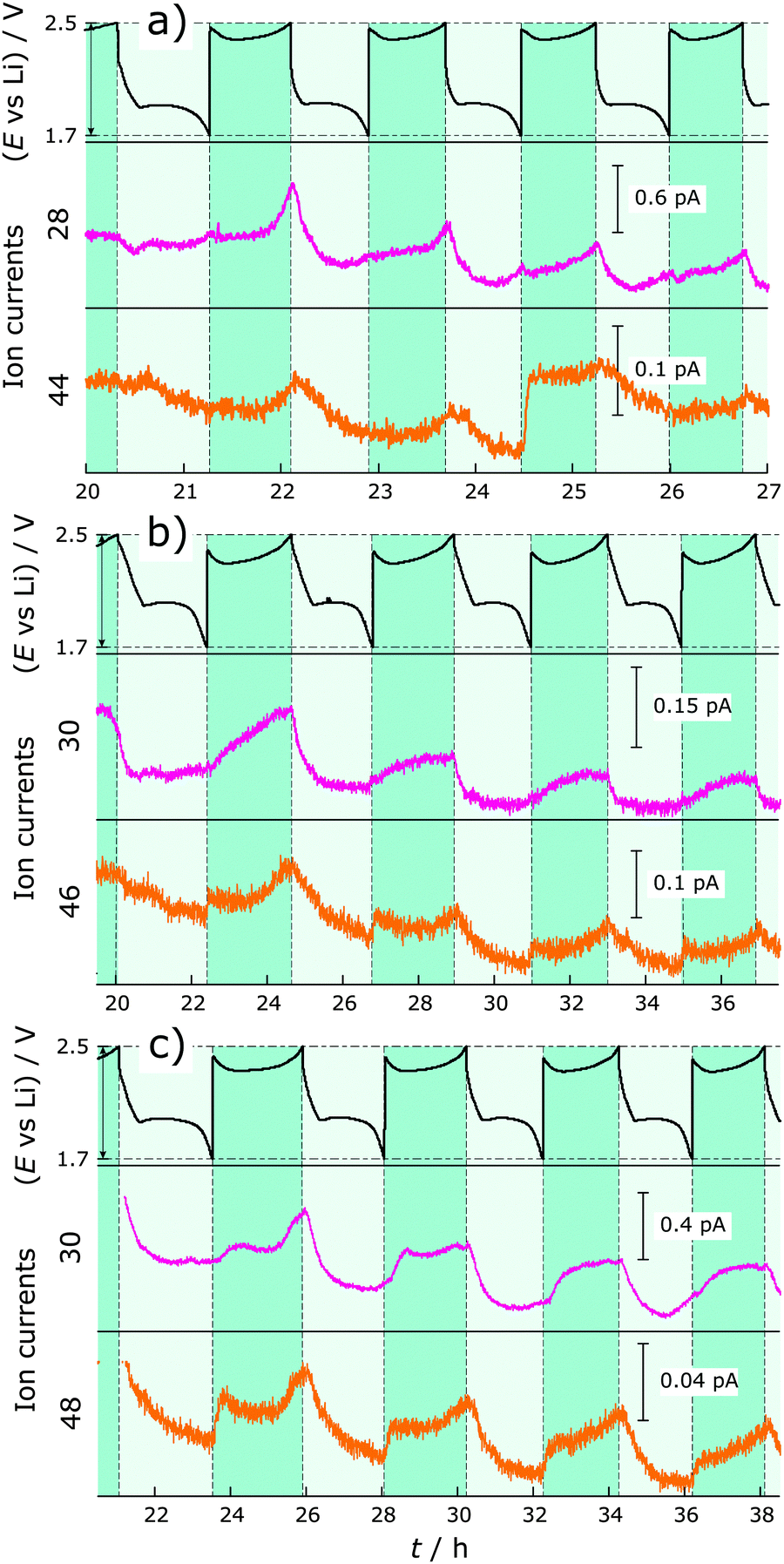 | ||
| Fig. 3 DEMS measurements on Li–S cells with additive: LiNO3 (a), Li15NO3 (b) and Li15N18O3 (c). Top: Voltage profile. Middle: MS ion current for N2 or 15N2 with m/z = 28 or 30, respectively. Bottom: MS ion current for N2O, 15N2O or 15N218O with m/z = 44, 46 or 48, respectively. Discharge and charge regions are highlighted in grey and cyan, respectively. | ||
In the experiments without LiNO3, the generation of H2 and CH4 is much more pronounced (higher signal to noise ratio of the signals). Fig. 4 shows the trend of the MS and FTIR signals. Similar to the Li–S cells with LiNO3, in the first cycle, H2 is produced at the beginning of discharge and during the entire charge. In subsequent cycles, H2 is evolved further and the amount seems to gradually exceed that which can effectively be purged out. There is a much steeper increase of H2 generation towards the end of charge than in cells containing additives. This might be because there is no protective layer on the freshly deposited lithium, unlike in the case with additive. Also different is the appearance of a H2 peak at the voltage dip of discharge. CH4 is again produced mainly during discharge, but in higher amounts. As is evident, its evolution contributes significantly to the pressure increase shown in Fig. 1. We also observe further increased CH4 production during the OCV period after cycling, which is consistent with the pressure trend for the cells without LiNO3. Without LiNO3 the SEI formed on Li (if at all) is less stable. Most probably, Li reacts instantaneously with the electrolyte when assembling the cell. During electrochemical cycling, Li becomes more "reactive'' due to continuous dissolution/deposition which increases the surface area. Without the protective SEI layer, diglyme readily reacts with Li, which is probably more reactive when charging the battery (Li deposition on the anode)—with that the signal of CH4 increases in the charge cycle. As evident from the pressure measurements and MS/IR data, the reduction of diglyme continues in the OCV period (increasing pressure and methane signal). Therefore, it must be a chemical process occurring on lithium. Eqn (1)–(3) below describe possible reactions of LiNO3 in the system—which are likely to be spontaneous according to thermodynamic calculations—and Scheme 1 summarizes the gassing behavior in cells with and without additive.
| LiNO3 + 2Li → Li2O + LiNO2 ΔrG = −472.56 kJ mol−1 | (1) |
| 8LiNO2 + S8 → 4Li2S2O3 + 4N2O | (2) |
| N2O + 2Li → N2 + Li2O ΔrG = −664.90 kJ mol−1 | (3) |
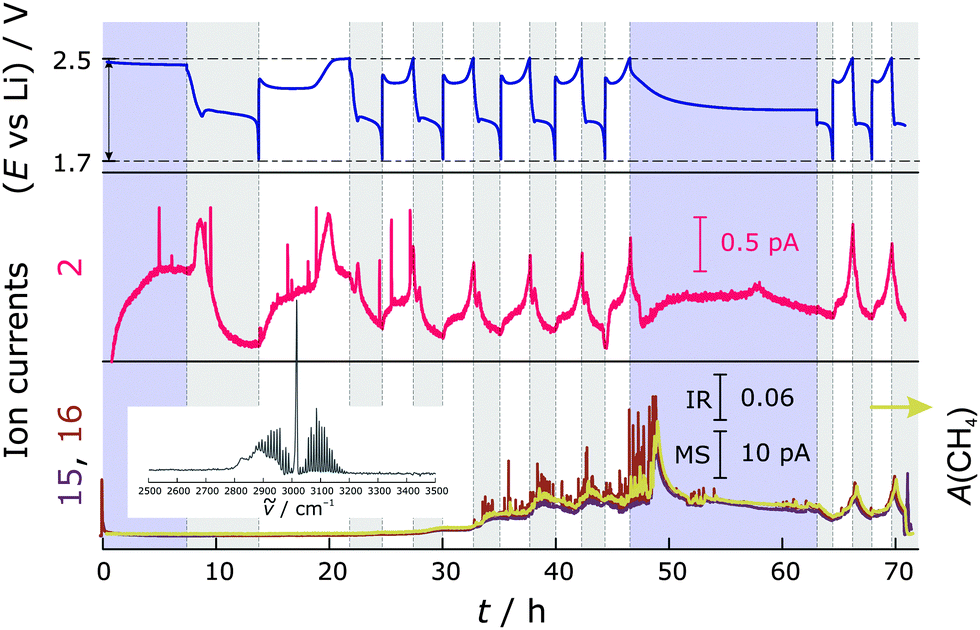 | ||
| Fig. 4 Top: Voltage profile of a Li–S cell without LiNO3. Middle: MS ion current for m/z = 2. Bottom: MS ion current for m/z = 15 and 16 as well as the integrated FTIR signal (A(CH4)) of the Q-branch of CH4 (yellow) at 3015 cm−1. Inset: FTIR spectrum of CH4 measured with the DEIRS setup at maximum concentration. Purple shaded areas represent OCV periods, while discharge and charge regions are highlighted in grey and white, respectively. | ||
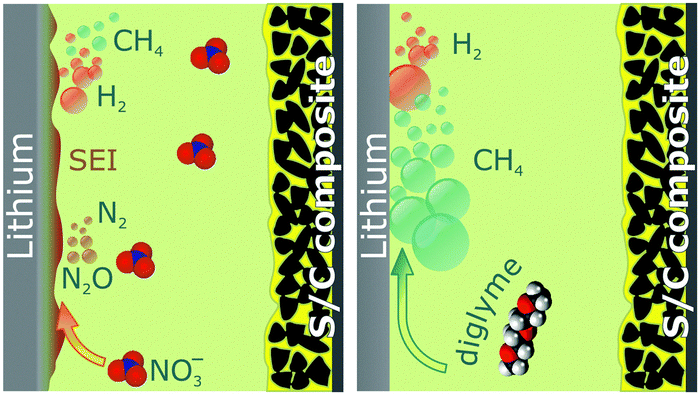 | ||
| Scheme 1 Overview of the reactions producing gases in Li–S batteries with the diglyme-based electrolyte with (left) and without LiNO3 (right). | ||
According to eqn (2), sulfur reacts with LiNO2. Given that sulfur is formed in the charge cycle, the formation of N2O, which reacts further with Li to give N2 (eqn (3)), is expected to occur during charge, in line with our findings. A reaction similar to that in eqn (2) has already been reported by Schmidt et al.25 They studied the conversion of NaNO2 to Na2S2O3 through reaction with S8 in a non-aqueous medium (see also gas evolution in symmetric Li–Li cells with and without the LiNO3 additive in Fig. S3, ESI†). In addition, we hypothesize the formation of methyl radicals that can further react with the electrolyte to produce CH4. However, the mechanism of this reaction might be very complex. Unveiling goes beyond the scope of the paper and further experiments are needed. Because H2 formation is observed during the entire cycling process, it does not originate solely from water residues. It is more likely that a reaction product—e.g., an alcohol—forms during discharge (e.g., beneath CH4) and further reacts to form H2 during charge.
Overall, the DEMS-DEIRS results are in good agreement with the pressure measurements. However, care should be taken when comparing the details. The system for pressure measurements is closed and gases can potentially further react to form other products, which may influence the pressure change. On the other hand, the flow-through cell used for DEMS-DEIRS is continuously purged and all gases, once generated, have little time for follow-up reactions.
Besides CH4 and H2, we were able to find other gaseous decomposition products for some experiments independent of LiNO3 addition, like H2S (m/z = 32, 33 and 34) and SO2 (m/z = 64 and 48 and characteristic vibrations in the FTIR spectrum). However, there was no clear pattern or trend on why these gases occasionally show up. It is very likely that the generation of H2S and SO2 is related to the total amount of H2O contamination in the battery cell and further experiments are needed to investigate this relationship.
3 Conclusions
This study reveals, for the first time, gas evolution in operating lithium–sulfur batteries. CH4 and H2 were identified as the major gaseous decomposition products, particularly during charging when a fresh lithium surface is created at the anode. We show that gassing is suppressed when LiNO3 is used as an electrolyte additive. Especially, the amount of CH4 is dramatically decreased and either very little or no H2 is generated during discharge. Further N2 and N2O evolve in the charge cycles when LiNO3 is present. Their generation is probably related to the formation of a relatively stable SEI on lithium and with that, suppression of the polysulfide shuttle. From our pressure measurements, it seems that these gases are consumed and possibly built into the SEI. Therefore, it would be interesting to test whether just simple addition of N2 and/or N2O, instead of LiNO3, will give the same positive or even a better effect during cycling. Some measurements also revealed H2S evolution independent of LiNO3 addition. Its appearance could be related to the H2O traces in the battery cell, but further work is needed to confirm this hypothesis.4 Experimental
4.1 Materials and electrode preparation
For the preparation of the positive electrode, sulfur (Aldrich, reagent grade), Super C65 (Timcal) and Printex-XE2 (Orion) in a ratio of 1.7:0.5:0.5 were ground. Poly(vinyl alcohol) Selvol 425 (Sekisui) dissolved in a mixture of water, isopropanol and 1-methoxy-2-propanol was added to the blend and the mixture was ball-milled for 20 h to form a slurry. The slurry was then coated onto 8 μm-thick primed aluminum and dried in a vacuum at 40 °C for 16 h. The sulfur content in the electrode was 60%, with sulfur loadings of 1.7–1.8 mg cm−2. Coin-type cells were assembled in an argon-filled glovebox by stacking a lithium anode (China Lithium Ltd, size: 600 μm × 40 mm), a GF/A separator (GE Healthcare Life Sciences, Whatman, 42 mm diameter) and a cathode (40 mm diameter with a 4 mm hole in the center) using 600 μL of electrolyte. The electrolyte was either a solution of 0.325 M lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, Aldrich, 99.95%) and 0.675 M lithium nitrate (Merck, 99.995%) or just 1 M lithium bis(trifluoromethanesulfonyl)imide in bis(2-methoxyethyl)ether (diglyme, Aldrich, 99.95%). The water content of all electrolytes used was <20 ppm, except that the electrolyte prepared with Li15N18O3 had 120 ppm, as determined by Karl Fischer titration. Li15NO3 was prepared by addition of 1.1 eq. of Li2CO3 to a diluted aqueous solution of H15NO3 (Aldrich, 98 at% 15N). Once gas evolution was completed, solid byproducts were removed from the reaction solution by filtration and the solvent was removed under reduced pressure. The remaining white solid (Li15NO3) was dried at 180 °C for 24 h yielding 81–96%. Li15N18O3 was prepared from H15N18O3 (Aldrich, 95 at% 18O, 98 at% 15N) according to the procedure for Li15NO3, yielding 61%.
4.2 Electrochemical testing
Electrochemical cycling was performed in the potential range of 1.7–2.5 V with respect to Li/Li+ using a BioLogic VSP-300 potentiostat. The batteries were kept at rest (OCV) for several hours prior to cycling to allow for equilibration. This resulted in a similar initial potential of each cell and therefore, comparable conditions. The OCV period was set to 7 h for DEMS-DEIRS experiments and 48 h for pressure measurements (to guarantee full equilibration by temperature). Pressure measurements were performed in a temperature-controlled cabinet (ESPEC Corp. PU-1KP) set to 25 °C. After one cycle at C/10 (with 1C = 1672 mA gsulfur−1) was completed, the cells were cycled at charge and discharge rates of C/5. For cells without lithium nitrate, a time limit in addition to the potential limit was set to terminate the charge step and avoid shuttling.264.3 Instrumentation
Both the DEMS setup and battery design have been described elsewhere.21,22 The same cell was used for pressure and DEMS-DEIRS measurements. A commercially available quadrupole mass spectrometer with a scan range of 1–200 Da and detection limits down to 10 ppb and a secondary electron multiplier detector was used for gas analysis (GSD 320 O2, OmniStar Gasanalysesystem, Pfeiffer Vacuum GmbH, Germany). DEIRS measurements were performed using an FTIR spectrophotometer (Bruker Tensor II) equipped with a heatable gas cell module.22 The FTIR signal in Fig. 2 and 4 is the integrated area of the Q-branch of CH4 at 3010–3021 cm−1. For pressure measurements, one of the cell lid holes was closed with a screw, while the other one was equipped with a pressure sensor (Omega PAA33X-V-3).Acknowledgements
We thank Sven Leuthner for preparation of the materials and electrolytes. This study is a part of the projects being funded within the BASF International Network for Batteries and Electrochemistry.References
- S. Evers and L. F. Nazar, Acc. Chem. Res., 2013, 46, 1135–1143 CrossRef CAS PubMed.
- M. Wild, L. O'Neill, T. Zhang, R. Purkayastha, G. Minton, M. Marinescu and G. J. Offer, Energy Environ. Sci., 2015, 8, 3477–3494 Search PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 50, 5904–5908 CrossRef CAS PubMed.
- J. T. Lee, Y. Zhao, S. Thieme, H. Kim, M. Oschatz, L. Borchardt, A. Magasinski, W.-I. Cho, S. Kaskel and G. Yushin, Adv. Mater., 2013, 25, 4573–4579 CrossRef CAS PubMed.
- J. Song, T. Xu, M. L. Gordin, P. Zhu, D. Lv, Y.-B. Jiang, Y. Chen, Y. Duan and D. Wang, Adv. Funct. Mater., 2014, 24, 1243–1250 CrossRef CAS.
- A. Schneider, C. Weidmann, C. Suchomski, H. Sommer, J. Janek and T. Brezesinski, Chem. Mater., 2015, 27, 1674–1683 CrossRef CAS.
- G. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Nat. Commun., 2015, 6, 7760 CrossRef CAS PubMed.
- R. Xu, J. Lu and K. Amine, Adv. Energy Mater., 2015, 5, 1500408 CrossRef.
- Y. Diao, K. Xie, S. Xiong and X. Hong, J. Power Sources, 2013, 235, 181–186 CrossRef CAS.
- D. Moy, A. Manivannan and S. R. Narayanan, J. Electrochem. Soc., 2015, 162, A1–A7 CrossRef CAS.
- M. Hagen, P. Fanz and J. Tübke, J. Power Sources, 2014, 264, 30–34 CrossRef CAS.
- M. Barghamadi, A. S. Best, A. I. Bhatt, A. F. Hollenkamp, M. Musameh, R. J. Rees and T. Ruther, Energy Environ. Sci., 2014, 7, 3902–3920 Search PubMed.
- D. Aurbach, E. Pollak, R. Elazari, G. Salitra, C. S. Kelley and J. Affinito, J. Electrochem. Soc., 2009, 156, A694–A702 CrossRef CAS.
- S. S. Zhang and J. A. Read, J. Power Sources, 2012, 200, 77–82 CrossRef CAS.
- A. Jozwiuk, H. Sommer, J. Janek and T. Brezesinski, J. Power Sources, 2015, 296, 454–461 CrossRef CAS.
- M. Hagen, D. Hanselmann, K. Ahlbrecht, R. Maça, D. Gerber and J. Tübke, Adv. Energy Mater., 2015, 5, 1401986 CrossRef.
- D. Eroglu, K. R. Zavadil and K. G. Gallagher, J. Electrochem. Soc., 2015, 162, A982–A990 CrossRef CAS.
- Y. Han, X. Duan, Y. Li, L. Huang, D. Zhu and Y. Chen, Mater. Res. Bull., 2015, 68, 160–165 CrossRef CAS.
- S. Xiong, K. Xie, Y. Diao and X. Hong, J. Power Sources, 2014, 246, 840–845 CrossRef CAS.
- B. B. Berkes, A. Jozwiuk, M. Vračar, H. Sommer, T. Brezesinski and J. Janek, Anal. Chem., 2015, 87, 5878–5883 CrossRef CAS PubMed.
- B. B. Berkes, A. Jozwiuk, H. Sommer, T. Brezesinski and J. Janek, Electrochem. Commun., 2015, 60, 64–69 CrossRef CAS.
- K. Kumaresan, Y. Mikhaylik and R. E. White, J. Electrochem. Soc., 2008, 155, A576–A582 CrossRef CAS.
- T. Zhang, M. Marinescu, L. O'Neill, M. Wild and G. Offer, Phys. Chem. Chem. Phys., 2015, 17, 22581–22586 RSC.
- M. Schmidt and R. R. Wägerle, Angew. Chem., 1958, 70, 594–595 CrossRef CAS.
- A. F. Hofmann, D. N. Fronczek and W. G. Bessler, J. Power Sources, 2014, 259, 300–310 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ee00789a |
| This journal is © The Royal Society of Chemistry 2016 |