
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Morphological and electrical control of fullerene dimerization determines organic photovoltaic stability†
Thomas
Heumueller
ab,
William R.
Mateker
b,
Andreas
Distler
c,
Urs F.
Fritze
d,
Rongrong
Cheacharoen
b,
William H.
Nguyen
b,
Markus
Biele
a,
Michael
Salvador
a,
Max
von Delius
d,
Hans-Joachim
Egelhaaf
e,
Michael D.
McGehee
*b and
Christoph J.
Brabec
*aef
aMaterials Science and Engineering, FAU Erlangen-Nürnberg, Germany. E-mail: christoph.brabec@fau.de
bMaterials Science and Engineering, Stanford University, California, USA. E-mail: mmcgehee@stanford.edu
cBelectric GmbH, Germany
dDepartment of Chemistry and Pharmacy, FAU Erlangen-Nürnberg, Germany
eEnergy Campus Nürnberg, Germany
fBavarian Center for Applied Energy Research, Erlangen, Germany
First published on 25th November 2015
Abstract
Fullerene dimerization has been linked to short circuit current (Jsc) losses in organic solar cells comprised of certain polymer–fullerene systems. We investigate several polymer–fullerene systems, which present Jsc loss to varying degrees, in order to determine under which conditions dimerization occurs. By reintroducing dimers into fresh devices, we confirm that the photo-induced dimers are indeed the origin of the Jsc loss. We find that both film morphology and electrical bias affect the photodimerization process and thus the associated loss of Jsc. In plain fullerene films, a higher degree of crystallinity can inhibit the dimerization reaction, as observed by high performance liquid chromatography (HPLC) measurements. In blend films, the amount of dimerization depends on the degree of mixing between polymer and fullerene. For highly mixed systems with very amorphous polymers, no dimerization is observed. In solar cells with pure polymer and fullerene domains, we tune the fullerene morphology from amorphous to crystalline by thermal annealing. Similar to neat fullerene films, we observe improved light stability for devices with crystalline fullerene domains. Changing the operating conditions of the investigated solar cells from Voc to Jsc also significantly reduces the amount of dimerization-related Jsc loss; HPLC analysis of the active layer shows that more dimers are formed if the cell is held at Voc instead of Jsc. The effect of bias on dimerization, as well as a clear correlation between PL quenching and reduced dimerization upon addition of small amounts of an amorphous polymer into PC60BM films, suggests a reaction mechanism via excitons.
Broader contextOrganic photovoltaics (OPV) enable light weight, flexible and semi-transparent devices with the potential for large scale power production. After a recently established world record of 11.5% power conversion efficiency for single junction organic bulk heterojunction solar cells and amplified interest in applications like building integrated OPV, the device stability is turning into key factor for the success of this technology. While highly crystalline donor polymers with relatively pure fullerene phases currently achieve the highest efficiencies, these pure fullerene phases can be prone to dimerization that causes performance losses. We investigate a wide range of polymer:fullerene blend systems and demonstrate how dimerization can be controlled by tuning the morphology of fullerene domains, leading to improved solar cell stability. |
Introduction
Light induced dimerization of PC60BM1,2 has been shown to cause severe short circuit current (Jsc) losses of organic photovoltaic (OPV) cells during operation in a few cases.3,4 In apparent contradiction the most studied OPV system,5 optimized P3HT:PC60BM devices, do not suffer significant dimerization-related Jsc losses.6 To understand why fullerene dimerization causes severe performance losses in some cases, but is not significant in other cases, we study the factors that govern fullerene dimerization and the related short circuit current losses in multiple bulk heterojunction solar cell blends.Photoinduced transformation of fullerenes into dimers, oligomers and polymers was first studied by Rao et al. in 1993.7 Using laser desorption mass spectrometry, clusters of cross-linked fullerene molecules (C60)N with N up to 20 were found in photoirradiated fullerene films.7,8 Raman spectroscopy of cross-linked fullerenes shows a shift of the characteristic “pentagonal pitch” mode from 1469 to 1460 cm−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7,9–11 and high resolution infrared spectroscopy measurements suggest that dimers12,13 are the main reaction product for photo-crosslinking of fullerenes.14–16 After dimerization, a reduced room temperature electron mobility of C60 films was observed.10,17
7,9–11 and high resolution infrared spectroscopy measurements suggest that dimers12,13 are the main reaction product for photo-crosslinking of fullerenes.14–16 After dimerization, a reduced room temperature electron mobility of C60 films was observed.10,17
More recently, photo-induced dimerization of PC60BM has been observed in organic photovoltaic (OPV) devices. Dimers in plain PC60BM can be detected using Raman1,18,19 or FTIR20 spectroscopy. In polymer–fullerene blends, overlapping peaks21 complicate the analysis and make liquid chromatography3,18 the method of choice. A less direct, but more facile method to monitor dimerization is UV-vis absorption where a characteristic feature around 320 nm can be observed in dimerized fullerenes.3,4 Dimerization of fullerenes22 and crosslinking23,24 in general is a pathway to prevent morphological changes25–27 and stabilize the bulk heterojunction (BHJ) morphology. Durrant18 and Manca19,28 independently showed that illumination of polymer–fullerene blends significantly reduced large scale phase separation upon thermal annealing. Morphological control using dimers was also observed to improve performance in some cases.29,30 Conversely, dimerization was found to deteriorate the electronic properties of organic solar cells during operation in the light.3 In polymer–fullerene bulk heterojunctions3,31 and planar small molecule–fullerene devices,32 significant Jsc losses were observed due to photo-induced dimerization in the absence of oxygen. Reduced charge carrier mobility due to trap formation3,33 and reduced exciton lifetimes20 are proposed to explain these performance losses. Dimerization was shown to be negligible for bis-PC60BM due to steric hindrance by the substituents.3 Steric hindrance is also suggested to be the reason why PC60BM mainly forms dimers and not higher oligomers.1
Herein, we report an investigation of the amount of light-induced dimers formed in multiple polymer–fullerene blends processed and aged under different conditions. After directly linking fullerene dimerization with light induced Jsc losses, the Jsc losses are investigated in several polymer–fullerene systems. While optimized P3HT:PC60BM devices cast from chlorobenzene do not show photo-induced Jsc losses, we observe significant dimerization in P3HT:PC60BM solar cells cast from chloroform. The possibility to turn light induced Jsc losses on and off by changing only the processing conditions demonstrates the effect of morphology on fullerene dimerization. Quantifying the amount of dimers formed in crystalline and amorphous fullerene films with high performance liquid chromatography (HPLC) reveals a morphological control of fullerene dimerization that is also observed in solar cells annealed for different times. Besides morphology, the electrical bias during illumination and the degree of polymer–fullerene mixing are identified as critical factors controlling fullerene dimerization. These observations allow us to understand under which circumstances fullerene dimerization will affect OPV stability and how stability can be improved.
Dimerization induced Jsc losses in different polymer–fullerene systems
Before discussing the factors determining fullerene dimerization, we first show that fullerene dimers are able to directly cause severe short circuit current losses in organic solar cells. Adding fullerene dimer in fresh solar cells during cell production reproduces the Jsc losses observed in aged solar cells. We isolated PC60BM dimers from photo-irradiated PC60BM films using semi-preparative high performance liquid chromatography (HPLC) and added the material to a solution of KP115 (PDTSTzTz)3,33,34 blend with PC60BM before spin coating active layers for solar cells. In such cells, we observed a Jsc reduction of 26% when adding 16% of dimer to the fullerene content, compared to a reference solar cell fabricated with only fresh PC60BM (Fig. 1).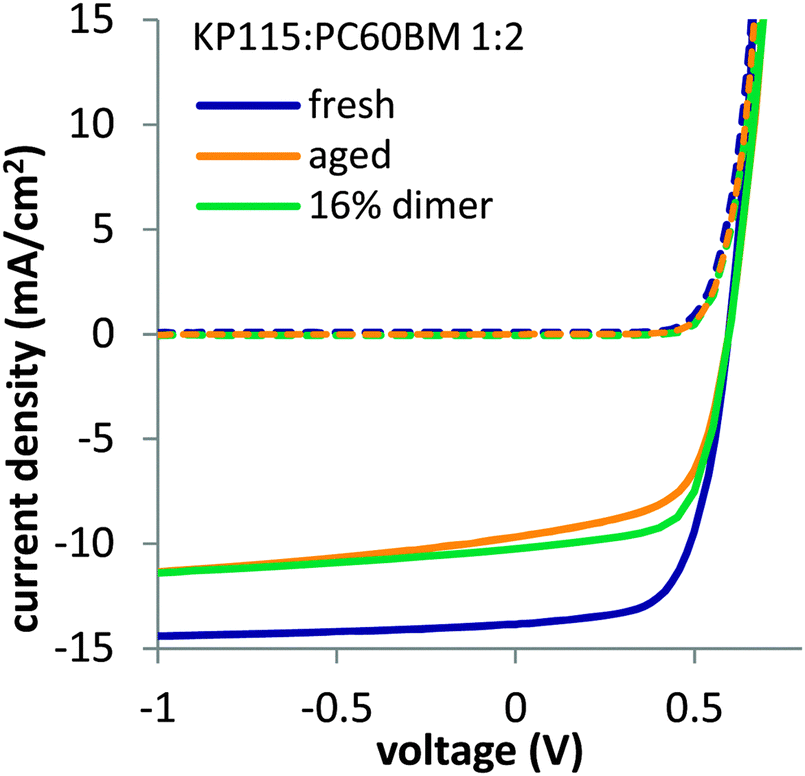 | ||
| Fig. 1 Light (solid lines) and dark (dashed lines) JV curves of fresh and aged (120 h) KP115:PC60BM solar cells along with KP115:PC60BM solar cells containing 16% PC60BM dimers, added during cell production. PC60BM dimers were isolated using HPLC from photo-irradiated PC60BM films. The structure of KP115 (PDTSTzTz)3,33,34 is given in the ESI.† | ||
The shape of the IV curve for the device with added dimer is very similar to the IV curve of a solar cell aged under one sun equivalent LED illumination for 120 hours. There is no significant Voc loss observed when using dimerized PC60BM, suggesting that the charge transfer energy is not significantly altered, which is consistent with sensitive photocurrent spectroscopy measurements (FTPS, see ESI†). Averaged values of more than 10 devices are shown in the ESI.† Also when adding dimer to P3HT:PC60BM solar cells, we observed a strong Jsc loss, while Voc was barely affected (see ESI†). We thus conclude that fullerene dimerization has no significant effect on the open circuit voltage but directly impacts the Jsc of organic solar cells.
To investigate which factors affect the dimerization related Jsc losses, we performed burn-in tests for a variety of polymer–fullerene systems under inert conditions with a one sun equivalent white light source. The solar cells were held at open circuit conditions between periodic IV measurements and were cast from 1,2-dichlorobenzene (DCB) unless noted otherwise. A comparison of the amount of Jsc loss after 40 hours reveals significant differences between similar systems (Fig. 2).
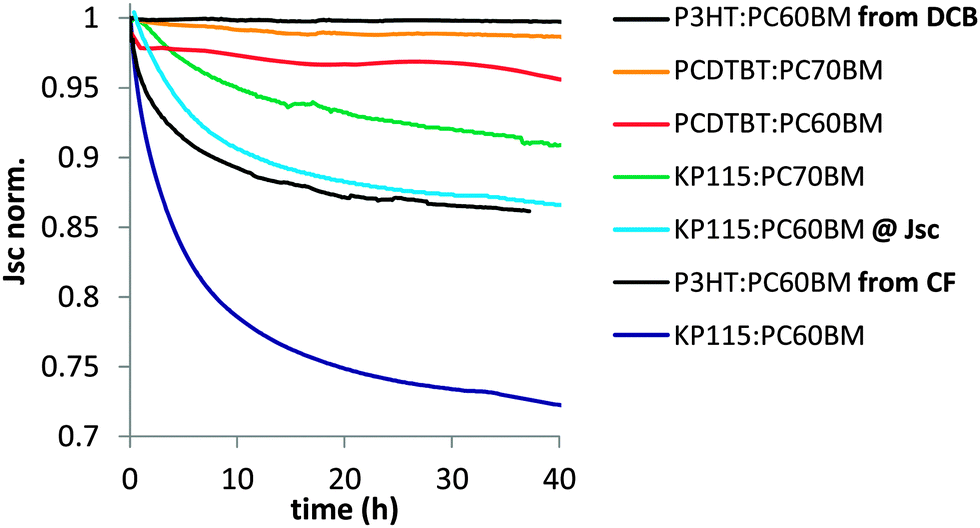 | ||
| Fig. 2 Normalized Jsc losses during illumination for a variety of polymer–fullerene solar cells. P3HT:PC60BM loses much more Jsc when cast from chloroform instead of dichlorobenzene. All other devices were cast from dichlorobenzene. For PCDTBT35 and KP115 reduced Jsc losses for PC70BM compared to PC60BM are observed. Operating a KP115:PC60BM solar cell at Jsc instead of Voc significantly reduces the Jsc losses. One representative device is shown for each system/condition. | ||
There is a wide spread of dimerization related Jsc loss among different material systems and even for the same material systems when processed or aged in different ways. We make several important observations that are listed here and will be discussed in the following sections. First, when casting P3HT:PC60BM devices from chloroform instead of chlorobenzene, we do observe substantial photo-induced Jsc losses (Fig. 2) that are not present in optimized devices. As the used solvent affects film formation, this observation suggests that the fullerene morphology plays an important role in the dimerization process. Second, changing the electrical bias from Voc to Jsc significantly reduces the Jsc losses in KP115:PC60BM solar cells. This is also true for all other systems investigated and suggests that the dimerization reaction proceeds via excited species that are present in higher concentrations at Voc conditions. Third, there is a wide spread of Jsc losses between solar cells that contain the same fullerene, PC60BM, but are made from different polymers. As the crystallinity of the used polymers and the consequent film morphology is very different, the degree of polymer–fullerene mixing seems to be critical for fullerene dimerization. Finally, a clear reduction of the Jsc losses is observed for KP115 when changing the fullerene from PC60BM to PC70BM. This is consistent with a reduced tendency of the C70 framework to dimerize2,4 and is also observable in PCDTBT. The PCDTBT device made with PC70BM shows barely any Jsc losses, which partially explains the high photovoltaic stability6,36,37 reported for this material system.
Effect of fullerene packing on dimerization in PC60BM films
Since the strong dependence of Jsc losses on the casting solvent in P3HT suggests a morphological effect, we study as cast and crystalline fullerene films as a model system for amorphous and crystalline fullerene domains. Recently, it was observed that the morphology of organic films has a strong impact on the photostability.38,39 Amorphous films of PC60BM photobleach four times faster than crystalline films of the same material, most likely since geometric constraints in dense ordered films reduce the reactivity.39 We propose that such morphological effects on chemical reactivity are also at play for photo-induced fullerene dimerization, causing crystalline fullerene to dimerize less than amorphous fullerene. To test this hypothesis, we used high performance liquid chromatography (HPLC) to investigate the amount of photo-induced dimers in amorphous and crystalline PC60BM films (Fig. 3). An amorphous film as cast from chloroform and a film that was annealed for 10 minutes at 180 °C to crystallize PC60BM were illuminated simultaneously under a white light LED with 1 sun light intensity for 40 hours. The experiments were conducted under nitrogen atmosphere since oxygen is known to prevent photo-induced dimerization.40 A third film was kept in the dark as reference.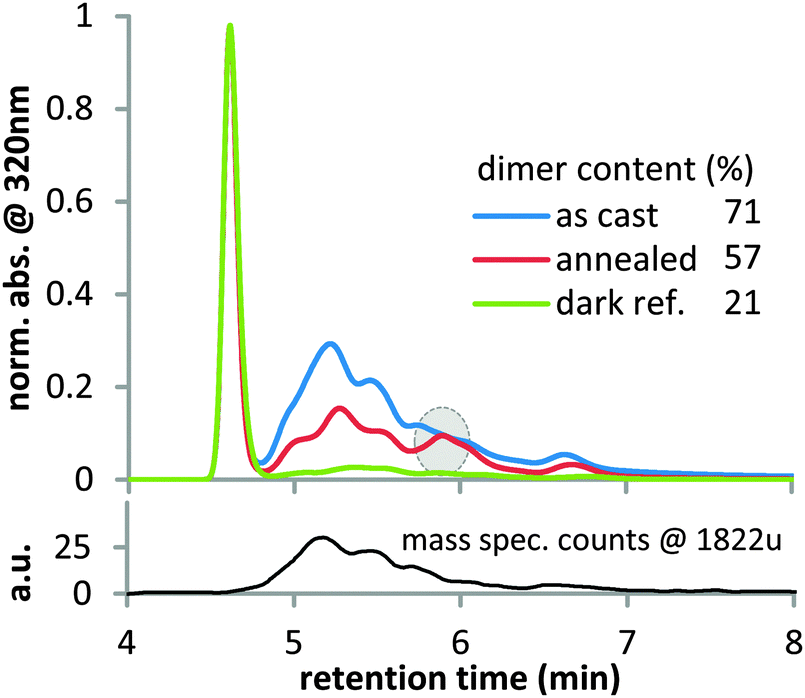 | ||
| Fig. 3 Top: High performance liquid chromatography (HPLC) traces from UV-vis detection at 320 nm of illuminated and re-dissolved PC60BM films normalized to the monomer peak at 4.6 min. A significant amount of dimer is observed in the as cast film (blue) between 5 and 7 min retention time (herein referred to as ‘dimer region’). Annealing at 180 °C for 10 min before illumination (red) reduces dimerization. Films were illuminated 40 hours under a one sun equivalent white light LED. The dark reference (green) was exposed to a small amount of ambient light. Bottom: For an aged as cast sample, a mass spectrometry trace (HPLC-MS in APCI mode) was recorded using single-ion-monitoring (SIM) at an atomic mass of 1822 u, which represents the PC60BM dimer mass. The excellent correlation between the SIM and the UV trace confirms that the signal between 5 and 7 minutes corresponds to PC60BM dimers. Multiple isomers have different geometries and HPLC retention times, which explains the presence of multiple dimer peaks between 5 and 7 minutes. | ||
HPLC analysis of the reference sample shows a sharp peak at 4.6 min retention time, which is associated with monomeric PC60BM (Fig. 3). A special HPLC column, Cosmosil Buckyprep, was chosen for this study, because on this type of column one generally observes a good separation between fullerene monomers and dimers, with the former eluting first and the latter eluting later from the column. In the amorphous illuminated film, we observe a range of new peaks between 4.8 and 7.0 min retention time. Using HPLC-mass spectrometry (HPLC-MS), we could demonstrate that these new peaks indeed correspond to PC60BM dimers. The selected-ion-monitoring trace for m/z 1822 u, which is twice the PC60BM molecular mass of 911 u, (Fig. 3) clearly confirms that these compounds are PC60BM dimers and not other potential photodegradation products. The broad shape of the HPLC trace in the ‘dimer region’ (5 to 7 minutes retention time) can be attributed to the presence of multiple regioisomers3 of the PC60BM dimer, which all have slightly different shapes and are thus retained to a varying degree by the HPLC column. In the illuminated crystalline film, the dimer content is significantly reduced compared to the amorphous film. Furthermore a distinct isomer peak at 5.9 min is visible for the crystalline film that is at best present as a shoulder in the HPLC trace of the amorphous film. We propose that the ordering due to crystallization favors the formation of specific dimers with a configuration similar to the structure of the crystal and impedes the formation of isomers that do not align with the crystal structure.41 This suggests that the reduced dimerization in the crystalline film is due to geometric restrictions that are imposed by solid state packing. A comparatively small amount of dimer is also found in the reference film that was only exposed to ambient light in the glovebox. This finding could suggest that already small amounts of light can induce dimerization of PC60BM.18
Effect of fullerene morphology on light-induced Jsc losses
Having shown the impact of crystallinity on fullerene dimerization, we now want to study how fullerene domains with different degrees of crystallinity affect the stability of full solar cells. We chose P3HT:PC60BM cast from chloroform as a system that allows us to control the fullerene morphology in a full solar cell and study its impact on the Jsc losses. Annealing at 140 °C resulted in fast crystallization of the polymer and after only 2 min annealing, strong lamellar (q = 0.35 Å−1) and π–π stacking (q = 1.65 Å−1) peaks were observed in X-ray diffraction patterns (Fig. 4a). The fullerene is still amorphous as seen from the diffuse halo at q = 1.4 Å−1 which represents diffraction from small amorphous PC60BM clusters.42 After 10 min annealing (Fig. 4b), multiple peaks were visible inside the fullerene halo indicating increased fullerene ordering or crystallization. For even longer annealing times, more crystallization of the fullerene is expected.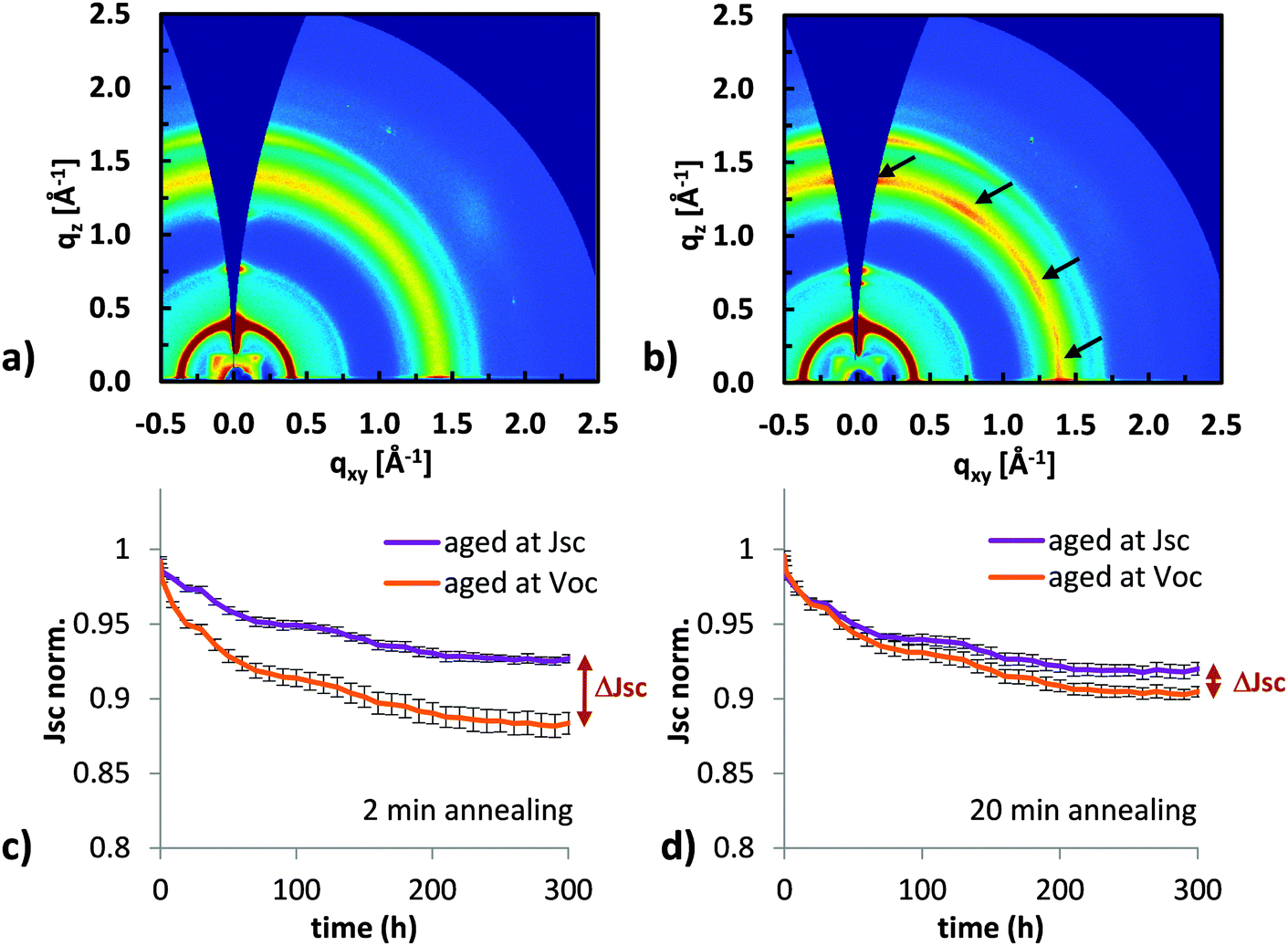 | ||
| Fig. 4 (a) Grazing incidence X-ray diffraction (GIXD) pattern of a P3HT:PC60BM film annealed at 140 °C for 2 min, (b) annealed for 10 min. The polymer crystallizes already after two minutes, while the fullerene is still amorphous and shows clear crystalline peaks only after 10 min (indicated by arrows). Light induced short circuit current losses of devices made under the same conditions as the GIXD films and annealed for (c) 2 min and (d) 20 min. Three devices for each annealing time were held at Jsc during degradation and three at Voc. Average values are shown. Devices annealed for only 2 min with amorphous fullerene show a clear dependence of the Jsc losses on the electrical bias. | ||
Importantly, P3HT:PC60BM solar cells cast from chloroform and annealed for 2 min will still have amorphous fullerene domains, while solar cells annealed for 10 min or longer will have crystalline fullerene domains. By fabricating solar cells annealed for different periods of time (Fig. 4c and d), we compared the behavior of devices containing amorphous fullerene with devices containing more ordered or crystalline fullerene. To ensure good crystallization of the fullerene in solar cells, we annealed the active layer for 20 min. Data for 10 min annealing can be found in the ESI.† All solar cells in Fig. 4c and d were fabricated in the same batch and aged side by side in a highly pure36 nitrogen atmosphere with an oxygen content of less than 0.1 ppm. The cells were illuminated with a sulfur plasma lamp that has no UV component in the spectrum and IV curves were taken automatically every hour. After 300 hours, the solar cells with crystalline fullerene showed significantly less Jsc losses for samples degraded at Voc.
Two important points can be observed from the degradation curves shown in Fig. 4c and d. First, the amount of Jsc loss depends on the electrical bias during degradation. And second, the difference in Jsc loss between samples degraded at Voc and Jsc conditions (ΔJsc between the orange and blue curve in Fig. 4c and d at 300 h) is three times larger for the solar cell annealed for 2 min, which has amorphous fullerene domains, compared to the solar cell annealed for 20 min with more crystalline fullerene domains.
From the measurements of solar cells made with fullerenes that were previously dimerized, we concluded that PC60BM dimers lead to Jsc losses in organic solar cells, which is in agreement with previous reports.3,33 The correlation of reduced Jsc losses with a higher degree of fullerene ordering (Fig. 4) suggests that fewer dimers are formed in solar cells with more ordered fullerene domains. This is fully consistent with the observation of reduced dimerization in crystalline films of plain fullerene (Fig. 3). Besides the reduced tendency of crystalline fullerenes to dimerize, a high degree of crystallinity could also play another beneficial role. A high mobility in crystalline domains would make such devices less sensitive to energetic traps which are likely to be caused by fullerene dimerization.
Bias dependence of light induced dimerization
In order to further investigate the impact of electrical bias on degradation (Fig. 4), the amount of dimer in cells aged at Voc and Jsc is measured with HPLC. We illuminated two P3HT:PC60BM solar cells cast from chloroform on the same substrate with one sun equivalent white LED illumination for 65 h, while one is held at Jsc and the other at Voc. Since both solar cells are on the same substrate directly next to each other, the only difference in this experiment is the electrical bias. After illumination, we scraped off the active area of both solar cells, dissolved the collected material in toluene and performed HPLC analyses. We observed fullerene dimers, as seen from the characteristic peaks between 5 and 7 min in Fig. 5. An increased amount of dimer with respect to the monomer peak is observed in the solar cell aged at Voc conditions. This finding is consistent with the increased performance losses observed in Fig. 4 and suggests that dimerization is affected by the electrical bias during degradation. For KP115:PC60BM solar cells aged for 15 h under one sun equivalent LED illumination, we also observed an increased dimer content for the device aged at Voc conditions, Fig. 5b. More HPLC data for devices aged under different conditions can be found in the ESI.† A consistently higher amount of dimer is found in all solar cells aged at Voc conditions. Generally, the difference in the amount of dimers measured in HPLC is smaller than the differences in Jsc losses observed for solar cells aged at Voc or Jsc. It could be that only a part of all dimers formed – maybe the ones within transport channels or near the electrodes – affect device performance.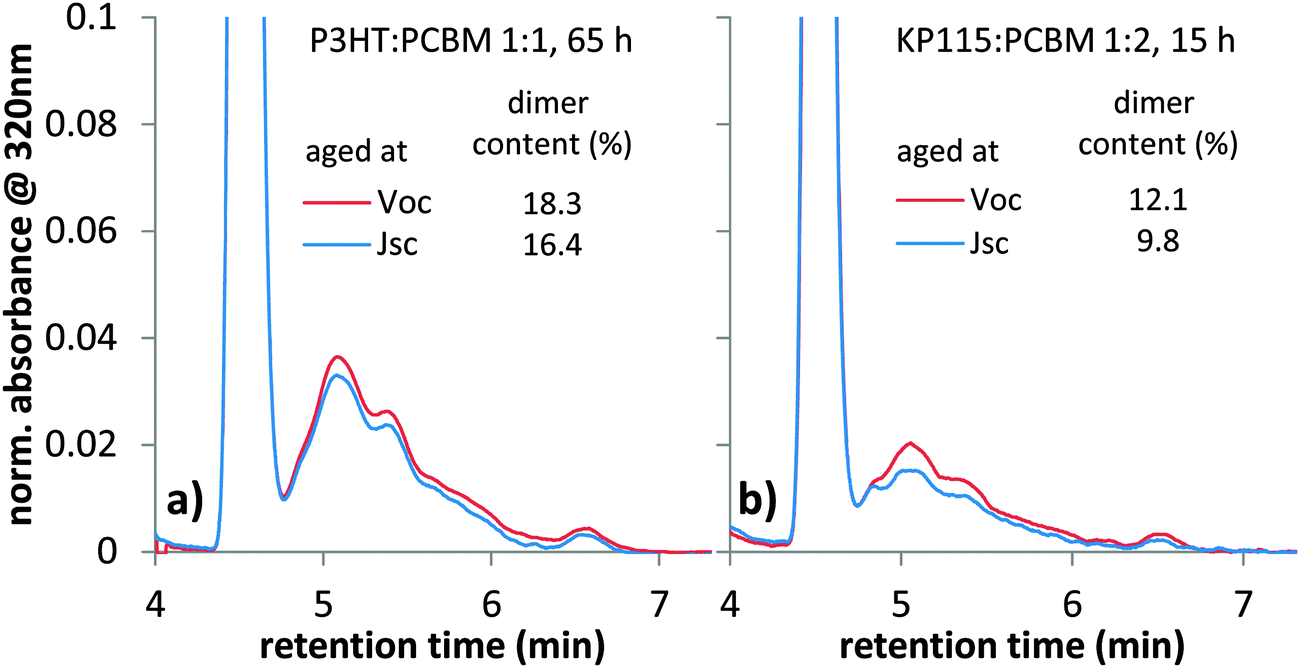 | ||
| Fig. 5 HPLC analyses of re-dissolved material from the active layer of (a) P3HT:PC60BM solar cells cast from chloroform and (b) KP115:PC60BM solar cells. An increased amount of dimers is observed in solar cells aged at Voc. Degradation was performed in inert conditions under one sun equivalent LED illumination. The data is normalized to the monomer peak and the dimer content is determined by integration of the monomer and dimer peaks. | ||
In order to investigate the generality of the bias dependence of dimerization, we also studied PCDTBT:PC60BM devices and KP115:PC60BM devices with inverted architecture (see ESI†). A dependence of photo-induced performance losses on the electrical bias during degradation was observed in both systems and is independent of the device architecture. This highlights the generality of the observed degradation effects.
Effect of polymer–fullerene mixing on fullerene dimerization
Besides the electrical bias and the fullerene morphology, also the degree of polymer–fullerene mixing,4 which in polymer:PC60BM devices is largely correlated to polymer crystallinity, can impact dimerization. To study the influence of polymer morphology on dimerization we investigated a larger number of polymer: PC60BM systems and used UV-vis absorption measurements to identify dimers. This technique can be readily performed on plain films and allows for multiple measurements on the same sample while for HPLC only one measurement is possible since the film has to be dissolved.Using diode array UV-vis detection during an HPLC run of illuminated fullerene, we obtained the absorption spectra of the PC60BM monomer and dimer at 4.6 min and 5.3 min retention time, respectively (Fig. 6a and b). The dimer shows significantly increased absorption around 320 nm and can thus be distinguished from the monomer by the spectral shape of the absorption profile. This allows monitoring the temporal progression of fullerene dimerization upon illumination (Fig. 6c).
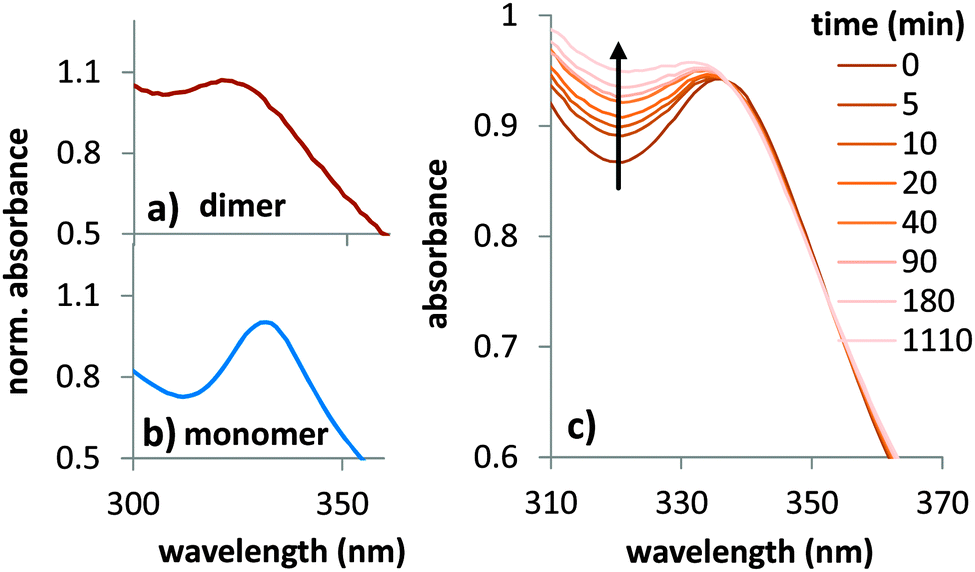 | ||
| Fig. 6 UV-vis absorption of (a) PC60BM dimer and (b) PC60BM monomer as obtained from HPLC data at 4.6 min and 5.3 min retention time, see also Fig. 3. (c) Spectral changes during illumination of a PC60BM film indicate the formation of dimers. | ||
After an initially fast dimerization rate, the increase in absorption at 320 nm becomes slower with time and levels out on long timescales, which is consistent with the time evolution of Jsc losses observed during burn in. We used several polymers that have a stronger or weaker tendency to aggregate. Polymers that do not aggregate very well usually have a high solubility for fullerene in the amorphous phase and consist of only mixed domains.43 For polymers that aggregate well, like P3HT, a high degree of polymer crystallinity44 leads to the formation of relatively pure fullerene domains. Besides P3HT, KP115 has also been shown to have a high degree of aggregation.45 Si-PCPDTBT is less ordered but still shows clear diffraction peaks,46 while PCPDTBT is largely amorphous.47
The highly aggregated polymers P3HT and KP115 show the highest amount of fullerene dimerization in blend films during illumination (Fig. 7a). In contrast, the amorphous polymer PCPDTBT shows negligible fullerene dimerization. We propose that for fully amorphous systems with good mixing between polymer and fullerene, the probability of two fullerenes being in close proximity and in the right alignment40 for dimerization is low. Thus, dimerization is preferred for polymers with high aggregation. Polymer aggregation drives fullerenes out of the polymer phase and results in the formation of fullerene clusters. In such pure fullerene clusters dimerization is likely to occur. Besides geometric restrictions, an increased triplet lifetime in pure fullerene clusters48 seems to play an important role for dimerization. Assuming that photo-induced fullerene dimerization proceeds via the triplet state,4,40,49–51 the dimerization reaction is competing with charge transfer to the polymer.
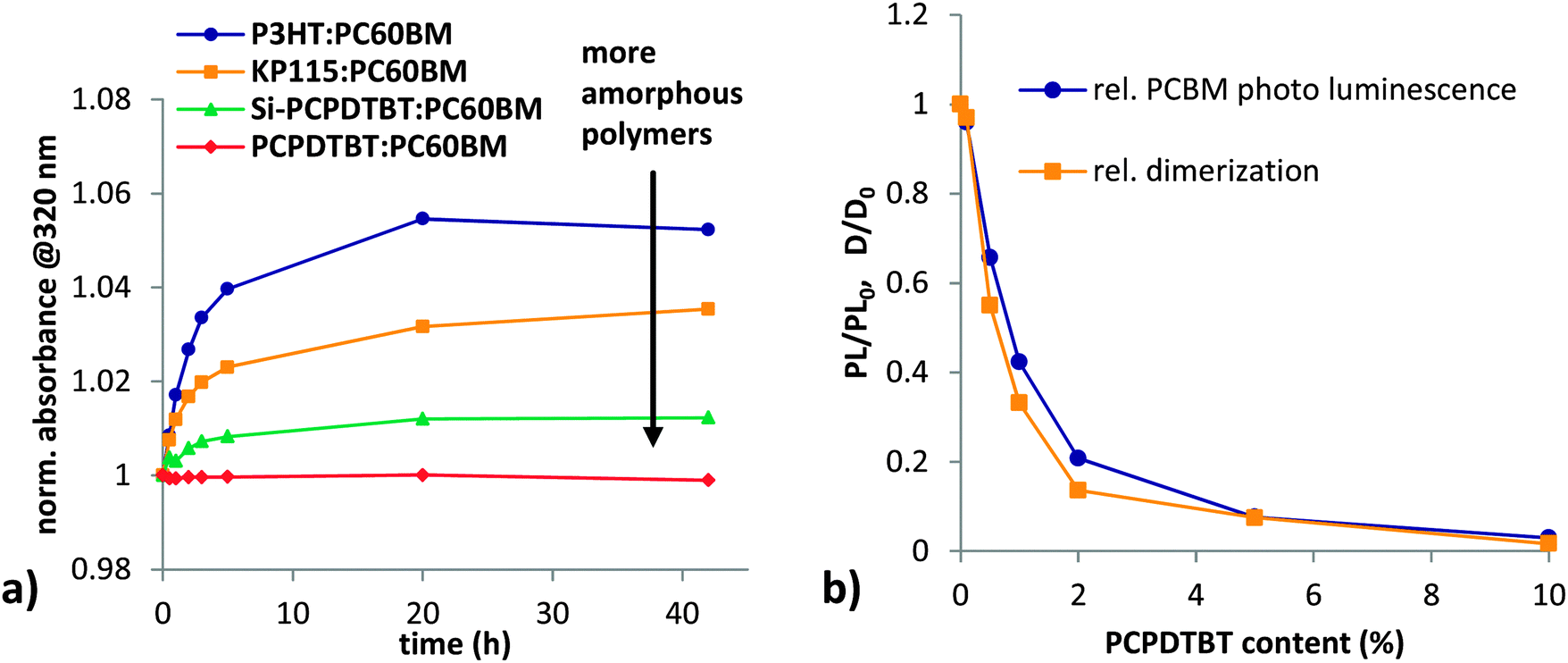 | ||
| Fig. 7 (a) Photo-induced dimerization measured with UV-vis absorption in polymer–fullerene (1:2) blends with different degrees of polymer aggregation. (b) Relative PC60BM photoluminescence (PL/PL0), which is obtained by integration over the emission band and normalization to the value in neat PCBM, and the relative degree of dimerization compared to a plain PC60BM film (D/D0) as measured by absorption is plotted for a series of fullerene films with increasing content of PCPDTBT. | ||
We investigated the influence of fullerene exciton dissociation on dimerization in the PCPDTBT:PC60BM system by monitoring fullerene photoluminescence quenching. A series of fullerene films with increasing content of PCPDTBT was illuminated for 100 hours in inert conditions and dimerization was monitored using absorption measurements (Fig. 7b). Addition of only 0.5% polymer reduces the change in absorption at 320 nm to half the value of a plain PC60BM film. For more than 5% polymer content, no significant dimerization is occurring. The corresponding PL quenching data shows the same trend as the absorption measurements. This direct correlation of PL-quenching data with fullerene dimerization upon addition of PCPDTBT to PC60BM suggests that excitons on the fullerene drive dimerization, and that an efficient mixing of polymer and fullerene reduces dimerization. This is consistent with the observations of Morse et al.31 who showed a reduction of light induced Jsc losses when lowering the fullerene content.
Further discussion
Considering the strong dependence of dimerization on the electrical bias that was observed in all investigated systems, a dimerization mechanism via excitons on the fullerene would also imply that under Voc more triplets are present on the fullerene than if the cell is held at Jsc. Further investigation will be needed to verify this. Besides a reaction mechanism via triplets, also dimerization reactions via polarons or charged fullerenes have been predicted by quantum chemical calculations.52 This reaction mechanism would also be consistent with our data, since the concentration of polarons is higher at Voc where no carriers are extracted, than at Jsc. While a detailed study of the photophysical reaction mechanism for dimerization is beyond the scope of this study, we and others3,20 have directly linked photo-induced Jsc losses with dimerization.More insight on the underlying mechanisms for dimerization induced Jsc losses can be gained from EQE measurements. The EQE data for the aged device (ESI†) shows current losses across the whole spectral range. Nevertheless, those losses are higher between 350 nm and 450 nm where most of the absorption is contributed by the fullerene domains. This can be seen more clearly in normalized EQE curves, see Fig. S9 (ESI†). A significant change of absorption after degradation or when adding fullerene is not indicated from reflectance measurements on full devices (see ESI†). The observed current losses from fullerene domains suggest that excitons on larger fullerene clusters cannot be harvested efficiently in the presence of dimers. This result is consistent with the observations of Forrest et al.20 who showed that the exciton diffusion length in the 50 nm C60 layer of a planar architecture SubPc/C60 solar cell is significantly reduced upon light induced dimerization. We suggest that in bulk heterojunction polymer–fullerene solar cells, exciton harvesting from domains on the length scale of several tens of nanometers is also reduced by light-induced dimerization.
Nevertheless, those effects cannot account for all of the Jsc loss that is observed after degradation. Polymer photo-luminescence (PL) quenching data on KP115:fullerene blends showed that with a PC60BM monomer:dimer mixture of 1:1, the PL quenching is reduced compared to blends with pristine PC60BM, see ESI.† While a detailed study is beyond the scope of this work, this finding may suggest that exciton splitting at the polymer–dimer interface is less efficient than at the polymer–monomer interface. Further, we propose that another part of the current losses in aged KP115:PC60BM solar cells is caused by inefficient extraction of charge carriers. EQE measurements with and without light bias, see ESI,† show a significant increase of the EQE under light bias for aged solar cells. This result suggests that those cells are transport limited and a partial trap filling due to bias illumination can improve the transport properties. We measured photo-CELIV (charge extraction by linearly increasing voltage) for fresh and aged devices, see ESI,† and observed a much more dispersive transport in aged solar cells, as well as in solar cells with dimers added. Dispersive transport means that there is a large fraction of charge carriers with a lower mobility. This transport can be well described with a multiple trapping and release model.53 Reduced electron mobility was also observed by McCulloch et al. when adding synthetic fullerene dimers with alkyl bridges to PC60BM films.22 The presence of traps is further supported by the fact that after a delay time of 100 μs in photo-CELIV, more charge can be extracted from aged cells and cells with dimers than from the reference cells. This observation is consistent with findings from Mozer et al.33 who observed trapped carriers in aged KP115 solar cells with transient absorption and charge extraction. It is surprising that the fill factor is only slightly affected by those traps and that in other systems traps are observed that don't affect Jsc but Voc.45 Besides the energetic level and the lifetime of a trapped carrier, also the positon of traps with respect to transport paths,54 the polymer–fullerene interface or the electrode interface could play a crucial role in determining how a trap will affect device performance. In bulk heterojunctions with three phase morphologies,54,55 charge carrier generation and transport occur in different regions and the presence of traps in one or the other region is expected to influence the impact on device performance.
Conclusions
Fullerene dimerization and correlated performance losses of organic solar cells can be inhibited by an intimately mixed BHJ with highly amorphous polymers. But for highly efficient solar cells pure fullerene domains that promote dimerization seem necessary since pure domains in a three phase morphology have been shown to improve charge extraction, efficiency and stability in organic solar cells. Consequently, different measures to inhibit fullerene dimerization are necessary for high efficiency materials with pure fullerene domains. We show that crystallization of the fullerene domains by annealing at high temperatures reduces dimerization and provides a way to stabilize organic solar cells against performance losses during illumination. Using non-fullerene acceptors could be another pathway to improved OPV stability.The high sensitivity of fullerene dimerization on the electrical bias during degradation enables an easy measurement to characterize this degradation mechanism. As most known degradation mechanisms are independent of the electrical bias, differences in performance losses between ageing at Voc and Jsc are a strong indication for fullerene dimerization.
Experimental section
Device fabrication
All P3HT and KP115 devices are produced in standard architecture unless noted otherwise. A PEDOT:PSS (Clevios P VP AI 4083) hole extraction layer of about 30 nm thickness is spin coated on cleaned and UV-ozone treated ITO substrates at 4000 rpm and dried on a hotplate at 140 °C for 15 min. All active layers are spin coated inside a nitrogen filled glovebox from dichlorobenzene (DCB) or chloroform (CF) solutions. 7 nm of calcium (0.5 Å s−1) and 150 nm (1 to 5 Å s−1) aluminum are deposited in a thermal evaporator. P3HT(BASF P200):PC60BM (1:1) solar cells are spin coated with a total solution concentration of 30 mg ml−1 in CF or 50 mg ml−1 in DCB at 1500 rpm or 900 rpm, respectively, to 280 nm thickness. KP115(Konarka):PC60BM 1:2 devices are fabricated from a 20 mg ml−1 DCB solution heated to 100 °C at a spin speed of 1200 rpm resulting in a thickness of 250–300 nm. Fullerenes were purchased from Solenne BV. PC60BM films were spun at 900 rpm from a 40 mg ml−1 chlorobenzene solution. Inverted architecture (ITO/ZnO/active layer/PEDOT:PSS/Ag) KP115:PC60BM 1:2 solar cells are doctor bladed in air from a 40 mg ml−1 DCB solution. PCDTBT:PC60BM 1:4 solar cells are cast from a 16 mg ml−1 CF solution to a thickness of 70 nm. P3HT solar cells cast from CB are annealed at 110 °C for 10 min, P3HT devices cast from CF are annealed for 2–20 min to obtain various degrees of fullerene crystallinity. No thermal annealing is performed on PCDTBT and KP115 solar cells.
Device characterization
IV curves were measured with a Keithley 2400 under a Newport solar simulator calibrated to an AM 1.5G spectrum. The cells are measured without a mask, since no significant lateral conductivity is observed due to the relatively low conductivity of the used PEDOT:PSS (Clevios P VP AI 4083). For degradation experiments the cells were loaded into a sealed chamber inside the glovebox. This chamber was then continuously purged with nitrogen and illuminated with a sulphur plasma lamp for the 300 h test. For short term testing also one sun equivalent LED illumination was used. IV curves were taken periodically with a Keithley 2400. EQE and reflectance was measured with a QE-R3011 from Enlitech. UV/vis absorption spectra of single photoactive layers were recorded with a Perkin Elmer Lambda 35 UV/vis spectrometer. Photoluminescence (PL) measurements were performed with an Andor Shamrock SR 303i Spectrometer using a Si-detector for the visible spectral range and an InGaAs-detector for near-infrared measurements. PC60BM samples were excited with a 405 nm laser with an intensity of 0.95 mW. In order to exclude contributions of the lasers in the fluorescence spectra a 550 nm high pass cut-off filter was used.X-ray diffraction
Films were prepared on silicon substrates with the same spin coating parameters and solution concentrations as for solar cells. Grazing incidence X-ray scattering (GIXS) was obtained at beamline 11–3 at the Stanford Synchotron Radiation Lightsource using an X-ray energy of 12.7 eV, a MAR 345 image plate area detector, a helium-filled sample chamber, and an incident X-ray beam angle of 0.12°.Author contributions
M. D. M. and C. J. B. supervised the research. T. H. coordinated the experiments, performed device fabrication as well as characterization and wrote the manuscript. W. R. M. performed long term testing and contributed ideas. A. D. contributed photoluminescence, absorption measurements and ideas. U. F. F. performed chromatography and mass spectrometry. M. B. prepared inverted devices. R. C. measured GIXD. M. v. D., H. J. E., M. S., and W. N. assisted with data interpretation and contributed ideas.Acknowledgements
T. H. gratefully acknowledges support from the DFG under the SFB 953 Synthetic Carbon Allotropes and a DAAD Doktorandenstipedium. C. J. B. acknowledges funding from the Cluster of Excellence Engineering of Advanced Materials, the Bavarian SolTech initiative and the GRK 1896 in situ microscopy. M. D. M. acknowledges funding from the Office of Naval Research Award No. N00014-14-1-0580 and N00014-14-1-0280. U. F. F. is grateful to the Villigst Studienwerk for a doctoral scholarship. M. v. D. acknowledges financial support from the Daimler und Benz Stiftung (32-12/13) and the DFG (Emmy-Noether-Programme, DE 1830/2-1). M. S. acknowledges primary support from a fellowship by the Portuguese Fundação para a Ciência e a Tecnologia (SFRH/BPD/71816/2010). H.-J. E. acknowledges the “Solar Factory of the Future” as part of the Energy Campus Nuremberg (EnCN), which is supported by the Bavarian State Government (FKZ 20-3043.5).References
- A. Dzwilewski, T. Wågberg and L. Edman, J. Am. Chem. Soc., 2009, 131, 4006–4011 CrossRef CAS PubMed.
- P. C. Eklund, A. M. Rao, P. Zhou, Y. Wang and J. M. Holden, Thin Solid Films, 1995, 257, 185–203 CrossRef CAS.
- A. Distler, T. Sauermann, H. Egelhaaf, S. Rodman, D. Waller, K. Cheon, M. Lee and D. M. Guldi, Adv. Energy Mater., 2013, 4, 1300693 Search PubMed.
- N. Wang, X. Tong, Q. Burlingame, J. Yu and S. R. Forrest, Sol. Energy Mater. Sol. Cells, 2014, 125, 170–175 CrossRef CAS.
- M. T. Dang, L. Hirsch and G. Wantz, Adv. Mater., 2011, 23, 3597–3602 CrossRef CAS PubMed.
- C. H. Peters, I. T. Sachs-Quintana, J. P. Kastrop, S. Beaupré, M. Leclerc and M. D. McGehee, Adv. Energy Mater., 2011, 1, 491–494 CrossRef CAS.
- A. M. Rao, P. Zhou, K.-A. Wang, G. T. Hager, J. M. Holden, Y. Wang, W.-T. Lee, X.-X. Bi, P. C. Eklund, D. S. Cornett, M. A. Duncan and I. J. Amster, Science, 1993, 259, 955–957 CrossRef CAS.
- D. S. Cornett, I. J. Amster, M. a Duncan, A. M. Rao and P. C. Eklund, J. Phys. Chem., 1993, 97, 5036–5039 CrossRef CAS.
- P. Zhou, a. M. Rao, K. A. Wang, J. D. Robertson, C. Eloi, M. S. Meier, S. L. Ren, X. X. Bi, P. C. Eklund and M. S. Dresselhaus, Appl. Phys. Lett., 1992, 60, 2871–2873 CrossRef CAS.
- A. Dzwilewski, T. Wågberg and L. Edman, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 075203 CrossRef.
- K. Sinha, S. Guha, J. Menéndez, B. Ramakrishna, D. Wright and T. Karcher, Solid State Commun., 1993, 87, 981–986 CrossRef CAS.
- N. Matsuzawa, M. Ata, D. a Dixon and G. Fitzgerald, J. Phys. Chem., 1994, 98, 2555–2563 CrossRef CAS.
- J. L. Segura and N. Martín, Chem. Soc. Rev., 2000, 29, 13–25 RSC.
- J. Onoe and K. Takeuchi, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 6167–6171 CrossRef CAS.
- S. G. Stepanian, V. a Karachevtsev, A. M. Plokhotnichenko, L. Adamowicz and A. M. Rao, J. Phys. Chem. B, 2006, 110, 15769–15775 CrossRef CAS PubMed.
- G. B. Adams, J. B. Page, O. F. Sankey and M. O'Keeffe, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17471–17479 CrossRef CAS.
- Y. Chiba, S.-R. Chen, H. Tsuji, M. Ueno, N. Aoki and Y. Ochiai, J. Phys.: Conf. Ser., 2009, 159, 012017 CrossRef.
- Z. Li, H. C. Wong, Z. Huang, H. Zhong, C. H. Tan, W. C. Tsoi, J. S. Kim, J. R. Durrant and J. T. Cabral, Nat. Commun., 2013, 4, 2227 Search PubMed.
- F. Piersimoni, G. Degutis, S. Bertho, K. Vandewal, D. Spoltore, T. Vangerven, J. Drijkoningen, M. K. Van Bael, A. Hardy, J. D'Haen, W. Maes, D. Vanderzande, M. Nesladek and J. Manca, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1209–1214 CrossRef CAS.
- Q. Burlingame, X. Tong, J. Hankett, M. Slootsky, Z. Chen and S. R. Forrest, Energy Environ. Sci., 2015, 8, 1005–1010 CAS.
- S. Falke, P. Eravuchira, A. Materny and C. Lienau, J. Raman Spectrosc., 2011, 42, 1897–1900 CrossRef CAS.
- B. C. Schroeder, Z. Li, M. a. Brady, G. C. Faria, R. S. Ashraf, C. J. Takacs, J. S. Cowart, D. T. Duong, K. H. Chiu, C.-H. Tan, J. T. Cabral, A. Salleo, M. L. Chabinyc, J. R. Durrant and I. McCulloch, Angew. Chem., Int. Ed., 2014, 53, 12870–12875 CrossRef CAS PubMed.
- C. Y. Chang, C. E. Wu, S. Y. Chen, C. Cui, Y. J. Cheng, C. S. Hsu, Y. L. Wang and Y. Li, Angew. Chem., Int. Ed., 2011, 50, 9386–9390 CrossRef CAS PubMed.
- J. W. Rumer, R. S. Ashraf, N. D. Eisenmenger, Z. Huang, I. Meager, C. B. Nielsen, B. C. Schroeder, M. L. Chabinyc and I. McCulloch, Adv. Energy Mater., 2015, 5 DOI:10.1002/aenm.201401426.
- O. Synooka, K.-R. Eberhardt, C. R. Singh, F. Hermann, G. Ecke, B. Ecker, E. von Hauff, G. Gobsch and H. Hoppe, Adv. Energy Mater., 2014, 4, 1300981 Search PubMed.
- S. Bertho, B. Campo, F. Piersimoni, D. Spoltore, J. D'Haen, L. Lutsen, W. Maes, D. Vanderzande and J. Manca, Sol. Energy Mater. Sol. Cells, 2013, 110, 69–76 CrossRef CAS.
- B. Conings, S. Bertho, K. Vandewal, A. Senes, J. D'Haen, J. Manca and R. a. J. Janssen, Appl. Phys. Lett., 2010, 96, 163301 CrossRef.
- I. Cardinaletti, J. Kesters, S. Bertho, B. Conings, F. Piersimoni, J. D'Haen, L. Lutsen, M. Nesladek, B. Van Mele, G. Van Assche, K. Vandewal, A. Salleo, D. Vanderzande, W. Maes and J. V. Manca, J. Photonics Energy, 2014, 4, 040997 CrossRef.
- J. Liu, X. Guo, Y. Qin, S. Liang, Z.-X. Guo and Y. Li, J. Mater. Chem., 2012, 22, 1758–1761 RSC.
- H. C. Wong, Z. Li, C. H. Tan, H. Zhong, Z. Huang, H. Bronstein, I. McCulloch, J. T. Cabral and J. R. Durrant, ACS Nano, 2014, 8, 1297–1308 CrossRef CAS PubMed.
- G. E. Morse, A. Tournebize, A. Rivaton, N. Blouin and S. Tierney, Phys. Chem. Chem. Phys., 2015, 17, 11884–11897 RSC.
- X. Tong, N. Wang, M. Slootsky, J. Yu and S. R. Forrest, Sol. Energy Mater. Sol. Cells, 2013, 118, 116–123 CrossRef CAS.
- T. M. Clarke, C. Lungenschmied, J. Peet, N. Drolet, K. Sunahara, A. Furube and A. J. Mozer, Adv. Energy Mater., 2013, 3, 1473–1483 CrossRef CAS.
- J. Peet, L. Wen, P. Byrne, S. Rodman, K. Forberich, Y. Shao, N. Drolet, R. Gaudiana, G. Dennler and D. Waller, Appl. Phys. Lett., 2011, 98, 043301 CrossRef.
- S. H. Park, A. Roy, S. Beaupré, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–302 CrossRef CAS.
- W. R. Mateker, I. T. Sachs-Quintana, G. F. Burkhard, R. Cheacharoen and M. D. McGehee, Chem. Mater., 2015, 27, 404–407 CrossRef CAS.
- R. Roesch, K. R. Eberhardt, S. Engmann, G. Gobsch and H. Hoppe, Sol. Energy Mater. Sol. Cells, 2013, 117, 59–66 CrossRef CAS.
- Y. W. Soon, H. Cho, J. Low, H. Bronstein, I. McCulloch and J. R. Durrant, Chem. Commun., 2013, 49, 1291–1293 RSC.
- W. R. Mateker, T. Heumueller, R. Cheacharoen, I. T. Sachs-Quintana and M. D. McGehee, Chem. Mater., 2015, 27, 6345–6353 CrossRef CAS.
- P. Zhou, Z. Dong, A. Rao and P. Eklund, Chem. Phys. Lett., 1993, 2, 337–340 CrossRef.
- V. Ramamurthy and K. Venkatesan, Chem. Rev., 1987, 87, 433–481 CrossRef CAS.
- E. Verploegen, R. Mondal, C. J. Bettinger, S. Sok, M. F. Toney and Z. Bao, Adv. Funct. Mater., 2010, 20, 3519–3529 CrossRef CAS.
- H. W. Ro, B. Akgun, B. T. O'Connor, M. Hammond, R. J. Kline, C. R. Snyder, S. K. Satija, A. L. Ayzner, M. F. Toney, C. L. Soles and D. M. DeLongchamp, Macromolecules, 2012, 45, 6587–6599 CrossRef CAS.
- S. T. Turner, P. Pingel, R. Steyrleuthner, E. J. W. Crossland, S. Ludwigs and D. Neher, Adv. Funct. Mater., 2011, 21, 4640–4652 CrossRef CAS.
- T. Heumueller, W. R. Mateker, I. T. Sachs-Quintana, K. Vandewal, J. A. Bartelt, T. M. Burke, T. Ameri, C. J. Brabec and M. D. McGehee, Energy Environ. Sci., 2014, 7, 2974–2980 CAS.
- M. Morana, H. Azimi, G. Dennler, H. J. Egelhaaf, M. Scharber, K. Forberich, J. Hauch, R. Gaudiana, D. Waller, Z. Zhu, K. Hingerl, S. S. Van Bavel, J. Loos and C. J. Brabec, Adv. Funct. Mater., 2010, 20, 1180–1188 CrossRef CAS.
- M. C. Scharber, M. Koppe, J. Gao, F. Cordella, M. a Loi, P. Denk, M. Morana, H.-J. Egelhaaf, K. Forberich, G. Dennler, R. Gaudiana, D. Waller, Z. Zhu, X. Shi and C. J. Brabec, Adv. Mater., 2010, 22, 367–370 CrossRef CAS PubMed.
- J. C. Hummelen, J. Knol and L. Sánchez, Proc. SPIE-Int. Soc. Opt. Eng., 2001, 4108, 76–84 CrossRef CAS.
- J. Knol and J. C. Hummelen, J. Am. Chem. Soc., 2000, 122, 3226–3227 CrossRef CAS.
- Y. Wang, J. M. Holden, Z.-H. Dong, X.-X. Bi and P. C. Eklund, Chem. Phys. Lett., 1993, 211, 341–345 CrossRef CAS.
- J. Wang, J. Enevold and L. Edman, Adv. Funct. Mater., 2013, 23, 3220–3225 CrossRef CAS.
- E. F. Sheka, Chem. Phys. Lett., 2007, 438, 119–126 Search PubMed.
- M. Schubert, E. Preis, J. Blakesley, P. Pingel, U. Scherf and D. Neher, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 024203 CrossRef.
- W. Yin and M. Dadmun, ACS Nano, 2011, 5, 4756–4768 CrossRef CAS PubMed.
- J. A. Bartelt, Z. M. Beiley, E. T. Hoke, W. R. Mateker, J. D. Douglas, B. A. Collins, J. R. Tumbleston, K. R. Graham, A. Amassian, H. Ade, J. M. J. Fréchet, M. F. Toney and M. D. McGehee, Adv. Energy Mater., 2013, 3, 364–374 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ee02912k |
| This journal is © The Royal Society of Chemistry 2016 |