
Structural basis for differing electrocatalytic water oxidation by the cubic, layered and spinel forms of lithium cobalt oxides†
Graeme
Gardner
a,
Jafar
Al-Sharab
b,
Nemanja
Danilovic
c,
Yong Bok
Go
a,
Katherine
Ayers
c,
Martha
Greenblatt
a and
G.
Charles Dismukes
*ad
aDepartment of Chemistry and Chemical Biology, Rutgers, The State University of New Jersey, 610 Taylor Road, Piscataway, NJ 08854, USA. E-mail: martha@chem.rutgers.edu; dismukes@rutger.edu
bDepartment of Engineering Technology, Northwestern State University, 205 Williamson Hall, Natchitoches, LA 71497, USA
cProton OnSite, 10 Technology Drive, Wallingford, CT 06492, USA
dWaksman Institute of Microbiology, Rutgers, The State University of New Jersey, 190 Frelinghuysen Road, Piscataway, NJ 08854, USA
First published on 27th October 2015
Abstract
The two polymorphs of lithium cobalt oxide, LiCoO2, present an opportunity to contrast the structural requirements for reversible charge storage (battery function) vs. catalysis of water oxidation/oxygen evolution (OER; 2H2O → O2 + 4H+ + 4e−). Previously, we reported high OER electrocatalytic activity from nanocrystals of the cubic phase vs. poor activity from the layered phase – the archetypal lithium-ion battery cathode. Here we apply transmission electron microscopy, electron diffraction, voltammetry and elemental analysis under OER electrolysis conditions to show that labile Li+ ions partially deintercalate from layered LiCoO2, initiating structural reorganization to the cubic spinel LiCo2O4, in parallel with formation of a more active catalytic phase. Comparison of cubic LiCoO2 (50 nm) to iridium (5 nm) nanoparticles for OER catalysis (commercial benchmark for membrane-based systems) in basic and neutral electrolyte reveals excellent performance in terms of Tafel slope (48 mV dec−1), overpotential (η = ∼420 mV@10 mA cm−2 at pH = 14), faradaic yield (100%) and OER stability (no loss in 14 hours). The inherent OER activity of cubic LiCoO2 and spinel LiCo2O4 is attributed to the presence of [Co4O4]n+ cubane structural units, which provide lower oxidation potential to Co4+ and lower inter-cubane hole mobility. By contrast, the layered phase, which lacks cubane units, exhibits extensive intra-planar hole delocalization which entropically hinders the four electron/hole concerted OER reaction. An essential distinguishing trait of a truly relevant catalyst is efficient continuous operation in a real electrolyzer stack. Initial trials of cubic LiCoO2 in a solid electrolyte alkaline membrane electrolyzer indicate continuous operation for 1000 hours (without failure) at current densities up to 400 mA cm−2 and overpotential lower than proven PGM (platinum group metal) catalysts.
Broader contextThe basis for hydrogen economy requires the large-scale production of hydrogen, many times the level supported by steam reforming. It has been proposed that the electrolysis of water can yield supply on a par with the world's energy needs. However, efficient water splitting requires electrocatalysts that help to lower the total energy input closer to the thermodynamic minimum. These electrocatalysts are typically based on platinum group metals that cannot be produced at such a scale. In membrane-based electrolyzers, the implementation of catalysts based upon earth-abundant metals has not been realized because they mostly operate under highly acidic conditions afforded by the proton exchange membrane Nafion. With the development of anion exchange membranes, non-PGM metal catalysts can be used and the high-efficiency of membrane-based electrolyzers can be attained without non-scalable catalysts. In this study, we show that LiCoO2 transforms through in situ delithiation into cubic spinel LiCo2O4, forming a highly active and stable oxygen evolution reaction (OER) catalyst that works at both the lab scale and in a membrane-based electrolysis configuration relevant to large scale commercial applications of hydrogen production via water splitting. |
Introduction
Electrolytic storage of solar energy through water splitting chemistry, eqn (1) and (2), is recognized as indispensable for achieving widespread adoption of photovoltaic and wind electrical sources that displace fossil energy.1,2 The energy storage density of chemical bonds exceeds that of any battery yet conceived by 100 fold or more, is long-term stable, more scalable, and can be used for other applications (e.g. hydrogen).3 If done efficiently, it would transform both the electrical power and fuel industries. Presently, electrolyzer stacks operate at best case efficiencies of 74% (proton exchange membrane, PEM)4 and 68% (alkaline)5 due to fundamental limitations in the catalysts. The O2 evolution (OER) and H2 evolution (HER) half-reactions require catalysts at the respective electrodes to lower the intrinsic barriers to these multi-electron/proton reactions. Electrocatalysts allow reactions to proceed faster by lowering activation energy barriers and thus save energy otherwise wasted as heat.| Anode: 2H2O → O2 + 4H+ and 4e− | (1a) |
| 4OH− → O2 + 2H2O + 4e− | (1b) |
| Cathode: 2H2O + 2e− → 2H2 + 2OH− | (2a) |
| 2H+ + 2e− → H2 | (2b) |
It has been known for more than 30 years that first-row transition metal oxides, particularly the perovskite, spinel and pyrochlore structure types, can catalyze oxygen evolution from strongly alkaline solutions.6,8–12 However, the oxides of first-row transition metals have so far been relatively unsuitable because of two limitations: instability due to corrosion in acids – conditions used in present day PEM electrolyzers7,13 – and inability to generate the necessary current densities at low overpotentials (inefficiency). Commercial alkaline electrolyzers can use low-cost Ni electrodes as catalysts, but these require 30% KOH electrolyte and elevated temperature to achieve competitive efficiencies below PEM electrolyzers.14,15 This combination of high corrosion and need for thermal management is dangerous and costly. Commercial electrolyzers employing alkaline exchange membranes (AEM) do not yet exist.
OER and HER catalysts can also be prepared as amorphous oxide coatings generated in situ by controlled electrolysis of non-noble metal electrodes, or by electrodeposition of soluble precursors.16–23 Some films exhibit high initial activity, but typically are unstable over time at high current densities owing to dissolution and passivation of the surface.24,25 The amorphous forms have a high degree of defects.9,18,26,27 Overcoming these limitations is a challenge due to the lack of structural knowledge for films.
Of the first-row transition metals selected for the OER, nickel, iron, and especially cobalt have been front-runners as both homogeneous and heterogeneous catalysts due to low overpotentials, approaching those of the noble metal analogues. In contrast to noble metals however, not all such materials are stable as high surface area nanoparticles. The crystalline oxides tend to be spinel-type: Co3O4,28,29 NiCo2O4,30 LiCoO2,31 perovskite-type: LaCoO3,32 SrCoO3,6 or layered oxy-hydroxides.33,34 In particular, exceptional activity has been observed in mixed nickel and iron hydroxide/oxide catalysts.35 Under acidic electrolysis conditions, such as used in proton exchange membranes (PEM's), materials containing cobalt or nickel are highly susceptible to dissolution.
Since the discovery of lithium cobalt oxide as a highly active catalyst for water oxidation,31 there have been subsequent reports that have sought to further explain the nature of the apparent crystal structure dependence on catalysis.36–40 Our group was the first to report that cubic LiCoO2 (Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, also called lithiated spinel) can catalyze water oxidation near neutral pH using a photochemically driven-system, while the layered polymorph (trigonal space group, Rm) cannot, even after accounting for the effects of surface area. It has been reported that layered LiCoO2 is unstable after repeated electrochemical cycling in neutral and basic conditions and that this instability is linked to surface structural changes.36 In ref. 36, it was suggested that the active phase for catalysis is an amorphous surface similar to the CoOx of electrodeposited materials, and that a crystalline spinel phase only serves to passivate the catalyst. A separate study also found that the layered LiCoO2 is unstable (current decays) in long-term (∼20 hours) studies, but in contrast to the aforementioned study, they found both cubic LiCoO2 and chemically delithiated Li1−xCoO2 (0 ≤ x ≤ 1) are electrocatalytically active and stable.38
m, also called lithiated spinel) can catalyze water oxidation near neutral pH using a photochemically driven-system, while the layered polymorph (trigonal space group, Rm) cannot, even after accounting for the effects of surface area. It has been reported that layered LiCoO2 is unstable after repeated electrochemical cycling in neutral and basic conditions and that this instability is linked to surface structural changes.36 In ref. 36, it was suggested that the active phase for catalysis is an amorphous surface similar to the CoOx of electrodeposited materials, and that a crystalline spinel phase only serves to passivate the catalyst. A separate study also found that the layered LiCoO2 is unstable (current decays) in long-term (∼20 hours) studies, but in contrast to the aforementioned study, they found both cubic LiCoO2 and chemically delithiated Li1−xCoO2 (0 ≤ x ≤ 1) are electrocatalytically active and stable.38
Before any studies of water oxidation electrocatalysis with LiCoO2 were reported, the relative activity and stability of the two polymorphs had been a source of active discussion in the battery literature. The structural dynamics of the system in non-aqueous electrochemistry has been elucidated.41–43 The layered LiCoO2, which is superior for Li secondary battery applications, undergoes surface and sub-surface reconstruction to cubic and spinel phases after repeated charge/discharge cycles and at intermediate and low values of Li stoichiometry that results in reduced performance.41,42 This transformation can also be induced thermally above 1000 °C, where Li starts to evaporate, leaving behind a solid solution of cubic and layered phase LiCoO2 and Co3O4 as surface nodules.44 It has been suggested that the stability of the spinel-type structure (LiCo2O4) with Li occupying 8a tetrahedral sites, is actually greater than either cubic or layered forms in the intermediate Li content range, and the kinetic barrier for Li and Co migration between octahedral sites is a small penalty for enhanced stability.45 The connection between the electrochemistry of LiCoO2 in non-aqueous battery electrolytes versus oxygen evolution catalysis in aqueous electrolyte will be discussed in more detail below, as the subject of this study.
The origin and nature of the structural transformation of the layered LiCoO2 and the absence of it for cubic LiCoO2 during electrocatalysis in aqueous electrolyte is described herein. The outcome is the formation of a highly active catalytic phase present in both materials, namely, spinel LiCo2O4. The spinel LiCo2O4 and cubic LiCoO2 retain the same cubic space group built upon [Co4O4]n+ cubane subunits, unlike layered LiCoO2. We report different intrinsic electrocatalytic water oxidation activities of the two polymorphs of LiCoO2, and demonstrate performance approaching that of iridium nanoparticles, the best membrane-based OER catalyst to date.
Experimental
Synthesis of lithium cobalt oxide nanoparticles
Both cubic and layered-rhombohedral lithium cobalt oxides were synthesized via a previously reported sol–gel procedure (summarized in ESI†).31,46 Briefly, nitrate salts of the metals were mixed in stoichiometric amounts and dissolved in an aqueous solution with citric acid and urea. The solution was stirred and evaporated to form a gel at 80 °C, dried overnight at 170 °C to form the xerogel polymerized intermediate, and then crushed and calcined at 400 °C (denoted LT) or 700 °C (denoted HT) for 4 hours to form the final crystalline products. These are designated as LT-LiCoO2 and HT-LiCoO2. The product morphology and crystallinity were confirmed by powder X-ray diffraction (PXRD) and scanning electron microscopy (SEM). We purchased 20% Ir@C (vulcanized carbon) from Premetek.Electrochemical measurements
The electrode configuration used for studying the electrochemistry of LiCoO2 and 20% Ir@C was a film formed by drop-casting an ink of catalyst and conductive carbon suspended with Nafion® polymer as binder (0.7% by weight) onto a glassy carbon electrode (5 mm diameter, custom made, working electrode). Prior to use, the Nafion was cation exchanged with 0.1 M NaOH to neutralize the acid functional groups as described in earlier work.47 Catalyst loading was between 0.2–1.2 mg cm−2 for electrochemical measurements. Higher loading was used for HT-LiCoO2, as it was determined to have ∼3 fold lower surface area than LT-LiCoO2 based on BET N2 adsorption.31 We tested activity in both neutral (1 M phosphate buffer), and basic (1 M NaOH) conditions. Experiments were typically performed in a two-chamber cell separated by a glass frit with vigorous stirring. The reference electrode used in alkaline electrolyte was Hg/HgO (1 M NaOH) from CHI, and in neutral electrolyte a homemade Ag/AgCl (sat'd KCl) calibrated against NHE. Counter electrodes were either boron-doped diamond or titanium mesh. Details of the electrochemical measurements are given in the ESI.†Faradaic efficiency in basic solution was measured for each catalyst by gas chromatography. We used a two-electrode setup sealed in an electrochemical cell with a known headspace and electrolyte volume, as previously described.48 A chronopotentiometric experiment at 10 mA cm−2 in 1 M NaOH was run for 2 hours for all 3 samples in replicate.
For long-term stability measurements, pellet electrodes were fabricated using catalyst, conductive carbon, and PVDF in the ratio 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:10.49 Chronopotentiometric experiments were conducted over 14 hours at 10 mA cm−2. The reason pellet electrodes were used in place of films is that bubble formation in the film leads to instability and separation of the film from the glassy carbon substrate. This makes the assessment of true catalyst stability difficult.
:15:10.49 Chronopotentiometric experiments were conducted over 14 hours at 10 mA cm−2. The reason pellet electrodes were used in place of films is that bubble formation in the film leads to instability and separation of the film from the glassy carbon substrate. This makes the assessment of true catalyst stability difficult.
Transmission electron microscopy
Samples used for high-resolution transmission electron microscopy (HRTEM) were pristine powders of cubic and layered LiCoO2 synthesized by the aforementioned sol–gel method. For HRTEM analysis of “post-catalysis” samples, a modified procedure for electrochemical tests had to be used to avoid interference in the analysis by carbon and Nafion. These samples were prepared by dispersing only the catalyst onto a titanium grid with which we conducted chronopotentiometry at 1 mA cm−2 for 1 hour in 1 M NaOH. The Ti meshes were then sonicated in a small amount of ethanol and the suspensions were dispersed onto lacey carbon TEM grids. A JOEL 2010F TEM/STEM with EDS and EELS attachments was used for the high-resolution (HRTEM) and selected area electron diffraction (SAED) microscopy observations. The scope is operating at 200 keV with resolution better than 1.5 Angstrom in both imaging and scanning modes.Corrosion analysis
The (electro)chemical leaching of lithium was monitored by inductively-coupled plasma optical emission spectroscopy, on a Perkin Elmer Optima 7300 ICP-OES. Chronopotentiometry at 10 mA cm−2 was performed with drop-casted film electrodes for 2 hours, and the resulting electrolyte was analyzed for trace metals (e.g. Li, Co, Ni, Mn). Details on sample preparation and data analysis are given in the ESI.†Catalyst performance in AEMWE
The performance of cubic LiCoO2 as an anode catalyst for alkaline exchange membrane water electrolysis was tested in a single stack configuration and compared to a benchmark PGM AEM cell. The details of the cell hardware and catalyst loading can be found in the ESI.† Polarization curves, along with a long-term stability test are given in Fig. 6.Results
Morphology and crystallinity
The morphology and crystallinity of the metal oxides were examined by SEM/TEM, and powder X-ray diffraction (PXRD), respectively. It was confirmed that the as-synthesized materials had nanoparticulate morphology, with a range of particle sizes (Fig. 1 and 2). The PXRD patterns for each catalyst match well with the standards, with the expected splittings at 38 and 65° 2θ at higher temperatures, signifying the formation of layered LiCoO2. Low-temperature LiCoO2, synthesized at 400 °C, crystallizes in a cubic phase (Fdm) where Co and Li are at octahedral 16d and 16c sites, respectively. The particles were generally spherical with diameters between 20–100 nm. High-temperature LiCoO2, synthesized at 700 °C, crystallizes in a layered rhombohedral form (Rm) with alternating octahedral layers of Li–O and Co–O. Co and Li occupy 3a and 3b sites, respectively. The particle size was noticeably larger, but still nanoscopic, with generally more flattened crystallites ranging between 50–150 nm. Both phases have the same oxide sublattice and differ only in cation ordering. These morphological and structural properties are consistent with our previous work using the same synthetic procedure.31
 | ||
| Fig. 1 PXRD of LT- (blue) and HT- (black) LiCoO2 along with SEM images confirming nanoscopic morphology. The standard patterns for Fdm and Rm lithium cobalt oxide are in red. | ||
 | ||
| Fig. 2 TEM images: (a) and (c), as-synthesized LT-LiCoO2, (b) and (d), as-synthesized HT-LiCoO2. | ||
Because PXRD reflects bulk composition and the patterns are so similar to one another, some layered phase may begin to form and be present in the low-temperature sample (<2% limit of detection). TEM performed on pristine samples of both low- and high-temperature synthesized LiCoO2 demonstrates a highly crystalline single phase product extending from the interior to the surface of all nanoparticles. There are well-faceted crystallites that are generally smaller and more spherically symmetric in cubic, than in layered LiCoO2, which are more elongated. Additional HRTEM studies corroborated the phase purity of the as-prepared sample (Fig. 2 and Fig. S6, ESI†). We designate these two samples by their synthesis temperatures, as LT and HT LiCoO2, respectively.
OER electrocatalytic activity
The electrochemical activity of cubic and layered LiCoO2 is illustrated for both basic and neutral conditions. The figures of merit important for OER catalysis are given in Table 1. We normalized the loading of electrocatalyst to the surface area obtained from N2 gas absorption (BET) as this eliminated possible artifacts from diffusion limitation of reactants and products. For control, the electrocatalytic activity of 5 nm iridium nanoparticles on Vulcan carbon (20%) (Premetek) is reported as well.| Sample (particle size) | BET surface area, m2 g−1 | Cat. loading mg cm−2 electrode | pHa | Tafel slope, mV dec−1 | η@10 mA cm−2 | Mass activity, A g−1 (η = 400 mV) | Surface activity, A m−2 (η = 400 mV) | %O2 faradaic efficiency, pH 14 | AEMWE cell potential (400 mA cm−2)d |
|---|---|---|---|---|---|---|---|---|---|
| a Electrolytes: pH 7 = NaH2PO4/K2PO4, pH 14 = 1 M NaOH. b Amps (cat surface area)−1 following surface reconstruction. c 5 nm Ir specific surface area (Premetek). d Values obtained from polarization curve (Fig. 6a), Pt cathode catalyst. | |||||||||
| LT-LiCoO220–100 nm | 56 | 0.32 | 7 | 75 ± 11 | 570 ± 12 | 2.32 | 0.041b | 105 ± 11 | 1.91 V |
| 14 | 48 ± 3 | 430 ± 14 | 5.24 | 0.094 | |||||
| HT-LiCoO250–150 nm | 14 | 1.2 | 7 | 81 ± 6 | 588 ± 27 | 0.39 | 0.028b | 96 ± 11 | n/a |
| 14 | 49 ± 3 | 430 ± 8 | 1.71 | 0.12 | |||||
| 20% Ir@C 5 nm | 80c | 0.21 | 7 | 61 ± 9 | 460 ± 13 | 29.1 | 0.36 | 98 ± 9 | 2.22 V |
| 14 | 40 ± 8 | 330 ± 4 | 151.2 | 1.89 | |||||
The working electrode film containing catalyst was characterized by cross-sectional SEM (Fig. S1, ESI†), and it was determined to be 5–10 μm thick. There is substantial porosity, but also very uniform dispersion of catalyst, binder, and carbon, with no distinguishable segregation of any component. The average polarization curves for each sample are shown in Fig. 3. They were generated by averaging the forward and reverse sweeps of slow scan rate cyclic voltammograms (iR corrected, 10 mV s−1). 20% Ir on carbon performed the best in both neutral and basic conditions, with specific activity and Tafel slope values matching closely those reported in the literature6,50 (Table 1). Both phases of LiCoO2 show highest activity in alkaline solution, and substantial decreases in activity in phosphate buffer at pH 7. This non-Nernstian behavior is typical of first-row transition metal oxide catalysts, and indicates a larger activation energy at pH 7 relative to alkaline pH. In base, LT-LiCoO2 exhibits mass specific activity of ∼5 A g−1 (η = 400 mV), with a Tafel slope of ∼48 mV dec−1. Fig. 4 shows that HT-LiCoO2 deactivates from its initial state in ∼200 s. Once in steady state it has a Tafel slope value ∼49 mV dec−1 and mass activity of ∼2 A g−1.
 | ||
| Fig. 3 Averaged cyclic voltammograms of electrocatalysts in two electrolytes, pH 14 and 7. Correction for capacitance charging done by averaging forward and reverse sweeps. Scan rate 10 mV s−1. The potential is iR-corrected. | ||
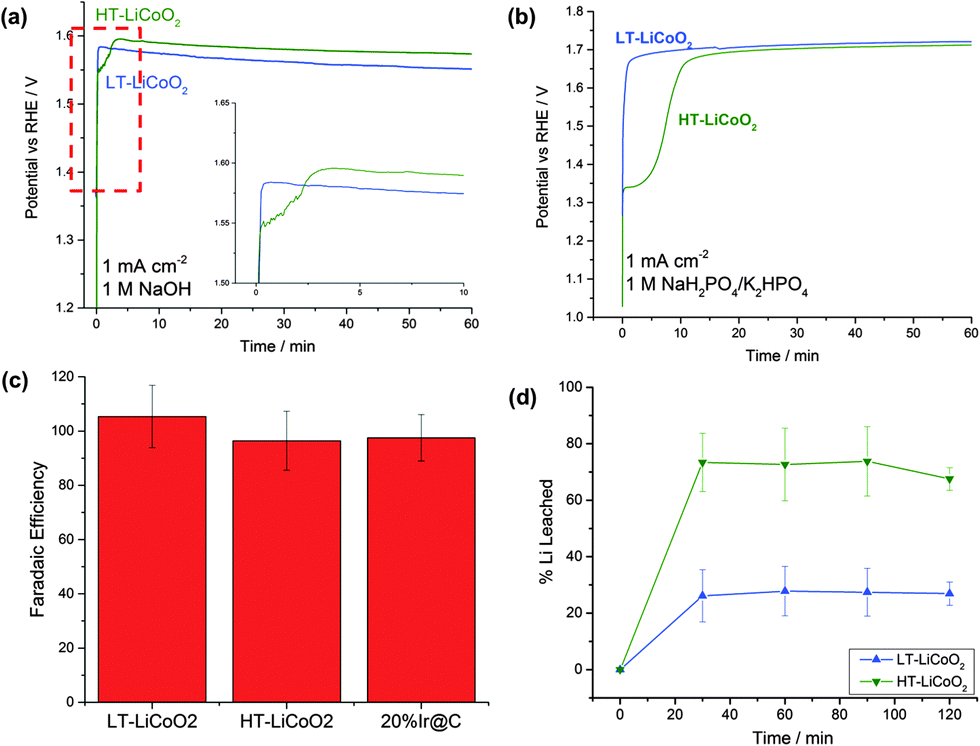 | ||
| Fig. 4 (a and b) Comparison of the operating potential at 1 mA cm−2 for HT- and LT-LiCoO2 at pH 14 (a) and at pH 7 (b). Inset in (a) is region in dotted line showing initial activation process seen in HT-LiCoO2. (c) Faradaic efficiency (O2 yield) measured from experiments at 10 mA cm−2 and after 2 hours (Fig. S4, ESI†), and (d) Li+ leaching into solution over the course of 2 hours at 10 mA cm−2 plotted as %Li in the starting material. | ||
The cyclic voltammograms (CV) of pristine LT- and HT-LiCoO2 electrodes are presented in Fig. S2 (ESI†), which highlights their different redox features below the water oxidation wave. The layered HT-LiCoO2 CV shows more complex features and a much higher yield for the Co3/4+ peak in both neutral and alkaline conditions. This has been previously ascribed to both lithium removal into electrolyte and Li reordering within the surface/bulk.51 However, both materials show marked decreases in the non-OER redox peaks after only one cycle, indicating some irreversible process occurring upon oxidation.
To quantify this electrochemical/charging effect, we measured the scan rate dependence of the double layer capacitance (Cdl) for both materials below the OER potential (Fig. S3, ESI†). The values obtained are 2.5 mF for LT, and 11 mF for HT-LiCoO2. These values reflect differences in access to the electrochemically active material. The greater capacitance of layered LiCoO2 is known to be due to its larger accessible volume for Co oxidation and Li deintercalation.52,53 This outcome is also consistent with the larger pre-OER peaks in the CV's. This will be discussed further with respect to the Li leaching data presented below.
In order to understand how the catalytic activity changes over time we performed chronopotentiometric experiments at 1 and 10 mA cm−2. The 1 mA cm−2 traces for both LT- and HT-LiCoO2 in alkaline and neutral solution are shown in Fig. 4a and b. Most visibly, in the 1 mA cm−2 trace, pristine electrodes of HT-LiCoO2 undergo a ∼3 minute activation process. Instead of the typical logarithmic potential rise to steady-state expected for chronopotentiometry, there is an inflection indicative of another possible reaction taking place. After this time point it starts to drop, and very nearly approaches the same overpotential as for LT-LiCoO2. At pH 7, the same behavior is seen; LT-LiCoO2 is immediately active compared to an even longer (10 min) lag phase for HT-LiCoO2 (Fig. 4b). This activation phenomenon is also observed on the 10 mA cm−2 traces over a much shorter time scale and, at that current density, the two polymorphs maintain the same overpotential (within error) over the 2 hour time scale (Fig. S4, ESI†). Attempts to separate the intrinsic O2 evolution activity of nascent HT-LiCoO2 from the electrolytically driven capacitive charging before appreciable Li+ deintercalation and phase change were unsuccessful.
Fig. S4 (ESI†) also shows a comparison to the control, 5 nm particles of Ir@C, which exhibits a 100 mV lower overpotential at 10 mA cm−2. As the overpotential can be particle size dependent, we compared the specific activity normalized to catalyst surface area in Table 1. This shows that following restructuring HT- and LT-LiCoO2 (100 nm and 50 nm, respectively) have the same specific activity and this is about 10–20 fold lower than 5 nm Ir@C.
Faradaic efficiency
Important to any catalysis is the measurement of products, and we have measured the faradaic yield of oxygen using gas chromatography (see Fig. 4c and Table 1). Within expected error due to crossover reduction of O2 and the inherent sensitivity limits for the technique, the samples all yield high, nearly 100% faradaic efficiency over the course of two hour 10 mA cm−2 experiments.Corrosion analysis
Since both polymorphs of lithium cobalt oxide have been established as intercalation compounds, we decided to analyze the electrolyte solutions of the chronopotentiometric experiments. We used ICP-OES to quantify lithium or cobalt ions that may have leached during the electrolysis. The results are presented in Fig. 4d. Although we did not see any Co in the electrolyte, we did see significant amounts of Li. We found that ∼30% of the original Li content in the LT sample had been removed and ∼70% of that in HT-LiCoO2. The results are consistent with the observed larger measured Cdl for HT-LiCoO2. The layered phase is the more easily delithiated compound, because of the lability of Li. For both materials, after an initial period of activation, no further Li removal is observed, indicative of a stable endpoint for catalyst composition.Surface analysis by HRTEM
To monitor atomic structure following catalysis, we performed HRTEM analysis on the two LiCoO2 polymorphs after being subjected to prolonged electrolysis at 1 mA cm−2. Because the layered polymorph of lithium cobalt oxide has previously been shown to undergo surface restructuring during charge/discharge cycling in battery applications, we decided to conduct this type of analysis for both LT and HT-LiCoO2 samples in OER electrolytic conditions. The results are shown in Fig. 5 and additional images, including the analysis of pristine samples can be found in the ESI† (Fig. S5–S7). | ||
| Fig. 5 HRTEM images of (a) LT-LiCoO2 and (b) HT-LiCoO2 after electrolysis in 1 M NaOH at 1 mA cm−2. The FFT patterns of the interior and edges can both be assigned to the spinel LiCo2O4. Reflection assignments are labeled for 220, 111, and 311 peaks of cubic phase. | ||
Both “post-catalysis” LiCoO2 samples have nanocrystallite morphology, exhibiting well-defined lattice fringes in both the bulk and surface of the particles examined. LT-LiCoO2 has smaller crystallite domains, as indicated by the difference in SAED patterns – rings, as opposed to spots seen in the HT-LiCoO2 pattern (Fig. S6, ESI†). In the analysis of the HRTEM images, we took the Fast-Fourier Transform (FFT) from different regions of the post-catalysis particles. We found that for both samples, the patterns are best ascribed to a cubic phase of LiCoO2 (Fig. 5), specifically viewed along the [110] and [112] zone axes (ZA). Importantly, we see reflections originating from the 220 plane for both materials, indicative of Li occupancy at tetrahedral sites.42 The peak was not assignable to Co tetrahedral site occupancy (Co3O4 spinel) based upon the d-spacings and the generally weaker intensity. In addition, XPS of the post-catalysis samples reveals broadening of the Co2p3/2 peak to higher binding energy, consistent with the increased oxidation state expected for a delithiated sample (Fig. S7, ESI†).54 It is also apparent that the phase transformation in the case of HT-LiCoO2 extends beyond just the first few layers since the FFT pattern from the interior matches well that from the surface.
For long-term electrolysis stability tests, a different electrode configuration was employed to avoid failure due to film instability. The drop-cast films are only transiently stable, and can peel off or oxygen bubbles can block the surface, leading to false indications of catalyst instability during cycling or fixed electrolysis experiments (see Fig. S8A, ESI†). Instead, we used a pellet electrode made from the catalyst embedded in epoxy to conduct 14 hour experiments at 10 mA cm−2. The results, given in Fig. S8B (ESI†), show that both HT and LT-LiCoO2 are long-term stable in basic and neutral electrolyte.
Discussion
Pristine HT-LiCoO2 (layered) and LT-LiCoO2 (cubic) exhibit distinctly different OER electrocatalytic activity and electrochemical responses, confirming that these structurally distinct compounds of identical composition have electronically distinct Co environments and inherently different rates for water oxidation. However, upon close inspection of the surface structure with HRTEM, a structural convergence between the two phases occurs upon Li removal and oxidation of the bulk, which produces the spinel LiCo2O4. Starting from cubic LiCoO2, this transformation requires relatively minor rearrangement of the remaining Li+ cation from octahedral to tetrahedral site at the four O corners of the cubane [Co4O4]4+. The same cubic cobalt oxide sublattice is shared by cubic LiCoO2 and spinel LiCo2O4 phases. The cubic phase can tolerate the electron loss associated with oxidation/OER catalysis without large structural rearrangement of the Co–O sublattice. By contrast, layered LiCoO2 is built around incomplete cubanes, [LiCo3O4]2+, and requires major rearrangements to form the stable spinel phase.As established for lithium ion batteries, layered LiCoO2 has the ideal structure type for facile cation deintercalation.55,56 However, multiple reports45,57,58 have now established that the relative thermodynamic stability of layered LiCoO2 is affected by lithium removal and Co oxidation and thus the structure can become kinetically determined. These phenomena have been mostly observed in organic electrolyte42,43 (ethylene/propylene carbonate) and partly in aqueous electrolyte when used for oxygen evolution reaction catalysis.36,39 It is observed in the majority of cases that after overcharging or repeated cycling, layered LiCoO2 particles have substantial surface and subsurface restructuring, with the resulting phase being assigned to either spinel LiCo2O4 or cubic LiCoO2. This leads to degradation in performance. In support of this finding is the observation that the low-temperature synthesized cubic LiCoO2 and delithiated LixCoO2 (0 < x < 1) have poorer performance as intercalation compounds, with a lower amount of extractable Li and higher operating voltages with multiple voltage plateaus. In fact, with lithium extraction it is observed that some O2 is liberated and the transformation of layered to spinel is assisted by such a reaction.42,59,60 By comparison, we observe, in aqueous electrolytes under electrolytic O2 evolution conditions, removal of 70% of the Li content from HT-LiCoO2 in conjunction with the formation of the spinel phase at the surface and partly in the interior, where the stoichiometry at the bulk level is close to Li0.3CoO2. XPS also shows broadening of the Co2p3/2 peak toward greater binding energy, indicative of higher valence of Co. The resulting phase is an active and compositionally stable catalyst nearly mirroring that which forms from LT-LiCoO2, in which less than half as much Li can be removed, and its starting structure is already a more active catalyst than HT-LiCoO2.
Theoretical calculations have predicted that for LixCoO2 (0 ≤ x ≤ 1) at intermediate lithium compositions (i.e. Li0.5CoO2), the thermodynamically favored structure is the spinel LiCo2O4, where Li would migrate to tetrahedral sites and 25% of the Co would be in the Li layers.45,61 We observe that this is the preferred structure that forms during OER catalysis, evidenced both by the structural change and by the loss of Li content.
A minimal requirement for catalysis of O2 evolution from water by these materials is oxidation of Co3+ to Co4+ at a potential at or above eqn (1a) or (1b). Recent electronic structure calculations along with in situ X-ray spectroscopy have shown that the process of Li deintercalation from layered LiCoO2 is charge balanced by not only Co3/4+ oxidation, but also electron loss from a lower lying oxide band in the most highly charged samples of LiCoO2.62 Some reports suggest that this larger intermixing of Co 3d states and O 2p states yields higher conductivity and that is the source of greater OER activity.38,40 This condition alone is insufficient for the four-electron concerted oxidation reaction. We suggest an alternative explanation.
By comparison, among molecular Co oxide clusters, it has been found that only in molecules containing [Co4O4]4+ cubes is the Co4+ oxidation state accessible at reasonable potentials, while the di- and tri-nuclear analogues (containing [Co2O2]2+ and [Co3O4]+ cores, respectively) cannot be oxidized in this same window (>1 V difference) and actually decompose at higher potentials where water oxidation by cubanes begins.63 The explanation for the accessible potential of [Co4O4]4+/5+ is due to resonance delocalization of electron holes around the cube, as established by ESR spectroscopy which found comparable spin density on Co and O centers.64 Among these molecular Co clusters only the cubes were found to be active water oxidation catalysts at potentials just above the [Co4O4]4+/5+ couple.63,65
Taken together with the observations of this study, it is apparent that the individual cubical [Co4O4]4+ core, such as present in cubic LiCoO2, spinel LiCo2O4 and molecular clusters provides exceptional energetic stabilization of the HOMO which facilitates oxidation from electronic levels comprised of mixed O 2p and Co 3d(t2g) orbitals. This stabilization arises from hole delocalization in the symmetrical cube that is absent in incomplete cubes comprised of [Co3O4]+ cores and in layered LiCoO2 where [LiCo3O4]2+ incomplete cubes exist. Instead, hole delocalization in layered LiCoO2 occurs macroscopically within the individual CoO layers, due in part to favorable entropy. Macroscopic charge delocalization is necessary for charge storage devices such as batteries, but detrimental to water oxidation catalysis which requires concerted four-electron oxidation and O–O bond formation localized on two water molecules bound to one or two adjacent Co centers. These considerations rationalize the distinct catalytic performance of the two polymorphs of LiCoO2.
Validation of the unique water oxidation capability of the cubical metal–oxo cluster motif has been verified across multiple examples of molecular63,66–68 and solid-state transition metal–oxo compounds.31,69,70 Prediction of this potential originated from consideration of the water oxidation site found in photosynthetic organisms where a similar core type is found.71
Finally, to test actual commercial relevance, we have tested whether the observed activity in a flooded electrochemical cell can be realized in a solid electrolyte membrane-based electrolyzer stack, where operating current densities far exceed those normally probed at the lab scale. In Fig. 6a, the polarization curve of a cell using cubic LiCoO2 as the anode catalyst illustrates higher electrochemical efficiency than a cell using an iridium oxide catalyst at comparable loading. This performance improvement extends up to 500 mA cm−2 (at least), and the activity is reproducible across different batches of catalyst. Fig. 6b shows the operation of such a cell at various current densities for 1000 hours without failure. These experiments demonstrate that the LiCoO2 is a highly efficient and robust catalyst for alkaline water electrolysis and has potential for displacement of noble metal catalysts used in commercial electrolyzers.
 | ||
| Fig. 6 LiCoO2 performance as anode catalyst in alkaline exchange membrane-based electrolysis. (a) Polarization curve showing favorable efficiency as compared to an IrOx-based anode (0.3 V lower@500 mA cm−2), as well as reproducibility of activity. (b) Run time-averaged operating potential for a single cell stepped to different current densities. | ||
Conclusions
In combination with electrochemistry, corrosion analysis, and surface sensitive HRTEM, we were able to probe precisely the distinguishing characteristics of the complex LiCoO2 system during aqueous OER electrocatalysis. Starting from the pristine structures, both the layered and the cubic phases of LiCoO2 form the same structure type during catalysis, the cubic spinel phase, LiCo2O4. We have temporally resolved this transformation and identified its origin stemming from Li+ deintercalation. In addition, we have demonstrated excellent electrocatalytic efficiency for the OER and long-term stability of the resulting spinel phase in comparison to the best noble metal catalyst used in commercial membrane-based electrolyzers (5 nm iridium). This was realized at both the lab-scale and in a membrane-based electrolyzer relevant for commercial applications. Further reduction in particle size of LiCoO2 (20–100 nm) is possible and additional efficiency benefits can be anticipated.Acknowledgements
Research jointly funded by the National Science Foundation, Division of Chemical, Bioengineering, Environmental, and Transport Systems (CBET), and the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Fuel Cell Technologies Office, award number CBET-1433492. Research was also funded by DOE Small Business Innovation Research (SBIR), award number DE-SC0013179. We would like to thank Shinjae Hwang for obtaining XPS data, and Ryan Bu for assistance in obtaining the ICP-OES data.References
- J. Turner, Science, 1999, 285, 687–689 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- M. Ball and M. Wietschel, The Hydrogen Economy: Oppurtunities and Challenges, Cambridge University Press, New York, NY USA, 2010 Search PubMed.
- U. S. D. D. R. a. I. f. V. e. a. E. sustainability, ed. DOE, 2013.
- D. o. Energy, 2008, Advanced Alkaline Electrolysis, pp. 110–112.
- Y. Matsumoto and E. Sato, Mater. Chem. Phys., 1986, 14, 397–436 CrossRef CAS.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- J. O. Bockris and T. Otagawa, J. Phys. Chem., 1983, 87, 2960–2971 CrossRef CAS.
- B. S. Brunschwig, M. H. Chou, C. Creutz, P. Ghosh and N. Sutin, J. Am. Chem. Soc., 1983, 105, 4832–4833 CrossRef CAS.
- P. Rasiyah and A. C. C. Tseung, J. Electrochem. Soc., 1983, 130, 365–368 CrossRef CAS.
- J. O. M. Bockris and T. Otagawa, J. Electrochem. Soc., 1984, 131, 290–302 CrossRef CAS.
- P. Rasiyah and A. C. S. Tseung, J. Electrochem. Soc., 1984, 131, 803–808 CrossRef CAS.
- D. Seley, K. Ayers and B. A. Parkinson, ACS Comb. Sci., 2013, 15, 82–89 CrossRef CAS PubMed.
- Ø. Ulleberg, Int. J. Hydrogen Energy, 2003, 28, 21–33 CrossRef.
- J. O. Jensen, V. Bandur, N. J. Bjerrum, S. H. Jensen, S. Ebbesen, M. Mogensen, N. Tophøj, L. Yde, K. L. Technical Univ. of Denmark and R. N. L. f. S. E. Technical Univ. of Denmark, Pre-investigation of Water Electrolysis, 2008 Search PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prevot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- S. E. S. E. Wakkad and A. Hickling, Trans. Faraday Soc., 1950, 46, 820–824 RSC.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- M. E. G. Lyons and M. P. Brandon, Int. J. Electrochem. Sci., 2008, 3, 1425–1462 CAS.
- M. E. G. Lyons and M. P. Brandon, Int. J. Electrochem. Sci., 2008, 3, 1386–1424 CAS.
- M. E. G. Lyons and M. P. Brandon, Int. J. Electrochem. Sci., 2008, 3, 1463–1503 CAS.
- M. E. G. Lyons and M. P. Brandon, Phys. Chem. Chem. Phys., 2009, 11, 2203–2217 RSC.
- M. Risch, A. Grimaud, K. J. May, K. A. Stoerzinger, T. J. Chen, A. N. Mansour and Y. Shao-Horn, J. Phys. Chem. C, 2013, 117, 8628–8635 CAS.
- J. B. Gerken, J. G. McAlpin, J. Y. C. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14431–14442 CrossRef CAS PubMed.
- M. Risch, V. Khare, I. Zaharieva, L. Gerencser, P. Chernev and H. Dau, J. Am. Chem. Soc., 2009, 131, 6936–6937 CrossRef CAS PubMed.
- M. W. Kanan, J. Yano, Y. Surendranath, M. Dinca, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 13692–13701 CrossRef CAS PubMed.
- A. Harriman, I. J. Pickering, J. M. Thomas and P. A. Christensen, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2795–2806 RSC.
- A. J. Esswein, M. J. McMurdo, P. N. Ross, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2009, 113, 15068–15072 CAS.
- B. Cui, H. Lin, J. B. Li, X. Li, J. Yang and J. Tao, Adv. Funct. Mater., 2008, 18, 1440–1447 CrossRef CAS.
- G. P. Gardner, Y. B. Go, D. M. Robinson, P. F. Smith, J. Hadermann, A. Abakumov, M. Greenblatt and G. C. Dismukes, Angew. Chem., Int. Ed., 2012, 51, 1616–1619 CrossRef CAS PubMed.
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 3, 2439 Search PubMed.
- B. M. Hunter, J. D. Blakemore, M. Deimund, H. B. Gray, J. R. Winkler and A. M. Muller, J. Am. Chem. Soc., 2014, 136, 13118–13121 CrossRef CAS PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CAS.
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- S. W. Lee, C. Carlton, M. Risch, Y. Surendranath, S. Chen, S. Furutsuki, A. Yamada, D. G. Nocera and Y. Shao-Horn, J. Am. Chem. Soc., 2012, 134, 16959–16962 CrossRef CAS PubMed.
- Z. Lu, H. Wang, D. Kong, K. Yan, P.-C. Hsu, G. Zheng, H. Yao, Z. Liang, X. Sun and Y. Cui, Nat. Commun., 2014, 5, 4345 CAS.
- T. Maiyalagan, K. A. Jarvis, S. Therese, P. J. Ferreira and A. Manthiram, Nat. Commun., 2014, 5, 3949 CAS.
- N. Colligan, V. Augustyn and A. Manthiram, J. Phys. Chem. C, 2015, 150126162959004 Search PubMed.
- H. Liu, Y. Zhou, R. Moré, R. Müller, T. Fox and G. R. Patzke, ACS Catal., 2015, 3791–3800 CrossRef CAS.
- H. Gabrisch, R. Yazami and B. Fultz, J. Power Sources, 2003, 119–121, 674–679 CrossRef CAS.
- H. Gabrisch, R. Yazami and B. Fultz, J. Electrochem. Soc., 2004, 151, A891–A897 CrossRef CAS.
- S. Hwang, W. Chang, S. M. Kim, D. Su, D. H. Kim, J. Y. Lee, K. Y. Chung and E. A. Stach, Chem. Mater., 2014, 26, 1084–1092 CrossRef CAS.
- N. Pereira, J. F. Al-Sharab, F. Cosandey, F. Badway and G. G. Amatucci, J. Electrochem. Soc., 2008, 155, A831–A838 CrossRef CAS.
- C. Wolverton and A. Zunger, J. Electrochem. Soc., 1998, 145, 2424–2431 CrossRef CAS.
- S. Vivekanandhan, M. Venkateswarlu and N. Satyanarayana, J. Alloys Compd., 2007, 441, 284–290 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1263–B1268 CrossRef CAS.
- A. B. Laursen, K. R. Patraju, M. J. Whitaker, M. Retuerto, T. Sarkar, N. Yao, K. V. Ramanujachary, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2015, 8, 1027–1034 CAS.
- Y.-I. Jang, B. Huang, H. Wang, D. R. Sadoway, G. Ceder, Y.-M. Chiang, H. Liu and H. Tamura, J. Electrochem. Soc., 1999, 146, 862–868 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- N. Pereira, C. Matthias, K. Bell, F. Badway, I. Plitz, J. Al-Sharab, F. Cosandey, P. Shah, N. Isaacs and G. G. Amatucci, J. Electrochem. Soc., 2005, 152, A114–A125 CrossRef CAS.
- G. G. Amatucci, J. M. Tarascon and L. C. Klein, J. Electrochem. Soc., 1996, 143, 1114–1123 CrossRef CAS.
- E. Antolini, Solid State Ionics, 2004, 170, 159–171 CrossRef CAS.
- J. C. Dupin, D. Gonbeau and H. Benqlilou-Moudden, Thin Solid Films, 2001, 384, 23–32 CrossRef CAS.
- R. J. Gummow, D. C. Liles and M. M. Thackeray, Mater. Res. Bull., 1993, 28, 235–246 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4301 CrossRef CAS PubMed.
- Y. Shao-Horn, S. A. Hackney, C. S. Johnson, A. J. Kahaian and M. M. Thackeray, J. Solid State Chem., 1998, 140, 116–127 CrossRef CAS.
- Y. Shao-Horn, S. A. Hackney, A. J. Kahaian and M. M. Thackeray, J. Solid State Chem., 2002, 168, 60–68 CrossRef CAS.
- S. Choi and A. Manthiram, J. Solid State Chem., 2002, 164, 332–338 CrossRef CAS.
- Y. Ozawa, R. Yazami and B. Fultz, Electrochem. Solid-State Lett., 2004, 8, A38–A41 CrossRef.
- A. V. d. Ven and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 742–749 CrossRef.
- D. Ensling, G. Cherkashinin, S. Schmid, S. Bhuvaneswari, A. Thissen and W. Jaegermann, Chem. Mater., 2014, 26, 3948–3956 CrossRef CAS.
- P. F. Smith, C. Kaplan, J. E. Sheats, D. M. Robinson, N. S. McCool, N. Mezle and G. C. Dismukes, Inorg. Chem., 2014, 53, 2113–2121 CrossRef CAS PubMed.
- J. G. McAlpin, T. A. Stich, C. A. Ohlin, Y. Surendranath, D. G. Nocera, W. H. Casey and R. D. Britt, J. Am. Chem. Soc., 2011, 133, 15444–15452 CrossRef CAS PubMed.
- G. La Ganga, F. Puntoriero, S. Campagna, I. Bazzan, S. Berardi, M. Bonchio, A. Sartorel, M. Natali and F. Scandola, Faraday Discuss., 2012, 155, 177–190 RSC.
- R. Brimblecombe, G. F. Swiegers, G. C. Dismukes and S. Spiccia, Angew. Chem., Int. Ed., 2008, 47, 7335–7338 CrossRef CAS PubMed.
- M. Yagi, K. V. Wolf, P. J. Baesjou, S. L. Bernasek and G. C. Dismukes, Angew. Chem., Int. Ed., 2001, 40, 2925–2928 CrossRef CAS.
- W. Ruettinger, M. Yagi, K. Wolf, S. Bernasek and G. C. Dismukes, J. Am. Chem. Soc., 2000, 122, 10353–10357 CrossRef CAS.
- D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2013, 135, 3494–3501 CrossRef CAS PubMed.
- D. M. Robinson, Y. B. Go, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2010, 132, 11467 CrossRef CAS PubMed.
- G. C. Dismukes, R. Brimblecombe, G. A. N. Felton, J. R. S. Pryadun, E. Sheats, L. Spiccia and G. F. Swiegers, Acc. Chem. Res., 2009, 42, 1935–1943 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ee02195b |
| This journal is © The Royal Society of Chemistry 2016 |