
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, structure and reactivity of Pd and Ir complexes based on new lutidine-derived NHC/phosphine mixed pincer ligands†
Práxedes
Sánchez
,
Martín
Hernández-Juárez
,
Eleuterio
Álvarez
,
Margarita
Paneque
*,
Nuria
Rendón
and
Andrés
Suárez
 *
*
Instituto de Investigaciones Químicas (IIQ), Departamento de Química Inorgánica and Centro de Innovación en Química Avanzada (ORFEO-CINQA), CSIC and Universidad de Sevilla, Avda Américo Vespucio 49, 41092, Sevilla, Spain. E-mail: paneque@iiq.csic.es; andres.suarez@iiq.csic.es
First published on 27th September 2016
Abstract
Coordination studies of new lutidine-derived hybrid NHC/phosphine ligands (CNP) to Pd and Ir have been performed. Treatment of the square-planar [Pd(CNP)Cl](AgCl2) complex 2a with KHMDS produces the selective deprotonation at the CH2P arm of the pincer to yield the pyridine-dearomatised complex 3a. A series of cationic [Ir(CNP)(cod)]+ complexes 4 has been prepared by reaction of the imidazolium salts 1 with Ir(acac)(cod). These derivatives exhibit in the solid state, and in solution, a distorted trigonal bipyramidal structure in which the CNP ligands adopt an unusual C(axial)–N(equatorial)–P(equatorial) coordination mode. Reactions of complexes 4 with CO and H2 yield the carbonyl species 5a(Cl) and 6a(Cl), and the dihydrido derivatives 7, respectively. Furthermore, upon reaction of complex 4b(Br) with base, selective deprotonation at the methylene CH2P arms is observed. The, thus formed, deprotonated Ir complex 8b reacts with H2 in a ligand-assisted process leading to the trihydrido complex 9b, which can also be obtained by reaction of 7b(Cl) with H2 in the presence of KOtBu. Finally, the catalytic activity of Ir–CNP complexes in the hydrogenation of ketones has been briefly assessed.
Introduction
Metal complexes based on lutidine-derived PNP pincer ligands have gained considerable attention due to their applications in organometallic chemistry and catalysis (Fig. 1a).1 In these derivatives metal–ligand cooperativity, triggered by deprotonation of the methylene arms of the ligand accompanied by dearomatisation of the pyridine ring, has led to unique reactivity in the activation of a diversity of X–H (X = H, C, O, N) bonds. While significantly less studied, analogous complexes based on CNC pincers (C stands for a N-heterocyclic carbene, NHC) have also been described (Fig. 1b).2–4 As shown with Ru–CNC complexes,3,4 these derivatives can also be deprotonated at the methylene CH2N bridges, and participate in metal–ligand cooperation processes. Furthermore, the larger Py–CH2–NHC linkage, which forms 6-membered metallacycles upon coordination, confers a greater flexibility on the ligand in comparison to 5-membered rings formed in PNP pincers. This flexibility should permit stabilizing metal complexes in a variety of coordination geometries, a relevant issue in catalysis where the intermediates in the catalytic cycle may need to adopt different structural arrangements. For example, while PNP ligands have only exhibited meridional coordination modes, facial coordination of CNC ligands in Ru complexes has been observed.3 More remarkable, as demonstrated by the Pidko's group, is that the increased flexibility of the CNC pincer results in enhanced reactivities towards H2 and CO2 in comparison to Ru–PNP systems.4b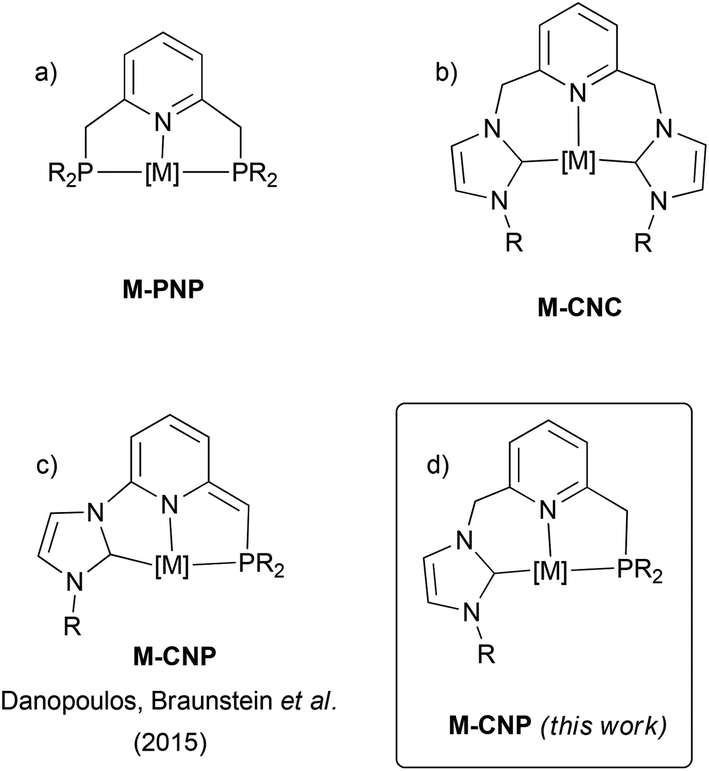 | ||
| Fig. 1 General structure of metal complexes with (a) lutidine-derived PNP ligands, (b) lutidine-derived CNC ligands, (c) deprotonated picoline-derived CNP ligands, and (d) lutidine-derived CNP ligands (this work). | ||
In addition, non-symmetric pincer ligands, i.e. having two inequivalent flanking donor groups, allow for a larger electronic and steric diversity derived from the potential tuning of two different side donors.5 With respect to lutidine-derived pincer complexes, some examples of PNP′6 and CNC′4a derivatives have been reported. Unsymmetrical PNX and CNX pincer complexes have also been described, although these derivatives are usually of the type PNN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 and CNN7 where hemilabile coordination of the N-donor flanking group has been proposed. In marked contrast, complexes based on CNP ligands having a pyridine central moiety and in which the two side functionalities are two significantly different strong σ-donors, such as a phosphine and a NHC, have not been investigated. In fact, a limited number of hybrid tridentate ligands possessing both phosphine and NHC donors have been reported, and these have either a ligand backbone based on a 1,3-disubstituted phenyl ring,8a or a different arrangement of the donor moieties, where the NHC group is the central unit of the pincer ligand.8b–f Also, recently Danopoulos, Braunstein et al. have prepared Co and Cr complexes based on deprotonated NHC/phosphine mixed pincer ligands having a central picoline motif (Fig. 1c).9
1 and CNN7 where hemilabile coordination of the N-donor flanking group has been proposed. In marked contrast, complexes based on CNP ligands having a pyridine central moiety and in which the two side functionalities are two significantly different strong σ-donors, such as a phosphine and a NHC, have not been investigated. In fact, a limited number of hybrid tridentate ligands possessing both phosphine and NHC donors have been reported, and these have either a ligand backbone based on a 1,3-disubstituted phenyl ring,8a or a different arrangement of the donor moieties, where the NHC group is the central unit of the pincer ligand.8b–f Also, recently Danopoulos, Braunstein et al. have prepared Co and Cr complexes based on deprotonated NHC/phosphine mixed pincer ligands having a central picoline motif (Fig. 1c).9
Based on these precedents, we aimed to develop a new class of ligands having NHC and phosphine side donors and a lutidine central fragment (CNP, Fig. 1d). A fundamental difference of these ligands with the previous picoline-based pincer derivatives reported by Danopoulos, Braunstein et al. resides in the presence of a methylene linker between the pyridine and the NHC functionalities, which could also be susceptible to deprotonation and should enhance pincer flexibility. In this contribution, we report on the synthesis of the precursors of these ligands as well as their coordination to Pd and Ir complexes. In particular, the ability of CNP ligands to adapt to different coordination geometries and participate in ligand-assisted processes has been assessed.
Results and discussion
Syntheses of imidazolium salts 1
Syntheses of imidazolium salts 1a(Cl), 1b(Cl) and 1b(Br) were effected as shown in Scheme 1. Derivative 1a(Cl) was prepared by reaction of the corresponding 2-chloromethyl-6-imidazolylmethyl-pyridine with diphenyl phosphine in the presence of KOtBu. Alternatively, in the case of salts 1b(Cl) and 1b(Br), higher product yields were obtained when diphenyl phosphine–borane adduct was used for the introduction of the P-donor fragment, followed by phosphine deprotection by simple treatment with refluxing MeOH. The CNP ligands precursors were obtained with moderate to good yields (55–85%) as white to brown solids. | ||
| Scheme 1 Syntheses of CNP ligands precursors. | ||
Synthesis and deprotonation of Pd–CNP complex 2a
For an evaluation of the coordination capabilities of these new CNP ligands, we initially studied the formation of Pd derivatives. Thus, salt 1a(Cl) was reacted with Ag2O in CH2Cl2, followed by addition of PdCl2(cod) to yield complex 2a (Scheme 2).10 The spectroscopic data support the formation of a complex in which the CNP ligand is coordinated to the metal centre as a pincer. For example, its 1H NMR spectrum shows distinctly two signals for the bridging methylenes. The CH2P protons appear at 4.32 ppm (d, 2JHP = 11.3 Hz), whereas the NCH2 hydrogens produce a singlet at 6.05 ppm. The 13C{1H} NMR spectrum exhibits a doublet at 166.4 ppm with a large 2JCP (183 Hz, carbene carbon, C2, of the NHC moiety), indicating the trans disposition of the NHC and phosphine moieties. | ||
| Scheme 2 Synthesis and reactivity of Pd complex 2a. | ||
A single crystal X-ray diffraction study of 2a confirmed the proposed structure (Fig. 2). Thus, complex 2a in the solid state is comprised of a Pd atom in an square-planar coordination geometry, with the carbene and phosphine fragments of the pincer disposed trans to each other (C2(NHC)–Pd–P = 168.65°), and the chloride ligand trans to the pyridine (N(Py)–Pd–Cl = 175.03°). The NHC–Pd–Py chelate ring has a boat conformation as determined by the torsion angle C(14)–N(3)–Pd(1)–C(1) of 32.4°, whereas the 5-membered ring involving the phosphine donor has an envelope conformation with a C(18)–N(3)–Pd(1)–P(1) torsion angle of 28.4°.
 | ||
| Fig. 2 ORTEP drawing at 30% ellipsoid probability of the cationic component of complex 2a. Hydrogen atoms and solvent molecules have been omitted for clarity. Selected bond lengths [Å] and angles [°]: Pd(1)–C(1) 2.038(6), Pd(1)–N(3) 2.071(4), Pd(1)–P(1) 2.2623(15), Pd(1)–Cl(1) 2.2758(14), C(1)–Pd(1)–P(1) 168.65(16), N(3)–Pd(1)–Cl(1) 175.03(14), P(1)–Pd(1)–Cl(1) 93.97(5), P(1)–Pd(1)–N(3) 81.34(14), C(1)–Pd(1)–Cl(1) 95.93(16), C(1)–Pd(1)–N(3) 88.6(2). | ||
Since it can be expected that CNP ligands can be deprotonated in either the CH2P or CH2–NHC arms, the acid/base responsiveness of 2a was tested by adding KHMDS to a suspension of the complex in THF.11 In the 1H NMR spectrum of the resulting product 3a, a significant up-field shift for the signals of the pyridine protons (5.55–6.46 ppm) in accord with the dearomatisation of this moiety is observed. Meanwhile, the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHP fragment produces a singlet signal at 3.41 ppm (integrating to 1H) in the 1H NMR spectrum and a doublet at 63.3 ppm (1JCP = 66 Hz) in the 13C{1H} NMR experiment, evidencing the selective deprotonation of the CNP ligand at the CH2P arm.
CHP fragment produces a singlet signal at 3.41 ppm (integrating to 1H) in the 1H NMR spectrum and a doublet at 63.3 ppm (1JCP = 66 Hz) in the 13C{1H} NMR experiment, evidencing the selective deprotonation of the CNP ligand at the CH2P arm.
Synthesis and structural features of Ir–CNP complexes
Reaction of imidazolium salts 1 with Ir(acac)(cod) provided cationic olefin complexes 4, isolated as yellow to orange solids in moderate to good yields (30–80%) (Scheme 3). These derivatives are stable in the solid state to the atmospheric agents, and have been fully characterised by NMR. For example, in the 1H NMR spectrum of 4b(Br), the CH2P protons are diastereotopic and appear as doublet of doublets at δ 3.36 (2JHP = 2.1 Hz) and 4.17 ppm (2JHH = 15.5 Hz and 2JHP = 11.6 Hz). Similarly, protons for the CH2N bridge produce two doublets at δ 5.65 and 6.91 (2JHH = 14.1 Hz). The 13C{1H} NMR spectrum shows a doublet at 164.8 ppm for the C2 NHC carbon with a very small 2JCP coupling constant of 8 Hz. These data suggest a cis coordination of the phosphine and NHC donors, also confirmed in the solid state by a single crystal X-ray diffraction study of 4b(BArF) (Fig. 3), obtained by anion exchange in complex 4b(Br) with NaBArF. | ||
| Scheme 3 Synthesis and reactivity of Ir–CNP complexes 4. | ||
 | ||
| Fig. 3 ORTEP drawing at 30% ellipsoid probability of the cationic component of complex 4b(BArF). Hydrogen atoms and solvent molecules have been omitted for clarity. Selected bond lengths [Å] and angles [°]: Ir(1)–C(1) 2.029(4), Ir(1)–N(3) 2.253(3), Ir(1)–P(1) 2.3412(9), Ir(1)–C(31) 2.263(4), Ir(1)–C(32) 2.217(4), Ir(1)–C(35) 2.140(3), Ir(1)–C(36) 2.101(4), C(1)–Ir(1)–P(1) 95.61(10), N(3)–Ir(1)–P(1) 75.98(8), N(3)–Ir(1)–C(1) 81.07(13). | ||
The structure of complex 4b(BArF) is best described as adopting a distorted trigonal bipyramidal geometry despite the acute P–Ir–N(Py) bond angle of 75.98(8)°. The CNP ligand exhibits a facial coordination, with the NHC donor in the apical position (P–Ir–C(NHC) angle of 95.61(10)°). In addition, the six-membered chelate ring involving the NHC and pyridine donors adopts a boat-like conformation as defined by the dihedral angle C(13)–N(3)–Ir(1)–C(1) of −50.6°, while the chelate ring containing the phosphine fragment exhibits an envelope conformation with a C(17)–N(3)–Ir(1)–P(1) angle of 29.5°. This facial coordination mode is unprecedented in M–PNP complexes and may be ascribed to the larger flexibility of the six-membered Py–M–NHC chelate ring, as previously observed in Ru–CNC complexes.3 In addition, the C(axial)–N(equatorial)–P(equatorial) coordination mode of the pincer differs significantly from previously reported pentacoordinated d8 pincer complexes,12 for which an eq–ax–eq distribution is usually observed.13 Finally, as observed with other pentacoordinated diolefin Ir complexes,13 the distance from the Ir atom to the centroid of the CC bonds is slightly longer for the alkene coordinated trans to the NHC than for the olefin placed in the meridional position (Δd(Ir-centroid CC) = 0.14 Å).
As exemplified with complex 4a(Cl), a dynamic behaviour in solution for complexes 4 has been evidenced by NMR. VT-1H NMR spectra of 4a(Cl) registered in the temperature range between 50 and −80 °C show sharp signals for the resonances attributable to the CNP ligand. In contrast, broad signals at 2.98 and 3.49 ppm, integrating for two protons each, are observed for the olefinic protons at 25 °C. 1H–1H COSY and 1H–1H NOESY experiments indicate that each signal is produced by protons of different olefinic moieties; i.e. Hb1,Hb2 and Ha1,Ha2 produce signals at 2.98 and 3.49 ppm, respectively (Fig. 4a). In addition, these signals broaden upon lowering the temperature and eventually split at temperatures below −25 °C into two sets of two signals each, appearing at δ 2.23 (Hb2) and 3.39 (Hb1), and 2.86 (Ha2) and 3.90 (Ha1), respectively. An approximate value of ΔG‡ = 10.9 kcal mol−1 at the coalescence temperature (244 K) can be estimated for the fluxional process. This dynamic behaviour can be ascribed to alkene site exchange allowed by the decoordination of the CC fragment trans to the NHC moiety to produce the distorted tetrahedral intermediate A, followed by re-coordination of the free olefin moiety to the opposite side without a net change of the fac coordination mode of the CNP ligand (Fig. 4a).
 | ||
| Fig. 4 Proposed dynamic processes in solution operating in the cationic part of complexes 4 (positive charges have been supressed for clarity; PR2 = PPh2, Ar = mesityl or 3,5-xylyl). | ||
In addition, the 1H,1H-exchange spectroscopy (EXSY) experiment of 4a(Cl) registered at 50 °C demonstrates the existence of additional dynamic processes with higher energy barriers. Thus, intense exchange cross-peaks between the signals for the olefinic protons appearing at 2.98 and 3.49 ppm are observed, which can be explained by the formal rotation of the diolefin ligand allowed by the decoordination of one of the Ir–alkene bonds (Fig. 4a). Furthermore, the observation of strong correlation peaks between the resonances of the o-, m- and p-protons of one of the PPh groups with the aromatic protons of the other phenyl group, as well as between the signals of the methylene protons in each of the CH2P and CH2N arms, indicates that the CNP pincer in complexes 4 also undergoes structural changes, which could be assigned to a slow interconversion between the two enantiomeric forms of the complex. Previously, mirror-image isomer exchange involving a pseudo-Berry rotation has been observed for pentacoordinated pincer Ir complexes containing diolefin ligands.13c However, this process should not be possible in complexes 4 due to the C(axial)–N(equatorial)–P(equatorial) coordination mode of the pincer. Since, as discussed above, olefin decoordination seems facile, the observed fluxional process could likely involve the intermediacy of the square-planar structure B (Fig. 4b).
To evaluate the donating properties of the CNP ligands, we prepared the carbonyl derivative 5a(Cl) by bubbling CO through a CH2Cl2 solution of complex 4a(Cl) (Scheme 3). Signals of the bridging CH2P and CH2N protons in the 1H NMR spectrum support a planar coordination of the CNP pincer. Thus, the CH2P protons produce a doublet signal at 4.18 ppm (2JHP = 10.0 Hz) while the CH2N hydrogens appear as a singlet at 6.11 ppm. In the IR spectrum, the carbonyl ligand absorbs at 1985 cm−1, which is a higher frequency than that corresponding to the (tBu–PNP)–Ir analogue (1964 cm−1),14 suggesting a lower electron density at the metal centre in the Ir–CNP system. The CO ligand is detected in the 13C{1H} NMR spectrum by the appearance of a doublet signal at 177.2 ppm (2JCP = 10 Hz), while the C2 NHC carbon appears at 178.1 ppm (2JCP = 99 Hz).
Interestingly, complex 5a(Cl) reversibly coordinates a new CO molecule yielding complex 6a(Cl), as inferred from the absorption bands corresponding to the CO ligands, which appear in the IR spectrum at 1946 and 2021 cm−1.15 In the 1H NMR spectrum of 6a(Cl), the presence of a singlet signal at 6.09 ppm (2H) for the CH2N arm and a doublet at 4.29 ppm (2H, 2JHP = 10.9 Hz) attributable to the CH2P moiety suggests the existence of a symmetry plane containing the CNP–Ir coordination plane and points out to a meridional coordination of the pincer ligand,13b at variance with the coordination geometry in the also pentacoordinated compounds 4.
As determined by VT-1H NMR spectroscopy (see ESI†), carbonyl complexes 5a(Cl) and 6a(Cl) exhibit a dynamic behaviour in solution, which equilibrates the two otherwise diastereotopic hydrogens of both methylene bridges. In square-planar Pd16 and octahedral Ru complexes incorporating CNC ligands,3b similar dynamic processes have been ascribed to a slow interconversion between the two twisted conformations adopted by both C2(NHC)–N(Py)–M chelate rings of the pincer ligand. Similarly, the observed fluxionality in derivatives 5a(Cl) and 6a(Cl) can be attributed to the fast atropoisomerism between the two limiting enantiomeric forms shown in Fig. 5.
 | ||
| Fig. 5 Interconversion between the limiting conformations of 5a(Cl) and 6a(Cl). | ||
Complexes 4a(Cl) and 4b(Cl) react with H2 producing the dihydrido derivatives 7 and cyclooctene (Scheme 3). At room temperature, the 1H NMR spectrum of complex 7a(Cl) shows two doublets of doublets at −20.19 (2JHP = 13.8 Hz, 2JHH = 7.0 Hz) and −23.30 ppm (2JHP = 18.9 Hz) due to the hydrido ligands placed trans to the pyridine and trans to the chloride, respectively. In the 13C{1H} NMR spectrum, the C2 NHC appears at 172.9 ppm as a doublet signal (2JCP = 119 Hz). Exposure of a sample of 7a(Cl) in CD2Cl2 to deuterium gas (2 bar) or addition of CD3OD causes fast H/D exchange of the hydrido ligands.
Furthermore, the structural features of complex 7a(Cl) have been studied in the solid state by single crystal X-ray diffraction (Fig. 6). This derivative has an octahedral geometry with the two hydrido ligands occupying mutually cis positions and the CNP ligand adopting a meridional coordination, as defined by the C(1)–Ir(1)–P(1) angle value of 166.5(3)°. The chelate ring incorporating the NHC fragment has a boat-like conformation as shown by the C(14)–N(3)–Ir(1)–C(1) torsion angle of −26.5(9)°, whereas an envelope conformation for the N(Py)–Ir–P ring is observed with a C(18)–N(3)–Ir(1)–P(1) dihedral angle of −14.9(8)°.
 | ||
| Fig. 6 ORTEP drawing at 30% ellipsoid probability of complex 7a(Cl). Hydrogen atoms, except hydrido ligands, and solvent molecules have been omitted for clarity. Selected bond lengths [Å] and angles [°]: Ir(1)–C(1) 2.037(10), Ir(1)–N(3) 2.192(9), Ir(1)–P(1) 2.262(3), Ir(1)–H(1)Ir 1.600, Ir(1)–H(2)Ir 1.599, Ir(1)–Cl(1) 2.509(3), C(1)–Ir(1)–P(1) 166.5(3), N(3)–Ir(1)–H(1)Ir 173.9, P(1)–Ir(1)–H(1)Ir 92.7, P(1)–Ir(1)–N(3) 82.2(3), C(1)–Ir(1)–H(1)Ir 95.4, C(1)–Ir(1)–N(3) 89.0(4). | ||
Deprotonation and ligand-assisted H2 activation
We have also explored the deprotonation of the Ir–CNP complexes 4.17 Treatment of 4b(Br) with KOtBu produces the selective deprotonation of the CH2P arm (Scheme 4). The resulting complex 8b is characterised in the 1H NMR spectrum by the presence of significantly high-field shifted signals for the pyridine protons (5.6–6.4 ppm), evidencing the dearomatisation of the pyridine ring. TheCHP proton appears as a singlet at 3.86 ppm, while the CH2–NHC hydrogens generate two doublets at 4.93 and 5.29 ppm (2JHH = 13.6 Hz). In the 13C{1H} NMR spectrum, the resonance caused by the C2 NHC carbon appears as an overlapped doublet at 170.6 ppm. Although the JCP value cannot be unambiguously calculated, a value of 2 to 20 Hz is estimated, suggesting a cis coordination of the phosphine and NHC fragments. This coordination mode is further supported by the existence of strong NOE contacts between the protons of the xylyl and PPh2 groups.
 | ||
| Scheme 4 Selective deprotonation of complex 4b(Br), and formation of complex 9b. | ||
Deprotonated complex 8b reacts with H2 at 0 °C to produce the trihydrido derivative 9b in a ligand-assisted process leading to the re-aromatisation of the pyridine fragment. Complex 9b is only stable under an atmosphere of H2 and can be also obtained by reaction of 7b(Cl) with KOtBu followed by exposure to H2. The trihydrido complex 9b shows in the 1H NMR spectrum a doublet of doublets at −9.98 ppm (2H, 2JHP = 18.2 Hz, 2JHH = 4.8 Hz) due to the apical hydrido ligands and a doublet of triplets at −19.64 ppm (1H, 2JHP = 14.4 Hz) produced by the hydride trans to the pyridine N. The C2 of the NHC fragment appears in the 13C{1H} NMR spectrum as a doublet at 176.9 ppm with a large 2JCP (121 Hz), in agreement with a trans disposition of the NHC and phosphine donors of the CNP ligand.
Hydrogenation of ketones catalysed by Ir–CNP complexes
In order to assess the catalytic potential of these Ir–CNP complexes, their performance in the hydrogenation of ketones was studied (Table 1).18 In the presence of KOtBu, complex 4a(Cl) smoothly catalysed the hydrogenation of acetophenone under 4 bar of H2 at 30 °C in 2-methyltetrahydrofuran, using a S/C/B ratio of 100/1/15 (entry 1). By using the same pressure, catalyst loading could be decreased to a S/C ratio of 250 after heating to 60 °C (entry 2). Also, at this temperature, a lower H2 pressure (1 bar) could be employed (entry 3). Under the latter conditions, complex 4b(Cl) was found to be slightly less active, whereas a significantly lower catalytic activity was obtained with the carbonyl derivative 5a(Cl) (entries 4 and 5). Finally, the hydrogenation of a series of ketones was performed with complex 4a(Cl). High conversions were obtained in the case of acetophenone derivatives substituted with p-methoxi, p-chloro and o-bromo substituents (entries 6–8). Alternatively, the presence of fluoro substituents seems somewhat detrimental since 2-fluoroacetophenone was reduced with a slightly lower yield (entry 9). Also, the hydrogenation of a cyclic ketone, α-tetralone, proceeded with a high conversion (entry 10).| Entry | Ketone | Ir–CNP | T (°C) | Conv. (%) |
|---|---|---|---|---|
| a Reaction conditions, unless otherwise noted: 1 bar of H2, 2-methyltetrahydrofuran, S/C/B = 100/1/15, base: KOtBu, 16 h. [S] = 0.13 M. Conversion was determined by 1H NMR spectroscopy. b 4 bar of H2. c S/C/B = 250/1/15. | ||||
| 1b | Acetophenone | 4a(Cl) | 30 | 95 |
| 2b,c | 60 | 93 | ||
| 3 | 60 | >99 | ||
| 4 | 4b(Cl) | 60 | 98 | |
| 5 | 5a(Cl) | 60 | 43 | |
| 6 | 4′-Methoxiacetophenone | 4a(Cl) | 80 | 96 |
| 7 | 4′-Chloroacetophenone | >99 | ||
| 8 | 2′-Bromoacetophenone | 99 | ||
| 9 | 2′-Fluoroacetophenone | 82 | ||
| 10 | α-Tetralone | 89 | ||
Conclusions
In summary, Pd and Ir complexes based on novel lutidine-derived CNP pincer ligands have been synthesised. The flexibility of the chelating Py–CH2–NHC fragment of the ligands allows for both facial and meridional coordination modes in five- and six-coordinated Ir–CNP complexes. Furthermore, selective deprotonation of the CH2P arm in Ir–CNP complexes promotes ligand-assisted H–H activation, leading to active species in ketone hydrogenation. Further studies involving the application of metal complexes based on CNP ligands in X–H (X = H, C, heteroatom) bond activation and as catalysts in the (de)hydrogenation of polar substrates are currently in progress in our laboratory.Experimental
General procedures
All reactions and manipulations were performed under nitrogen or argon, either in a Braun Labmaster 100 glovebox or using standard Schlenk-type techniques. All solvents were distilled under nitrogen with the following desiccants: sodium-benzophenone-ketyl for diethyl ether (Et2O) and tetrahydrofuran (THF); sodium for hexane, pentane and toluene; CaH2 for dichloromethane (CH2Cl2) and acetonitrile (CH3CN); and NaOMe for methanol (MeOH). 1-(3,5-Dimethylphenyl)-1H-imidazole and 1-(2,4,6-trimethylphenyl)-1H-imidazole were prepared as previously described.19 Ir(acac)(cod)20 and NaBArF21 were synthesized according to literature procedures. All other reagents were purchased from commercial suppliers and used as received. NMR spectra were obtained on Bruker DPX-300, DRX-400, AVANCEIII/ASCEND 400R or DRX-500 spectrometers. 31P{1H} NMR shifts were referenced to external 85% H3PO4, while 13C{1H} and 1H shifts were referenced to the residual signals of deuterated solvents. All data are reported in ppm downfield from Me4Si. All NMR measurements were carried out at 25 °C, unless otherwise stated. NMR signal assignations were confirmed by 2D NMR spectroscopy (1H–1H COSY, 1H–1H NOESY, 1H–13C HSQC and 1H–13C HMBC). HRMS data were obtained on a JEOL JMS-SX 102A mass spectrometer at the Instrumental Services of Universidad de Sevilla (CITIUS). ESI-MS experiments were carried out in a Bruker 6000 apparatus by the Mass Spectrometry Service of the Instituto de Investigaciones Químicas. Elemental analyses were run by the Analytical Service of the Instituto de Investigaciones Químicas in a Leco TrueSpec CHN elemental analyzer. IR spectra were acquired on a Bruker Tensor 27 instrument.
In subsequent step, to a solution of PPh2H (1.08 g, 5.80 mmol) in THF (10 mL) was added a solution of KOtBu (0.650 g, 5.80 mmol) in THF (10 mL). The resulting mixture was stirred for 5 min, and added to a solution of [2-chloromethyl-6-(3-mesitylimidazolium-1-yl)methyl]pyridine chloride (2.00 g, 5.52 mmol) in MeCN (40 mL). The suspension was stirred overnight, and MeOH (15 mL) was added to quench the reaction. Solvent was evaporated, and the residue was extracted with CH2Cl2 (2 × 15 mL). The solid obtained after removal of the solvent was washed with diethyl ether (3 × 20 mL) and pentane (3 × 20 mL). Imidazolium salt 1a(Cl) was isolated as a light brown solid (2.40 g, 85%). 1H NMR (500 MHz, CDCl3): δ 10.30 (s, 1H, H arom Imid), 7.85 (s, 1H, H arom Imid), 7.60 (d, 3JHH = 7.5 Hz, 1H, H arom Py), 7.54 (dd, 3JHH = 7.6 Hz, 3JHH = 7.6 Hz, 1H, H arom Py), 7.36 (m, 4H, 4 H arom PPh), 7.28 (m, 6H, 6 H arom PPh), 7.12 (s, 1H, H arom Imid), 7.03 (d, 3JHH = 7.7 Hz, 1H, H arom Py), 7.01 (s, 2H, 2 H arom Mes), 5.94 (s, 2H, CH2N), 3.59 (s, 2H, CH2P), 2.33 (s, 3H, CH3), 2.01 (s, 6H, 2 CH3). 31P{1H} NMR (202 MHz, CD2Cl2): δ −11.7. 13C{1H} NMR (126 MHz, CD2Cl2): δ 158.8 (d, JCP = 8 Hz, Cq arom), 152.7 (Cq arom), 141.4 (Cq arom), 138.7 (d, JCP = 3 Hz, CH arom), 138.5 (d, JCP = 15 Hz, 2 Cq arom), 137.9 (CH arom), 134.7 (2 Cq arom), 133.0 (d, JCP = 19 Hz, 4 CH arom), 131.2 (Cq arom), 129.9 (2 CH arom), 129.0 (2 CH arom), 128.7 (d, JCP = 7 Hz, 4 CH arom), 124.0 (d, JCP = 5 Hz, CH arom), 123.9 (CH arom), 122.7 (CH arom), 121.3 (d, JCP = 2 Hz, CH arom), 53.7 (CH2N), 38.3 (d, JCP = 17 Hz, CH2P), 21.1 (CH3), 17.7 (2 CH3). HRMS (ESI): m/z 476.2243 [(M − Cl)+] (exact mass calculated for C31H31N3P: 476.2256).
In a subsequent step, to a solution of Ph2P(BH3)H (0.288 g, 1.44 mmol) in THF (10 mL) was added a solution of KOtBu (0.161 g, 1.44 mmol) in THF (5 mL). The mixture was stirred for 10 min, and added to a suspension of [2-chloromethyl-6-(3-(3,5-xylyl)imidazolium-1-yl)methyl]pyridine chloride (0.500 g, 1.44 mmol) in MeCN (10 mL). The resulting suspension was stirred overnight, and MeOH (10 mL) was added to quench the reaction. The solvent was evaporated under vacuum, and the solid was extracted with CH2Cl2 (3 × 10 mL). Solvent removal followed by washings with Et2O (2 × 10 mL) yields a light orange solid which should correspond to the borane adduct of 1b(Cl). This solid was dissolved in MeOH (10 mL), and the solution was transferred to a Fisher–Porter vessel and heated to 75 °C for 24 h. Volatiles were removed under vacuum, and MeOH (10 mL) was newly added and the previous procedure repeated. The resulting solid was washed with toluene (2 × 5 mL) and Et2O (3 × 5 mL) to give an off-white solid (0.444 g, 62%). 1H NMR (400 MHz, CD2Cl2): δ 11.28 (s, 1H, H arom Imid), 7.74 (d, 3JHH = 7.6 Hz, 1H, H arom Py), 7.64 (dd, 3JHH = 7.7 Hz, 3JHH = 7.7 Hz, 1H, H arom Py), 7.55 (d, 3JHH = 1.1 Hz, 1H, H arom Imid), 7.44 (m, 4H, 4 H arom), 7.36 (m, 7H, 7 H arom), 7.29 (s, 2H, 2 H arom Xyl), 7.21 (s, 1H, H arom Xyl), 7.12 (d, 3JHH = 7.8 Hz, 1H, H arom Py), 5.91 (s, 2H, CH2N), 3.69 (s, 2H, CH2P), 2.45 (s, 6H, 2 CH3). 31P{1H} NMR (121 MHz, CD2Cl2): δ −11.8. 13C{1H} NMR (101 MHz, CD2Cl2): δ 159.0 (d, JCP = 8 Hz, Cq arom), 152.6 (Cq arom), 141.0 (2 Cq arom), 138.5 (d, JCP = 15 Hz, 2 Cq arom), 138.0 (CH arom), 136.6 (CH arom), 134.9 (Cq arom), 133.0 (d, JCP = 19 Hz, 4 CH arom), 131.9 (CH arom), 129.1 (2 CH arom), 128.7 (d, JCP = 7 Hz, 4 CH arom), 124.2 (d, JCP = 5 Hz, CH arom), 123.7 (CH arom), 121.8 (CH arom), 120.2 (CH arom), 119.7 (2 CH arom), 54.0 (CH2N), 38.3 (d, JCP = 16 Hz, CH2P), 21.3 (2 CH3). HRMS (ESI): m/z 462.2082 [(M − Cl)+] (exact mass calculated for C30H29N3P: 462.2094).
In a subsequent step, to a solution of Ph2P(BH3)H (0.483 g, 2.42 mmol) in THF (10 mL) was added a solution of KOtBu (0.271 g, 2.42 mmol) in THF (10 mL). The resulting mixture was stirred for 10 min, and added to a solution of [2-bromomethyl-6-(3-(3,5-xylyl)imidazolium-1-yl)methyl]pyridine bromide (1.01 g, 2.39 mmol) in MeCN (20 mL). The suspension was stirred overnight, and MeOH (15 mL) was added to quench the reaction. The solvent was evaporated under vacuum, and the resulting solid was extracted with CH2Cl2 (3 × 10 mL). Solvent removal followed by washings with Et2O (2 × 10 mL) yields a light orange solid which should correspond to the borane adduct of 1b(Br). This solid was dissolved in MeOH (10 mL), and the solution was transferred to a Fisher–Porter vessel and heated to 75 °C for 24 h. Volatiles were removed under vacuum, and MeOH was newly added and the previous procedure was repeated. The resulting solid was washed with toluene (10 mL) and Et2O (10 mL) to give an off-white solid (0.693 g, 55% yield). 1H NMR (500 MHz, CD2Cl2): δ 10.77 (s, 1H, H arom Imid), 7.65 (d, 3JHH = 7.6 Hz, 1H, H arom Py), 7.61 (t, 3JHH = 1.2 Hz, 1H, H arom Imid), 7.56 (m, 2H, H arom Py + H arom Imid), 7.38 (m, 4H, 4 H arom PPh), 7.32 (s, 2H, 2 H arom Xyl), 7.29 (m, 6H, 6 H arom PPh), 7.13 (s, 1H, H arom Xyl), 7.07 (d, 3JHH = 7.6 Hz, 1H, H arom Py), 5.84 (s, 2H, CH2N), 3.62 (s, 2H, CH2P), 2.37 (s, 6H, 2 CH3). 31P{1H} NMR (202 MHz, CD2Cl2): δ −11.7. 13C{1H} NMR (125 MHz, CD2Cl2): δ 159.0 (d, JCP = 8 Hz, Cq arom), 152.3 (Cq arom), 141.0 (2 Cq arom), 138.4 (d, JCP = 15 Hz, 2 Cq arom), 137.9 (CH arom), 135.9 (CH arom), 134.8 (Cq arom), 132.9 (d, JCP = 19 Hz, 4 CH arom), 131.9 (CH arom), 129.1 (2 CH arom), 128.7 (d, JCP = 7 Hz, 4 CH arom), 124.2 (d, JCP = 5 Hz, CH arom), 123.9 (CH arom), 121.6 (CH arom), 120.3 (CH arom), 119.7 (2 CH arom), 54.0 (CH2N), 38.3 (d, JCP = 16 Hz, CH2P), 21.3 (2 CH3). HRMS (ESI): m/z 462.2089 [(M − Br)+] (exact mass calculated for C30H29N3P: 462.2099).
Synthesis of Pd–CNP complexes 2a and 3a
Synthesis of Ir–CNP complexes 4–9


In a J. Young valved NMR tube, a suspension cooled to −20 °C of 7b(Cl) (0.012 g, 0.017 mmol) in THF-d8 (0.7 mL) was treated with KOtBu (0.002 g, 0.018 mmol). Immediately, the NMR tube was charged with 5 bar of H2 and kept to 0 °C to avoid thermal decomposition of the product. After 1 h, the resulting solution was analysed by NMR spectroscopy.
1H NMR (400 MHz, THF-d8): δ 7.78 (dd, 3JHP = 8.5 Hz, 3JHH = 8.5 Hz, 4H, 4 H arom PPh), 7.82 (s, 2H, 2 H arom), 7.50 (dd, 3JHH = 7.6 Hz, 3JHH = 7.6 Hz, 1H, H arom Py), 7.35 (m, 2H, 2 H arom), 7.24 (m, 6H, 6 H arom), 7.11 (m, 1H, H arom), 7.07 (s, 1H, H arom NHC), 6.94 (s, 1H, H arom NHC), 5.18 (s, 2H, NCH2), 3.98 (d, 2JPH = 10.0 Hz, 2H, PCH2), 2.38 (s, 6H, 2 CH3), −9.98 (dd, 2JHP = 18.2 Hz, 2JHH = 4.8 Hz, 2H, IrH cis to Py), −19.64 (dt, 2JHP = 14.4 Hz, 2JHH = 4.8 Hz, 1H, IrH trans to Py). 31P{1H} NMR (162 MHz, THF-d8): δ 30.9. 13C{1H} NMR (101 MHz, THF-d8): δ 176.9 (d, JCP = 121 Hz, C-2 NHC), 164.7 (d, JCP = 6 Hz, Cq arom), 155.9 (Cq arom), 143.0 (Cq arom), 139.1 (d, JCP = 42 Hz, 2 Cq arom), 137.2 (2 Cq arom), 134.3 (d, JCP = 13 Hz, 4 CH arom), 134.1 (CH arom), 129.4 (2 CH arom), 128.2 (CH arom), 127.8 (d, JCP = 10 Hz, 4 CH arom), 125.5 (2 CH arom), 121.3 (CH arom), 121.1 (d, JCP = 9 Hz, CH arom), 120.5 (CH arom), 120.1 (d, JCP = 4 Hz, CH arom), 59.9 (CH2N), 49.2 (d, JCP = 34 Hz, CH2P), 21.2 (2 CH3).
Representative procedure for ketone hydrogenation
In a glovebox, a Fischer–Porter vessel was charged with a solution of complex 4a(Cl) (2.0 mg, 2.5 μmol), KOtBu (2.7 mg, 37 μmol) and acetophenone (30 μL, 0.26 mmol) in 2-methyltetrahydrofuran (2.0 mL). The reactor was purged three times with H2, and finally pressurized to 1 bar and heated to 60 °C. After 16 h, the reactor was slowly cooled down to room temperature, the reaction solution was evaporated, and conversion was determined by 1H NMR spectroscopy using mesitylene as internal standard.Acknowledgements
Financial support (FEDER contribution) from the Spanish MINECO (CTQ2013-45011-P, CTQ2016-80814-R and CTQ2014-51912-REDC) is gratefully acknowledged. M. H. J. thanks SECITI-DF for a postdoctoral fellowship.Notes and references
-
(a) J. I. van der Vlugt and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 8832–8846 CrossRef CAS PubMed
; (b) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed
-
(a) R. E. Andrew and A. B. Chaplin, Inorg. Chem., 2015, 54, 312–322 CrossRef CAS PubMed
-
(a) M. Hernández-Juárez, M. Vaquero, E. Álvarez, V. Salazar and A. Suárez, Dalton Trans., 2013, 42, 351–354 RSC
-
(a) G. A. Filonenko, E. Cosimi, L. Lefort, M. P. Conley, C. Copéret, M. Lutz, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2014, 4, 2667–2671 CrossRef CAS
-
(a) M. Asay and D. Morales-Morales, Dalton Trans., 2015, 44, 17432–17447 RSC
-
(a) E. Kinoshita, K. Arashiba, S. Kuriyama, Y. Miyake, R. Shimazaki, H. Nakanishi and Y. Nishibayashi, Organometallics, 2012, 31, 8437–8443 CrossRef CAS
-
(a) Y. Sun, C. Koehler, R. Tan, V. T. Annibale and D. Song, Chem. Commun., 2011, 47, 8349–8351 RSC
-
(a) X. Liu and P. Braunstein, Inorg. Chem., 2013, 52, 7367–7379 CrossRef CAS PubMed
-
(a) T. Simler, A. A. Danopoulos and P. Braunstein, Chem. Commun., 2015, 51, 10699–10702 RSC
- N. Selander and K. J. Szabó, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS PubMed
-
(a) J. I. van der Vlugt, M. A. Siegler, M. Janssen, D. Vogt and A. L. Spek, Organometallics, 2009, 28, 7025–7032 CrossRef CAS
- G. A. Silantyev, O. A. Filippov, S. Musa, D. Gelman, N. V. Belkova, K. Weisz, L. M. Epstein and E. S. Shubina, Organometallics, 2014, 33, 5964–5973 CrossRef CAS
-
(a) D. M. Roddick and D. Zargarian, Inorg. Chim. Acta, 2014, 422, 251–264 CrossRef CAS
-
(a) S. M. Kloek, D. M. Heinekey and K. I. Goldberg, Organometallics, 2006, 25, 3007–3011 CrossRef CAS
-
(a) L. Vaska, Science, 1966, 152, 769–771 CAS
-
(a) S. Gründemann, M. Albrecht, J. A. Loch, J. W. Faller and R. H. Crabtree, Organometallics, 2001, 20, 5485–5488 CrossRef
-
(a) E. Ben-Ari, G. Leitus, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2006, 128, 15390–15391 CrossRef CAS PubMed
- For selected examples of ketone hydrogenation catalysed by Ir complexes with proton-responsive ligands:
(a) L. Dahlenburg and R. Götz, Eur. J. Inorg. Chem., 2004, 888–905 CrossRef CAS
- M. C. Perry, X. Cui, M. T. Powell, D.-R. Hou, J. H. Reibenspies and K. Burgess, J. Am. Chem. Soc., 2003, 125, 113–123 CrossRef CAS PubMed
- M. Rueping, R. M. Koenigs, R. Borrmann, J. Zoller, T. E. Weirich and J. Mayer, Chem. Mater., 2011, 23, 2008–2010 CrossRef CAS
- N. A. Yakelis and R. G. Bergman, Organometallics, 2005, 24, 3579–3581 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: VT-1H NMR spectra, 1H and 13C{1H} NMR spectra of selected derivatives and X-ray crystallography data. CCDC 1486492–1486495. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03652j |
| This journal is © The Royal Society of Chemistry 2016 |