
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssessment of the nematocidal activity of metallocenyl analogues of monepantel†
Jeannine
Hess
a,
Malay
Patra
a,
Abdul
Jabbar
b,
Vanessa
Pierroz
ac,
Sandro
Konatschnig
a,
Bernhard
Spingler
a,
Stefano
Ferrari
c,
Robin B.
Gasser
*b and
Gilles
Gasser‡
 *a
*a
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: gilles.gasser@chimie-paristech.fr; Web: http://www.gassergroup.com
bFaculty of Veterinary and Agricultural Sciences, The University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: robinbg@unimelb.edu.au; Web: http://www.gasserlab.org/ Tel: +61 3 9731 2283
cInstitute of Molecular Cancer Research, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland
First published on 4th October 2016
Abstract
In this study, we present the design, synthesis, characterization and biological evaluation of structurally new ferrocenyl and ruthenocenyl derivatives of the organic anthelmintic monepantel (Zolvix®). All seven metallocenyl derivatives prepared (4a/b, 5a/b, 6a/b and 7) were isolated as racemates and characterized by 1H, 13C and 19F NMR spectroscopies, mass spectrometry, IR spectroscopy and elemental microanalysis. The molecular structures of four compounds (4a/b, 6a and 7) were further confirmed by X-ray crystallography. The biological activities of the organometallic intermediates (4a/b) and organometallic derivatives of monepantel (5a/b, 6a/b and 7) were evaluated in vitro using parasitic nematodes of major importance in livestock, namely Haemonchus contortus and Trichostrongylus colubriformis. Two ferrocenyl compounds (4a and 6a) showed nematocidal activity, while the analogous ruthenocenyl compounds (4b and 6b) were not active at the highest concentration tested (10 μg mL−1). In order to obtain insight into the difference in activity between ferrocenyl and ruthenocenyl derivatives, the potential of the compounds for reactive oxidative species (ROS) production in live cells was assessed. Interestingly, neither the ferrocenyl nor the ruthenocenyl compounds (4a/b and 6a/b) produced significant ROS in HeLa cells when checked after 22 h, potentially indicating a redox-independent activity of 4a and 6a on the parasites. The selectivity of the compounds on parasites was confirmed by investigating their cytotoxicity profiles. None of these compounds was toxic either to HeLa or MRC-5 cells. Thus, 4a and 6a could be considered as interesting leads for further development of new classes of anti-parasitic agents.
Introduction
The impact of parasitic diseases on animals and humans is substantial around the world. Controlling parasites of domesticated animals is a major issue, and usually relies exclusively on chemotherapy with an arsenal of broad-spectrum anthelmintics.2,3 The excessive use of these drugs has resulted in the emergence of resistance, and multi-drug resistant parasites are now widespread.5–8 Apart from the classical groups of anthelmintics, such as the benzimidazoles, imidazothiazoles and macrocyclic lactones, a new synthetic anthelmintic class, the amino-acetonitrile derivatives (AADs, see Scheme 1), has been discovered, and a promising candidate of this class, called monepantel (AAD 1566), was recently commercialized.1,9,10 | ||
| Scheme 1 Schematic representation of previously (green) and newly (red) designed organometallic derivatives of monepantel (AAD 1566) using different strategies. OM = ferrocene, ruthenocene or cymantrene; FG = SCF3, F, Cl, Br, I, SCH3, CF3, OCF3, S(O)CF3, S(O)2CF3. | ||
Investigations have shown that the safety profile of monepantel relates to its target, a nematode specific nicotinic acetylcholine receptor (nAChr) subunit, which is absent in host mammals.11–13 Although monepantel now represents a new class of anthelmintic drug, unfortunately, some years after its introduction and use in the field, nematodes with reduced sensitivity to monepantel have been detected in several countries including Uruguay, New Zealand and Brazil.14–18 Given this rapid emergence of resistance to monepantel, there is an urgent need to develop novel and superior control strategies to ensure the sustainability of parasite control. With this in mind, our group recently started to derivatize monepantel with various organometallic moieties using different strategies (Scheme 1).19,20 The derivatization of a known organic drug with organometallic moieties has proven to be extremely successful in various fields of medicinal chemistry.21–35 Ferroquine, a ferrocenyl analogue of the antimalarial drug chloroquine, is one of the best examples of such a derivatization. Thanks to the presence of a ferrocenyl moiety, Ferroquine is active against chloroquine-resistant strains of the malaria parasite, Plasmodium falciparum (P. falciparum), where the original organic drug chloroquine is inactive.29,36,37
In our initial study, we replaced the aryloxy unit of monepantel with organometallic moieties and modified the benzamide part with various functional groups (Scheme 1, Strategy I/II).19 Subsequently, we replaced the chiral C2 spacer of monepantel by a ferrocenyl moiety that comprised planar chirality with a 1,2-unsymmetric substitution on a cyclopentadienyl ring (Scheme 1, Strategy III).20
Extending this work to develop organometallic monepantel derivatives, we report here a new strategy for achieving structurally distinct organometallic-containing monepantel derivatives. First, we kept the aryloxy unit of monepantel unperturbed and substituted the benzamide unit with metallocenes, namely ferrocene and ruthenocene (Scheme 1, Strategy IV). Then, we replaced simultaneously both the aryloxy and the benzamide units using two metallocenyl fragments (Scheme 1, Strategy IV). During the course of present study, we first prepared ferrocenyl monepantel derivatives, since the presence of the ferrocenyl unit might result in additional metal-specific modes of action, such as the production of ROS under physiological conditions. In order to investigate if these redox reactions contribute to the antiparasitic activity of our novel organometallic compounds, we designed ruthenocenyl analogous of the ferrocenyl monepantel derivatives. The replacement of the iron(II) by a ruthenium(II) center is expected to prevent redox mediated ROS production under physiologically relevant conditions.36,37
Results and discussion
Synthesis and characterization
The ferrocenyl and ruthenocenyl analogues of monepantel corresponding to Strategy IV were prepared in a two-step reaction procedure as presented in Scheme 2. In order to obtain initial insights into their potential as anthelmintic agents, we focused on the isolation of the organometallic derivatives as racemates rather than as their enantiomerically pure forms. | ||
| Scheme 2 Reagents and conditions: (i) (a) oxalyl chloride, dry CH2Cl2, r.t.; (b) 2-amino-2-hydroxymethylproprionitrile (1), NEt3, dry THF, r.t., o.n., 4a (50%), 4b (31%), 5a (11%), 5b (19%); (ii) NaH, 3-fluoro-4-(trifluoromethyl)benzonitrile, dry THF, overnight, 0 °C→r.t., 6a (26%), 6b (11%); (iii) (ferrocenylmethyl)trimethylammonium iodide, K2CO3, 18-crown-6, dry CH3CN, reflux, 144 h (120 h plus overnight), 7 (43%). | ||
The syntheses of desired organometallic analogues commenced with the preparation of a 2-amino-2-hydroxymethylproprionitrile (1) synthon, following a literature procedure by Gauvry et al.38 The intermediate N-(2-cyano-1-hydroxypropan-2-yl)ferroceneamide (4a) was obtained in a moderate yield by reacting 1 with activated ferrocencarboxylic acid (2) under basic conditions. Moreover, the same reaction allowed the isolation of a di-ferrocenyl analogue of monepantel, 2-ferroceneamido-2-cyanopropyl ferroceneoate (5a), with 11% yield. Compound 4a was then converted to the desired final compound N-(2-cyano-1-(5-cyano-2-(trifluoromethyl)phenoxy)propan-2-yl)ferroceneamide (6a) using a Williamson ether synthesis in the presence of NaH and commercially available 3-fluoro-4-(trifluoromethyl)benzonitrile. Compound 6a was isolated in 26% yield. In addition to the di-ferrocenyl analogue 5a, another disubstituted organometallic analogue, namely N-(1-(ferrocenyloxy)-2-cyanopropan-2-yl)ferroceneamide (7) was also prepared by reacting 4a with (ferrocenylmethyl)trimethylammonium iodide under basic conditions. By contrast to 5a, where one of the ferrocenyl units is linked to C2 spacer by an ester functionality, the similar ferrocene unit in 7 is attached to the C2 spacer by an ether functionality. Moreover, we synthesized three ruthenocenyl analogues (4b, 5b and 6b), which are structurally identical to the ferrocenyl analogues 4a, 5a and 6a. Despite iso-structural, ferrocene and ruthenocene have distinct redox properties, therefore comparison of the biological activity of ferrocenyl and the corresponding ruthenocenyl analogues might shed light on the possible involvement of the redox properties in their activity. The synthetic sequences to obtain the desired ruthenocenyl analogues of monepantel are similar to those of ferrocenyl compounds (see Scheme 2). All novel ferrocenyl and ruthenocenyl analogues of monepantel described here were isolated as racemic mixtures and characterized by 1H, 13C, 19F NMR spectroscopies, ESI-mass spectrometry and IR spectroscopy, and their purities were analyzed by elemental microanalysis.
X-ray crystallography
The molecular structures of four compounds (4a/b, 6a and 7) were further confirmed by X-ray crystallography. Details of the crystallization procedure were given in the Experimental section. The amide unit is coplanar with the cyclopentadienyl ring. The alcohol of compound 4a is part of a cyclic and a linear hydrogen-bridge network. The R22 (14) cycle39 is formed by an alcohol hydrogen, making contact with a symmetry (−x, 1 − y, −z)-related carbonyl oxygen; the corresponding alcohol donates back to the original carbonyl oxygen. Additionally, the same alcohol is an acceptor of a hydrogen bridge from a symmetry related amide nitrogen (−x, 1/2 + y, 1/2 − z). The structures of 4a and 4b are essentially isostructural, with the major difference being that the Fe–C bond lengths in 4a are between 2.0293(14) Å and 2.0619(17) Å, whereas the Ru–C bond lengths in 4b are between 2.155(3) Å and 2.192(3) Å (see Fig. 1). Compound 6a does not form any “typical” hydrogen bonds, despite the presence of an amide unit (see Fig. 1). | ||
| Fig. 1 Molecular structures of 4a, 4b and 6a with atoms shown as thermal ellipsoids (drawn at 50% probability; hydrogen atoms are omitted for clarity). | ||
Compound 7 was measured on a synchrotron; it crystallized with two molecules in the asymmetric unit. All cyclopentadienyl rings of the 4 ferrocene units are in an almost perfect eclipsed conformation. One linker starting from the quarternary carbon to the methylene unit next to the cyclopentadienyl ring is disordered in a ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (see ESI† for structure).
:2 (see ESI† for structure).
Biological evaluation
The anthelmintic potentials of our ferrocenyl and ruthenocenyl precursors (4a/b) and final derivatives 5a/b, 6a/b and 7 were first evaluated on two common parasites of small ruminants, Haemonchus contortus (H. contortus) and Trichostrongylus colubriformis (T. colubriformis) using a larval development assay (LDA) (see Experimental section for details). The results are summarized in Table 1. The organic derivatives AAD85, AAD96 and ivermectin were included as controls.1| Compound | EC60 value | EC50 value | IC50 value | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| H. contortus | T. colubriformis | D. immitis | D. immitis | HeLa | MRC-5 | |||||
| 24 hours | 48 hours | |||||||||
| [μg mL−1] | [μM] | [μg mL−1] | [μM] | [μg mL−1] | [μM] | [μg mL−1] | [μM] | [μM] | [μM] | |
| a EC100 value1. n.d.i. = non-disclosable information4 and n.d. = not determined. EC values are given in μg mL−1 as well as in μM. EC values are calculated as a mean of 2 series of triplicated dose responses. Experimental errors are not included as they are too low to influence the overall EC values. | ||||||||||
| 4a | 4.90 | 15.70 | 8.00 | 25.63 | >10.00 | >32.04 | >10.00 | >32.04 | >100 | >100 |
| 5a | >10.00 | >19.08 | >10.00 | >19.08 | >10.00 | >19.08 | >10.00 | >19.08 | >100 | >100 |
| 6a | 4.70 | 9.77 | 7.00 | 14.55 | >10.00 | >20.78 | >10.00 | >20.78 | >100 | >100 |
| 4b | >10.00 | >27.98 | >10.00 | >27.98 | >10.00 | >27.98 | >10.00 | >27.98 | >100 | >100 |
| 5b | >10.00 | >16.27 | >10.00 | >16.27 | >10.00 | >16.27 | 6.60 | 10.74 | >100 | >100 |
| 6b | >10.00 | >18.99 | >10.00 | >18.99 | 6.60 | 12.54 | 6.60 | 12.54 | >100 | >100 |
| 7 | >10.00 | >19.60 | >10.00 | >19.60 | >10.00 | >19.60 | >10.00 | >19.60 | >100 | >100 |

|
0.01a | 0.022a | 0.032a | 0.07a | 2.40 | 5.25 | 2.20 | 4.81 | n.d. | n.d. |

|
0.01a | 0.021a | 0.032a | 0.068a | n.d.i. | n.d.i. | n.d.i. | n.d.i. | n.d. | n.d. |
| Ivermectine | 0.001a | 0.001a | 0.01a | 0.011a | 1.00–3.00 | 1.14–3.43 | 1.00–3.00 | 1.14–3.43 | n.d. | n.d. |
| Cisplatin | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 9.6 ± 1.2 | 17.6 ± 4.3 |
Two of seven metal-based monepantel derivatives tested displayed moderate activities against H. contortus and T. colubriformis. The final ferrocenyl derivative of Strategy IV (6a) and its corresponding ferrocene precursor (4a) showed EC60 values in a similar range when tested against H. contortus 4.90 μg mL−1 (15.70 μM) (4a) and 4.70 μg mL−1 (9.77 μM) (6a) and T. colubriformis 8.00 μg mL−1 (25.63 μM) (4a) and 7.00 μg mL−1 (14.55 μM) (6a). Interestingly, the potencies of both active derivatives (4a, 6a) are approximately twice higher against H. contortus compared with T. colubriformis. However, the potencies of 4a and 6a are lower when compared with the organic controls AAD85 and AAD96, which displayed EC100 values of 0.01 μg mL−1 (0.022 μM/0.021 μM) and 0.032 μg mL−1 (0.07 μM/0.068 μM) against H. contortus and T. colubriformis, respectively.1 Importantly, the ruthenocenyl analogues (4b, 5b) of the active ferrocenyl derivatives 4a, 5a displayed no activity against H. contortus and T. colubriformis at the highest concentration evaluated (10 μg mL−1). At this point, it is reasonable to speculate that the iron(II) center in 4a and 6a contributes to the production of toxic ROS which could be the reason for the observed anti-parasitic activity of these compounds. Therefore, we evaluated ROS generation using the ferrocene and ruthenocene-containing derivatives in live cells (Fig. 2). Although an assessment of ROS generation in the parasitic nematodes would have been the ideal, however for technical reasons we used a mammalian cervical cancer cell line (HeLa) as model. The ROS production in the cells treated with 4a/b and 6a/b (25 μM for 22 h) was quantified using the fluorescent indicator 2′,7′-dichlorofluorescein diacetate (H2DCFDA). As positive control, tert-butyl hydroperoxide (TBH) was included in the same assay. Surprisingly, as shown in Fig. 1, the ROS levels in cells treated with the ferrocenyl compounds (4a, 6a) and the ruthenocenyl compounds (4b, 6b) did not display a significant difference. Moreover, the ROS levels of the cells treated with compounds are comparable to that of the untreated cells.19
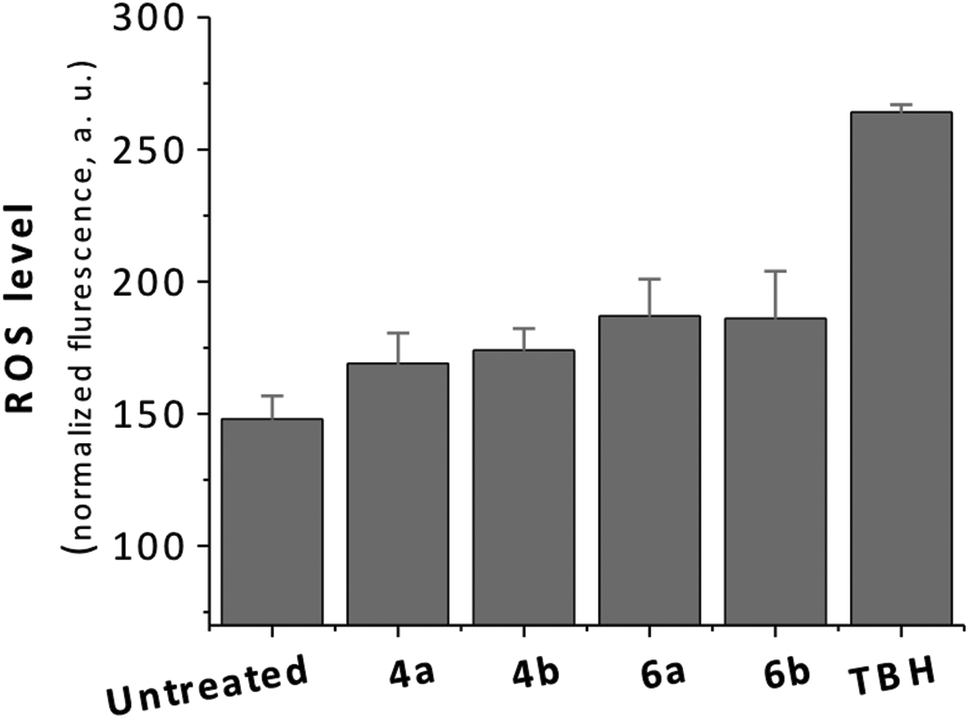 | ||
| Fig. 2 Level of ROS production in HeLa cells untreated or treated with 4a, 4b, 6a and 6b after 22 h. THB = tert-butyl hydroperoxide. | ||
The results from ROS assay suggest that the differences in anti-parasitic activity between the ferrocene- and ruthenocene-containing organometallic compounds are not related to the production of ROS in cells. However, it has to be pointed out that this is for a timepoint only and that increase in ROS production can be faster or can take longer. The potency of the organometallic monepantel derivatives synthesized using Strategy IV against H. contortus and T. colubriformis is lower than for the organometallic derivatives from Strategy I/II (best EC60 of 1.80 μg mL−1), but superior to those from Strategy III (best EC60 of 6.60 μg mL−1).19,20 Overall, based on knowledge obtained from these structure–activity relationships of organometallic monepantel derivatives, it can be concluded that it is preferable to keep the benzamide part of monepantel unperturbed, while structural modifications can be made to the aryloxy portion for the designing of new derivatives of monepantel to retain anti-parasitic activity against H. contortus and T. colubriformis.
Previous studies have shown that organometallic derivatization modulates the spectrum of activity of a known organic drug through the addition of metal-specific mode of actions.37,40 Therefore, we decided to evaluate these compounds further against other parasites.41,42 The activity of our organometallic-analogues (4a/b, 5a/b, 6a/b and 7) of monepantel was evaluated on the canine heartworm, Dirofilaria immitis (D. immitis). Two ruthenocenyl compounds (5b, 6b) showed moderate activity against microfilariae of this nematode species in a 48 h motility assay with EC50 values of 6.60 μg mL−1 (10.74 μM (5b), 12.54 μM (6b)). Interestingly, the corresponding ferrocenyl derivatives 5a and 6a are inactive against D. immitis. However, once again, 5b and 6b are less potent compared to the organic control AAD85, which displays an EC50 value of 2.20 μg mL−1 (4.81 μM).4 In addition to this, we further investigated the activity of the ferrocenyl and ruthenocenyl precursors (4a/b) and final derivatives 5a/b, 6a/b and 7 on three arthropods Ctenocephalides felis (cat flea), Lucilia cuprina (blow fly) and Rhipicephalus sanguineus (brown dog tick). Unfortunately, none of the compounds synthesized displayed potency at the highest concentration tested on C. felis (100 μg mL−1), L. cuprina (32 μg mL−1) or R. sanguineus (100 μg mL−1 and 640 μg mL−1) (Table S1†).
For development of new anti-parasitic compounds, the selectivity of the compounds towards parasites is an important parameter. An ideal anthelmintic should be non-toxic to the mammalian host, while being efficient in killing the parasites. For this purpose, we evaluated the cytotoxicity of the compounds using a cervical cancer cell line (HeLa) and a non-cancerous human lung fibroblast cell line (MRC-5). None of the compounds tested were toxic up to 100 μM (the highest concentration assayed), indicating selectivity of our compounds (4a, 6a) to the nematodes H. contortus, T. colubriformis and D. immitis over mammalian cells.
Conclusion
The extensive use of commercially available broad-spectrum organic anthelmintics has led to a situation where parasites develop resistant to at least one or more available drug classes. With the view of developing a new class of metal-based anthelmintic agents, we synthesized and characterized a series of ferrocene- and ruthenocene-containing organometallic analogues of the anthelmintic drug monepantel. The biological efficacy of all newly synthesized intermediates as well as final analogues (4a/b, 5a/b, 6a/b, 7) was assessed in a LDA against H. contortus and T. colubriformis. Two ferrocenyl derivatives 4a and 6a showed moderate efficacy with EC60 values between 4.70–8.00 μg mL−1 against both nematode species, while the corresponding ruthenocenyl derivatives displayed no activity. The ROS production of both types of organometallic derivatives (4a/b and 6a/b) was evaluated in an in vitro model using the HeLa cervical cancer cell line. However, no significant difference in ROS levels after 22 h was observed between the ferrocenyl (4a and 6a) and ruthenocenyl (4b and 6b) analogues. This unexpected finding might suggest a redox independent mode of action for the anti-parasitic activity of the ferrocenyl derivatives (4a and 6a) although more investigations will need to be performed to confirm this hypothesis. The selectivity of the compounds towards parasites was demonstrated by assessment of their cytotoxicity using a cancer (HeLa) and a non-cancer (MRC-5) cell line.Experimental section
Materials
All chemicals were of reagent grade quality or better, obtained from commercial suppliers and used without further purification. Solvents were used as received or distilled using standard procedures.43 All preparations were carried out using standard Schlenk techniques. Thin layer chromatography (TLC) was performed using silica gel 60 F-254 (Merck) plates with detection of spots being achieved by exposure to UV light. Column chromatography was performed using Silica gel 60 (0.040–0.063 mm mesh, Merck). Eluent mixtures are expressed as volume to volume (v/v) ratios. Chlorocarbonyl ferrocene and chlorocarbonyl ruthenocene were synthesized according to literature procedures.44Instrumentation and methods
1H, 13C and 19F NMR spectra were recorded in deuterated solvents on Bruker 400 or 500 MHz NMR at room temperature. The chemical shifts, δ, are reported in ppm (parts per million). The signals from the residual protons of deuterated solvent have been used as an internal reference.45,46 The abbreviations for the peak multiplicities are as follows: s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), and br (broad). ESI mass spectrometry was performed using a Bruker Esquire 6000 spectrometer. In the assignment of the mass spectra, the most intense peak is listed. UPLC-ESI-MS was performed on a Waters Acquity UPLC System coupled to a Bruker HCTTM, using an Acquity UPLC BEH C18 1.7 μm (2.1 × 50 mm) as a reverse phase column with a flow rate of 0.6 mL min−1. The UV absorption was measured at 254 nm. The runs were performed with a linear gradient of A (acetonitrile (Sigma-Aldrich HPLC grade) and B (distilled water containing 0.02% TFA and 0.05% HCOOC)): t = 0–0.5 min, 5% A; t = 4.0 min, 100% A; t = 5 min, 100% A. High-resolution mass spectrometry were performed on a Bruker ESQUIRE-LC quadrupole ion trap instrument (Bruker Daltonik GmbH, Bremen, Germany), equipped with a combined Hewlett-Packard Atmospheric Pressure Ion (API) source (Hewlett-Packard Co., Palo Alto, CA, USA). The solutions (about 0.1–1 μmol ml−1) were continuously introduced through the electrospray interface with a syringe infusion pump (Cole-Parmer 74900-05, Cole-Parmer Instrument Company, Vernon Hills, Illinois, USA) at a flow rate of 5 μl min−1. The MS-conditions were: nebulizer gas (N2) 15 psi, dry gas (N2) 7 min−1, dry temperature 300 °C, capillary voltage 4000 V, end plate 3500 V, capillary exit 100 V, skimmer1 30 V, and trap drive 70. The MS acquisitions were performed at normal resolution (0.6 u at half peak height), under ion charge control (ICC) conditions (10000) in the mass range from m/z 100 to 2000. To get representative mass spectra, 8 scans were averaged. Infrared spectra were recorded on Perkin-Elmer FTIR spectrometer using KBr pellets. Peak intensities are given as broad (b), very strong (vs), strong (s), medium (m) and weak (w). Elemental microanalyses were performed on a LecoCHNS-932 elemental analyser.
Synthesis
:ethyl acetate (4:1) as eluent (Rf (5a) = 0.69, hexane:ethyl acetate 1:1, Rf (4a) = 0.30, hexane:ethyl acetate 1:1) to afford 2-ferroceneamido-2-cyanopropyl ferroceneoate (5a) and N-(2-cyano-1-hydroxypropan-2-yl)ferroceneamide (4a) as orange solids, respectively. Yield: 11% (5a, 0.075 g, 0.143 mmol) and 50% (4a, 0.41 g, 1.31 mmol). Data 4a: IR (KBr, cm−1): 3467s, 3412s, 3103w, 2941w, 2862w, 1635s, 1534m, 1454w, 1377w, 1312m, 1267w, 1201w, 1160w, 1099w, 1056m, 1037w, 1023w, 998w, 911w, 826w, 772w, 710w, 620m, 528w, 499w, 483w, 464w. 1H NMR (400 MHz, d6-acetone): δ/ppm = 7.12 (s, 1H, NH), 5.01 (t, 3J = 6.4 Hz, 1H, OH), 4.85–4.84 (m, 2H, C5H4), 4.39–4.38 (m, 2H, C5H4), 4.24 (s, 5H, C5H5), 4.0–3.93 (m, 1H, CH2), 3.90–3.86 (m, 1H, CH2), 1.71 (s, 3H, CH3). 13C NMR (125 MHz, d6-acetone): δ/ppm = 170.9, 121.1, 76.1, 71.5, 70.5, 69.5, 69.4, 67.0, 66.9, 53.3, 22.6. ESI-MS: m/z (%) = 351.02 [M + K]+ (8), 335.04 [M + Na]+ (100), 312.06 [M]+ (52). HR ESI-MS: m/z (%) = 312.05557, calcd for C15H16FeN2O2 (M+) m/z (%) = 312.05508. Elemental analysis: calcd for C15H16N2O2Fe = C, 57.72; H, 5.17; N, 8.97. Found = C, 57.48; H, 5.13; N, 8.69. Data 5a: IR (golden gate, cm−1): 3368w, 1690m, 1656m, 1520m, 1453w, 1378w, 1288m, 1264w, 1214w, 1164m, 1144m, 1104w, 1027w, 999w, 918w, 827m, 814m, 770m, 741w. 1H NMR (500 MHz, d6-acetone): δ/ppm = 7.53 (s, 1H, NH), 4.88–4.87 (m, 4H, C5H4), 4.77 (d, 2J = 10.8 Hz, 1H, CH2), 4.82–4.50 (m, 3H, C5H4, CH2), 4.44–4.41 (m, 2H, C5H4), 4.27 (s, 5H, C5H5), 4.25 (s, 5H, C5H5), 1.90 (s, 3H, CH3). 13C NMR (125 MHz, d6-acetone): δ/ppm = 171.7, 170.7, 120.21, 75.8, 72.7, 71.7, 71.0, 70.8, 70.6, 69.5, 69.4, 66.9, 51.5, 23.1. ESI-MS: m/z (%) = 524.1 [M]+ (100). Elemental analysis: calcd for C26H24N2O3Fe2 = C 59.58; H 4.62; N 5.34. Found = C 59.51; H 4.52; N 5.25.
:ethyl acetate 7:1 → 1:7, and the methanol as eluent (Rf (4b) = 0.05, hexane:ethyl acetate 7:1, Rf (5b) = 0.2, methanol) to afford N-(2-cyano-1-hydroxypropan-2-yl)ruthenocenamide (4b) and 2-cyano-2-(ruthenocenecarboxamido)propyl ruthenocnecarboxylate (5b) as pale yellow solids, respectively. Yield: 31% (4b, 0.77 g, 2.06 mmol) and 19% (5b, 0.81 g, 1.32 mmol). Data 4b: IR (KBr, cm−1): 3248br, 31122s, 3056w, 2943w, 2887w, 2641w, 2324w, 2241w, 2050w, 1981w, 1720w, 1633s, 1531s, 1455s, 1376s, 1308s, 1130s, 823s. 1H NMR (500 MHz, DMSO): δ/ppm = 7.51 (s, 1H, NH), 5.64 (t, 3J = 6.46 Hz, 1H, OH), 5.23–5.22 (m, 2H, C5H4), 4.73–4.73 (m, 2H, C5H4), 4.59 (s, 5H, C5H5), 3.80–3.76 (m, 1H, CH2), 3.53–3.50 (m, CH2), 1.54 (s, 3H, CH3). 13C NMR (125 MHZ, DMSO): δ/ppm = 168.1, 120.5, 79.1, 72.4, 72.3, 71.6, 70.4, 70.3, 64.8, 51.9, 21.7. ESI-MS: m/z (%) = 359.1 [M + H]+ (100), 259.0 [M − C4H4N2OH]+ (17). Elemental Analysis: calcd for C15H16O2N2Ru = C, 50.41; H, 4.51; N, 7.84. Found = C, 50.85; H, 4.44; N, 7.41. Data 5b: IR (KBr, cm−1): 3320m, 3104w, 2956w, 2651w, 2322w, 2161s, 2053s, 1976s, 1690s, 1656s, 1521s, 1450s, 1373s, 1267s, 1135s, 1035m, 997m, 808s, 759m. 1H NMR (500 MHz, DMSO): δ/ppm = 7.93 (s, 1H, NH), 5.27–5.25 (m, 2H, C5H3), 5.15–5.13 (m, 2H, C5H3), 4.85–4.84 (m, 2H, C5H4), 4.76–4.75 (m, 2H, C5H4), 4.65 (s, 5H, C5H3), 4.59 (s, 5H, C5H3), 4.51 (d, 2J = 10.8, 2H, CH2), 4.27 (d, 2J = 10.4, 2H, CH2), 1.66 (s, 3H, CH3). 13C NMR (125 MHZ, DMSO): δ/ppm = 168.6, 168.2, 119.2, 78.7, 74.1, 73.3, 72.5, 71.9, 71.6, 71.4, 70.5, 70.3, 64.9, 49.7, 22.1. ESI-MS: m/z (%) = 639.3 [M + Na]+ (100). Elemental analysis: calcd for C26H24O3N2Ru2 = C 50.81; H 3.94; N 4.56. Found = C 50.92; H 3.96; N 4.53.
:ethyl acetate (6:1) as the eluent (Rf = 0.36, hexane:ethyl acetate 2:1) to give N-(2-cyano-1-(5-cyano-2-(trifluoromethyl)phenoxy)propan-2-yl)ferroceneamide (6a) as an orange solid. Yield: 26% (0.041 g, 0.085 mmol). IR (KBr, cm−1): 3478s, 3414s, 3355s, 2925s, 2851m, 2358m, 2336m, 2240s, 1653s, 1613s, 1574w, 1527m, 1510w, 1465m, 1415m, 1409w, 1373w, 1309m, 1281m, 1261w, 1211w, 1180m, 1141m, 1131m, 1037m, 895w, 841w, 822w, 632w, 606w, 531w, 503w, 486w. 1H NMR (400 MHz, d6-acetone): δ/ppm = 7.90 (d, 3J = 8.4 Hz, 1H, arom.), 7.84 (s, 1H, arom.), 7.61 (d, 3J = 7.6 Hz, 1H, arom.), 7.52 (s, 1H, NH), 4.88–4.83 (m, 2H, C5H4; 1H, CH2), 4.67 (d, 2J = 9.2 Hz, 1H, CH2), 4.41–4.40 (m, 2H, C5H4), 4.22 (s, 5H, C5H5), 1.93 (s, 3H, CH3). 13C NMR (125 MHz, d6-acetone): δ/ppm = 170.9, 157.0, 129.3, 129.2, 129.2, 129.1, 126.0, 125.1, 123.2, 123.0, 122.9, 119.7, 118.4, 118.0, 75.6, 71.9, 71.7, 71.6, 70.6, 69.6, 69.4, 51.2, 23.0. 19F NMR (282.23 MHz, d6-acetone): δ/ppm = −63.6. ESI-MS: m/z (%) = 504.0 [M + Na]+ (100), 985.1 [2M + Na]+ (12). HR ESI-MS: m/z (%) = 504.05927 calcd for C23H18F3FeN3NaO2 ([M + Na]+) m/z (%) = 504.05958. Elemental analysis: calcd for C23H18N3O2F3Fe = C 57.40; H 3.77; N 8.73. Found = C 57.62; H 4.01; N 8.38.
:ethyl acetate 2:1 as the eluent (Rf = 0.60) to give N-(2-cyano-1-(5-cyano-2-(trifluoromethyl)phenoxy)propan-2-yl)ruthenoceneamide (6b) as white solid. Yield: 11% (0.024 g, 0.046 mmol). IR (KBr, cm−1): 3341br, 3076br, 2952w, 2234s, 2165w, 1977s, 1630s, 1575s, 1528s, 1416s, 1285s, 1264s, 1186s, 1132s, 1039s, 816s, 815s, 735s. 1H NMR (500 MHz, d6-acetone): δ/ppm = 7.89 (d, 3J = 8.0 Hz, 1H, arom. H), 7.80 (s, 1H, arom. H), 7.61 (d, 3J = 8.0 Hz, 1H, arom. H), 7.34 (s, 1H, NH), 5.20–5.19 (m, 2H, C5H4), 4.77 (d, 3J = 9.5 Hz, 1H, CH2), 4.71 (s, 2H, C5H4), 4.60–4.58 (m, 6H, CH2 and C5H5), 1.87 (s, 3H, CH3). 13C NMR (125 MHZ, d6-acetone): δ/ppm = 169.6, 169.5, 157.0, 156.9, 129.3, 129.3, 129.2, 129.2, 127.2, 126.1, 125.1, 123.5, 123.3, 123.0, 122.9, 122.8, 120.7, 119.6, 118.4, 118.4, 118.1, 118.0, 79.9, 79.9, 73.2, 73.2, 72.5, 72.0, 72.0, 71.2, 71.1, 51.2, 51.1, 22.9, 22.9. 19F NMR (470.59 MHz, d6-acetone): δ/ppm = −63.4. ESI-MS: m/z (%) = 550.0 [M + Na]+ (100). HR ESI-MS: m/z = 528.04751, calcd for C23H18F3N3O2Ru ([M]) m/z = 528.04733; m/z (%) = 550.02942, calcd for C23H18F3N3NaO2Ru ([M + Na]) m/z (%) = 550.02928. Elemental analysis: calcd for C23H21F3N3O3Ru(H2O) = C 50.73; H 3.70; N 7.72. Found = C 50.61; H 3.40; N 7.51.
:ethyl acetate 3:1 as the eluent (Rf = 0.29) to afford N-(1-(ferrocenyloxy)-2-cyanopropan-2-yl)ferroceneamide (7) as a yellow solid. Yield: 43% (0.021 g, 0.041 mmol). IR (KBr, cm−1): 3469s, 2929w, 2852w, 1637s, 1518s, 1378w, 1339w, 1305w, 1280w, 1101m, 1008m, 825m, 523m, 502m, 485m. 1H NMR (500 MHz, CD3CN): δ/ppm = 6.48 (s, 1H, NH), 4.73–4.70 (m, 2H, C5H4), 4.46 (s, 2H, RCH2OR), 4.39–4.38 (m, 2H, C5H4), 4.32–4.31 (m, 2H, C5H4), 4.20 (s, 7H, C5H5, C5H4), 4.17 (s, 5H, C5H5), 3.80 (d, 3J = 9.3 Hz, 1H, CH2), 3.70 (d, 3J = 9.3 Hz, 1H, CH2), 1.68 (s, 3H, CH3). 13C NMR (125 MHz, CD3CN): δ/ppm = 207.9, 171.2, 121.2, 83.9, 75.6, 73.5, 71.9, 70.7, 70.6, 70.5, 69.6, 69.5, 69.5, 69.4, 51.7, 22.9. ESI-MS: m/z (%) = 484.03 [M − CN]+ (12), 510.04 [M]+ (100), 533.00 [M + Na]+ (7). HR ESI-MS: m/z (%) = 510.06882, calcd for C26H26Fe2N2O2 ([M + Na]+) m/z (%) = 510.06864. Elemental analysis: calcd for C26H26N2O2Fe2 = C 61.21; H 5.14; N 5.49. Found = C 61.06; H 5.20; N 5.31.
Crystallographic studies
Single crystals of 4a and 7 were grown by slow evaporation of acetonitrile solutions of 4a or 7 respectively. Single crystals of 4b were grown by slow evaporation of an dichloromethane solution of 4b. Single crystals of 6a were grown by slow evaporation of an acetone solution of 6a.Crystallographic data of 4a, 4b and 6a were collected at 183(2) K with Mo Kα radiation (λ = 0.7107 Å) that was graphite-monochromated on an Oxford Diffraction CCD Xcalibur system with a Ruby detector. Suitable crystals were covered with oil (Infineum V8512, formerly known as Paratone N), placed on a nylon loop that is mounted in a CrystalCap Magnetic™ (Hampton Research) and immediately transferred to the diffractometer. The program suite CrysAlisPro was used for data collection, multi-scan absorption correction and data reduction.47 Crystallographic data of 7 were collected at 100(2) K at the PXIII beamline of the SLS synchrotron with a radiation wavelength of 0.71255 Å. The data was integrated with the XDS software48 and further processed with the CCP449 and POINTLESS50 software. The data has a low completeness because of the one-circle geometry at the beamline and the low symmetry. All structures were solved with direct methods using SIR9751 and were refined by full-matrix least-squares methods on F2 with SHELXL-2014.52 CCDC entries 1501422–1501425 contain the X-ray data of compounds 4a, 4b, 6a and 7.
Bioassay/s to assess anti-parasitic activity
Some parasites were produced in vivo in or on animals. Haemonchus contortus and Trichostrongylus colubriformis (strongylid nematodes) were maintained in sheep, and Dirofilaria immitis (filarial nematodes) in dogs. Rhipicephalus sanguineus (tick) was maintained on dogs. All animal experiments were approved by the State of Fribourg, Switzerland, and supervised by the Animal Welfare Officer of Novartis Animal Health. Other parasites, that is, Ctenocephalides felis (flea) and Lucilia cuprina (fly), were produced in vitro and maintained on defibrinated cattle blood. All bioassays were performed by Novartis Animal Health employing industry-standard operating procedures.Assay to test activity in vitro against Haemonchus contortus and Trichostrongylus colubriformis
This method was conducted as described by Kaminsky et al. (2008).10 In brief, freshly harvested and cleaned nematode eggs were seeded into a 96-well plate containing the test substances to be evaluated for anthelmintic activity. Each compound was tested by serial dilution in order to determine its minimum effective dose. The test compounds were embedded in an agar-based nutritive medium allowing the full development of eggs through to third stage larvae (L3). The plates were incubated for 6 days at 28 °C and 80% relative humidity. Egg hatching and ensuing larval development were recorded to identify a possible nematocidal activity. Efficacy was expressed as a percentage of reduced egg hatch, reduced development of L3, or paralysis and death of larvae of all stages.Activity in vitro against Dirofilaria immitis
Microfilariae present in blood from donor dogs chronically infected with D. immitis were seeded into 96-well microplates. Individual test compounds were tested by serial dilution to determine their minimum effective doses. The plates were incubated for 48 h at 26 °C and 60% relative humidity. The motility of microfilariae was then recorded to identify any anti-filarial activity. Efficacy was expressed as the percentage of reduced motility compared to the control (untreated) and standards. In these assays, a compound needed to exhibit a nematodicidal efficacy of >60% at a concentration of 32 μg ml−1 (32 ppm) to qualify for further testing.Activity in vitro against Ctenocephalides felis
Activity in vitro against Rhipicephalus sanguineus (dog tick)
Determination of ROS level
The assay was performed following a procedure published by our group with slight modification.57 Briefly, HeLa cells (8000 in 100 μL medium per well) were seeded in a 96 well plate (black, clear flat bottom from corning). Next day, the media was aspirated and 200 μM fresh media containing 4a, 4b, 6a or 6b (freshly prepared stock solution in DMSO, after dilution with culture medium final concentration of DMSO ≤0.3%, 25 μM exposure concentration) was added. Cells were then incubated at 37 °C incubator for 22 h. The medium was then removed, washed with 150 μL PBS and then 150 μL solution of H2DCFDA (final concentration 20 μM) in serum free medium was added and incubated for 40 min in dark. The fluorescence generated by intracellular ester cleavage followed by oxidation of H2DCFDA by intracellular ROS was quantified at 528 nm emission with 485 nm excitation wavelength in a SpectraMax M5 microplate reader. For the positive control TBH, 100 μM concentration and 6 h incubation time was used. After quantification of the ROS, cell viability on HeLa cells was determined to quantify the potential decrease in cellular mass due to treatment with different compounds. The media was aspirated and cells were washed with 150 μL PBS. Afterwards, 100 μL 0.4% formaldehyde in PBS was added and cells were allowed to fix for 20 min at room temperature. The formaldehyde solution was aspirated and cells were washed with 150 μL PBS. 0.02% crystal violet (CV) solution in PBS was then added (100 μL per well) and incubated at room temperature for 30 min. The CV solution was then aspirated, washed with 150 μL of distilled water and dried overnight. Next day, 180 μL 80% ethanol was added to each well and the plates were gently shaken on a rocker for 2–3 h and were read at 570 nm in a SpectraMax M5 microplate reader. The results expressed as mean and standard error of six replicates, corrected for the viable cell population.Cell culture
Human cervical carcinoma cells (HeLa) were cultured in DMEM (Gibco) supplemented with 5% fetal calf serum (FCS, Gibco), 100 U ml−1 penicillin, 100 μg ml−1 streptomycin at 37 °C and 5% CO2. The normal human fetal lung fibroblast MRC-5 cell line was maintained in F-10 medium (Gibco) supplemented with 10% FCS (Gibco), penicillin (100 U ml−1), and streptomycin (100 μg ml−1).Cytotoxicity studies
Cytotoxicity studies were performed on two different cell lines, namely HeLa, and MRC-5, by a fluorometric cell viability assay using Resazurin (Promocell GmbH). Briefly, one day before treatment, cells were seeded in triplicates in 96-well plates at a density of 4 × 103 cells per well for HeLa and 7 × 103 for MRC-5 in 100 μl growth medium. Upon treating cells with increasing concentrations of organometallic-monepantel derivatives for 48 h, the medium was removed, and 100 μl complete medium containing Resazurin (0.2 mg ml−1 final concentration) were added. After 4 h of incubation at 37 °C, fluorescence of the highly red fluorescent product Resorufin was quantified at 590 nm emission with 540 nm excitation wavelength in a SpectraMax M5 microplate reader.Abbreviations
| AADs | Amino-acetonitrile derivatives |
| C. felis | Ctenocephalides felis |
| D. immitis | Dirofilaria immitis |
| ESI-MS | Electrospray ionisation-mass spectrometry |
| H. contortus | Haemonchus contortus |
| H2DCFDA | 2′,7′-Dichlorofluorescein diacetate |
| LDA | Larval development assay |
| L. cuprina | Lucilia cuprina |
| nAChR | Nicotinic acetylcholine receptor |
| o.n. | Overnight |
| P. falciparum | Plasmodium falciparum |
| ROS | Reactive oxygen species |
| R. sanguineus | Rhipicephalus sanguineus |
| r.t. | Room temperature |
| SAR | Structure activity relationship |
| TBH | tert-Butyl hydroperoxide |
| T. colubriformis | Trichostrongylus colubriformis. |
Acknowledgements
This work was financially supported by the Swiss National Science Foundation (Professorships No. PP00P2_133568 and PP00P2_157545 to G. G.), the University of Zurich (G. G., S. F.), the Stiftung für wissenschaftliche Forschung of the University of Zurich (G. G., S. F.), the Novartis Jubilee Foundation (G. G.), the Kurt u. Senta Hermann Stiftung (S. F.) and the Swiss Government Excellence Scholarship for Postdoctoral Researcher (R. L.). R. B. G.'s research program is supported by the Australian Research Council (ARC), the National Health and Medical Research Council (NHMRC), Melbourne Water Corporation, Yourgene Bioscience and the Alexander von Humboldt Foundation and The University of Melbourne. R. B. G. is a grateful recipient of Professorial Humboldt Research Awards. The authors would like to thank Dr Jacques Bouvier (Novartis Animal Health, St-Aubin, Switzerland) and Dr Noëlle Gauvry (Novartis Animal Health, Basel, Switzerland) for their help with the biological assays.References
- P. Ducray, N. Gauvry, F. Pautrat, T. Goebel, J. Fruechtel, Y. Desaules, S. S. Weber, J. Bouvier, T. Wagner, O. Froelich and R. Kaminsky, Bioorg. Med. Chem. Lett., 2008, 18, 2935–2938 CrossRef CAS PubMed.
- R. Kaminsky, L. Rufener, J. Bouvier, R. Lizundia, S. Schorderet Weber and H. Sager, Vet. Parasitol., 2013, 195, 286–291 CrossRef PubMed.
- C. P. Gordon, L. Hizartzidis, M. Tarleton, J. A. Sakoff, J. Gilbert, B. E. Campbell, R. B. Gasser and A. McCluskey, MedChemComm, 2014, 5, 159–164 RSC.
- For legal issues the efficacy of AAD96 cannot be disclosed.
- R. M. Kaplan and A. N. Vidyashankar, Vet. Parasitol., 2012, 186, 70–78 CrossRef PubMed.
- R. M. Kaplan, Trends Parasitol., 2004, 20, 477–481 CrossRef CAS PubMed.
- S. B. Howell, J. M. Burke, J. E. Miller, T. H. Terrill, E. Valencia, M. J. Williams, L. H. Williamson, A. M. Zajac and R. M. Kaplan, J. Am. Vet. Med. Assoc., 2008, 233, 1913–1919 CrossRef PubMed.
- L. L. Mortensen, L. H. Williamson, T. H. Terrill, R. A. Kircher, M. Larsen and R. M. Kaplan, J. Am. Vet. Med. Assoc., 2003, 23, 495–500 CrossRef.
- C. Epe and R. Kaminsky, Trends Parasitol., 2013, 29, 129–134 CrossRef CAS PubMed.
- R. Kaminsky, P. Ducray, M. Jung, R. Clover, L. Rufener, J. Bouvier, S. S. Weber, A. Wenger, S. Wieland-Berghausen, T. Goebel, N. Gauvry, F. Pautrat, T. Skripsky, O. Froelich, C. Komoin-Oka, B. Westlund, A. Sluder and P. Maser, Nature, 2008, 452, 176–180 CrossRef CAS PubMed.
- L. Rufener, J. Keiser, R. Kaminsky, P. Mäser and D. Nilsson, PLoS Pathog., 2010, 6, e1001091 Search PubMed.
- R. Baur, R. Beech, E. Sigel and L. Rufener, Mol. Pharmacol., 2015, 87, 96–102 CrossRef PubMed.
- L. Rufener, N. Bedoni, R. Baur, S. Rey, D. A. Glauser, J. Bouvier, R. Beech, E. Sigel and A. Puoti, PLoS Pathog., 2013, 9, e1003524 CAS.
- L. Rufener, P. Maeser, I. Roditi and R. Kaminsky, PLoS Pathog., 2009, 5, e1000380 Search PubMed.
- R. Van den Brom, L. Moll, C. Kappert and P. Vellema, Vet. Parasitol., 2015, 209, 278–280 CrossRef CAS PubMed.
- A. Mederos, Z. Ramos and G. Banchero, Parasites Vectors, 2014, 7, 598 CrossRef PubMed.
- A. S. Cezar, G. Toscan, G. Camillo, L. A. Sangioni, H. O. Ribas and F. S. F. Vogel, Vet. Parasitol., 2010, 173, 157–160 CrossRef PubMed.
- I. Scott, W. E. Pomroy, P. R. Kenyon, G. Smith, B. Adlington and A. Moss, Vet. Parasitol., 2013, 198, 166–171 CrossRef CAS PubMed.
- J. Hess, M. Patra, L. Rangasamy, S. Konatschnig, O. Blacque, A. Jabbar, P. Mac, E. M. Jorgensen, R. B. Gasser and G. Gasser, Chem. – Eur. J., 2016 DOI:10.1002/chem.201602851.
- J. Hess, M. Patra, V. Pierroz, B. Spingler, A. Jabbar, S. Ferrari, R. B. Gasser and G. Gasser, Organometallics, 2016, 35, 3369–3377 CrossRef CAS.
- G. Gasser and N. Metzler-Nolte, Curr. Opin. Chem. Biol., 2012, 16, 84–91 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- G. Jaouen and N. Metzler-Nolte, Topics in Organometallic Chemistry, Springer, 2010 Search PubMed.
- C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC.
- P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS PubMed.
- C. Biot, G. Glorian, L. A. Maciejewski, J. S. Brocard, O. Domarle, G. Blampain, P. Millet, A. J. Georges, H. Abessolo, D. Dive and J. Lebibi, J. Med. Chem., 1997, 40, 3715–3718 CrossRef CAS PubMed.
- C. Biot and D. Dive, in Medicinal Organometallic Chemistry, ed. G. Jaouen and N. Metzler-Nolte, Springer-Verlag, Heidelberg, 2010, vol. 32, pp. 155–193 Search PubMed.
- C. Biot, W. Castro, C. Y. Botte and M. Navarro, Dalton Trans., 2012, 41, 6335–6349 RSC.
- D. Dive and C. Biot, ChemMedChem, 2008, 3, 383–391 CrossRef CAS PubMed.
- R. Rubbiani, O. Blacque and G. Gasser, Dalton Trans., 2016, 45, 6619–6626 RSC.
- M. Patra, G. Gasser, M. Wenzel, K. Merz, J. E. Bandow and N. Metzler-Nolte, Organometallics, 2010, 29, 4312–4319 CrossRef CAS.
- M. Patra, G. Gasser, A. Pinto, K. Merz, I. Ott, J. E. Bandow and N. Metzler-Nolte, ChemMedChem, 2009, 4, 1930–1938 CrossRef CAS PubMed.
- M. Patra, K. Ingram, V. Pierroz, S. Ferrari, B. Spingler, R. B. Gasser, J. Keiser and G. Gasser, Chem. – Eur. J., 2013, 19, 2232–2235 CrossRef CAS PubMed.
- M. Patra, K. Ingram, V. Pierroz, S. Ferrari, B. Spingler, J. Keiser and G. Gasser, J. Med. Chem., 2012, 55, 8790–8798 CrossRef CAS PubMed.
- M. Patra, K. Ingram, A. Leonidova, V. Pierroz, S. Ferrari, M. N. Robertson, M. H. Todd, J. Keiser and G. Gasser, J. Med. Chem., 2013, 56, 9192–9198 CrossRef CAS PubMed.
- F. Dubar, T. J. Egan, B. Pradines, D. Kuter, K. K. Ncokazi, D. Forge, J.-F. O. Paul, C. Pierrot, H. Kalamou, J. Khalife, E. Buisine, C. Rogier, H. Vezin, I. Forfar, C. Slomianny, X. Trivelli, S. Kapishnikov, L. Leiserowitz, D. Dive and C. Biot, ACS Chem. Biol., 2011, 6, 275–287 CrossRef CAS PubMed.
- N. Chavain, H. Vezin, D. Dive, N. Touati, J.-F. Paul, E. Buisine and C. Biot, Mol. Pharmaceutics, 2008, 5, 710–716 CrossRef CAS PubMed.
- N. Gauvry, T. Goebel, P. Ducray, F. Pautrat, R. Kaminsky and M. Jung, WO05/044784, 2005; Chem. Abstr. 2005, 142, 481750 Search PubMed.
- J. Bernstein, R. E. Davis, L. Shimoni and N. L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- S. Top, A. Vessières, G. Leclercq, J. Quivy, J. Tang, J. Vaissermann, M. Huché and G. Jaouen, Chem. – Eur. J., 2003, 9, 5223–5236 CrossRef CAS PubMed.
- C. Biot, G. Glorian, L. A. Maciejewski and J. Brocard, J. Med. Chem., 1997, 40, 3715–3718 CrossRef CAS PubMed.
- G. Jaouen, S. Top, A. Vessières, G. Leclercq and M. J. McGlinchey, Curr. Med. Chem., 2004, 11, 2505–2517 CrossRef CAS PubMed.
- W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, Butterworth-Heinemann, Oxford, UK, 4th edn, 1996 Search PubMed.
- D. P. Cormode, A. J. Evans, J. J. Davis and P. D. Beer, Dalton Trans., 2010, 39, 6532–6541 RSC.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- Agilent Technologies, Journal, 2014, Xcalibur CCD system.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- S. Bailey, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1994, 50, 760–763 CrossRef PubMed.
- P. R. Evans, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 282–292 CrossRef CAS PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- M. Zakson-Aiken, L. M. Gregory, P. T. Meinke and W. L. Shoop, J. Med. Entomol., 2001, 38, 576–580 CrossRef CAS PubMed.
- S. E. Wade and J. R. Georgi, J. Med. Entomol., 1988, 25, 186–190 CrossRef CAS PubMed.
- L. Lovis, J. L. Perret, J. Bouvier, J. M. Fellay, R. Kaminsky, B. Betschart and H. Sager, Vet. Parasitol., 2011, 182, 269–280 CrossRef CAS PubMed.
- L. Lovis, M. C. Mendes, J. L. Perret, J. R. Martins, J. Bouvier, B. Betschart and H. Sager, Vet. Parasitol., 2013, 191, 323–331 CrossRef CAS PubMed.
- R. Rubbiani, T. N. Zehnder, C. Mari, O. Blacque, K. Venkatesan and G. Gasser, ChemMedChem, 2014, 9, 2781–2790 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 13C and 19F NMR spectra (4a/b, 5a/b, 6a/b and 7) (Fig. S1–S16), ORTEP plot of 7 (Fig. S17), anti-parasitic activity against C. felis, L. cuprina and R. sanguineus of organometallic precursors and derivatives (4a/b, 5a/b, 6a/b and 7) (Table S1). CCDC 1501422–1501425. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03376h |
| ‡ Present address: Chimie ParisTech, PSL Research University, 11, rue Pierre et Marie Curie, F-75005 Paris, France. |
| This journal is © The Royal Society of Chemistry 2016 |