
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a lithium–cyclopentadienide complex by addition of LiNTf2 to a zwitterionic fulvalene†‡
Dominic
Schmid
,
Alexander
Seyboldt
,
Klaus
Eichele
and
Doris
Kunz
 *
*
Institut für Anorganische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: Doris.Kunz@uni-tuebingen.de
First published on 29th November 2016
Abstract
The synthesis of a η5-coordinated LiCp complex by simple addition of a Li-salt in benzene is presented. A strongly zwitterionic fulvalene serves as the Cp-precursor. Evidence for the coordination of Li+ was obtained by the characterisitic 7Li NMR chemical shifts, variable temperature experiments in solution and by X-ray structure analysis in the solid state.
Lithium cyclopentadienide derivatives are important Cp-transfer reagents in the synthesis of metallocenes and half-sandwich complexes of transition metals.1 Usually, their preparation follows one of the three major routes (Scheme 1)2: (a) deprotonation of cylcopentadiene with a strong base, (b) reduction of cyclopentadiene with lithium metal (dehydrogenation), or (c) nucleophilic carbolithiation at the exocyclic double bond of a fulvene or its enolisation (deprotonation), wherein the five-membered ring plays the role of the carbonyl group.3 Usually, all these reactions are carried out in coordinating solvents, for example THF, diethylether or liquid NH3.2b
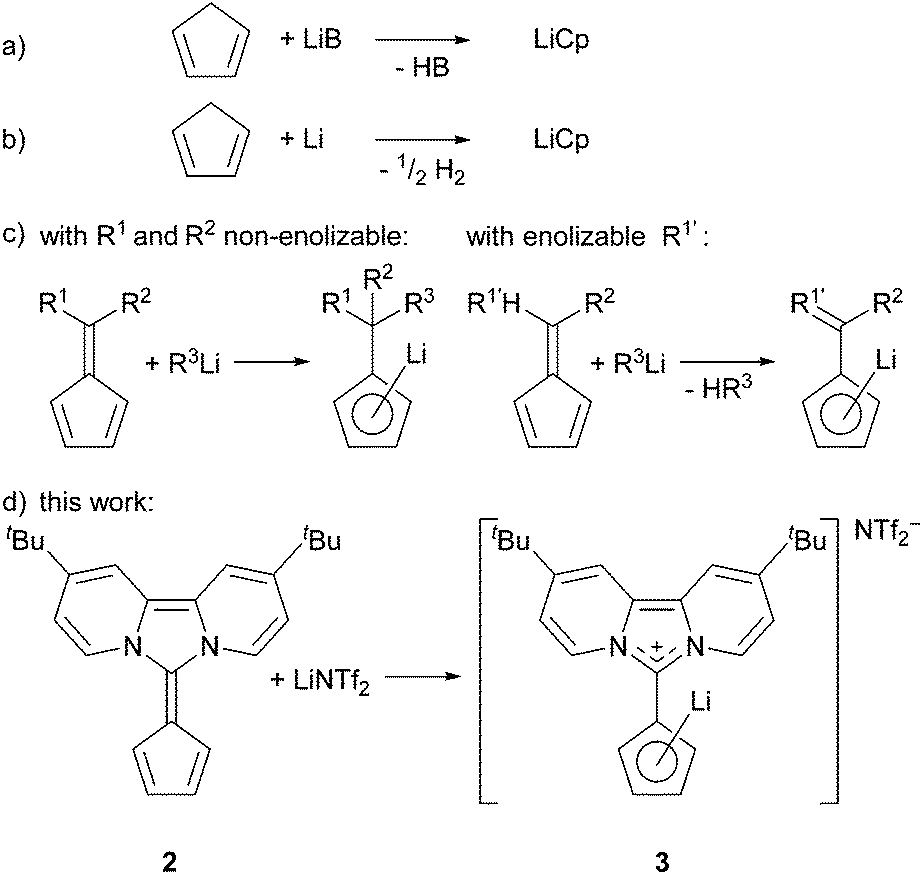 | ||
| Scheme 1 Methods for the synthesis of LiCp compounds. | ||
In 1972 Müller–Westerhoff showed that dipolar 6,6-bis(dimethylamino)fulvene can serve as a Cp-precursor upon reaction with FeCl2,4a a reaction that takes advantage of the high stability of ferrocenes and the dipolar character of the aminofulvene.
Over the past few years, we reported zwitterionic fulvalenes 1 and 2 (Fig. 1, Scheme 1d) and their straightforward conversion to imidazolium-substituted metallocenes4b and half-sandwich complexes.5 Together with their structural features, we concluded that they can be regarded as organic cyclopentadienide equivalents and are best depicted in their zwitterionic resonance forms 1′ and 2′.6 Thus, they are similar to phosphonium cyclopentadienylides,7 for which the addition to transition metal complexes has been shown in the past,8 and only one case of a neutral Li-adduct was reported for the solid state.9 As fulvalenes provide a high versatility in metallocene chemistry, we set out to test their potentials and limitations in coordination chemistry. For this purpose, we wanted to probe whether Li+ ions are able to form stable cyclopentadienide complexes by simple addition of lithium salts to fulvalene 2 (Scheme 1d) that bears the strongest ylidic character of all 6,6-diaminofulvenes or -fulvalenes.
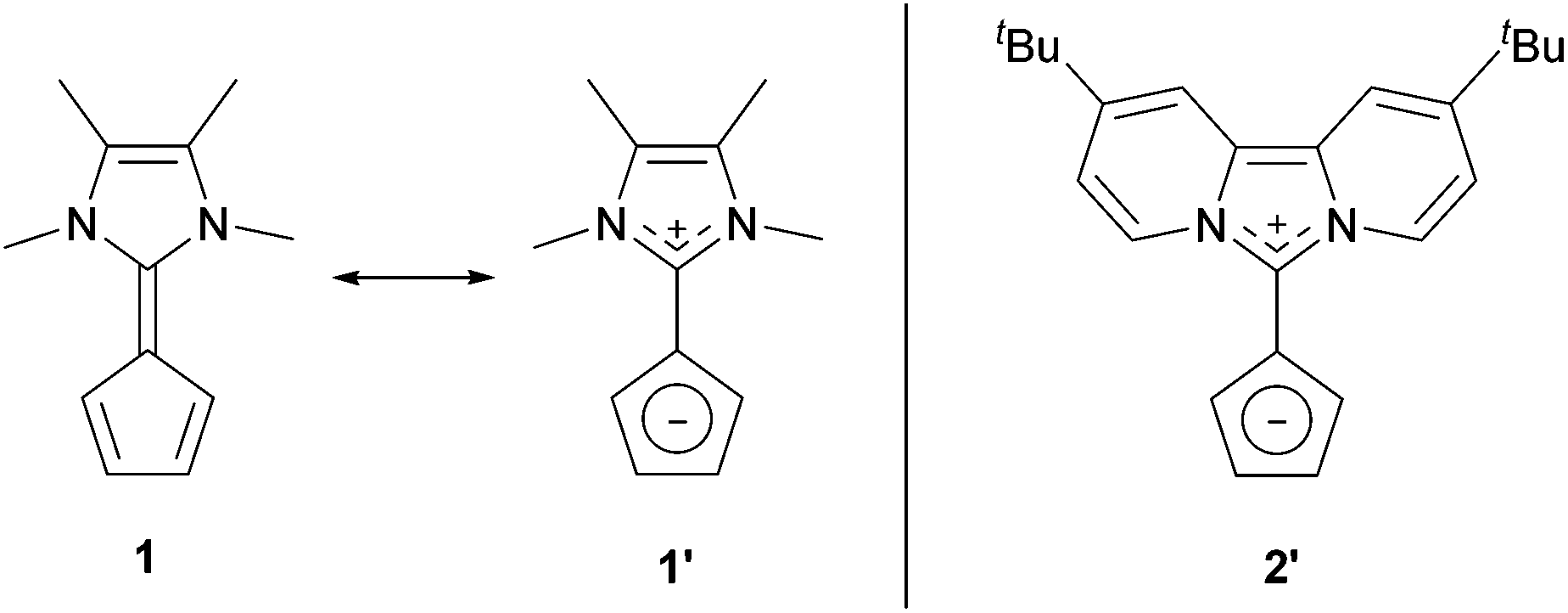 | ||
| Fig. 1 Zwitterionic diazafulvalenes 1′ and 2′ are organic Cp analogues. | ||
At first, we reacted 2 and LiOTf in acetonitrile-d3 as well as 2 and LiNTf2 in THF-d8, but a prolonged reaction time neither at room temperature nor at 60 °C caused any significant shift of the peaks in the 1H NMR and 7Li NMR spectra. To avoid any competing coordination of the solvent to the Li+ cation, we chose benzene-d6 as a solvent. Heating 2 and LiOTf at 60 °C in benzene-d6 did not show any reaction, possibly due to the extremely low solubility of lithium triflate. However, keeping a mixture of 2 and LiNTf2 for 16 h at room temperature leads to differences in the chemical shift of all but the tert-butyl signals of 0.05 to 0.25 ppm in the 1H NMR spectrum. Heating the mixture up to 60 °C for 16 h increased these differences to 0.10–0.41 ppm (Fig. 2).
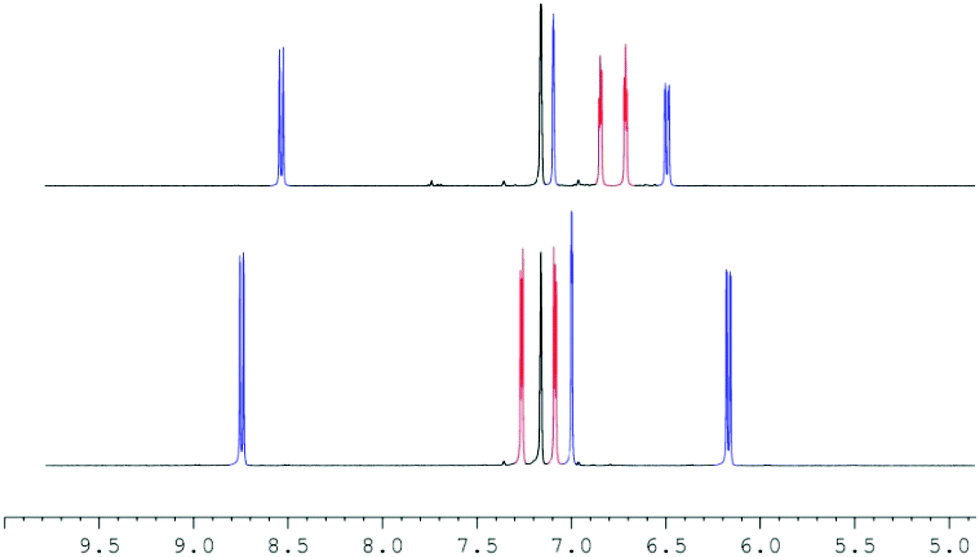 | ||
| Fig. 2 Detail of the 1H NMR spectrum (400.1 MHz) of 2 in C6D6 before (bottom) and after addition of LiNTf2 and at 16 h at 60 °C (top) showing the imidazolium (blue), the Cp signals (red) as well as the solvent peak (C6HD5) (black). | ||
The Cp–H signals are shifted upfield by 0.4 ppm, while the signals of the dipyridoimidazolinylidene moiety are shifted partly both upfield and downfield. These shifts indicate the formation of the coordination complex 3 rather than a mere change in the polarity of the solution upon addition of LiNTf2. The 7Li NMR spectrum of 3 shows a singlet at −5.6 ppm in toluene-d8 as well as in benzene-d6. In both cases, no signal was detected at the beginning of the reaction due to the poor solubility of LiNTf2 in both solvents. This chemical shift is characteristic of a contact ion pair (−5 to −8.5 ppm)10 in which the Li resides above the centre of an aromatic ring, and has also been observed for Cp-substituted Li-phosphoniumylides recently.11 In contrast, solvent separated ion pairs show the Li signal typically between 1 and −1 ppm.10 To gain more insight into the bonding situation in solution, we carried out variable temperature NMR experiments. Cooling down the sample in 10 °C steps reveals a significant broadening of the 1H NMR signals at −50 °C down to −80 °C (Fig. 3, left). Below −70 °C an additional broad signal set appears. Due to the different shapes of the t-Bu signals, a hindered rotation about the exocyclic C–C bond that leads to two non-equivalent sides of the dipyrido moiety cannot explain this result. For comparison, we also cooled a sample of fulvalene 2 in toluene-d8 to −80 °C and did not observe any significant broadening of the signals. A 7Li NMR experiment shows not only the broadening of the Li signal at −50 °C but also the formation of two additional peaks in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at −0.5 and −11 ppm at −80 °C (Fig. 3, right). This behavior was reported in the literature before and is diagnostic for the formation of a lithiocene species (−10 to −12 ppm) and a solvent separated Li-cation.12 By applying these results to our case, we conclude that a mono-cationic lithiocene 4 and the negative counterion 513 that consists of a lithium cation coordinated by two NTf2− ligands are formed (Scheme 2). Moreover, an EXSY experiment at −80 °C reveals lithium exchange of all three species (see the ESI‡).
:1 ratio at −0.5 and −11 ppm at −80 °C (Fig. 3, right). This behavior was reported in the literature before and is diagnostic for the formation of a lithiocene species (−10 to −12 ppm) and a solvent separated Li-cation.12 By applying these results to our case, we conclude that a mono-cationic lithiocene 4 and the negative counterion 513 that consists of a lithium cation coordinated by two NTf2− ligands are formed (Scheme 2). Moreover, an EXSY experiment at −80 °C reveals lithium exchange of all three species (see the ESI‡).
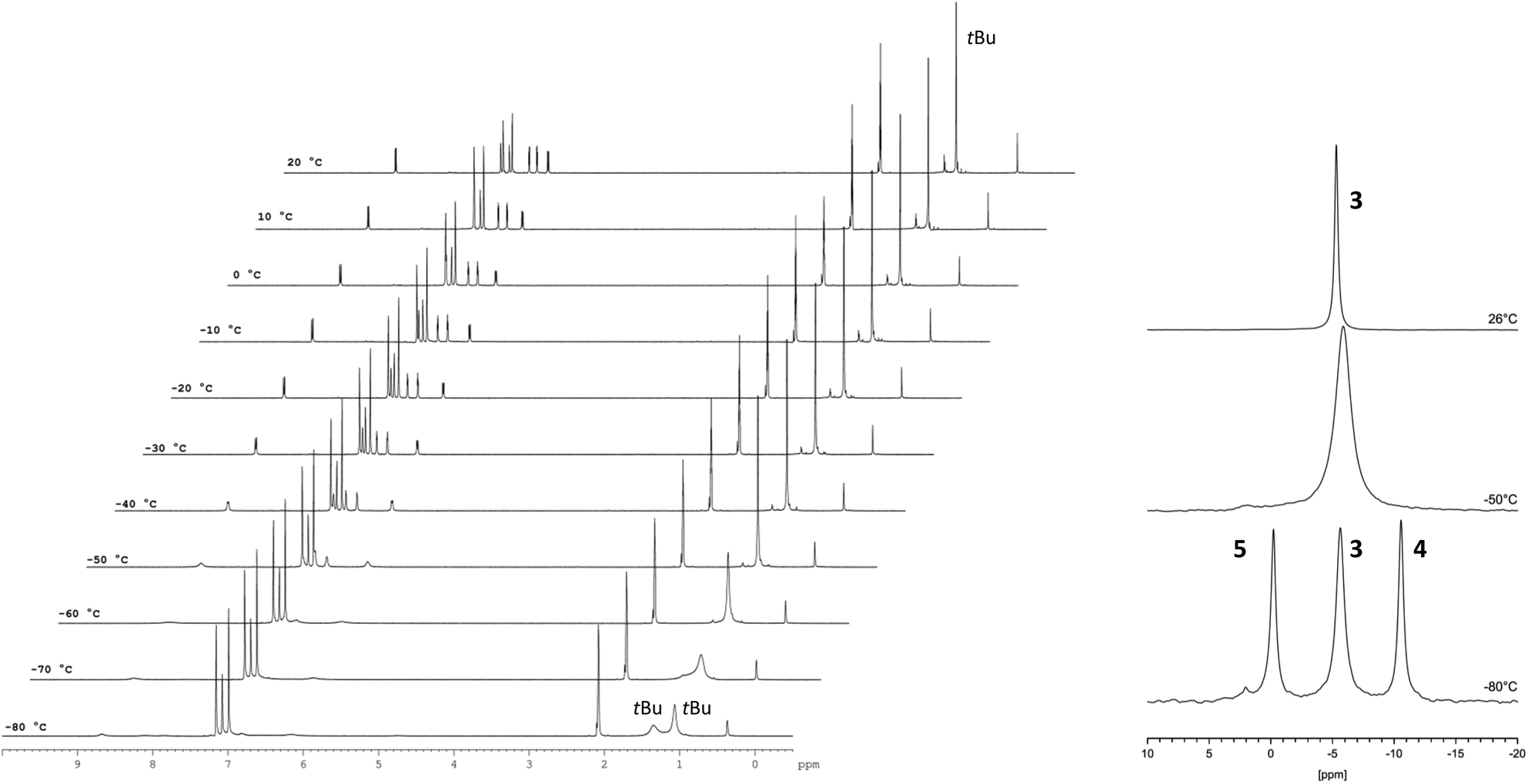 | ||
| Fig. 3 VT NMR experiments of 3 in toluene-d8. In the 1H NMR spectrum (400.1 MHz) (left) an additional (broader) signal set indicating the presence of a second species at −80 °C is observed. In the 7Li NMR spectrum (194.4 MHz) two new peaks in a 1:1 ratio at −0.5 ppm and −11 ppm are observed in addition to the signal at −5.8 ppm at −80 °C. The respective lithium atoms exchange at this temperature as revealed by an EXSY experiment (see the ESI‡). | ||
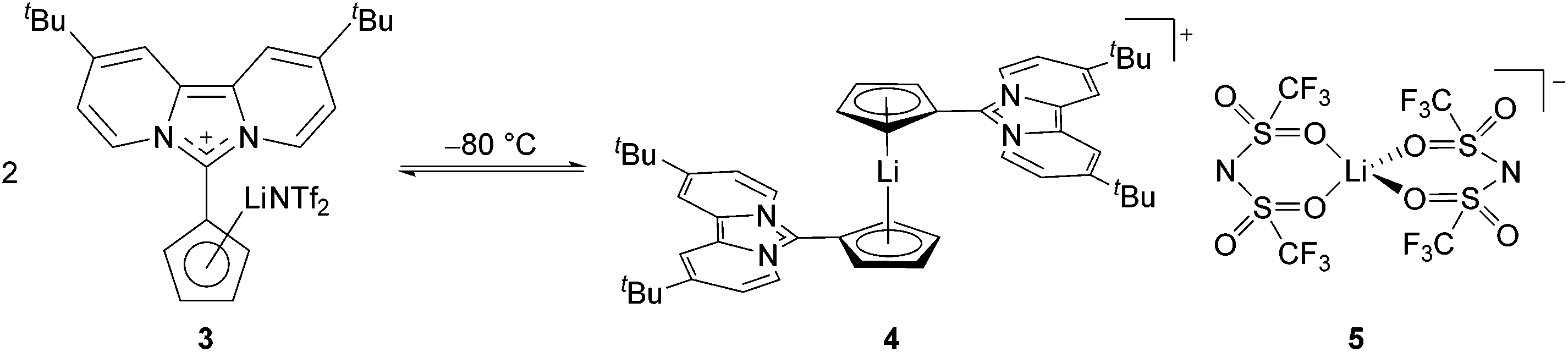 | ||
| Scheme 2 Fast equilibrium between the LiCp complex 3 and a solvent separated salt that consists of monocationic lithiocene 4 and lithiate 5 at −80 °C. | ||
By slow diffusion of dichloromethane into a solution of 3 in C6D6 at room temperature, we obtained single crystals suitable for X-ray diffraction analysis.
The molecular structure (Fig. 4) clearly shows a η5-coordination between the Li+ cation and the cyclopentadienide ring. The Li–CpCentroid distance of 2.057 Å is in complete agreement with those of other LiCp complexes (1.910 Å to 2.086 Å).14 The Li–carbon distances range from 2.354(4) Å to 2.422(4) Å, which is a typical range for η5-coordinated LiCpLn compounds.14
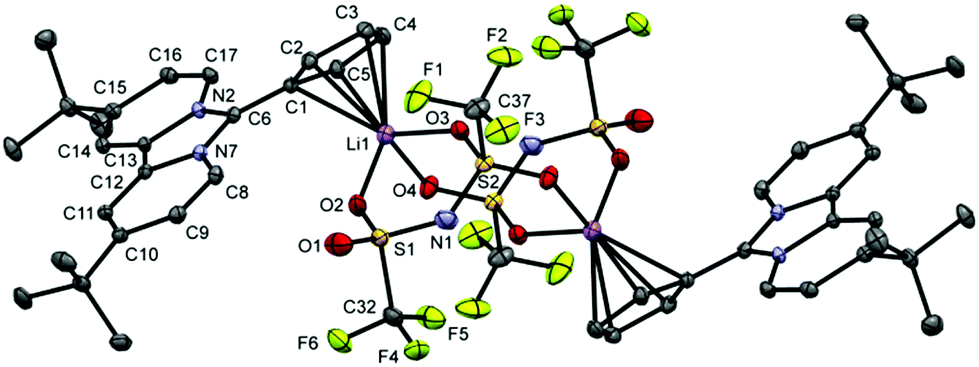 | ||
| Fig. 4 X-ray crystal structure of complex 3 with anisotropic atomic displacement parameters at the 50% probability level. Hydrogen atoms and three benzene molecules (1.5 per asymmetric unit) are omitted for clarity. | ||
The remaining coordination sphere of the Li-cation is occupied by three oxygen atoms of the two bridging bis(trifluoromethanesulfonyl)imide anions with Li–O distances between 2.033(4) Å and 2.057(4) Å. Complex 3 crystallises as a dimeric variation of the “donor ligand stabilised neutral monomer” motif of alkali metal cyclopentadienides.2 Following the nomenclature established for LiNTf2 crystalline solvates, complex 3 shows an aggregate coordination (AGG) in the transoid form (C2) regarding the position of the CF3 groups relative to the S–N–S plane.15 As three oxygen atoms are coordinated to two Li+ cations, the so called AGG-Ib-C2 coordination mode is realised in the solid state of complex 3.15
Although the coordination of the lithium cation to the Cp ring could be demonstrated clearly for 3 both in solution and in solid states, this bond formed is rather weak. This can be deduced from the fact that during monitoring the course of the reaction of 2 and LiNTf2 only one species is observed in the 1H NMR spectra, which implies a fast equilibrium between the free fulvalene and the coordinated species. This leads to the continuous shifting of the signals until the final values for compound 3 (fulvalene:Li = 1:1) are reached. Using two equivalents of Li(NTf)2 does not lead to further shifting of the signals. Another argument is the fast Li+ exchange at −80 °C revealed by the EXSY spectrum. Furthermore, already one equivalent of added THF leads to a recognisable shift of the signals into the direction of the free fulvalene 2 in the 1H NMR spectrum. With 8 equivalents of THF, the signals of pure 2 are almost obtained back. This is also corroborated by the ESI+ mass spectrum of complex 3 in which only the signal of the decoordinated, protonated fulvalene 2 [M + H]+ is detected.
In conclusion, we have presented a simple method for the preparation of a η5-cyclopentadienide lithium complex by addition of LiNTf2 to a highly zwitterionic fulvalene. While a dimeric structure forms in the solid state, the complex undergoes fast exchange in solution at room temperature. Evidence for an equilibrium between 3 and a monocationic lithiocene species 4 together with the [Li(NTf2)2]− counter ion (5) at −80 °C was obtained by the characteristic 7Li NMR chemical shifts. An EXSY experiment at −80 °C revealed Li exchange between all of these three species.
We will now put our efforts in applying this method to other group 1 and group 2 metal salts, and also on probing the utility of 3 as a Cp-transfer reagent in non-polar solvents.
We thank Kristina Strohmaier for help with the VT NMR experiments as well as Karl W. Törnroos and Eva Jürgens for help with X-ray structure analysis. We also thank a referee for suggesting the titration as well as the VT 7Li NMR experiment.
Notes and references
- P. Merino, Sci. Synth., 2009, 45a, 109–156 Search PubMed.
- (a) C. Elschenbroich, Organometallchemie, B. G. Teubner/GWV, Wiesbaden, 2008 Search PubMed; (b) P. Jutzi, W. Leffers, S. Pohl and W. Saak, Chem. Ber., 1989, 122, 1449–1456 CrossRef CAS; (c) G. Erker, G. Kehr and R. Fröhlich, Organometallics, 2008, 27, 3–14 CrossRef CAS.
- Review on the reactivity of fulvenes: (a) P. Zeller, Methoden Org. Chem. (Houben-Weyl), 1985, 5/2c, 504–684 Search PubMed; (b) M. Neuenschwander, in The Chemistry of Double-bonded Functional Groups, ed. S. Patai, Wiley & Sons, Chichester, 1989, vol. 2/2, suppl. A Search PubMed.
- (a) U. Mueller-Westerhoff, Tetrahedron Lett., 1972, 13, 4639–4642 CrossRef; (b) D. Kunz, E. Ø. Johnsen, B. Monsler and F. Rominger, Chem. – Eur. J., 2008, 14, 10909–10914 CrossRef CAS PubMed.
- D. Schmid, A. Seyboldt and D. Kunz, Z. Anorg. Allg. Chem., 2015, 641, 2228–2232 CrossRef CAS.
- D. Schmid, A. Seyboldt and D. Kunz, Z. Naturforsch., B: Chem. Sci., 2014, 69, 580–588 CrossRef CAS.
- F. Ramirez and S. Levy, J. Am. Chem. Soc., 1957, 79, 67–69 CrossRef.
- (a) J. H. Brownie and M. C. Baird, Coord. Chem. Rev., 2008, 252, 1734–1754 CrossRef CAS; (b) F. G. Schröder, C. Lichtenberg, M. Elfferding and J. Sundermeyer, Organometallics, 2013, 32, 5082–5091 CrossRef.
- In this case, the complex was formed by deprotonation of phosphoniumcyclopentadiene iodide with BuLi: F. G. Schröder, Dissertation, Philipps-Universität, Marburg, 2014.
- (a) R. H. Cox and H. W. Terry, J. Magn. Reson., 1974, 14, 317–322 CAS; (b) D. Johnels, A. Boman and U. Edlund, Magn. Reson. Chem., 1998, 36, S151–S156 CrossRef CAS.
- C. Lichtenberg, N. S. Hillesheim, M. Elfferding, B. Oelkers and J. Sundermeyer, Organometallics, 2012, 31, 4259–4266 CrossRef CAS.
- (a) M. M. Exner, W. Waack and E. C. Steiner, J. Am. Chem. Soc., 1973, 95, 7009–7018 CrossRef CAS; (b) L. A. Paquette, W. Bauer, M. R. Sivik, M. Bühl, M. Feigel and P. v. R. Schleyer, J. Am. Chem. Soc., 1990, 112, 8776–8789 CrossRef CAS; (c) K. Kunz, G. Erker, G. Kehr and R. Fröhlich, Organometallics, 2001, 20, 392–400 CrossRef CAS.
- The existence of anion 5 was demonstrated for ionic liquids. (a) A. Shirai and Y. Ikeda, Inorg. Chem., 2011, 50, 1619–1627 CrossRef CAS PubMed; (b) Q. Zhou, K. Fitzgerald, P. D. Boyle and W. A. Henderson, Chem. Mater., 2010, 22, 1203–1208 CrossRef CAS.
- R. Michel, R. Herbst-Irmer and D. Stalke, Organometallics, 2011, 30, 4379–4386 CrossRef CAS.
- D. M. Seo, P. D. Boyle, R. D. Sommer, J. S. Daubert, O. Borodin and W. A. Henderson, J. Phys. Chem. B, 2014, 118, 13601–13608 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Rolf Gleiter on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedure and analytical data of 3. CCDC 1485216. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03355e |
| This journal is © The Royal Society of Chemistry 2017 |